

Abstract
The current studies were undertaken to determine the susceptibility of human alveolar macrophages (AMs) to influenza A virus (IAV) infection in comparison with autologous peripheral blood–derived monocytes-macrophages (PBMs). AMs and PBMs were exposed to IAV in vitro and examined for their ability to bind and internalize IAV, and synthesize viral proteins and RNA. PBMs but not AMs demonstrated binding and internalization of the virus, synthesizing viral proteins and RNA. Exposure of AMs in the presence of a sialidase inhibitor or anti-IAV antibody resulted in viral protein synthesis by the cells. Exposure of AMs to fluorescein isothiocyanate–labeled IAV in the presence of anti–fluorescein isothiocyanate antibody also resulted in viral protein synthesis. Thus, human AMs are apparently not susceptible to direct infection by a human IAV but are likely to be infected indirectly in the setting of exposure in the presence of antibody that binds the challenging strain of IAV.
Keywords: human alveolar macrophages, influenza virus, monocytes, macrophages
Human influenza A virus (IAV) predominantly infects the upper airways [1]. In the vast majority of infected individuals, pneumonia does not occur. In the ferret model, infection is confined largely to airway epithelium and is rare in the alveoli [2]. However, isolated ferret alveolar tissues seem to be as susceptible to infection as the airways, suggesting that host defenses prevent viral infection at the alveolar level [2]. Alveolar macrophages (AMs) generally have been considered to be the first leukocyte-based line of defense against respiratory pathogens such as influenza virus [3–6]. AMs must protect the lungs not only from pathogens but also from constant immunopathological processes due to all types of inhaled material [6], and elimination of AMs in immunized mice can enhance inflammatory responses to antigen [7], suggesting that AMs play a predominantly suppressive role during inflammatory responses in vivo.
Our early studies showed that human AMs from healthy volunteer donors differed substantially from autologous peripheral blood blood–derived monocytes-macrophages (PBMs) in support of lymphocyte proliferative responses to both mitogens and antigens, including inactivated IAV [3]. In contrast to significant accessory cell support demonstrated by PBMs, lymphocytes exposed to the inactivated virus in the presence of AMs showed no difference in proliferation compared with the lymphocytes cultured in the absence of any accessory cell. Furthermore, when AMs were added to PBMs plus lymphocytes, with appropriate controls for cell numbers, the AMs suppressed lymphocyte proliferative responses to antigen [8]. Others have reported that human IAV infection of human monocyte and macrophage subpopulations showed increased susceptibility associated with cell differentiation [9]. As differentiated macrophages likely to first encounter IAV, AMs have generally been assessed for infection using immunofluorescent staining for IAV antigens [10–12]. van Riel and colleagues have suggested that PBMs are not a good model for studying the interaction between AMs and human IAV [12]. Using immunofluorescent staining for IAV antigens 24 hours after exposure of ex vivo lung cultures, H1N1 and H3N2 viruses readily infected alveolar epithelial cells but not AMs, with approximately ≤1% of AMs appearing to be infected, unlike PBM cell preparations. Such observations raise the question of whether human AMs are susceptible to direct infection by human IAV, and the current studies were undertaken to address that question, with comparisons to PBMs that might be recruited to alveoli in response to IAV challenge.
MATERIALS AND METHODS
Subjects
Donors of AMs and PBMs were healthy men and women aged 20–40 years who met the following requirements: no pulmonary disease by history and physical examination, no present or past history of smoking, absence of upper respiratory illness for ≥6 weeks before the study, and normal spirometry. Informed consent was obtained from the subjects, and these studies were approved by the institutional investigational review committees at both the University of Rochester and the University of Texas Medical Branch. All comparisons in these studies were made between autologous cells, which are described further in the Supplemental Material.
Collection of PBMs
Phlebotomy was performed 7 days and 1 day before bronchoalveolar lavage (BAL) for each subject. Mononuclear leukocytes were obtained from heparinized blood by Ficoll-Hypaque sedimentation and incubated in plastic Petri dishes for 1 day (PBMs-1) or 7 days (PBMs-7) at 37°C in Medium 199 (Gibco) supplemented with glutamine, aqueous penicillin G (100 U/mL), streptomycin (50 μg/mL), and 20% autologous human serum, as described elsewhere [3, 13]. Nonadherent cells were removed by washing the plates gently 5 times; adherent cells were harvested with a rubber policeman. The cells were counted, viability was assessed by exclusion of Trypan blue dye, and the concentration of viable cells was adjusted to 1 × 106/mL. Using these methods, recovered cells were >90% monocytes-macrophages by morphology and nonspecific esterase staining [3]. After 7 days, the cells exhibited the light microscopic morphological appearance of tissue macrophages.
Collection of AMs
BAL was performed as described elsewhere [14]. Premedication was limited to atropine 0.75 mg given intravenously. After topical anesthesia of the upper airway with 2% lidocaine, a fiberoptic bronchoscope (Pentax FB-19H; outer diameter, 6.3 mm) was inserted orally and gently wedged in a subsegmental airway of the inferior segment of the lingula. Three 50-mL aliquots of sterile normal saline solution were sequentially instilled and immediately withdrawn under gentle suction. Lavage return using this technique averaged 65% of the volume instilled. In some subjects, a subsegment of the right middle lobe was similarly lavaged to provide additional cells. The cells were pelleted, washed 3 times in phosphate-buffered saline, and resuspended in antibiotic-supplemented M199. Viability was determined by exclusion of Trypan blue dye, and exceeded 95%. Differential counts were performed by assessing 500–1000 cells on a cytospin smear stained with Diff-Quick stain (American Scientific Products). A mean (standard deviation) of 89.2% (4.6%) alveolar cells were AMs by morphology, and the rest were predominantly lymphocytes.
In Vitro Exposure to Influenza Virus (IAV)
Influenza A/AA/Marton/43 H1N1 virus, originally a clinical isolate [15], was grown in allantoic cavities of 10-day-old embryonated hens' eggs, stored at −70°C, and titered at 108.0 plaque-forming units/mL when assayed on Madin-Darby canine kidney cells [13, 16]. Equivalent aliquots of viable autologous AMs, PBMs-1, or PBMs-7 were exposed or sham-exposed to virus at a multiplicity of infection (MOI) of 0.1, 1.0, 3 (where not reported otherwise), or 10 in serum-free Medium 199 for 1 hour at 37°C in a humidified 5% carbon dioxide atmosphere [13, 16]. Additional information regarding infection of the cells is provided in the Supplemental Material.
In 1 subset of experiments, AMs and PBMs were exposed to fluorescein isothiocyanate (FITC)–labeled IAV at an MOI of 3 [16]. The virus was labeled using methods described elsewhere [17, 18] and remained fully infectious for the cells [17]. Cells were collected and analyzed by means of light and fluorescence microscopy after exposure to the FITC-labeled virus. In another subset of experiments, AMs were exposed to IAV in the absence or presence of the potent sialidase inhibitor 2-Deoxy-2,3-dehydro-N-acetylneuraminic acid [19]. In a third subset of experiments, AMs were exposed to IAV in the presence of nonneutralizing anti-IAV antibody or exposed to FITC-labeled IAV in the presence of anti-FITC antibody. An anti–yellow fever virus (YFV) antibody was used as an isotype control for the exposures to virus in the presence of antibody. Cells were collected and analyzed with light and fluorescence microscopy and for protein synthesis after exposure to the FITC-labeled virus.
Analyses of Protein Synthesis
Cells were suspended in methionine-free medium and pulse labeled with 100 μCi of 35S-methionine for 2 hours. Cells were then washed and lysed by sodium dodecyl sulfate–containing detergent buffer with subsequent analysis by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and autoradiography [20, 21]. Lysates of equal numbers of cells were placed in each lane of the gels. Immunoprecipitation of synthesized viral proteins was performed using National Institutes of Health reference antisera directed against influenza hemagglutinin (anti-H1; V-314-511-517), neuraminidase (NA) (anti-N1; V-308-513-157), and matrix protein (anti-M; V-306-510-157), and murine monoclonal antibody directed against the viral nucleoprotein [20, 22].
Analyses of IAV NA Transcription
Total cellular RNA was obtained from sham-exposed and IAV-exposed cells using methods described elsewhere [23, 24]. After prehybridization, Northern blots were hybridized with a phosphorus 32–labeled cRNA strand complementary to the 1413 nucleotides of the influenza virus N1 NA-positive strand (messenger and template RNA) for 18 hours. The radiolabeled cRNA was transcribed using T7 RNA polymerase and a linearized (Hind III digest) template of a pGEM (Promega) plasmid containing the influenza A/WSN NA complementary DNA (kind gift from Louis Markoff, National Institute of Allergy and Infectious Diseases). Neither the N2 NA nor input virus (negative-strand) RNA was detected using this probe. Each blot was also hybridized subsequently using a probe for the human cellular gene product β-actin, derived from a complementary DNA cloned into the pBluescript II KS (−) vector (Stratagene), as a control for analyzing cell lysates.
RESULTS
Characteristics of Cell Preparations
Most cells retrieved by BAL were AMs (>90%), as expected, with most of the other cells being small resting lymphocytes. However, other cells were present in low (usually <1%) frequency, including polymorphonuclear leukocytes and cells that resembled morphologically mature monocytes [25]: large with an indented nucleus such as illustrated by the far left, lower cell in the selected high-power cytospin field that illustrates the different cell types in Figure 1. PBMs cultured for 7 days (PBMs-7) appeared larger and less dense than those cultured for 1 day (PBMs-1) and appeared morphologically similar to AMs on stained cytospin preparations, as described elsewhere [3]. Virus-exposed cells, observed in culture for as long as 7 days, showed no cytopathic effects or changes in viability compared with sham-infected cells and no production of progeny virus, as reported elsewhere [4].
Figure 1.
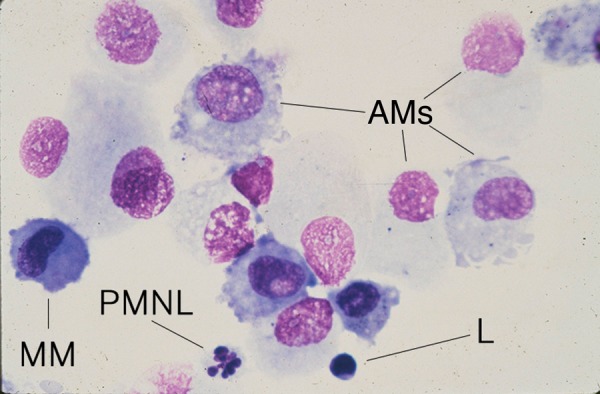
Photomicrograph of Wright-Giemsa-stained bronchoalveolar lavage cells, selected to illustrate the presence of alveolar macrophages (AMs) as the predominant cell, but also a small resting lymphocyte (L), the second most common type of cell, and the less common (usually <1%) polymorphonuclear leukocyte (PMNL) and a cell with the morphological characteristics of a mature monocyte (MM).
Binding and Uptake of IAV by AMs and PBMs
We have shown elsewhere that influenza virus binds to virtually all PBMs and lymphocytes, but such binding does not necessarily indicate that all of the cells express a true receptor for the virus that would lead to receptor-mediated endocytosis [17]. In fact, binding and internalization were quite different for lymphocytes (few internalized the virus) and PBMs (the majority internalized the virus). Those studies used FITC-labeled IAV and fluorescence resonant energy transfer, with addition of ethidium bromide to the cells, to distinguish between internalized virus, which maintained green fluorescence, and virus bound to the cell surface, which emitted red fluorescence based on fluorescence resonant energy transfer with the ethidium bromide [17].
For the current studies, we exposed autologous AMs and PBMs to FITC-IAV and examined the cells with light and fluorescent microscopy. In marked contrast to PBMs (Figure 2E and 2F), most of which bound and then internalized the virus, AMs neither bound (Figure 2A–2D) nor internalized the virus. Only the rare smaller cells that resembled monocytes morphologically (Figure 2A and 2B) or the occasional epithelial cells (Figure 2C and 2D) exhibited green fluorescence from bound or internalized FITC-IAV. The fields shown in Figure 2 are representative of 4 such experiments and show the cells before addition of ethidium bromide. This addition produced no change in the results: the PBMs retained almost all the green fluorescence, reflecting internalized virus, whereas the AMs remained nonfluorescent. Although FITC could be quenched at later time points by the acidic environment of the endocytic compartment, these experiments used measurements at earlier time points, and experiments in the presence of monensin or ammonium chloride, even at such early time points after exposure to the virus, did not lead to detectable FITC-IAV in AMs. The results suggested that AMs from healthy volunteers neither bind nor internalize IAV.
Figure 2.

Photomicrographs of alveolar macrophages (AMs) and peripheral blood–derived monocytes-macrophages (PBMs) exposed to fluorescein isothiocyanate (FITC)–labeled influenza A virus (IAV). A, C, E, Light photomicrographs. B, D, F, Corresponding fluorescence detection. Left-side panels are light images with reduced lighting to diminish potential quenching of fluorescence in the field. A–D, Bronchoalveolar lavage (BAL) preparations containing predominantly AMs. E, F, Autologous PBM preparation. Arrowhead in A indicates the cell infected by FITC-labeled IAV. Cells are shown before addition of ethidium bromide, and the results indicate binding and/or internalization of the virus. The addition of ethidium bromide did not detectably alter the findings, suggesting that the virus had internalized.
Assessment of IAV Replication In AMs and PBMs
We sham-exposed (Figure 3A, lanes 1 and 3) and exposed (Figure 3A, lanes 2 and 4) autologous AMs and PBMs to IAV and examined pulse-labeled cell lysates for evidence of synthesis of viral gene products. There was clear evidence of viral infection of the PBMs, with a commonly observed decrease in general protein synthesis by the cells coupled with synthesis of viral gene products, such as the hemagglutinin, the comigrating NA and nucleoprotein, and the matrix protein. The AMs were more synthetically active overall, and there was no decrease in their synthetic activity on exposure to the virus, nor was there evidence of synthesis of viral gene products.
Figure 3.

A, Autoradiograms examining synthesis of influenza A virus (IAV) proteins by alveolar macrophages (AMs; lanes 1 and 2) and autologous peripheral blood–derived monocytes-macrophages (PBMs; lanes 3 and 4). Cells were sham-exposed (lanes 1 and 3) or exposed (lanes 2 and 4) to IAV, pulse labeled 4–6 hours after exposure and analyzed with polyacrylamide gel electrophoresis. B, Viral protein synthesis by PBMs cultured for 1 day (PBMs-1; lanes 1–5), PBMs cultured for 7 days (PBMs-7; lanes 6–10), and AMs (lanes 11–15) exposed in vitro to influenza A/AA/Marton/43 H1N1 at a multiplicity of infection (MOI) of 0 (sham-exposed cells; lanes 1, 6, and 11), 0.1 (lanes 2, 7, and 12), 1.0 (lanes 3, 8, and 13), and 10 (lanes 4, 9, and 14). Lanes 5, 10, and 15 represent immunoprecipitation of lysates of virus-exposed (MOI, 10) PBMs-1, PBMs-7, and AMs respectively, using goat polyclonal antiserum directed against subtype-specific hemagglutinin (HA), neuraminidase (NA; which comigrates with nucleoprotein [NP]), matrix protein (M), and murine monoclonal antibody to NP. Lysates were derived from equal numbers of cells pulsed 4–6 hours after exposure to the virus.
We investigated this observation more carefully in additional experiments by using increasing MOIs and immunoprecipitation for the viral gene products (Figure 3B). We noted that both PBMs-1 and PBMs-7 were susceptible to an abortive infection by IAV, with synthesis of viral gene products but no release of new infectious progeny virions, whereas AMs again were not infected by such criteria. To determine whether failure to demonstrate viral protein synthesis by AMs was due to altered kinetics of infection, PBMs and AMs were pulsed at later time points, up to 24 hours after infection (data not shown). Viral protein synthesis by PBMs was maximal at 4–6 hours and declined thereafter. No viral protein synthesis was seen by AMs at any time when assays were performed. We then performed Northern blot analyses for IAV replication, using a probe for the NA gene (Figure 4), and found no evidence of IAV transcription in the AMs, but evidence of NA RNA synthesis was noted for both PBMs-1 and PBMs-7.
Figure 4.

Northern blot autoradiograms of lysates of alveolar macrophages (AMs) and autologous peripheral blood–derived monocytes-macrophages cultured for 1 (PBMs-1) or 7 (PBMs-7) days that were sham-exposed or exposed to influenza A virus (IAV). Lysates were probed for influenza virus neuraminidase (NA) and the same lanes were probed subsequently for β-actin. All lanes are from the same Northern blot.
IAV Infection of AMs Exposed in the Presence of Sialidase Inhibitor
The data raised the possibility that AMs do not constitutively or persistently express molecules on their surface with the appropriate sialic acid residues for binding by the influenza virus hemmagglutinin, or that such residues were under constant pressure of AM-derived sialidases. Studies by other investigators have suggested that there is modification of the glycosylation of surface molecules on murine macrophages that have been activated, with decreased sialic acid residues on the surface [26], and that there is modification of sialidase activity as human cells differentiate from monocytes to macrophages [27]. Because macrophage maturation is associated with increased enzymatic activity overall, and with increased sialidase activity specifically, we exposed the AMs to IAV in the absence or presence (lanes 2 and 3, respectively, in Figure 5) of the potent sialidase inhibitor NeuAc2en [19]. Treatment with the sialidase inhibitor resulted in infection and synthesis of IAV gene products by the exposed AMs.
Figure 5.
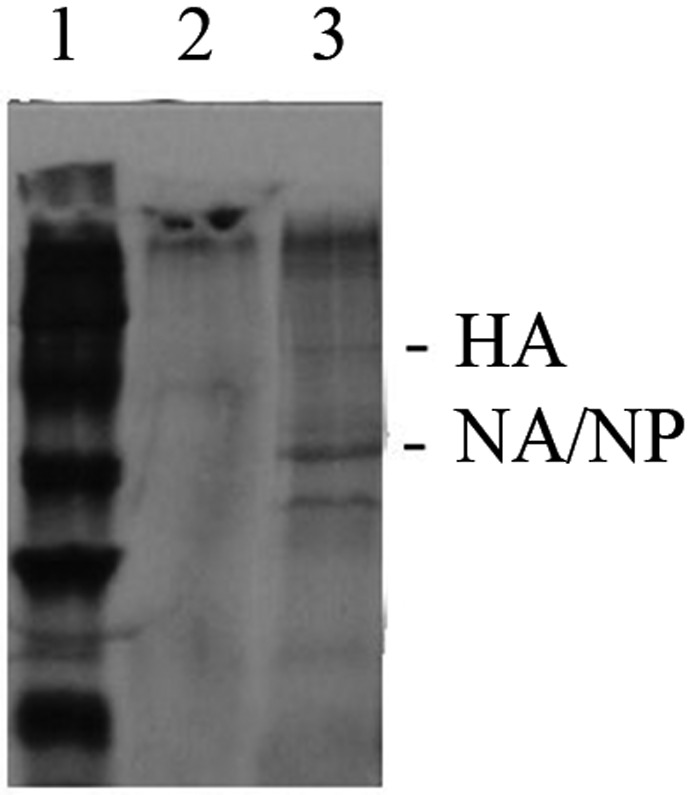
Autoradiograms of immunoprecipitated influenza A virus (IAV) proteins in lysates of alveolar macrophages (AMs). Lanes 2 and 3, Lysates from AMs exposed to IAV in the absence or presence of the neuraminidase (NA) inhibitor 2-Deoxy-2,3-dehydro-N-acetylneuraminic acid, respectively. Lane 1, Relative molecular mass markers. Lysates were derived from equal numbers of cells pulsed 4–6 hours after exposure to the virus and were immunoprecipitated, using the antibodies listed in Figure 3. Abbreviations: HA, hemagglutinin; NP, nucleoprotein.
IAV Infection of AMs Exposed in the Presence of Anti-IAV Antibody
In earlier studies, we demonstrated that each of the known types of Fc receptor (FcR) is expressed and functional on human AMs [28, 29]. The FcR could be expected to play a role in anti-IAV defense in the setting where an individual has been exposed previously to the strain (or to a cross-reactive strain) of IAV and would have immunoglobulin G available for binding to IAV in the respiratory tract, allowing uptake by AMs via FcR-mediated endocytosis. Therefore, we exposed AMs to IAV in the presence of antibodies that would bind to but not neutralize the virus.
We used the FITC-labeled IAV and anti-FITC antibody (Figure 6A and 6B) or anti-YFV virus antibody (isotype control, Figure 6C and 6D), and demonstrated virus binding (Figure 6) by AMs exposed to the anti-FITC antibody-coated virus. Addition of ethidium bromide to the culture did not quench the FITC-emitted green fluorescence evident in Figure 6, consistent with internalization of the virus by the AMs.
Figure 6.

Photomicrographs of alveolar macrophages (AMs) exposed to fluorescein isothiocyanate (FITC)–labeled influenza A virus (IAV). A, C, Light photomicrographs. B, D, Corresponding fluorescence detection. Left panels are light images with reduced lighting to diminish potential quenching of fluorescence in the field. A, B, AMs exposed to the virus in the presence of anti-FITC antibody. C, D, AMs exposed to virus in the presence of the control anti–yellow fever virus antibody. Cells are shown before addition of ethidium bromide, and the results indicate binding and/or internalization of the virus. Addition of ethidium bromide did not detectably alter the findings, suggesting that the virus had internalized.
Immunoprecipitation of IAV-exposed AM cell lysates with anti-IAV antibodies in a series of experiments showed existence of newly synthesized viral gene products when the AMs were exposed to IAV that was complexed with anti-IAV (Figure 7A, lane 4). No such viral gene product synthesis was evident in lysates from sham-exposed AMs (lane 2) or when the AMs were exposed in the presence of the control anti-YFV antibody (lane 3). Synthesis of IAV gene products by AMs was observed when we exposed the cells to FITC-labeled IAV in the presence of anti-IAV antibody (Figure 7B, lane 2) or anti-FITC antibody (lane 3) but not anti-YFV antibody (lane 4). The data from these experiments suggest that AMs can bind IAV via the FcR and, as a result, can internalize the virus and become infected.
Figure 7.

Autoradiograms of immunoprecipitated influenza A virus (IAV) proteins in lysates of alveolar macrophages (AMs). A, Relative molecular mass markers (lane 1), lysates from sham-exposed AMs and AMs exposed to virus in the presence of anti–yellow fever virus antibody (lanes 2 and 3, respectively), and lysate from AMs exposed to IAV in the presence of nonneutralizing anti-IAV antibody (lane 4). Lysates were derived from equal numbers of cells pulsed 4–6 hours after exposure to the virus and were immunoprecipitated using the antibodies listed in Figure 3. B, Relative molecular mass markers (lane 1), lysates from AMs exposed to virus in the presence of anti-IAV and anti–fluorescein isothiocyanate antibody (lanes 2 and 3, respectively), and lysate from AMs exposed to IAV in the presence of anti–yellow fever virus antibody (lane 4). Lysates were derived from equal numbers of cells pulsed 4–6 hours after exposure to the virus and were immunoprecipitated using the antibodies listed in Figure 3. Abbreviations: HA, hemagglutinin; M, matrix protein; NA, neuraminidase; NP, nucleoprotein.
DISCUSSION
The current studies were undertaken to help clarify the role of AMs in defending alveolar spaces against IAV infection. Monocytes and macrophages in general may contribute to control of seasonal or pandemic human IAV infection by direct inactivation or ingestion of virus (because the infection is abortive in the cells), release of interferon, lysis of infected cells, release of factors chemotactic for inflammatory cells, and support of lymphocyte proliferation and generation of cytotoxic T-lymphocyte effector cells [30]. Limited data have been available regarding the susceptibility of human AMs to seasonal and pandemic human IAV infection, using histopathology with antibodies against the virus [31, 32] or against FITC for tissues exposed to FITC-labeled IAV [33]. In the latter studies [33], the viruses were formalin inactivated before addition to the tissues. In the lung, alveolar epithelial cells are the primary targets for IAV, but study findings have suggested that a small percentage of AMs can be infected [11, 12], in 1 study using AMs that had been cultured in vitro for >48 hours before infection [11, 34]. We compared AMs with autologous PBMs to determine whether cells that may be recruited to the alveolus during an inflammatory response to challenge differ in their susceptibility and response to IAV from resident AMs.
The vast majority (>85%–96%) of cells obtained from healthy human volunteers by BAL are AMs [3, 5, 35, 36]. Most of the other cells are lymphocytes, with few polymorphonuclear leukocytes and few cells that appear to be mature monocytes at histopathology [25]. The latter cells are not generally cited in tables indicating BAL cell compositions. Furthermore, in addition to the small percentage of mature monocytes already present in the healthy alveoli, PBMs are recruited to the lungs, along with lymphocytes and polymorphonuclear leukocytes, in response to IAV challenge [37, 38], and they would also encounter the virus and potentially become infected [17, 20, 39].
To our knowledge, this is the first report regarding synthesis of IAV RNA and proteins by human AMs. None of the published studies that we have reviewed have shown clear synthesis of viral gene products by AMs. The studies have relied on histopathological detection of viral antigens [11, 31], staining cells clearly recognized as phagocytic and, as illustrated in those reports, often not clearly distinguishing between resident AMs and recruited PBMs, which in their mature macrophage form may resemble AMs histologically [3]. Studies that illustrated AMs stained for IAV antigens several hours to days after exposure have not shown staining soon (<1 hour) after exposure to determine input antigen that could persist. Notably, in the murine model of IAV infection, a large influx of PBMs occurs within a few days [37, 38], and alveolitis can persist for 6 months with detectable viral antigens but not infectious virus [40].
Virtually all PBMs and lymphocytes bind IAV through cell surface-expressed sialic acid residues, but only a majority (definitely not all, even at a very high MOI) of PBMs and a minority of lymphocytes endocytose the virus [17], the latter event being a more rigorous measurement of IAV receptor expression and function. The current data show that human AMs do not directly bind IAV, the usual first step in infection. The results provide an explanation for the previously reported absence of effects of IAV challenge on AM accessory cell function (for mitogen responses) and cytokine production [4]. The AMs did not bind or internalize IAV, even though studies by other investigators demonstrated that cells that are not expressing sialic acids, as well as desialylated cells, can be infected by IAV [41, 42].
Consistent with previous observations using PBMs [20], both PBMs-1 and PBMs-7 synthesized IAV proteins. However, synthesis of viral proteins by AMs was not detectable. Our findings contrast with those of Rodgers and Mims [31], who detected viral antigens in 15%–20% of AMs 24 hours after exposure to IAV in vitro, suggesting new viral protein synthesis. However, their studies did not completely exclude the possibility of reprocessing and expression of input viral protein. In addition, AMs (lavage cells) in their studies were obtained from patients undergoing diagnostic bronchoscopy for unspecified illnesses and may not be comparable with AMs from healthy subjects. The possibility that a significant proportion of cells were recently recruited to the alveoli could not be excluded. Such a possibility also exists for autopsy results showing histological macrophage-type cell infection in alveoli, related to the 1918 pandemic IAV, the most avianlike among the mammalian-adapted viruses [43]; all cases had histological evidence of severe acute bacterial pneumonia [44]. The current studies examined AMs from healthy individuals who were not infected with IAV. It is possible that AMs in infected patients augment their antiviral functions in response to activating signals from other cells, such as epithelial cells or T lymphocytes.
Although human AMs are apparently not directly infected by IAV, another way in which AMs may take up IAV proteins that can be detected by immunofluorescence is by phagocytosis of apoptotic IAV-infected cells [45, 46], such as alveolar epithelial cells that are more susceptible to infection [11, 12]. In the normal setting, with an initially low virus inoculum, and most likely in the IAV-experienced host, the presence of anti-IAV immunoglobulin G antibodies in the lower respiratory tract could lead to defense by FcR-mediated infection of the AMs by IAV. This might be a clearance mechanism for the virus because AMs are abortively infected, as are PBMs.
Supplementary Data
Supplementary materials are available at http://jid.oxfordjournals.org. Consisting of data provided by the author to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the author, so questions or comments should be addressed to the author.
Notes
Acknowledgments. We thank Molly Roth for help in preparing the manuscript.
Financial support. This work was supported by the National Institutes of Health (grants AI 15547, AI 23774, HL 34116, ES 02679, and RR00044-29S1), the Health Effects Institute (contract 84-3), and the Paul R. Stalnaker, MD, Endowment Fund.
Potential conflicts of interest. All authors: No potential conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1.van Riel D, den Bakker MA, Leijten LM et al. Seasonal and pandemic human influenza viruses attach better to human upper respiratory tract epithelium than avian influenza viruses. Am J Pathol 2010; 176:1614–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Husseini RH, Sweet C, Bird RA, Collie MH, Smith H. Distribution of viral antigen with the lower respiratory tract of ferrets infected with a virulent influenza virus: production and release of virus from corresponding organ cultures. J Gen Virol 1983; 64(Pt 3):589–98. [DOI] [PubMed] [Google Scholar]
- 3.Ettensohn DB, Roberts NJ Jr. Human alveolar macrophage support of lymphocyte responses to mitogens and antigens: analysis and comparison with autologous peripheral-blood-derived monocytes and macrophages. Am Rev Respir Dis 1983; 128:516–22. [DOI] [PubMed] [Google Scholar]
- 4.Ettensohn DB, Roberts NJ Jr. Influenza virus infection of human alveolar and blood-derived macrophages: Differences in accessory cell function and interferon production. J Infect Dis 1984; 149:942–9. [DOI] [PubMed] [Google Scholar]
- 5.Morales-Nebreda L, Misharin AV, Perlman H, Budinger GR. The heterogeneity of lung macrophages in the susceptibility to disease. Eur Respir Rev 2015; 24:505–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herold S, Becker C, Ridge KM, Budinger GR. Influenza virus-induced lung injury: pathogenesis and implications for treatment. Eur Respir J 2015; 45:1463–78. [DOI] [PubMed] [Google Scholar]
- 7.Thepen T, Van Rooijen N, Kraal G. Alveolar macrophage elimination in vivo is associated with an increase in pulmonary immune response in mice. J Exp Med 1989; 170:499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ettensohn DB, Lalor PA, Roberts NJ Jr. Human alveolar macrophage regulation of lymphocyte proliferation. Am Rev Respir Dis 1986; 133:1091–6. [DOI] [PubMed] [Google Scholar]
- 9.Hoeve MA, Nash AA, Jackson D, Randall RE, Dransfield I. Influenza virus A infection of human monocyte and macrophage subpopulations reveals increased susceptibility associated with cell differentiation. PLoS One 2012; 7:e29443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee SM, Dutry I, Peiris JS. Editorial: macrophage heterogeneity and responses to influenza virus infection. J Leukoc Biol 2012; 92:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Travanty E, Zhou B, Zhang H et al. Differential susceptibilities of human lung primary cells to H1N1 influenza viruses. J Virol 2015; 89:11935–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Riel D, Leijten LM, van der Eerden M et al. Highly pathogenic avian influenza virus H5N1 infects alveolar macrophages without virus production or excessive TNF-alpha induction. PLoS Pathog 2011; 7:e1002099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Roberts NJ Jr, Douglas RG Jr, Simons RL, Diamond ME. Virus-induced interferon production by human macrophages. J Immunol 1979; 123:365–9. [PubMed] [Google Scholar]
- 14.Frampton MW, Smeglin AM, Roberts NJ Jr, Morrow PE, Utell MJ. Nitrogen dioxide exposure in vivo and human alveolar macrophage inactivation of influenza virus in vitro. Environ Res 1989; 48:179–92. [DOI] [PubMed] [Google Scholar]
- 15.Kilbourne ED, Smith C, Brett I, Pokorny BA, Johansson B, Cox N. The total influenza vaccine failure of 1947 revisited: major intrasubtypic antigenic change can explain failure of vaccine in a post-World War II epidemic. Proc Natl Acad Sci U S A 2002; 99:10748–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts NJ Jr, Steigbigel RT. Effect of in vitro virus infection on response of human monocytes and lymphocytes to mitogen stimulation. J Immunol 1978; 121:1052–8. [PubMed] [Google Scholar]
- 17.Nichols JE, Mock DJ, Roberts NJ Jr. Use of FITC-labeled influenza virus and flow cytometry to assess binding and internalization of virus by monocytes-macrophages and lymphocytes. Arch Virol 1992; 130:441–55. [DOI] [PubMed] [Google Scholar]
- 18.Yoshimura A, Ohnishi SI. Uncoating of influenza virus in endosomes. J Virol 1984; 51:497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Usuki S, Hoops P, Sweeley CC. Growth control of human foreskin fibroblasts and inhibition of extracellular sialidase activity by 2-deoxy-2,3-dehydro-N-acetylneuraminic acid. J Biol Chem 1988; 263:10595–9. [PubMed] [Google Scholar]
- 20.Mock DJ, Domurat F, Roberts NJ Jr, Walsh EE, Licht MR, Keng P. Macrophages are required for influenza virus infection of human lymphocytes. J Clin Invest 1987; 79:620–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227:680–5. [DOI] [PubMed] [Google Scholar]
- 22.Van Wyck KL, Bean WJ Jr, Webster RG. Monoclonal antibodies to the influenza A virus nucleoprotein affecting RNA transcription. J Virol 1981; 39:313–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chomczynski P, Sacchi N. Single step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 1987; 162:156–9. [DOI] [PubMed] [Google Scholar]
- 24.Wahl GM, Meinkoth JL, Kimmel AR. Northern and Southern blots. In: Berger SL, Kimmel AR, eds. Guide to molecular cloning techniques: methods in enzymology. Vol 152. San Diego, CA: Academic Press, Inc., 1987:572–81. [DOI] [PubMed] [Google Scholar]
- 25.Goasguen JE, Bennett JM, Bain BJ, Vallespi T, Brunning R, Mufti GJ. Morphological evaluation of monocytes and their precursors. Haematologica 2009; 94:994–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Afroun S, Tenu JP, Lemaire G. Modifications of glycosylation patterns in macrophages upon activation. Biochim Biophys Acta 1988; 971:137–47. [DOI] [PubMed] [Google Scholar]
- 27.Lambre CR, Greffard A, Gattegno L, Saffar L. Modifications of sialidase activity during the monocyte-macrophage differentiation in vitro. Immunol Lett 1990; 23:179–82. [DOI] [PubMed] [Google Scholar]
- 28.Levy PC, Looney RJ, Shen L et al. Human alveolar macrophage FcR-mediated cytotoxicity: heteroantibody versus conventional antibody-mediated target cell lysis. J Immunol 1990; 144:3693–700. [PubMed] [Google Scholar]
- 29.Levy PC, Utell MJ, Fleit FB, Roberts NJ Jr, Ryan DH, Looney RJ. Characterization of human alveolar macrophage Fcγ receptor III: a transmembrane glycoprotein that is shed under in vitro culture conditions. Am J Respir Cell Mol Biol 1991; 5:307–14. [DOI] [PubMed] [Google Scholar]
- 30.Ada GL, Jones PD. The immune response to influenza virus infection. In: Kendal AP, Patriarca PA, eds. Options for the control of influenza. New York, NY: Liss, 1986:107–24. [Google Scholar]
- 31.Rodgers BC, Mims CA. Influenza virus replication in human alveolar macrophages. J Med Virol 1982; 9:177–84. [DOI] [PubMed] [Google Scholar]
- 32.Blusse van Oud Alblas A, van der Linden-Schrever B, van Furth R. Origin and kinetics of pulmonary macrophages during an inflammatory reaction induced by intraalveolar administration of aerosolized heat-killed BCG. Am Rev Respir Dis 1983; 128:276–81. [DOI] [PubMed] [Google Scholar]
- 33.van Riel D, Munster VJ, de Wit E et al. Human and avian influenza viruses target different cells in the lower respiratory tract of humans and other mammals. Am J Pathol 2007; 171:1215–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, Oberley-Deegan R, Wang S et al. Differentiated human alveolar type II cells secrete antiviral IL-29 (IFN-lambda 1) in response to influenza A infection. J Immunol 2009; 182:1296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heron M, Grutters JC, ten Dam-Molenkamp KM et al. Bronchoalveolar lavage cell pattern from healthy human lung. Clin Exp Immunol 2012; 167:523–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Meyer KC, Raghu G, Baughman RP et al. An official American Thoracic Society clinical practice guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med 2012; 185:1004–14. [DOI] [PubMed] [Google Scholar]
- 37.Wyde PR, Cate TR. Cellular changes in lungs of mice infected with influenza virus: characterization of the cytotoxic responses. Infect Immun 1978; 22:423–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wyde PR, Peavy DL, Cate TR. Morphological and cytochemical characterization of cells infiltrating mouse lungs after influenza infection. Infect Immun 1978; 21:140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts NJ Jr, Horan PK. Expression of viral antigens after infection of human lymphocytes, monocytes, and macrophages with influenza virus. J Infect Dis 1985; 151:308–13. [DOI] [PubMed] [Google Scholar]
- 40.Jakab GJ, Astry CL, Warr GA. Alveolitis induced by influenza virus. Am Rev Respir Dis 1983; 128:730–9. [DOI] [PubMed] [Google Scholar]
- 41.Stray SJ, Cummings RD, Air GM. Influenza virus infection of desialylated cells. Glycobiology 2000; 10:649–58. [DOI] [PubMed] [Google Scholar]
- 42.Kumari K, Gulati S, Smith DF, Gulati U, Cummings RD, Air GM. Receptor binding specificity of recent human H3N2 influenza viruses. Virol J 2007; 4:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Taubenberger JK. The origin and virulence of the 1918 “Spanish” influenza virus. Proc Am Philos Soc 2006; 150:86–112. [PMC free article] [PubMed] [Google Scholar]
- 44.Sheng ZM, Chertow DS, Ambroggio X et al. Autopsy series of 68 cases dying before and during the 1918 influenza pandemic peak. Proc Natl Acad Sci U S A 2011; 108:16416–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujimoto I, Pan J, Takizawa T, Nakanishi Y. Virus clearance through apoptosis-dependent phagocytosis of influenza A virus-infected cells by macrophages. J Virol 2000; 74:3399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hashimoto Y, Moki T, Takizawa T, Shiratsuchi A, Nakanishi Y. Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J Immunol 2007; 178:2448–57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.