

Abstract
Carbonic anhydrase (CA) IV is a glycosylphosphotidylinositol-anchored enzyme highly expressed on the plasma face of microcapillaries and especially strongly expressed in the choriocapillaris of the human eye. In collaboration with scientists at the University of Cape Town (Rondebosch, South Africa), we recently showed that the R14W mutation in the signal sequence of CA IV, which they identified in patients with the retinitis pigmentosa (RP) 17 form of autosomal dominant RP, results in accumulation of unfolded protein in the endoplasmic reticulum (ER), leading to ER stress, the unfolded protein response, and apoptosis in a large fraction of transfected COS-7 cells expressing mutant, but not wild-type, CA IV. Here we present experiments showing that several well characterized CA inhibitors largely prevent the adverse effects of expressing R14W CA IV in transfected COS-7 cells. Specifically, CA inhibitors prevent the accelerated turnover of the mutant protein, the up-regulation of Ig-binding protein, double-stranded RNA-regulated protein kinase-like ER kinase, and CCAAT/enhancer-binding protein homologous protein (markers of the unfolded protein response and ER stress), the inhibition of production of other secretory proteins expressed from COS-7-transfecting plasmids, and the induction of apoptosis, all characteristics of transfected cells expressing R14W CA IV. Furthermore, treatment with 4-phenylbutyric acid, a nonspecific chemical chaperone used in other protein-folding disorders, also dramatically reduces the apoptosis-inducing effect of expressing R14W CA IV cDNA in transfected COS-7 cells. These experiments suggest a promising approach to treatment of RP17 that might delay the onset or possibly prevent this autosomal dominant form of RP.
Keywords: protein-folding disease, unfolded protein response, endoplasmic reticulum-associated protein degradation, signal sequence mutation
Carbonic anhydrase (CA) IV is a glycosylphosphatidylinositol-anchored membrane protein, which is highly expressed at the plasma membrane of epithelial cells of kidney (1–3) and endothelial cells of pulmonary and other microcapillary beds (4–9). In the human eye, CA IV is highly expressed in the choriocapillaris but not in retina (10). In collaboration with Rajkumar Ramesar, George Rebello, and their coworkers in South Africa, we recently reported an apoptosis-inducing signal sequence mutation in the CA IV gene in patients with retinitis pigmentosa (RP) 17, an autosomal dominant form of RP (11). The R14W mutation is at position –5 relative to the signal cleavage site. We also showed that expression of R14W CA IV cDNA in COS-7 cells resulted in delayed maturation of newly synthesized CA IV and a decrease in the steady-state level of CA IV activity and protein, which reflected a combination of reduced synthesis and accelerated degradation. We observed that the R14W mutation in CA IV leads to accumulation of some of the CA IV as unfolded protein in the endoplasmic reticulum (ER), resulting in up-regulation of Ig-binding protein (BiP), double-stranded RNA-regulated protein kinase-like ER kinase (PERK), and CCAAT/enhancer-binding protein homologous protein (CHOP), markers of ER stress and the unfolded protein response (UPR). R14W CA IV also induced apoptosis, as evidenced by annexin V binding and terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) staining in a large fraction of the R14W CA IV-expressing transfected COS-7 cells.
In recent years, small molecule chemical chaperones have been shown to reverse misfolding or mislocalization of several mutant plasma membrane, lysosomal, and secretory proteins (12, 13). These chemical chaperones are of two kinds. The first, the specific chemical chaperones, binds to the active site of the mutant proteins and assists in protein folding. Assisted protein folding is the mechanism by which folding defects in mutant rhodopsins in several forms of RP were corrected in vitro by adding the ligand, 11-cis-7-ring retinal (14). Specific chemical chaperones have also been used for several lysosomal storage diseases, including Fabry disease (15), Gaucher disease (16), GM1 gangliosidosis (17), and adult Tay–Sachs and Sandhoff diseases (18). In each case, treatment with subinhibitory concentrations of a competitive inhibitor resulted in an increase in the enzyme activity in mutant cells. Similarly, functions of several mutant cell surface proteins, including V2 vasopressin receptor (19), human ether-a-gogo related K+ channel (20), δ-opioid receptor (21), and gonadotropin-releasing hormone receptor (22) have been rescued by using their specific ligands.
A second class of chaperones is called nonspecific chaperones, some of which are also known as osmolytes. The natural osmolytes, glycerol, trimethylamine N-oxide (TMO), and 4-phenylbutyric acid (PBA), are thought to work by altering the conditions of the ER to enhance correct folding of the mutant proteins (12). Rubenstein and Zeitlin (23) and Perlmutter and colleagues (24) have proposed PBA for treatment of ΔF508-homozygous cystic fibrosis patients, and α1-antitrypsin deficiency, respectively. PBA had previously been approved for clinical use in children with urea cycle disorders where it acts by a different mechanism as an ammonia scavenger (25).
In this study, we used the transfected COS-7 cell model to test several CA inhibitors as specific chemical chaperones for R14W CA IV. We also tested PBA as a nonspecific chemical chaperone. We examined their effects on the folding and stability of the expressed R14W CA IV. We also examined the consequences of expressing the mutant protein on the up-regulation of markers of ER stress and the UPR, and on the induction of apoptosis. The encouraging results suggest that chemical chaperones might prevent or delay the onset of this autosomal dominant form of RP.
Materials and Methods
Affinity-purified rabbit anti-human CA IV IgG was used (11, 26). Rabbit anti-human β-glucuronidase (GUS) was the same as described (27). Goat anti-rabbit IgG-peroxidase, rabbit anti-goat IgG-rhodamine, goat anti-rabbit IgG-FITC, and goat anti-mouse IgG-FITC conjugates were from Sigma–Aldrich. Goat anti-BiP, goat anti-PERK, and mouse anti-CHOP were from Santa Cruz Biotechnology. The TUNEL assay kit was obtained from Roche Applied Science. The CA inhibitors, acetazolamide and ethoxzolamide, were from Sigma–Aldrich. Azopt (Alcon Laboratories, Ft. Worth, TX) and Trusopt (Merck) were purchased from Walgreens Pharmacy. The PBA was from Sigma–Aldrich.
Construction of Mammalian Expression Vector. Full-length cDNAs of wild-type CA IV, R14W CA IV, or the truncated, secretory form of CA IV (G267X CA IV) (26, 28) were digested with EcoRI, and RI inserts were subcloned into the mammalian expression vector, pCXN, at the EcoRI site, as described (29).
Transfection of COS-7 Cells. Before transfection, COS-7 cells were transplanted on p60 or chamber slides. After 18 h, cells were incubated with 5 or 2 μg of cDNA, respectively, by using the DEAE–dextran procedure (30), followed by 1 h of incubation with 100 μM chloroquine (31). After chloroquine treatment, cells were fed normal DMEM containing 10% FBS with or without 10 μM acetazolamide, ethoxzolamide, Azopt, and Trusopt, or 2–5 mM PBA. After 72 h, the cells and media were collected. Cell lysates were prepared by sonication of cells in 25 mM Tris·HCl, pH 7.5, containing 1% Nonidet P-40 and protease inhibitors (11). When CA inhibitors were present as specific chemical chaperones, the cell membranes were collected after sonication in 25 mM Tris·HCl, pH 7.5, plus protease inhibitors without Nonidet P-40.
CA Assay. The cell lysates and media were measured for CA activity by using the end-point titration method of Maren (32). When cells were treated with CA inhibitors, the cell membranes were recovered after centrifugation at 40,000 × g for 30 min and washed three to five times with 0.1 M sodium acetate, pH 5.5, containing protease inhibitors to remove CA inhibitors before CA activity measurements. The protein concentration was determined by micro Lowry assay, with BSA as a standard (33).
GUS Assay. The GUS activity of the cell lysates was determined by using fluorogenic glucuronide substrate as described (27).
SDS/PAGE and Western Blot Analysis. The cell lysates containing 5–30 μg of cell protein or media containing the equivalent of 1–2 enzyme units [EU; 1 EU = the amount of enzyme that doubles the reaction rate in the Maren assay (32)] of G267X CA IV were analyzed on SDS/PAGE under nonreducing conditions according to Laemmli's procedure as described (11, 34).
Pulse–Chase Experiments to Measure Enzyme Turnover. After transfection, the COS-7 cells were pulse-labeled with Tran35S-label for 30 min in DMEM without methionine and cysteine and containing 5% dialyzed FBS. The cells were subjected to chase in normal DMEM containing 10% FBS and additional 10–20 mM methionine and cysteine for different times (35). Added chemical chaperones were present during both starvation and pulse–chase. The cells were harvested and lysed by sonication in 1 ml of lysis buffer, containing 10 mM Tris·HCl, pH 7.5, 150 mM NaCl, 0.1% Triton X-100, 1.5% deoxycholate, 0.1% SDS, and protease inhibitors. The cell lysates were subjected to immunoprecipitation by using anti-human CA IV IgG as described (11). The immunoprecipitates were analyzed by SDS/PAGE followed by fluorography. The gel pieces corresponding to precursor or mature polypeptides were excised, and radioactivity was determined by using scintillation fluid.
Immunohistochemical Staining. The COS-7 cells transfected with wild-type or R14W CA IV cDNA were treated with chemical chaperones for 72 h and fixed with warm 3% paraformaldehyde in PBS for 40 min at room temperature. The fixed cells were permeabilized with 0.3% Triton X-100. Primary antibodies were used at 1:100 dilutions in 0.2% gelatin in PBS for 1.5 h, and secondary antibodies were used at 1:200 dilutions for 1 h at room temperature. The slides were prepared for recording the images by Olympus microscope as described (11). TUNEL assay was performed according to manufacturer's protocol.
Immunofluorescent images were quantitated by counting the cells positive for CA IV, UPR and ER stress-responsive proteins, and TUNEL-positive cells from three to five fields of cells. The percent positive cells was calculated from the total number of CA IV-positive cells, which were positive for BiP, PERK, CHOP, or TUNEL reaction.
Results
Blocking of Accelerated Turnover of R14W CA IV by Chemical Chaperones. We previously reported (11) that a fraction of R14W CA IV precursor in transfected COS-7 cells was more slowly converted to mature enzyme and more rapidly degraded than the wild-type enzyme. Here, we tested the effects of acetazolamide on the accelerated degradation of the mutant CA IV. COS-7 cells were transfected with R14W CA IV cDNA in the presence and absence of 10 μM acetazolamide. After 72 h, transfected cells were pulse-labeled with Tran35S-label for 30 min and chased for various times in the presence and absence of inhibitor. The results presented in Fig. 1 show that in the absence of inhibitor, 60% of the R14W mutant enzyme was rapidly degraded over the first 30–40 min. However, 10 μM acetazolamide greatly inhibited this degradation, suggesting that this highly specific CA inhibitor acts as a chemical chaperone to prevent the accelerated degradation.
Fig. 1.

Effect of acetazolamide (AC) on accelerated degradation of R14W CA IV. COS-7 cells transfected with R14W CA IV cDNA were pulse-labeled with Tran35S-label for 30 min and chased for 15–180 min in the absence (–) and presence (+) of 10 μM acetazolamide. The CA IV immunoprecipitates were analyzed by SDS/PAGE under nonreducing conditions followed by fluorography. (Upper) The non-disulfide-bonded precursor (P) and sulfide-bonded mature (M) polypeptides are marked. (Lower) The percent residual protein remaining was calculated from the total radioactivity incorporated in both precursor and mature polypeptides.
We also studied the effect of PBA, a nonspecific chemical chaperone that has been used in several protein-folding diseases (12, 23). The results in Fig. 2 show that the rapid degradation of over half of the pulse-labeled CA IV was prevented by PBA. In addition, PBA accelerated the conversion of unfolded precursor to the mature (disulfide-bridged) form of the enzyme, which is probably more stable than the precursor. Thus, the mechanism of protection by PBA may be different from that by acetazolamide, which appears to stabilize both the precursor and mature enzyme.
Fig. 2.

Effect of PBA on the accelerated degradation of R14W CA IV. COS-7 cells transfected with R14W CA IV cDNA were pulse-labeled with Tran35S-label for 30 min and chased for 15–180 min in the absence (–) and presence (+) of 5 mM PBA. Analysis was as described for Fig. 1. (Upper) Precursor (P) and mature (M) polypeptides are marked. (Lower) The percent residual protein remaining was calculated from the total radioactivity incorporated in both precursor and mature polypeptides. The accelerated degradation of R14W CA IV was prevented by PBA, which also markedly accelerated the conversion of precursor to the faster-migrating, mature form.
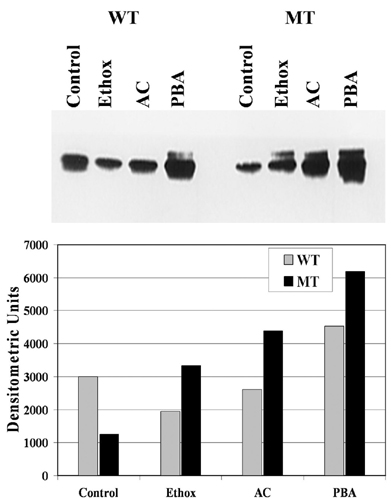
Table 1 presents additional data showing reversal of the effects of expressing R14W CA IV in transfected COS-7 cells by chemical chaperones. As reported (11), in the absence of chaperones, the R14W mutation reduces the amount of CA IV activity expressed by ≈30% compared with that seen in COS-7 cells expressing wild-type CA IV. However, the reduction in activity caused by the R14W mutation was largely prevented by acetazolamide (10 μM) and ethoxzolamide (10 μM), a related sulfonamide inhibitor. PBA nearly doubled the activity for the R14W mutant enzyme, but it also nearly doubled the activity of the wild-type CA IV. Fig. 6, which is published as supporting information on the PNAS web site, shows that the increases in activity by chaperones correlate directly with an increase in the steady-state level of protein, as judged by Western blot. The activity enhancing effects of the sulfonamides and PBA (at these doses) were not additive (data not shown).
Table 1. Effect of specific and nonspecific chemical chaperones on the enzyme activity of R14W CA IV expression in COS-7 cells.
| EU/mg*
|
||||
|---|---|---|---|---|
| Enzyme | Control | Acetazolamide (10 μM) | Ethoxzolamide (10 μM) | PBA (5 mM) |
| WT CA IV | 0.84 ± 0.06 | 0.78 ± 0.09 | 0.82 ± 0.04 | 1.62 ± 0.10 |
| R14W CA IV | 0.56 ± 0.08 | 0.74 ± 0.08 | 0.68 ± 0.08 | 1.02 ± 0.09 |
Mean ± SD (n = 8).
Protection from R14W CA IV ER Stress-Induced Disruption in Production of Other Secretory Proteins by Chemical Chaperones. Trans effects of expression of unfolded proteins on the production of other secretory or membrane proteins have been reported by others (36, 37). To test whether expressing the R14W CA IV has such an effect in transfected COS-7 cells, we compared R14W CA IV and wild-type CA IV for effects on the expression of two other secretory proteins expressed from cotransfecting plasmids. These plasmids expressed the full-length human GUS or the truncated, secretory form of CA IV (G267X CA IV) (38, 39). The results are summarized in Table 2. In the absence of chaperones, the GUS activity in the R14W-transfected cell lysate was reduced to 70% of the level seen in cells cotransfected with wild-type CA IV or vector only. The enzyme activity of G267X CA IV in the medium from R14W-transfected COS-7 cells was reduced to 50% of that secreted by cells expressing the cotransfecting wild-type CA IV or vector only. Fig. 7, which is published as supporting information on the PNAS web site, shows that the decreases in activity of GUS in R14W CA IV cotransfected cells, and of G267X CA IV in media of R14W cotransfected cells, correlate directly with a decrease in the protein levels evident on a Western blot.
Table 2. Effect of R14W CA IV expression on the activity of GUS and the human secretory form of CA IV (G267X CA IV) expressed from cotransfecting plasmids in COS-7 cells.
| Plasmid | GUS,* EU/mg | G267X CA IV,† EU/ml |
|---|---|---|
| Vector only | 459.0 ± 75.0 | 2.8 ± 0.1 |
| WT CA IV | 447.5 ± 185 | 2.7 ± 0.1 |
| R14W CA IV | 306.0 ± 85.6 | 1.2 ± 0.2 |
Cell lysates, mean ± SD (n = 6).
Media, mean ± SD (n = 6).
Tables 3, 4, and 5 summarize results showing that chemical chaperones reversed these negative trans effects of expressing the R14W CA IV on GUS production and G267X CA IV secretion. Table 3 shows that acetazolamide (10 μM) prevented the trans effect of expressing R14W CA IV on GUS expression. Table 4 shows that the presence of 5 mM PBA also completely corrected the reduction in GUS activity in the R14W CA IV-transfected cells. Table 5 shows that PBA also prevented the reduction of G267X CA IV secretion in the media of transfected COS-7 cells. Thus, both specific and nonspecific chemical chaperones corrected the impairment of synthesis and transport of other secretory proteins overexpressed from cotransfecting plasmids that was typical of R14W CA IV-expressing cells in the absence of chaperones.
Table 3. Reversal of the negative trans effect of R14W CA IV expression on GUS expression by acetazolamide (10 μM) in COS-7 cells.
| GUS,* EU/mg
|
||
|---|---|---|
| CA-expressing plasmid | - acetazolamide | + acetazolamide |
| WT CA IV | 321.8 ± 15.4 | 328.4 ± 16.2 |
| R14W CA IV | 215.9 ± 10.3 | 320.6 ± 16.5 |
Mean ± SD (n = 4).
Table 4. Reversal of the negative trans effect of R14W CA IV expression on GUS expression by PBA (2mM) in COS-7 cells.
| GUS,* EU/mg
|
||
|---|---|---|
| CA-expressing plasmid | - PBA | + PBA |
| WT CA IV | 151.0 ± 18.6 | 157.6 ± 16.5 |
| R14W CA IV | 105.9 ± 15.5 | 158.8 ± 18.2 |
Mean ± SD (n = 4).
Table 5. Reversal of the negative trans effect of R14W CA IV expression on G267X CA IV secretion by PBA (2mM) in COS-7 cells.
| G267X CA IV,* EU/ml
|
||
|---|---|---|
| CA-expressing plasmid | - PBA | + PBA |
| WT CA IV | 1.95 ± 0.15 | 1.91 ± 0.25 |
| R14W CA IV | 1.10 ± 0.2 | 2.06 ± 0.15 |
Mean ± SD (n = 4).
Chemical Chaperones Prevent the Up-Regulation of the Markers of UPR. We previously showed that expression of R14W CA IV resulted in up-regulation of BiP, PERK, and CHOP (11). Here we use immunohistochemical staining to ask whether chemical chaperones would prevent the up-regulation of markers of the UPR in R14W CA IV-expressing COS-7 cells. Fig. 3 A–C shows that, in the absence of chemical chaperones, the R14W CA IV-transfected COS-7 cells showed positive staining for BiP (A), PERK (B), and CHOP (C) in 82.5%, 69.0%, and 85.7% of the R14W CA IV expressing cells, respectively, whereas these markers were detectable in <5% of cells expressing wild-type CA IV. Expression of each of these markers in R14W-expressing cells was reduced to ≈20% by both acetazolamide (10 μM) and PBA (2 mM). Thus, both chemical chaperones greatly reduced the up-regulation of the markers of ER stress and the UPR in R14W CA IV-transfected COS-7 cells, presumably by enhancing folding and thereby reducing ER stress.
Fig. 3.

Prevention of up-regulation of UPR genes by chemical chaperones. COS-7 cells transfected with wild-type CA IV (WT) or R14W CA IV (MT) cDNA were incubated with normal medium (–), 10 μM acetazolamide (AC), or 2 mM PBA for 72 h after transfection. The results were quantitated by counting the cells positive for BiP (A), PERK (B), and CHOP (C) from three to five fields. The results indicate that up-regulation of the UPR genes in COS-7 cells expressing R14W CA IV was greatly reduced (but not totally prevented) by chemical chaperones.
In control experiments, when COS-7 cells were transfected with vector only and treated with or without 10 μM acetazolamide, activated PERK and CHOP were not detectable in the transfected cells. There were a few BiP-positive cells, but the expression was low (≈2%) and not affected by acetazolamide (data not shown).
Chemical Chaperones Protect R14W CA IV-Transfected Cells from Apoptosis. We previously showed that expression of R14W CA IV in transfected COS-7 cells leads to apoptosis in a large fraction of R14W CA IV-expressing cells. Fig. 4 Upper compares the R14W CA IV- and wild-type CA IV-expressing cells for the fraction of cells that show positive TUNEL staining in the absence and presence of acetazolamide and PBA. The TUNEL-positive R14W CA IV-expressing cells were reduced from ≈85% to 20% by acetazolamide. In a separate experiment, PBA reduced the TUNEL-positive cells from ≈65% to 25% (Fig. 4 Lower).
Fig. 4.

Protection from apoptosis by chemical chaperones. COS-7 cells were transfected with wild-type CA IV (WT) or R14W CA IV (MT) cDNA and incubated with 10 μM acetazolamide (+AC), 5 mM PBA (+PBA), or normal medium (–AC or –PBA) for 72 h after transfection. (Upper) The cells were fixed and processed for TUNEL staining to assess apoptosis. The percent of cells undergoing apoptosis was calculated from the number of TUNEL-positive cells divided by the number of CA IV-positive cells from three to five fields of each slide. (Lower) The percent TUNEL-positive cells is plotted. These results suggest that induction of apoptosis of COS-7 cells by overexpression of R14W CA IV was inhibited by acetazolamide and PBA.
In Fig. 5, we present results comparing the anti-apoptotic effects of acetazolamide with those of Azopt (brinzolamide) and Trusopt (dorzolamide), two proprietary CA inhibitors that are widely used for topical application to the eye to reduce intraocular pressure in glaucoma. Fig. 5 Upper compares the R14W CA IV- and wild-type CA IV-expressing cells for the fraction of cells that show positive TUNEL staining in the absence and presence of acetazolamide, Azopt, and Trusopt. Both Azopt and Trusopt appear at least as effective as acetazolamide at the same concentration (10 μM) in protecting from the cell death induced by R14W CA IV. Both reduced the TUNEL-positive, R14W-expressing cells from 80% to <10% (Fig. 5 Lower).
Fig. 5.

Comparison of acetazolamide with other commercially available CA-inhibitor drugs for protection of R14W-transfected cells from apoptosis. COS-7 cells were transfected with wild-type CA IV (WT) or R14W CA IV (MT) cDNA and incubated with medium alone (Untreated), 10 μM acetazolamide (+AC), 10 μM Azopt (+AZ) (brinzolamide), or 10 μM Trusopt (+TRU) (dorzolamide) for 72 h after transfection. (Upper) The cells were fixed and processed for TUNEL staining. Percent of cells undergoing apoptosis was determined by calculating the number of TUNEL-positive cells divided by the number of CA IV-positive cells from three to five fields of each slide. (Lower) The percent TUNEL-positive cells was plotted. These results suggest that apoptosis of COS-7 cells caused by overexpression of R14W CA IV was largely prevented by all three sulfonamide inhibitors. Un, untreated.
Discussion
Along with our collaborators in South Africa, we recently reported the identification of the R14W mutation in the signal sequence of human CA IV in patients with the RP17 form of autosomal dominant RP (11). Overexpression of the R14W CA IV protein in the COS-7 cells resulted in reduced steady-state levels of the enzyme activity and protein, which were due to a combination of reduced biosynthesis, reduced rate of maturation from precursor to mature enzyme, and accelerated degradation of the mutant enzyme by an MG-132-sensitive system (presumably the proteosome). Expression of the R14W CA IV also led to the UPR and ER stress in the COS-7 cells, as evidenced by up-regulation of BiP, PERK, and CHOP. Excessive activation of UPR in R14W CA IV-expressing cells induced cells to undergo apoptosis, as evidenced from annexin V binding at the cell surface and TUNEL staining (11). On this basis, we proposed a specific disease mechanism for the RP17 form of autosomal dominant RP. We suggested that apoptosis of the endothelial cells in the choriocapillaris of the eye, which normally highly expresses CA IV, leads to ischemia in the overlying retina and to eventual retinal cell death and secondary melanin deposition, the hallmarks of RP.
In most protein-folding diseases, a mutation in the primary sequence of the mature protein leads to the UPR and apoptosis of the cells producing the mutant protein (40). However, there are two clear examples where a mutation in the signal sequence of a secretory protein leads to an autosomal dominant disease. One affects the signal sequence in pre-pro-parathyroid hormone (41) and leads to hypoparathyrodism. The other affects the signal sequence of pre-pro-vasopressin (42, 43) and leads to neurohypophyseal diabetes insipidus. We presume that each of these hormone-deficiency diseases results because accumulation of unfolded hormone precursor in the ER produces ER stress and induction of the UPR, and that chronic up-regulation of the UPR leads to apoptosis of the hormone-producing cells (41, 43). We recently proposed that RP17 is a third example of an autosomal dominant protein-folding disease caused by a mutation in the signal sequence (11). We speculated that the delayed removal of signal sequence from the precursor and delayed formation of the folded mature enzyme lead to an accumulation of unfolded CA IV in the ER, induction of the UPR, and eventual apoptosis in the endothelial cells of the choriocapillaris, in which CA IV is normally highly expressed.
In the present studies, we report the additional finding that the R14W CA IV, when expressed together with either of two other secretory proteins, the lysosomal enzyme, GUS, and the secretory form of CA IV (G267X CA IV), exerts a negative trans effect on expression of these secretory proteins. The synthesis and secretion of each protein was reduced compared with that seen when wild-type CA IV cDNA was cotransfected with the respective cDNA. Similar results have been reported for a mutant growth hormone, which reduced the synthesis and secretion of other proteins (36), and for Δ508CFTR, the mutant cystic fibrosis protein (37), which reduced production of expressed CA IV. We demonstrate here that both specific and nonspecific chemical chaperones overcame the negative trans effect of R14W CA IV expression on production of the other secretory proteins.
Using several well characterized CA inhibitors as specific chemical chaperones, and PBA as a nonspecific chemical chaperone, we also showed by pulse–chase experiments, enzyme activity measurements, Western blot analyses, and immunocytochemistry that these agents prevented the accelerated turnover of the mutant CA IV, normalized the steady state level of the enzyme activity and protein, prevented the up-regulation of markers of UPR and ER stress, and reduced fraction of COS-7 cells transfected with R14W CA IV undergoing apoptosis.
Specific chemical chaperones have been shown in cell culture to correct protein processing and transport defects in other protein-folding diseases. Examples include 11-cis-7-ring retinal for correction of rhodopsin folding in RP (14), competitive inhibitors of α-galactosidase A in Fabry disease (15), inhibitors of β-glucosidase in Gaucher disease (16), and inhibitors of β-hexosaminidase A in adult Tay–Sachs and Sandhoff diseases (18). The mechanism by which most specific chemical chaperones are thought to work is by binding to the active site of the unfolded protein to either increase the rate of folding of the mutant protein or protect the unfolded protein from rapid degradation. Nonspecific chemical chaperones, such as PBA, have also been shown in cell culture or animal models to correct the protein-folding defects with several common mutations, including ΔF508CFTR and α1-antitrypsin (23, 24). The mechanism by which PBA corrects the misfolding of the mutant proteins is not known. However, it is speculated that PBA may stabilize the incorrect folding to reduce aggregation and prevent nonproductive interactions with resident chaperones, which facilitates the transport of the proteins from the intracellular compartment (12, 44).
The ability of the CA inhibitors acetazolamide, ethoxzolamide, brinzolamide, and dorzolamide and the nonspecific chaperone PBA to promote folding of the R14W CA IV and reduce the apoptotic effects of the R14W CA IV in transfected COS-7 cells suggests a pharmacological approach to treating this autosomal dominant form of RP. If these agents could achieve similar results in vivo, they might delay the onset or even prevent this form of hereditary loss of vision. The delayed onset of this disease and slow progression might make clinical trials to test this approach difficult. For this reason, development of a mouse or other animal model of this disorder for preclinical trials of therapy with chemical chaperones would be extremely valuable.
Supplementary Material
Acknowledgments
We thank Drs. Rajkumar Ramesar and George Rebello of the Division of Human Genetics and Institute of Infectious Diseases and Molecular Medicine at the University of Cape Town (Rondebosch, South Africa) for bringing this problem to our attention and for the fruitful collaboration leading to its description (see ref. 11). This research was supported by National Institutes of Health Grant DK40163 (to W.S.S.).
Abbreviations: CA, carbonic anhydrase; RP, retinitis pigmentosa; UPR, unfolded protein response; ER, endoplasmic reticulum; PBA, 4-phenylbutyric acid; BiP, Ig-binding protein; PERK, double-stranded RNA-regulated protein kinase-like ER kinase; CHOP, CCAAT/enhancer-binding protein homologous protein; TUNEL, terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling; GUS, β-glucuronidase; EU, enzyme units.
References
- 1.Zhu, X. L. & Sly, W. S. (1990) J. Biol. Chem. 265, 8795–8801. [PubMed] [Google Scholar]
- 2.Waheed, A., Zhu, X. L. & Sly, W. S. (1992) J. Biol. Chem. 267, 3308–3311. [PubMed] [Google Scholar]
- 3.Brown, D., Zhu, X. L. & Sly, W. S. (1990) Proc. Natl. Acad. Sci. USA 87, 7457–7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fleming, R. E., Crouch, E. C., Ruzicka, C. A. & Sly, W. S. (1993) Am. J. Physiol. 265, L627–L635. [DOI] [PubMed] [Google Scholar]
- 5.Ghandour, M. S., Langley, O. K., Zhu, X. L., Waheed, A. & Sly, W. S. (1992) Proc. Natl. Acad. Sci. USA 89, 6823–6827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parkkila, S., Parkkila, A. K., Kaunisto, K., Waheed, A., Sly, W. S. & Rajaniemi, H. (1993) J. Histochem. Cytochem. 41, 751–757. [DOI] [PubMed] [Google Scholar]
- 7.Sender, S., Gros, G., Waheed, A., Hageman, G. S. & Sly, W. S. (1994) J. Histochem. Cytochem. 42, 1229–1236. [DOI] [PubMed] [Google Scholar]
- 8.Fleming, R. E., Parkkila, S., Parkkila, A. K., Rajaniemi, H., Waheed, A. & Sly, W. S. (1995) J. Clin. Invest. 96, 2907–2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Parkkila, S., Parkkila, A. K., Juvonen, T., Waheed, A., Sly, W. S., Saarnio, J., Kaunisto, K., Kellokumpu, S. & Rajaniemi, H. (1996) Hepatology 24, 1104–1108. [DOI] [PubMed] [Google Scholar]
- 10.Hageman, G. S., Zhu, X. L., Waheed, A. & Sly, W. S. (1991) Proc. Natl. Acad. Sci. USA 88, 2716–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rebello, G., Ramesar, R., Vorster, A., Roberts, L., Ehrenreich, L., Oppon, E., Gama, D., Bardien, S., Greenberg, J., Bonapace, G., et al. (2004) Proc. Natl. Acad. Sci. USA 101, 6617–6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perlmutter, D. H. (2002) Pediatr. Res. 52, 832–836. [DOI] [PubMed] [Google Scholar]
- 13.Cohen, F. E. & Kelly, J. W. (2003) Nature 426, 905–909. [DOI] [PubMed] [Google Scholar]
- 14.Noorwez, S. M., Kuksa, V., Imanishi, Y., Zhu, L., Filipek, S., Palczewski, K. & Kaushal, S. (2003) J. Biol. Chem. 278, 14442–14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fan, J. Q., Ishii, S., Asano, N. & Suzuki, Y. (1999) Nat. Med. 5, 112–115. [DOI] [PubMed] [Google Scholar]
- 16.Sawkar, A. R., Cheng, W. C., Beutler, E., Wong, C. H., Balch, W. E. & Kelly, J. W. (2002) Proc. Natl. Acad. Sci. USA 99, 15428–15433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuda, J., Suzuki, O., Oshima, A., Yamamoto, Y., Noguchi, A., Takimoto, K., Itoh, M., Matsuzaki, Y., Yasuda, Y., Ogawa, S., et al. (2003) Proc. Natl. Acad. Sci. USA 100, 15912–15917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tropak, M. B., Reid, S. P., Guiral, M., Withers, S. G. & Mahuran, D. (2004) J. Biol. Chem. 279, 13478–13487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morello, J. P., Salahpour, A., Laperriere, A., Bernier, V., Arthus, M. F., Lonergan, M., Petaja-Repo, U., Angers, S., Morin, D., Bichet, D. G., et al. (2000) J. Clin. Invest. 105, 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ficker, E., Obejero-Paz, C. A., Zhao, S. & Brown, A. M. (2002) J. Biol. Chem. 277, 4989–4998. [DOI] [PubMed] [Google Scholar]
- 21.Petaja-Repo, U. E., Hogue, M., Bhalla, S., Laperriere, A., Morello, J. P. & Bouvier, M. (2002) EMBO J. 21, 1628–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janovick, J. A., Goulet, M., Bush, E., Greer, J., Wettlaufer, D. G. & Conn, P. M. (2003) J. Pharmacol. Exp. Ther. 305, 608–614. [DOI] [PubMed] [Google Scholar]
- 23.Rubenstein, R. C. & Zeitlin, P. L. (1998) Am. J. Respir. Crit. Care Med. 157, 484–490. [DOI] [PubMed] [Google Scholar]
- 24.Burrows, J. A., Willis, L. K. & Perlmutter, D. H. (2000) Proc. Natl. Acad. Sci. USA 97, 1796–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maestri, N. E., Brusilow, S. W., Clissold, D. B. & Bassett, S. S. (1996) N. Engl. J. Med. 335, 855–859. [DOI] [PubMed] [Google Scholar]
- 26.Okuyama, T., Waheed, A., Kusumoto, W., Zhu, X. L. & Sly, W. S. (1995) Arch. Biochem. Biophys. 320, 315–322. [DOI] [PubMed] [Google Scholar]
- 27.Islam, M. R., Grubb, J. H. & Sly, W. S. (1993) J. Biol. Chem. 268, 22627–22633. [PubMed] [Google Scholar]
- 28.Okuyama, T., Sato, S., Zhu, X. L., Waheed, A. & Sly, W. S. (1992) Proc. Natl. Acad. Sci. USA 89, 1315–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niwa, H., Yamamura, K. & Miyazaki, J. (1991) Gene 108, 193–199. [DOI] [PubMed] [Google Scholar]
- 30.Lopata, M. A., Cleveland, D. W. & Sollner-Webb, B. (1984) Nucleic Acids Res. 12, 5707–5717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Luthman, H. & Magnusson, G. (1983) Nucleic Acids Res. 11, 1295–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maren, T. H. (1960) J. Pharmacol. Exp. Ther. 130, 26–29. [PubMed] [Google Scholar]
- 33.Peterson, G. L. (1979) Anal. Biochem. 100, 201–220. [DOI] [PubMed] [Google Scholar]
- 34.Laemmli, U. K. (1970) Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- 35.Waheed, A., Parkkila, S., Zhou, X. Y., Tomatsu, S., Tsuchihashi, Z., Feder, J. N., Schatzman, R. C., Britton, R. S., Bacon, B. R. & Sly, W. S. (1997) Proc. Natl. Acad. Sci. USA 94, 12384–12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Graves, T. K., Patel, S., Dannies, P. S. & Hinkle, P. M. (2001) J. Cell Sci. 114, 3685–3694. [DOI] [PubMed] [Google Scholar]
- 37.Fanjul, M., Salvador, C., Alvarez, L., Cantet, S. & Hollande, E. (2002) Eur. J. Cell Biol. 81, 437–447. [DOI] [PubMed] [Google Scholar]
- 38.Waheed, A., Okuyama, T., Heyduk, T. & Sly, W. S. (1996) Arch. Biochem. Biophys. 333, 432–438. [DOI] [PubMed] [Google Scholar]
- 39.Stams, T., Nair, S. K., Okuyama, T., Waheed, A., Sly, W. S. & Christianson, D. W. (1996) Proc. Natl. Acad. Sci. USA 93, 13589–13594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rutkowski, D. T. & Kaufman, R. J. (2004) Trends Cell Biol. 14, 20–28. [DOI] [PubMed] [Google Scholar]
- 41.Karaplis, A. C., Lim, S. K., Baba, H., Arnold, A. & Kronenberg, H. M. (1995) J. Biol. Chem. 270, 1629–1635. [DOI] [PubMed] [Google Scholar]
- 42.Beuret, N., Rutishauser, J., Bider, M. D. & Spiess, M. (1999) J. Biol. Chem. 274, 18965–18972. [DOI] [PubMed] [Google Scholar]
- 43.Ito, M., Jameson, J. L. & Ito, M. (1997) J. Clin. Invest. 99, 1897–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rubenstein, R. C. & Lyons, B. M. (2001) Am. J. Physiol. 281, L43–L51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}