

Abstract
A polymorphism in the human serotonin transporter gene promoter (5-HTTLPR) is associated with anxiety and increased risk for developing depression in the face of adversity. Here, we report that among infant rhesus macaques, an orthologous polymorphism (rh5-HTTLPR) interacts with adversity in the form of peer rearing to influence adrenocorticotropic hormone (ACTH) response to stress and, further, that this interaction is sexually dichotomous. ACTH responses to separation are higher in l/s than in l/l males. In females, however, it is only among those with a history of adversity that the s allele is associated with increased ACTH responses to stress. Of interest, peer-reared animals, in particular females carrying the s allele, also exhibit lower cortisol responses to stress, a pattern that has been recognized in association with certain stress-related neuropsychiatric disorders. By extension, our findings suggest the intriguing possibility that human females carrying the 5-HTTLPR s allele could be more vulnerable to the effects of early adversity. This interactive effect may underlie the increased incidence of certain stress-related disorders in women.
Individuals exposed to adversity early in life are at increased risk for developing medical conditions, including diabetes, atherosclerosis, and heart disease, in addition to the stress-related neuropsychiatric disorders, such as posttraumatic stress disorder, depression, and alcoholism (1). Although an individual's early responses to adversity are believed to contribute to the pathogenesis and progression of such disorders, disease onset often does not occur until adulthood and, moreover, it is not the case that everyone who experiences adversity early in life eventually develops stress-related disease. There is accumulating evidence that early adversity and genetic vulnerability can interact, increasing one's susceptibility to stress-related disorders later in life (2). Although it is accepted that there are additional factors, such as gender, that may increase an individual's sensitivity to adversity (3–8), whether these factors modulate or contribute to the complex interactions between genes and early life stress has yet to be examined. Because of their genetic similarity to humans, non-human primates are particularly useful for genetic association studies and, more importantly, for studying gene by environment (G×E) interactions, because their rearing environments can be tightly controlled (9, 10). Of particular interest for the study of G×E interactions are gene variants that are likely to confer differential sensitivity to stress (2). By following animals from infancy, the rhesus macaque model affords the opportunity to study these interactions as they relate to vulnerability to challenge throughout early development. In this report, we examine whether there are sex differences in the interactive effects of early adversity and a functional variant in the transcriptional control region of the serotonin transporter gene (rhesus serotonin transporter-linked polymorphic region, rh5-HTTLPR) on reactivity to stress during early infant development in rhesus macaques.
Among humans, there is a common polymorphism involving an insertion/deletion in the transcriptional control region of the serotonin transporter gene (5-HTTLPR), which alters in vitro gene transcription (11), in vitro transporter activity (12), and in vivo serotonin transporter density (13). Individuals carrying the short (s) allele are more likely to demonstrate anxiety-related personality traits, such as neuroticism, harm avoidance, and disagreeableness, than are individuals homozygous for the long (l) allele (11, 14, 15). Studies have demonstrated increased activation of the right amygdala in response to stressful stimuli in carriers of the s allele (16) as well as an increase in the incidence of the stress-related neurospychiatric disorders, including alcoholism and depression (17). Recently, Caspi et al. (2) reported that the s allele is associated with depression but only among individuals exposed to childhood maltreatment or major life stressors as young adults.
In rhesus macaques, a 21-bp insertion/deletion polymorphism, rh5-HTTLPR, orthologous to the human serotonin transporter length variant (18), has been shown to alter transcriptional efficiency (19), resulting in decreased serotonin transporter mRNA levels in brains of macaques carrying the s rh5-HTTLPR allele (unpublished data). In our laboratory, we have demonstrated interactive effects between rearing experience and rh5-HTTLPR genotype on a number of phenotypes of interest (10), including sensitivity to alcohol (20), neuroendocrine stress reactivity (21), and infant affective responding (22).
In rodents, neonatal handling produces lasting changes in stress responsivity (4–8). Although the experience of postnatal handling increases males' capacities to face challenge, handled females demonstrate increased sensitivity to stress. This is interesting in light of the fact that women are more vulnerable to stress-related anxiety and mood disorders, such as depression. As stated above, the study by Caspi et al. (2) demonstrated that the 5-HTTLPR s allele is associated with depression but only among individuals exposed to adversity. Because of the increased incidence of depression among women, sex was included as a covariate in their analyses. However, the potential for interactions to occur among sex, life stress, and the 5-HTTLPR s allele was not addressed.
Central to physiological and behavioral adaptation to stress is the hypothalamic—pituitary–adrenal endocrine axis (HPA axis). In response to a stressful stimulus, corticotropin-releasing hormone is released from the hypothalamus (23), the site of convergence for stress signals in the brain. Adrenocorticotropic hormone (ACTH) is then released from the anterior pituitary gland, resulting in stimulation of glucocorticoid secretion from the adrenal cortex. We have obtained peripheral measures of both ACTH and the glucocorticoid, cortisol, from 190 infant rhesus macaques during the course of early development, at ages comparable to 9–24 months in humans. Three of these samplings occurred after 30 min of social separation, a stressor that, in macaques, is known to evoke a profound stress response (24). We wanted to determine whether rearing condition and rh5-HTTLPR genotype would interact to influence HPA axis output after acute separation stress during early infancy in rhesus macaques. Further, we wanted to examine whether sex would modify the interactive effect between maternal deprivation and rh5-HTTLPR genotype.
Materials and Methods
Animals. Infants (n = 190) from an outbred colony of rhesus macaques (Macaca mulatta) were obtained from seven birth-year cohorts and were randomly selected to be reared either with their mothers (mother-reared, MR) or in peer-only groups (peer-reared, PR), an established non-human primate model for early life stress (25). The peer-rearing condition deprives animals of parental input and the opportunity to learn appropriate social behaviors and context during early development and is a model for early life stress. These rearing conditions have been described in detail (26). Briefly, MR animals (n = 128, 62 females and 66 males) were reared in social groups composed of 8–12 females (about half of whom had same-age infants) and two adult males. PR animals (n = 62, 28 females and 34 males) were separated from their mothers at birth and hand-reared in a neonatal nursery for the first 37 days of life. For the first 14 days, they were kept in an incubator and hand-fed. From day 15 until day 37, they were placed alone in a nursery cage and provided a blanket and a terrycloth-covered rocking surrogate. A bottle from which the infants would feed was fixed to the surrogate. At 37 days of age, they were placed in a cage with three other age mates with whom they had continuous contact. Protocols for the use of experimental animals were approved by the Institutional Animal Care and Use Committee of the National Institute of Child Health and Human Development.
Separation Stress and Sample Collection. Blood samples were drawn from the femoral vein under ketamine anesthesia (10 mg/kg, i.m.) from infant rhesus macaques at exactly 2, 3, 4, 5, and 6 months of age. Nonstressed baseline samples were obtained at 60 days (2 months) and 6 months of age. At days 90, 120, and 150 (3, 4, and 5 months of age, respectively), subjects were removed from their mother (MR animals) or peer group (PR animals) and isolated in an unfamiliar single cage (64 × 61 × 76 cm) for 30 min. At the end of each 30-min separation, blood samples were obtained under anesthesia for determination of ACTH and total cortisol levels by radio immunoassay (27). All samples were collected between 1130 and 1430 h, within 15 min of investigators' entrance into the housing facility for capture and sampling.
Genotyping. By using standard extraction methods, DNA was isolated from whole blood, collected from the femoral vein under ketamine anesthesia (15 mg/kg, i.m.). The rh5-HTTLPR was amplified from 25 ng of genomic DNA with flanking oligonucleotide primers (stpr5, 5′-GGCGTTGCCGCTCTGAATGC; intl, 5′-CAGGGGAGATCCTGGGAGGG), as described (21). Amplicons were separated by gel electrophoresis on a 10% polyacrylamide gel, and the s (388-bp) and l (419-bp) alleles of the rh5-HTTLPR were identified by direct visualization after ethidium bromide staining.
Data Analyses. ANOVA was conducted to assess the effects of rh5-HTTLPR genotype, rearing condition, and sex on HPA axis output among infant macaques. Animals were assigned nominal independent variables according to rearing condition (MR or PR), rh5-HTTLPR genotype (l/l or l/s), and sex (male or female). To determine whether the between-subject factors would interact differentially after acute exposures to separation stress, testing condition was added as a within-subjects nominal independent variable, and data were analyzed by using a mixed-design ANOVA. Repeated-measures ANOVA demonstrated no differences or differential interactions among the three measures obtained after stress (months 3, 4, and 5). These were, therefore, collapsed into one “stress” measure. Because initial analyses indicated that average basal ACTH and cortisol levels differed between 2 and 6 months, these measures were entered as independent testing conditions (Basal-1 and Basal-2, respectively). The Komogorov–Smirnov Normality Test demonstrated ACTH and cortisol levels to be normally distributed.
For testing the effects of the between subjects variables on overall ACTH and cortisol output, areas under the curve with respect to ground (AUCG) were calculated by using the formula
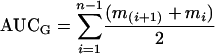 |
with mi denoting the individual endocrine measurement and n the total number of measurements (28). AUC were calculated only in instances in which ACTH or cortisol measures were available for every time point. Three-way ANOVA was then used to determine effects of sex, rearing condition, and rh5-HTTLPR genotype on AUCG for ACTH and cortisol.
The average identity by descent (IBD) for the 190 animals included in the study was 1.29%. This indicates that two randomly selected macaques would share only 1.29% of their genes by descent (equivalent to a degree of relationship observed between second cousins once removed and third cousins), thus demonstrating that most pairs of individuals have a low degree of relationship, approximating that observed in some human populations of study (29). To further verify that our effects were attributable to rh5-HTTLPR variation and not to general heritability of stress responsivity, we also repeated our analyses using a set of randomly selected biallelic genetic markers used for genotyping in our colony. Three genes with variant allele frequencies similar to the rh5-HTTLPR s allele were selected and, as with analyses performed with rh5-HTTLPR, animals homozygous for the rare allele were excluded. Because there were no effects of the other markers tested, and because the IBD was sufficiently low, standard statistical procedures were applied for testing the interactions among rh5-HTTLPR, sex, and rearing condition.
Genotype frequencies were distributed according to the Hardy –Weinberg equilibrium (X2 = 0.76, df = 2, P = 0.68). Because of the low frequency of the s allele in this colony, the number of animals with the s/s genotype (n = 3) was insufficient to perform three-way ANOVA. They were, therefore, excluded from all analyses. Numbers of animals indicated in Table 1 reflect these exclusions. In some instances, ACTH or cortisol values were not obtained due to the difficulty of collecting samples from infants, sample degradation, or inadequate sample volume. All analyses were performed by using statview (SAS Institute, Cary, NC) statistical software. The criterion for significance was set at P < 0.05.
Table 1. rh5-HTTLPR genotype count by sex and rearing condition.
| Sex | Rearing condition | l/l | l/s | Total |
|---|---|---|---|---|
| Female | MR | 42 | 20 | 62 |
| PR | 21 | 7 | 28 | |
| Male | MR | 48 | 18 | 66 |
| PR | 28 | 6 | 34 | |
| Total | 139 | 51 | 190 |
Results
Using a four-way mixed-design ANOVA, with rearing condition, genotype, and sex as between-group factors and testing condition (basal vs. stressed) as the within group factor, we found multiple main effects and interactions (Table 2). Because of the interactive effect on levels of ACTH among between-subject variables and testing condition, we performed follow-up ANOVA during the three main phases of testing (Fig. 1). During both basal samplings, PR animals had lower levels of ACTH than did MR animals [Fig. 1B, 2 months, F(1/146) = 14.5, P = 0.0002; Fisher's probable least-squares difference (PLSD), P < 0.0001; Fig. 1D, 6 months, F(1/148) = 6.4, P = 0.01; Fisher's PLSD, P < 0.0006], whereas, for samples taken after acute stress, the effect of rearing was absent. In response to stress, animals carrying the s allele had higher levels of ACTH than did l/l animals [Fig. 1C, F(1/154) = 14.3, P = 0.0002, Fisher's PLSD, P = 0.003], consistent with a role of rh5-HTTLPR variation in anxiety. In addition, there were interactions between rearing condition and genotype [Fig. 1C, F(1/154) = 4.7, P = 0.03] as well as among rearing, genotype, and sex [Fig. 1C, F(1/154) = 5.2, P = 0.02]. Post hoc analyses using Fisher's PLSD method demonstrated that PR infants carrying the s allele had higher levels of ACTH relative to PR l/l or MR infants (Fig. 1C, Fisher's PLSD, P < 0.05). When sex was also considered, this effect was observed exclusively among females (Fig. 1C), such that ACTH levels among PR l/s females were higher than in PR l/l or MR females (Fisher's PLSD, P < 0.05). Among males, the interaction between rearing and rh5-HTTLPR was not significant. Male carriers of the s allele had higher levels of ACTH after acute stress than did l/l males, independent of their rearing histories (Fig. 1C, Fisher's PLSD, P = 0.0013). Among PR l/s animals, ACTH levels after acute stress were higher among females than among males (Fisher's PLSD, P < 0.05). There were no interactions of either rearing or genotype with sex during the two baseline measures (Fig. 1 B and D).
Table 2. Main effects and interactive effects among rh5-HTTLPR genotype, rearing condition, and sex on HPA axis responses to acute stress in infant rhesus macaques.
| Variables | F | P value | Power |
|---|---|---|---|
| ACTH | |||
| Rearing | 18.1 | <0.0001 | 0.95 |
| rh5-HTTLPR | 5.3 | 0.02 | 0.63 |
| Testing condition | 243.6 | <0.0001 | 1 |
| Sex × rearing | 3.7 | 0.05 | 0.48 |
| Rearing × rh5-HTTLPR | 4.5 | 0.04 | 0.55 |
| Rearing × testing condition | 4.2 | 0.02 | 0.74 |
| rh5-HTTLPR × testing condition | 7.8 | 0.0005 | 0.96 |
| Sex × rearing × testing condition | 3.4 | 0.04 | 0.63 |
| Sex × rearing × rh5-HTTLPR × testing condition | 3.4 | 0.03 | 0.64 |
| Cortisol | |||
| Rearing | 4.0 | 0.05 | 0.5 |
| rh5-HTTLPR | 8.8 | 0.003 | 0.86 |
| Testing condition | 221.0 | <0.0001 | 1 |
| Sex × rearing × rh5-HTTLPR | 5.0 | 0.03 | 0.6 |
Fig. 1.

Effects of rh5-HTTLPR genotype (l/l or l/s), rearing condition (MR or PR), and sex (male or female) on ACTH responses to acute stress in infant rhesus macaques. (A) Values listed are mean plasma levels of ACTH in pg/ml ± SEM determined at baseline (Stress –, 2 and 6 months) and after stress (Stress +,3,4, and 5 months). (B–D) Shown are serum ACTH levels for Basal-1 (B), stress (C), and Basal-2 (D) testing conditions. During both baseline conditions, PR animals had lower levels of ACTH. After stress, there were interactions between rearing and genotype and among rearing, genotype, and sex. (*, P < 0.05; **, P < 0.01; ***, P < 0.005).
There were no interactive effects between any of the between-subjects variables and testing condition on plasma levels of cortisol (Table 2 and Fig. 2), and therefore follow-up ANOVA was not performed. Overall, there were main effects of rh5-HTTLPR genotype and rearing condition on cortisol levels and an interaction among rearing, rh5-HTTLPR, and sex. Rearing and rh5-HTTLPR genotype interacted to influence levels of cortisol among females (Fig. 2 A; Fisher's PLSD, P = 0.04) but not among males. PR females carrying the rh5-HTTLPR s allele exhibited lower cortisol levels overall.
Fig. 2.

Effects of rh5-HTTLPR genotype (l/l or l/s), rearing condition (MR or PR), and sex (male or female) on cortisol responses to acute stress in infant rhesus macaques. (A) Values listed are mean plasma levels of cortisol in μg/dl ± SEM determined at baseline (Stress –, 2 and 6 months) and after stress (Stress +,3,4, and 5 months). (B–D) Shown are serum cortisol levels for Basal-1 (B), stress (C), and Basal-2 (D) testing conditions. There were main effects of rearing condition and of rh5-HTTLPR genotype on cortisol. In addition, there was an interaction among sex, rearing condition, and rh5-HTTLPR, such that female PR l/s animals had lower levels of cortisol.
Because animals were represented multiple times in the dataset, the AUCG for samples taken at 2, 3, 4, 5, and 6 months of age were calculated for both cortisol and ACTH, so that the interactive effects on overall levels of ACTH and cortisol could be determined. Data were analyzed by using three-way ANOVA, with rearing, rh5-HTTLPR genotype, and sex as nominal independent variables. Although there was no main effect of rearing history on cumulative levels of ACTH, there was an effect of rh5-HTTLPR genotype [F(1/126) = 10.5, P = 0.0015, Fisher's PLSD, P = 0.007] and both rearing × genotype [F(1/126) = 3.9, P < 0.05] and rearing × genotype × sex [F(1/126) = 4.2, P = 0.04] interactions. Post hoc analyses using Fisher's PLSD method demonstrated significant differences between l/l and l/s males (Fisher's PLSD, P = 0.007). Males carrying the rh5-HTTLPR s allele had higher AUCG for ACTH. In females, s allele-associated increases in the AUCG for ACTH were observed only among those reared in peer-only groups (Fisher's PLSD, P < 0.05).
We found that PR animals had lower cumulative levels of cortisol than those reared with their mothers [F(1/160) = 4.7, P = 0.03, Fisher's PLSD, P = 0.01]. Neither rh5-HTTLPR genotype nor sex influenced cumulative cortisol levels, but there was a trend for an interaction among sex, rearing, and rh5-HTTLPR [F(1/160) = 3.6, P = 0.06].
Discussion
Studies in rodents have demonstrated that targeted disruption of the serotonin transporter gene produces increased ACTH, but not cortisol, responses to immobilization stress (30). The present study demonstrates an effect of naturally occurring rh5-HTTLPR variation on ACTH, but not cortisol, response to stress in infant macaques. Given the role of serotonin not only in sensitivity to stress but also in feedback control and activation of the HPA axis, a relationship between the low-functioning serotonin transporter gene variant and increased pituitary release of ACTH is not surprising. What is of particular interest, however, is the finding that rh5-HTTLPR variation and early adversity interact to influence this response. Our results are especially intriguing in light of those from the study by Caspi et al. (2), which indicate that, among human subjects, 5-HTTLPR moderates the influence of childhood maltreatment on the likelihood of later developing depression.
Although the genotype × rearing interaction demonstrated in the present study supports the findings of Caspi et al. (2), this report further suggests there may be increased vulnerability to early adversity among females in the presence of serotonin transporter gene promoter variation. Among males, there is a significant association between rh5-HTTLPR genotype and levels of ACTH in response to acute separation stress, indicating that the s allele does confer increased vulnerability to challenge among these subjects. Among females, however, the effect of the s allele is observed only among those that also have a history of early life stress in the form of maternal deprivation. Interactions among genetic differences, experience, and sex have also been noted in other species. In mice, HPA axis responses to repeated restraint are different in females from different strains, whereas in males there are no observable strain differences (31), and serotonin transporter gene disruption produces differential alterations in serotonin receptor densities in male and female brains (32). In adult Lewis rats, maternal deprivation during infancy results in increased pain sensitivity in females but not males. Interestingly, the direction of this effect is reversed in Long Evans rats (i.e., females subjected to early life stress demonstrate decreased nociception) (33, 34). In humans, certain gene variants are associated with neuropsychiatric diseases and anxiety-related intermediate phenotypes, but only among females (35, 36).
Consistent with findings from rodent studies, we demonstrated that cortisol levels are lower in infant macaques exposed to early adversity, a relationship particularly noticeable among l/s females (Fig. 1A). It has been noted that among clinically depressed or abused children, the morning rise in salivary cortisol levels is blunted (37). However, a review of the literature addressing HPA axis output anomalies in children subjected to maltreatment reveals paradoxical findings (38). Such inconsistencies have been posited to relate to experiential differences among subjects, and in particular, to whether subjects have prior histories of abuse or whether the abuse is ongoing at the time of testing (39). Our findings are in support of such variability, because the effects of rearing history are not observed in temporal proximity to a stressor. Instead, we observe interactions between rearing history and both sex and serotonin transporter gene promoter variation. It may be that individual differences (i.e., sex or genetic differences) also underlie the variability in the patterns observed among human populations of study, and that these individual differences may influence psychiatric disease progression and outcome.
High serum levels of ACTH and low levels of cortisol have been associated with several neuropsychiatric disorders, including chronic fatigue syndrome, posttraumatic stress disorder (PTSD), and atypical depression (40, 41). It has been proposed that lower cortisol responses to stress may be a predisposing factor in the development of PTSD, because low levels of glucocorticoids may compromise extinction of conditioned fear. Moreover, a diminished corticosteroid response could leave the HPA axis in a hyperresponsive facilitated state (42), making individuals more stress responsive. In this study, PR l/s females exhibit both increased stress-induced release of ACTH and decreased total cortisol levels. Of note, this pattern of HPA axis output is consistent with findings in serotonin transporter null mutant mice (43, 44), in which lower levels of corticosterone in the presence of high levels of ACTH have been proposed to relate in part to dysregulation of paracrine factors controlling adrenocortical sensitivity to ACTH. The ratio of ACTH to cortisol (ACTH:F) represents a crude index of adrenal sensitivity, with a lower ratio denoting higher sensitivity. In a set of follow-up analyses, we demonstrated that the ACTH:F ratio after stress is higher among carriers of the rh5-HTTLPR s allele, and that this effect is particularly marked among PR l/s females (Fig. 3, which is published as supporting information on the PNAS web site). Although the high ACTH:F ratio is suggestive of decreased adrenal sensitivity or reserve among these animals, whether other factors (i.e., insufficient time for cortisol levels to peak after initiation of the stressor, altered negative feedback control, induction of other ACTH secretagogues, adjustments in corticosteroid-binding globulin levels, or differential regulation of brain corticosteroid hormone receptor levels) could also be attributed to this pattern of HPA axis output has not been tested. Stress-related disorders in which low levels of circulating corticosteroids are observed have been shown to be associated with increased sensitivity to cortisol via increased glucocorticoid receptor (GR) efficiency and/or density (45). A relationship among increased levels of GR expression in the brain, high levels of ACTH, and low serum levels of cortisol has been confirmed in rodents overexpressing GR (46). Of interest, studies in our laboratory show that both PR and l/s macaques exhibit increased brain levels of GR mRNA (unpublished data), which would be expected to increase their response to corticosteroids during stress. Whether sex also modulates this effect, however, has yet to be tested.
There are several limitations to this study. First, when sex and rearing conditions are taken into consideration, there are only limited animals available in the PR l/s cells for each sex (males, n = 6; females, n = 7). This might be expected to result in loss of power, especially in light of the fact that this polymorphism would not be expected to contribute to a large portion of the variance in our data. Second, although the rh5-HTTLPR gene variant is known to be functional, it is possible that our result is due to the association of our phenotype with another gene variant that is in linkage disequilibrium with the rh5-HTTLPR locus. Finally, acute separation stress was used as a stressor because of the known effects of separation in this highly social species. The use of separation stress could be perceived as being potentially limiting, in that social separation may represent a different experience for MR than for PR animals. However, studies in our laboratory have demonstrated neither appreciable differences in social attachment between MR and PR animals nor any modulatory effect of sex (unpublished data). Still, it may be that sex modulates the effect of rearing in the presence of rh5-HTTLPR variation.
Anxiety and mood disorders are complex heritable conditions that are more common among women. Although individual differences are thought to contribute to the vulnerability and progression of stress-related neuropsychiatric disorders, environmental factors are also considered important. In the present report, we demonstrate that serotonin transporter gene variation affects stress reactivity and that the influence of rh5-HTTLPR on hormonal responses during stress is modulated by early experience, especially among female infant macaques. Male carriers of the rh5-HTTLPR allele, known to be weakly associated with anxiety in humans, are more responsive to acute stress than are l/l males, but, unlike in females, the long-lasting effects of early experience are absent. Among animals exposed to early adversity, we observe increased responsivity to acute challenge in l/s females relative to l/s males. On the other hand, the buffering effect of maternal contact that is observed among l/s females is absent among males. It has been posited that early stress (and, therefore, HPA axis activity) can influence the ongoing development of the HPA axis. It may be that early sensitivity to stress contributes to the pathogenesis of neuropsychiatric disease, such as depression, posttraumatic stress disorder (PTSD), or anxiety disorders. If our findings were to generalize to humans, then females with variation in the serotonin transporter gene promoter who had experienced childhood trauma (including parental loss or absence) might be especially predisposed to eventually developing alcoholism, depression, PTSD, or other related disorders. And, in fact, studies from our laboratory demonstrate there is an interactive effect between early rearing and the rh5-HTTLPR s allele on alcohol preference and consumption, but only among females (47). By learning more about genetic and environmental factors that contribute to reactivity to stress in non-human primates, we may be better able to identify risk factors and design combination therapies for predicting, treating, and ultimately preventing certain human neuropsychiatric disorders.
Supplementary Material
Acknowledgments
We thank Sue Higley, Pete Roma, Tami Gura, Kim Wojteczko, Kelli Chisholm, Ruth Woodward, Michelle Keawphalouk, and Clarissa Parker for assistance with data collection. This work was supported by the National Institute of Child Health and Human Development and National Institute on Alcohol Abuse and Alcoholism Intramural Research Programs and the Deutsche Forschungsgemeinschaft (SFB 581, KFO 125/1-1).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: 5-HTTLPR, serotonin transporter gene promoter; rh5-HTTLPR, rhesus 5-HTTLPR; s allele, short allele; l allele, long allele; HPA axis, hypothalamic–pituitary–adrenal endocrine axis; ACTH, adrenocorticotropic hormone; MR, mother-reared; PR, peer-reared; AUCG, area under the curve with respect to ground; PLSD, probable least-squares difference.
References
- 1.Heim, C. & Nemeroff, C. B. (2001) Biol. Psychiatry 49, 1023–1039. [DOI] [PubMed] [Google Scholar]
- 2.Caspi, A., Sugden, K., Moffitt, T. E., Taylor, A., Craig, I. W., Harrington, H., McClay, J., Mill, J., Martin, J., Braithwaite, A. & Poulton, R. (2003) Science 301, 386–389. [DOI] [PubMed] [Google Scholar]
- 3.McEwen, B. S. (1998) Ann. N.Y. Acad. Sci. 840, 33–44. [DOI] [PubMed] [Google Scholar]
- 4.Weinberg, J., Krahn, E. A. & Levine, S. (1978) Dev. Psychobiol. 11, 251–259. [DOI] [PubMed] [Google Scholar]
- 5.McCormick, C. M., Smythe, J. W., Sharma, S. & Meaney, M. J. (1995) Dev. Brain Res. 84, 55–61. [DOI] [PubMed] [Google Scholar]
- 6.Papaioannou, A., Gerozissis, K., Prokopiou, A., Bolaris, S. & Stylianopoulou, F. (2002) Behav. Brain Res. 129, 131–139. [DOI] [PubMed] [Google Scholar]
- 7.Park, M. K., Hoang, T. A., Bellussi, J. D. & Leslie, F. M. (2003) J. Neuroendocrinol. 15, 289–295. [DOI] [PubMed] [Google Scholar]
- 8.Panagiotaropoulos, T., Papaioannou A., Stavroula, P., Prokopiou, A., Stylianopoulou, F. & Gerozissis, K. (2004) Neuroendocrinology 79, 109–118. [DOI] [PubMed] [Google Scholar]
- 9.Gunzerath, L. & Goldman, D. (2003) Alcohol Clin. Exp. Res. 27, 540–562. [DOI] [PubMed] [Google Scholar]
- 10.Barr, C. S., Newman, T. K., Becker, M. L., Parker, C. C., Champoux, M., Lesch, K.-P., Goldman, D, Suomi, S. J. & Higley, J. D. (2003) Genes Brain Behav. 2, 336–340. [DOI] [PubMed] [Google Scholar]
- 11.Lesch, K.-P., Bengel, D., Heils, A., Sabol, S. Z., Greenberg, B. D., Petri, S., Benjamin, J., Muller, C. R., Hamer, D. H. & Murphy, D. L. (1996) Science 274, 1527–1531. [DOI] [PubMed] [Google Scholar]
- 12.Stoltenberg, S. F., Twitchell, G. R., Hanna, G. L., Cook, E. H., Fitzgerald, H. E., Zucker, R. A. & Little, K. Y. (2002) Am. J. Med. Genet. 114, 230–234. [DOI] [PubMed] [Google Scholar]
- 13.Heinz, A., Mann, K., Weinberger, D. R. & Goldman, D. (2001) Alcohol. Clin. Exp. Res. 25, 487–495. [PubMed] [Google Scholar]
- 14.Mazzanti, C. M., Lappalainen, J., Long, J. C., Bengel, D., Naukkarinen, H., Eggert, M., Virkkunen, M., Linnoila, M. & Goldman, D. (1998) Arch. Gen. Psychiatry 55, 936–940. [DOI] [PubMed] [Google Scholar]
- 15.Greenberg, B. D., McMahon, F. J. & Murphy, D. L. (1998) Mol. Psychiatry 3, 186–189. [DOI] [PubMed] [Google Scholar]
- 16.Hariri, A. R., Mattay, V. S., Tessitore, A., Kolachana, B., Fera, F., Goldman, D., Egan, M. F. & Weinberger, D. R. (2002) Science 297, 400–403. [DOI] [PubMed] [Google Scholar]
- 17.Lichtermann, D., Hranilovic, D., Trixler, M., Franke, P., Jernej, B., Delmo, C. D., Knapp, M., Schwab, S. G., Maier, W. & Wildenauer, D. B. (2000) Am. J. Psychiatry 157, 2045–2047. [DOI] [PubMed] [Google Scholar]
- 18.Lesch, K.-P., Meyer, J., Glatz, K., Flugge, G., Hinney, A., Hebebrand, J., Klauck, S. M., Poustka, A., Poustka, F., Bengel, D., et al. (1997) J. Neural Transm. 104, 1259–1261. [DOI] [PubMed] [Google Scholar]
- 19.Bennett, A. J., Lesch, K. P., Heils, A., Long, J. C., Lorenz, J. G., Shoaf, S. E., Champoux, M., Suomi, S. J., Linnoila, M. V. & Higley, J. D. (2002) Mol. Psychiatry 7, 118–122. [DOI] [PubMed] [Google Scholar]
- 20.Barr, C. S., Newman, T. K., Becker, M. L., Champoux, M., Lesch, K.-P., Suomi, S. J., Goldman, D. & Higley, J. D. (2003) Alcohol Clin. Exp. Res. 27, 812–817. [DOI] [PubMed] [Google Scholar]
- 21.Barr, C. S., Newman, T. K., Shannon, C., Parker, C., Dvoskin, R. L., Becker, M. L., Schwandt, M., Champoux, M., Lesch, K.-P., Goldman, D., et al. (2004) Biol. Psychiatry 55, 733–738. [DOI] [PubMed] [Google Scholar]
- 22.Champoux, M. B., Bennett, A., Shannon, C., Higley, J. D., Lesch, K.-P. & Suomi, S. J. (2002) Mol. Psychiatry 7, 1058–1063. [DOI] [PubMed] [Google Scholar]
- 23.Vale, W. W., Spiess, J., Rivier, C. & Rivier, J. (1981) Science 213, 1394–1397. [DOI] [PubMed] [Google Scholar]
- 24.McKinney, W., Suomi, S. J. & Harlow, H. F. (1972) Arch. Gen. Psychiatry 27, 200–203. [DOI] [PubMed] [Google Scholar]
- 25.Higley, J. D., Hasert, M. F., Suomi, S. J. & Linnoila, M. (1991) Proc. Natl. Acad. Sci. USA 88, 7261–7265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Higley, J. D., Suomi, S. J. & Linnoila, M. (1996) Alcohol. Clin. Exp. Res. 20, 643–650. [DOI] [PubMed] [Google Scholar]
- 27.Higley, J. D., Suomi, S. J. & Linnoila, M. (1992) Biol. Psychiatry 32, 127–145. [DOI] [PubMed] [Google Scholar]
- 28.Pruessner, J. C., Kirschbaum, C., Meinlschmid, G. & Hellhammer, D. H. (2003) Psychoneuroendocrinology 28, 916–931. [DOI] [PubMed] [Google Scholar]
- 29.Robin, R. W., Chester, B., Rasmussen, J. K., Jaranson, J. M. & Goldman, D. (1997) Am. J. Psychiatry 154, 1582–1588. [DOI] [PubMed] [Google Scholar]
- 30.Lanfumey, L., Mannoury La Cour, C., Froger, N. & Hamon, M. (2000) Neurochem. Res. 25, 1199–1206. [DOI] [PubMed] [Google Scholar]
- 31.Jones, B. C., Sarrieau, A., Reed, C. L., Azar, M. R. & Mormede, P. (1998) Psychoneuroendocrinology 23, 505–517. [DOI] [PubMed] [Google Scholar]
- 32.Li, Q., Wichems, C., Heils, A., Lesch, K.-P. & Murphy, D. L. (2000) J. Neurosci. 20, 7888–7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stephan, M., Helfritz, F., Pabst, R. & von Horsten, S. (2002) Behav. Brain Res. 133, 149–158. [DOI] [PubMed] [Google Scholar]
- 34.Smythe, J. W., McCormick, C. M., Rochford, J. & Meaney, M. J. (1994) Physiol. Behav. 55, 971–974. [DOI] [PubMed] [Google Scholar]
- 35.Enoch, M.-A., Xu, K., Ferro, E., Harris, C. R. & Goldman, D. (2003) Psychiatr. Genet. 13, 33–41. [DOI] [PubMed] [Google Scholar]
- 36.Deckert, J., Catalano, M., Syagailo, Y. V., Bosi, M., Okladnova, O., Di Bella, D., Nothen, M. M., Maffei, P., Franke, P., Fritze, J., et al. (1999) Hum. Mol. Genet. 8, 621–624. [DOI] [PubMed] [Google Scholar]
- 37.Gunnar, M. R. & Vazquez, D. M. (2001) Dev. Psychopathol. 13, 515–538. [DOI] [PubMed] [Google Scholar]
- 38.Cicchetti, D. & Rogosh, F. A. (2001) Dev. Psychopathol. 13, 677–693. [DOI] [PubMed] [Google Scholar]
- 39.Kaufman, J., Birmaher, B., Perel, J., Dahl, R. E., Moreci, P., Nelson, B., Wells, W & Ryan, N. D. (1997) Biol. Psychiatry 42, 669–679. [DOI] [PubMed] [Google Scholar]
- 40.Bremner, J. D. & Vermetten, E. (2001) Dev. Psychopathol. 13, 473–489. [DOI] [PubMed] [Google Scholar]
- 41.Oquendo, M., Exhavarria, G., Galfalvy, H. C., Grunebaum, M. F., Burke, A., Barrera, A., Cooper, T. B., Malone, K. M. & Mann, J. (2003) Neuropsychopharmacology 28, 591–598. [DOI] [PubMed] [Google Scholar]
- 42.Cassano W. J., Jr., & D'mello, A. P. (2001) Neuroendocrinology 74, 167–177. [DOI] [PubMed] [Google Scholar]
- 43.Armando, I., Tjurmina, O. A., Li, Q., Murphy, D. L. & Saavedra, J. M. (2003) Neuroendocrinology 78, 217–225. [DOI] [PubMed] [Google Scholar]
- 44.Tjurmina, O. A., Armando, I., Saavedra, J. M., Goldstein, D. S., Murphy, D. L. (2002) Endocrinology 143, 4520–4526. [DOI] [PubMed] [Google Scholar]
- 45.Liberzon, I., Lopez, J. F., Flagel, S. B., Vazquez, D. M. & Young, E. A. (1999) J. Neuroendocrinol. 11, 11–17. [DOI] [PubMed] [Google Scholar]
- 46.Reichardt, H. M., Umland, T., Bauer, A., Kretz, O. & Schutz, G. (2000) Mol. Cell. Biol. 20, 9009–9017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barr, C. S., Newman, T. K., Lindell, S. G., Shannon, C., Champoux, M., Lesch, K. P., Suomi, S. J., Goldman, D. & Higley, J. D. (2004) Arch. Gen. Psychiatry, in press. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}