

Abstract
Cdc25 phosphatases activate cyclin-dependent kinases (Cdks) and thereby promote cell cycle progression. In vertebrates, Chk1 and Chk2 phosphorylate Cdc25A at multiple N-terminal sites and target it for rapid degradation in response to genotoxic stress. Here we show that Chk1, but not Chk2, phosphorylates Xenopus Cdc25A at a novel C-terminal site (Thr504) and inhibits it from C-terminally interacting with various Cdk–cyclin complexes, including Cdk1–cyclin A, Cdk1–cyclin B, and Cdk2–cyclin E. Strikingly, this inhibition, rather than degradation itself, of Cdc25A is essential for the Chk1-induced cell cycle arrest and the DNA replication checkpoint in early embryos. 14-3-3 proteins bind to Chk1-phosphorylated Thr504, but this binding is not required for the inhibitory effect of Thr504 phosphorylation. A C-terminal site presumably equivalent to Thr504 exists in all known Cdc25 family members from yeast to humans, and its phosphorylation by Chk1 (but not Chk2) can also inhibit all examined Cdc25 family members from C-terminally interacting with their Cdk–cyclin substrates. Thus, Chk1 but not Chk2 seems to inhibit virtually all Cdc25 phosphatases by a novel common mechanism.
Keywords: Cdc25, cell cycle checkpoint, Chk1, Chk2, Xenopus
Introduction
In eukaryotic cells, genotoxic stress causes activation of checkpoints (Hartwell and Weinert, 1989), which leads to diverse cellular responses such as cell cycle arrest, DNA repair, and cell death (reviewed by Zhou and Elledge, 2000). In vertebrates, upstream elements of the checkpoint pathways include ATM and ATR kinases, which principally phosphorylate and activate the effector kinases Chk2 (also called Cds1) and Chk1, respectively (Abraham, 2001; Shiloh, 2001; Sørensen et al, 2003). While the ATM–Chk2 pathway is activated primarily by ionizing radiation (IR)-induced DNA damage and is nonessential for cell viability (Barlow et al, 1996; Matsuoka et al, 1998), the ATR–Chk1 pathway is induced primarily by stalled DNA replication (Guo et al, 2000; Shiloh, 2001) and is essential at least for early embryogenesis (Sibon et al, 1997; Liu et al, 2000; Shimuta et al, 2002). Although structurally very different (Xu et al, 2002; Katsuragi and Sagata, 2004), Chk1 and Chk2 phosphorylate many common substrates and have partly overlapping roles (reviewed by Bartek and Lukas, 2003).
The cell cycle checkpoint pathways ultimately inhibit cyclin-dependent kinases (Cdks), thereby delaying or arresting the cell cycle at specific stages (Nurse, 1997; Walworth, 2000). A major and crucial target for Chk1 and Chk2 in cell cycle checkpoints is the dual-specificity phosphatase Cdc25, which dephosphorylates and activates Cdks (Rhind and Russell, 2000; Bartek and Lukas, 2003). In vertebrates, there are three isoforms of Cdc25, that is, Cdc25A, Cdc25B, and Cdc25C (reviewed by Donzelli and Draetta, 2003). Cdc25B is thought to function at the G2/M transition of the cell cycle (Lammer et al, 1998) and Cdc25C acts during M phase to sustain the activity of Cdk1–cyclin B, a key regulator of the M phase (Takizawa and Morgan, 2000). Chk1 and Chk2 phosphorylate Cdc25C (as well as the fission yeast Cdc25) on overlapping N-terminal sites and either exclude it from the nucleus via the binding of 14-3-3 proteins (Peng et al, 1997; Sanchez et al, 1997; Kumagai and Dunphy, 1999; Lopez-Girona et al, 1999) or directly inhibit its phosphatase activity (Blasina et al, 1999; Furnari et al, 1999). Regulation of Cdc25B occurs in response to UV-induced DNA damage, but the role of Chk1 or Chk2 in this regulation is not known (Bulavin et al, 2001).
In contrast to Cdc25B and Cdc25C, Cdc25A functions throughout the cell cycle, activating both Cdk2–cyclin E (an S-phase kinase) and Cdk1–cyclin B (Hoffmann et al, 1994; Jinno et al, 1994; Molinari et al, 2000). In response to genotoxic stress, Chk1 and Chk2 phosphorylate Cdc25A on N-terminal sites and target it rapidly for ubiquitin-dependent degradation (Mailand et al, 2000, 2002; Molinari et al, 2000; Falck et al, 2001; Shimuta et al, 2002; Busino et al, 2003), which is thought to be central to the S and G2 cell cycle checkpoints (Bartek and Lukas, 2003; Donzelli and Draetta, 2003). Chk1 is also responsible for the instability of Cdc25A during an unperturbed cell cycle, probably contributing to genomic integrity in normal conditions (Zhao et al, 2002; Sørensen et al, 2003). Interestingly, a very recent study also reports that Chk1 phosphorylates human Cdc25A on a C-terminal 14-3-3 binding site and, thereby, may specifically inhibit its interaction with Cdk1–cyclin B but not with Cdk1/2–cyclin A or Cdk2–cyclin E (Chen et al, 2003).
In this study, we have investigated the mechanism of Chk1-mediated regulation of Cdc25A by using the Xenopus egg/embryo system. We show that Chk1, but not Chk2, phosphorylates Xenopus Cdc25A on a novel C-terminal site (equivalent to the 14-3-3 binding site in human Cdc25A; Chen et al, 2003) and inhibits it from C-terminally interacting not only with Cdk1–cyclin B but also with Cdk1–cyclin A and Cdk2–cyclin E. Strikingly, this inhibition, rather than degradation itself, of Cdc25A is essential for the DNA replication checkpoint in early embryos; however, the inhibition does not require 14-3-3 binding. Moreover, and importantly, Chk1 but not Chk2 can also phosphorylate all other examined Cdc25 family members (human Cdc25B, Xenopus Cdc25C, and Drosophila String) on their C-terminal site and inhibit their C-terminal interactions with their Cdk–cyclin substrates. Thus, inhibition of interactions with Cdk–cyclin complexes may be a general mechanism by which Chk1 (but not Chk2) inhibits virtually all Cdc25 phosphatases.
Results
Chk1 phosphorylation sites required for Cdc25A degradation
In Xenopus embryos, endogenous Chk1, but not Chk2, phosphorylates Cdc25A and targets it for rapid degradation at the physiological DNA replication checkpoint at the midblastula transition (MBT), and a persistently activated form of Chk1 (Δ60-Chk1; Oe et al, 2001) can induce similar events in artificially activated eggs (Shimuta et al, 2002). Xenopus Cdc25A protein has seven serine residues that lie in the consensus Chk1/Chk2 phosphorylation motif (Arg-X-X-Ser) (Figure 1A); among them, Ser73 is likely to be phosphorylated by a kinase distinct from Chk1 but is essential for the Chk1-induced degradation of Cdc25A (Shimuta et al, 2002). We first examined phosphorylation of the other six serine residues by Chk1 (and Chk2) in vitro and tested for their requirement for Chk1-induced Cdc25A degradation in Xenopus eggs. When analyzed by using glutathione S-transferase (GST)-Cdc25A peptide fusion proteins (each with or without a Ser → Ala substitution), four (Ser120, 137, 190, and 295) out of the six serine residues could be phosphorylated by Δ60-Chk1 in vitro (Chk2 could phosphorylate Ser137, 190, and 295) (Figure 1B). Previously, no single Ser → Ala mutations of the six serine residues had any appreciable effect on the stability of full-length Cdc25A in Δ60-Chk1-expressing eggs (Shimuta et al, 2002). As shown in Figure 1C, however, combined alanine mutations (6A or 4A) of the six or four (or Chk1-phosphorylatable) serine residues prevented the Chk1-induced degradation of Cdc25A, and reversion of any single Ala to Ser at the four Chk1-phosphorylatable sites (hence, 5A/S120, etc.) partially destabilized the 6A mutant. Moreover, when expressed in early embryos, the 4A mutant was very stable at the MBT, whereas wild-type Cdc25A was readily degraded (Figure 1D). Thus, Chk1 seemed to phosphorylate Xenopus Cdc25A at (at least) four N-terminal serine residues (while Chk2 phosphorylated it at three overlapping sites), each phosphorylation additively affecting the stability of the phosphatase. Similar but somewhat different results have recently been reported for human Cdc25A (Sørensen et al, 2003).
Figure 1.
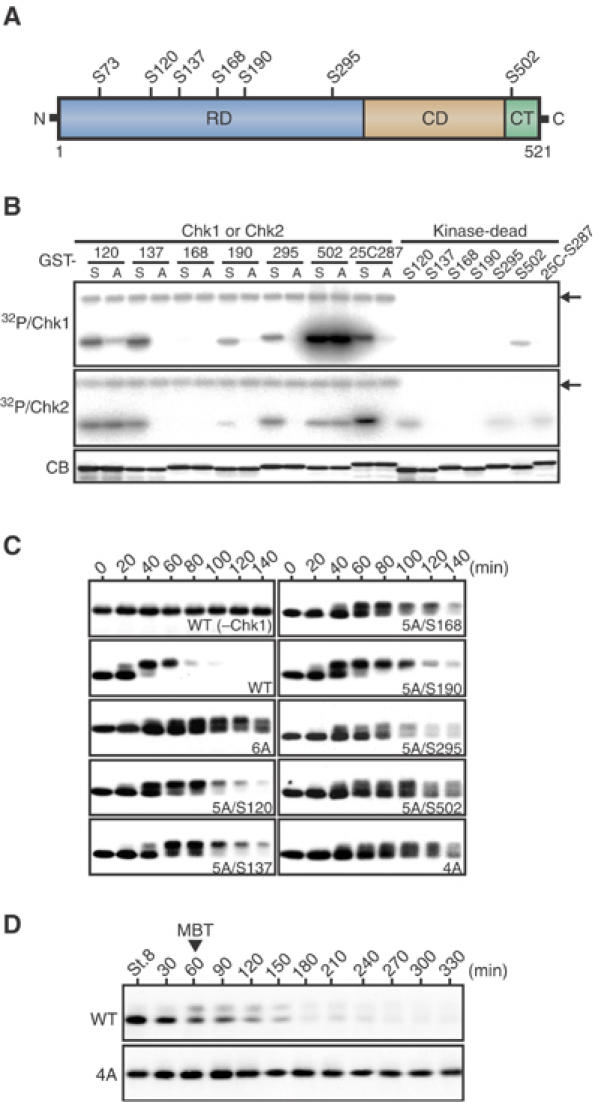
Chk1 phosphorylation sites required for Cdc25A degradation. (A) A schematic representation of Xenopus Cdc25A protein. Seven serine residues that lie in the consensus Chk1/Chk2 phosphorylation motif (Arg-X-X-Ser) are shown. RD, regulatory domain; CD, catalytic domain; CT, C-terminal tail. (B) GST-Cdc25A peptide fusion proteins (GST-S120, GST-A120, etc., each named after the relevant Ser or substituted Ala residue numbers) or GST-Cdc25C peptide fusion proteins (GST-S287 and GST-A287), 2 μg each, were incubated with [γ-32P]ATP and either Δ60-Chk1 protein, wild-type Chk2 protein or their kinase-dead forms, subjected to SDS–PAGE, stained with Coomassie blue (CB), and then autoradiographed (32P/Chk1 and 32P/Chk2). Because of the very low activity of Chk2, 32P/Chk2 was exposed substantially longer than 32P/Chk1. The arrows indicate autophosphorylated Δ60-Chk1 or Chk2. (C) Activated eggs were injected with 1 ng of mRNA encoding Myc-tagged wild-type Cdc25A (WT) or indicated Myc-tagged Cdc25A mutants (see text for the naming of the mutants), reinjected 2.5 h later with 2 ng of Δ60-Chk1 mRNA, and analyzed at 20 min intervals by immunoblotting using anti-Myc antibody. For WT Cdc25A, eggs expressing no Δ60-Chk1 (−Chk1) were also analyzed. (D) Fertilized eggs were injected with 1 ng of mRNA encoding Myc-tagged wild-type or 4A Cdc25A and, at 30 min intervals after the early blastula stage 8, analyzed as in (C). The MBT occurred 1 h after stage 8.
Phosphorylation of Thr504 by Chk1 but not Chk2
Intriguingly, in the above in vitro kinase assays, Δ60-Chk1 could phosphorylate a C-terminal Ser502-containing peptide of Xenopus Cdc25A extremely well, albeit not at Ser502 (Figure 1B). Alanine substitutions of the other conserved serine or threonine residues in this C-terminal peptide (Figure 2A) showed clearly that Thr504 was the Chk1 phosphorylation site in this region (Figure 2B). Thr504 lies in a motif (Lys-X-X-Thr) similar to the consensus Chk1/Chk2 phosphorylation motif (Arg-X-X-Ser) (Figure 2A), but Chk2 could not appreciably phosphorylate the Thr504 residue although it could phosphorylate a well-characterized Chk1/Chk2 phosphorylation site (Ser287) of Xenopus Cdc25C (Kumagai et al, 1998) (Figure 2B; see also Figure 1B). Thus, phosphorylation of Thr504 was specific to Chk1.
Figure 2.
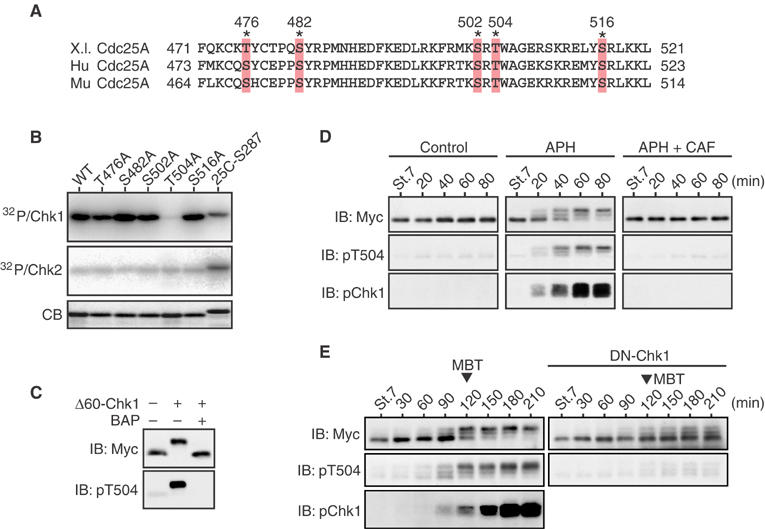
Phosphorylation of Cdc25A Thr504 by Chk1 but not Chk2. (A) Alignment of the C-terminal regions of Xenopus (X.l.), human (Hu), and mouse (Mu) Cdc25A proteins. The asterisks show conserved Ser/Thr residues. (B) GST-Cdc25A C-terminal peptide fusion proteins (each with a Ser/Thr → Ala substitution) were phosphorylated by Δ60-Chk1 or Chk2 in vitro and analyzed as in Figure 1B. (C) Extracts from the eggs expressing Myc-tagged S73A Cdc25A together with or without Δ60-Chk1 were mock-treated or treated with bacterial alkaline phosphatase (BAP) and analyzed by immunoblotting (IB) using anti-Myc or anti-phospho-Thr504 antibodies. (D) One-cell embryos were injected with 1 ng of mRNA encoding Myc-tagged S73A Cdc25A. At the early blastula stage 7, the embryos were mock-treated or treated with aphidicolin (APH) together with or without caffeine (CAF) and, at 20 min intervals, analyzed by immunoblotting using anti-Myc antibody, anti-phospho-Thr504 antibody, or anti-human Chk1 phospho-Ser345 antibody (pChk1, which can recognize well the activating phosphorylation of Xenopus Chk1; Shimuta et al, 2002). (E) One-cell embryos were coinjected with 1 ng of mRNA encoding Myc-tagged S73A Cdc25A and 15 ng of mRNA encoding either control GST (left panel) or a dominant-negative (or kinase-dead) form of Chk1 (DN-Chk1), and then analyzed as in (D).
Chk1 could also phosphorylate full-length Cdc25A on Thr504 not only in vitro (see Figure 4A) but also in vivo. Thus, when coexpressed with Δ60-Chk1 in eggs, a Myc-tagged Ser73 → Ala Cdc25A mutant (S73A, which can avoid Chk1-induced degradation; Shimuta et al, 2002) became strongly recognized by anti-phospho-Thr504 antibody, this recognition being abolished by alkaline phosphatase treatment of the Cdc25A protein (Figure 2C). These analyses identify the C-terminal Thr504 residue as a novel and major Chk1-phosphorylatable site in Xenopus Cdc25A.
Figure 4.
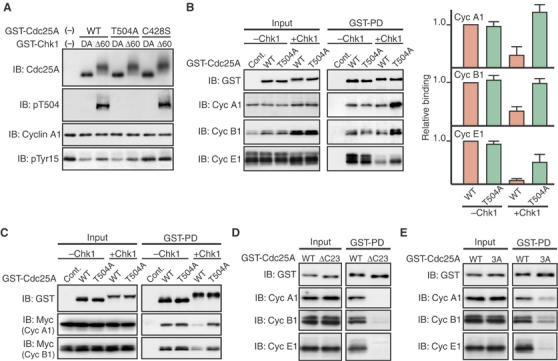
Inhibition of Cdc25A/Cdk–cyclin interactions by Thr504 phosphorylation. (A) GST-Cdc25A fusion proteins (wild type (WT), T504A, or C428S) were first incubated with GST-Δ60-Chk1 (Δ60) or its kinase-dead form (DA) and then with Tyr15-phosphorylated Cdk1–cyclin A1 complexes. The reaction mixture was then analyzed by immunoblotting using anti-Cdc25A, anti-phospho-Thr504, anti-cyclin A1, or anti-Cdk1 phospho-Tyr15 antibodies. (For exact experimental conditions, see Supplementary data.) (B) Activated eggs were injected with 2 ng of mRNA encoding GST (Cont.) or GST-Cdc25A (WT or T504A), incubated for 2.5 h, reinjected or not with 2 ng of Δ60-Chk1 mRNA, and then incubated further for 1.5 h. Egg extracts (Input; equivalent to half an egg) and GST-pulled down proteins (GST-PD; equivalent to 10 eggs) were then immunoblotted for GST-Cdc25A or endogenous cyclins A1, B1, or E1 (left panel). (The blot of GST-Cdc25A in GST-PD is short-exposed.) The levels of cyclins (in both Input and GST-PD) were quantified using the NIH Image program from four independent experiments, and the levels of cyclins bound to GST-Cdc25A proteins were normalized to the input of cyclins; values obtained for WT Cdc25A (−Chk1) were set at 1.0 (right panel). (C) Extracts from the eggs expressing GST-Cdc25A proteins (together with or without Δ60-Chk1) as in (B) were mixed with extracts from the eggs expressing Myc-tagged cyclins A1 or B1 (together with Xe-Wee1B (Okamoto et al, 2002) to induce Cdk1 Tyr15 phosphorylation), incubated for 1 h at 4°C, and analyzed for GST-Cdc25A or ectopic cyclins A1 or B1 as in (B, left). (D, E) Activated eggs were injected with 2 ng of mRNA encoding wild-type GST-Cdc25A, GST-Cdc25A-ΔC23 (D) or GST-Cdc25A-3A (E), incubated for 2.5 h, and analyzed as in (B). In (B–E), all the GST-Cdc25A constructs had both S73A and C428S mutations (see text).
We then determined whether Cdc25A was phosphorylated on Thr504 in an endogenous Chk1-dependent manner at the DNA replication checkpoint induced in early embryos. For this, we ectopically expressed the stable S73A mutant of Cdc25A in just fertilized embryos and then treated the embryos (at the early blastula stage 7) with aphidicolin, a replication checkpoint inducer (Shimuta et al, 2002). This treatment caused rapid mobility shifts and Thr504 phosphorylation of S73A Cdc25A, concurrently with an activating phosphorylation of endogenous Chk1 (Figure 2D, left and middle panels). Cotreatment of the embryos with caffeine, an inhibitor of ATR (Sarkaria et al, 1999), prevented both Cdc25A Thr504 and Chk1 phosphorylations (Figure 2D, right panel), consistent with endogenous Chk1 being involved in the aphidicolin-induced phosphorylation of Cdc25A Thr504. Moreover, even in the absence of aphidicolin treatment, S73A Cdc25A became phosphorylated on Thr504 at the physiological DNA replication checkpoint at the MBT, and this phosphorylation was inhibited by overexpression of a dominant-negative mutant of Chk1 (DN-Chk1) (Figure 2E). Thus, at both the experimental and physiological DNA replication checkpoints, Cdc25A was phosphorylated on Thr504 in an endogenous Chk1-dependent manner. These results, together with the above results, strongly indicate that Chk1, but not Chk2, phosphorylates Cdc25A on a novel C-terminal site, or Thr504.
Requirement of Thr504 phosphorylation for the DNA replication checkpoint
We next examined whether Chk1 phosphorylation of Cdc25A on Thr504 was required for Chk1-induced cell cycle arrest. For this, we ectopically expressed either wild-type Cdc25A or a Thr504 → Ala Cdc25A mutant (each with a Myc tag) in eggs and, after expression of Δ60-Chk1, monitored the inhibitory Tyr15 phosphorylation of endogenous Cdk1 (a hallmark of G2 arrest). Interestingly, the expression of the T504A mutant inhibited Chk1-induced Cdk1 Tyr15 phosphorylation significantly more strongly than the expression of wild-type Cdc25A (Figure 3A, right). In these experiments, however, the T504A mutant was (slightly) more stable than the wild-type Cdc25A after expression of Δ60-Chk1 (Figure 3A, left), raising the possibility that the stronger effect of the T504A mutant might have been due to its stability. However, the T504A mutant could inhibit Cdk1 Tyr15 phosphorylation more strongly even than the much more stable S73A mutant (Figure 3A). Indeed, such an inhibitory effect of Cdc25A T504A mutation was even clearer when the comparison was made between the S73A mutant and an S73A/T504A double mutant, which were comparably stable after Δ60-Chk1 expression (Figure 3B). Thus, these results strongly suggest that Chk1 phosphorylation of Cdc25A on Thr504 is required, and is more important than its stability, for Chk1-induced cell cycle arrest.
Figure 3.
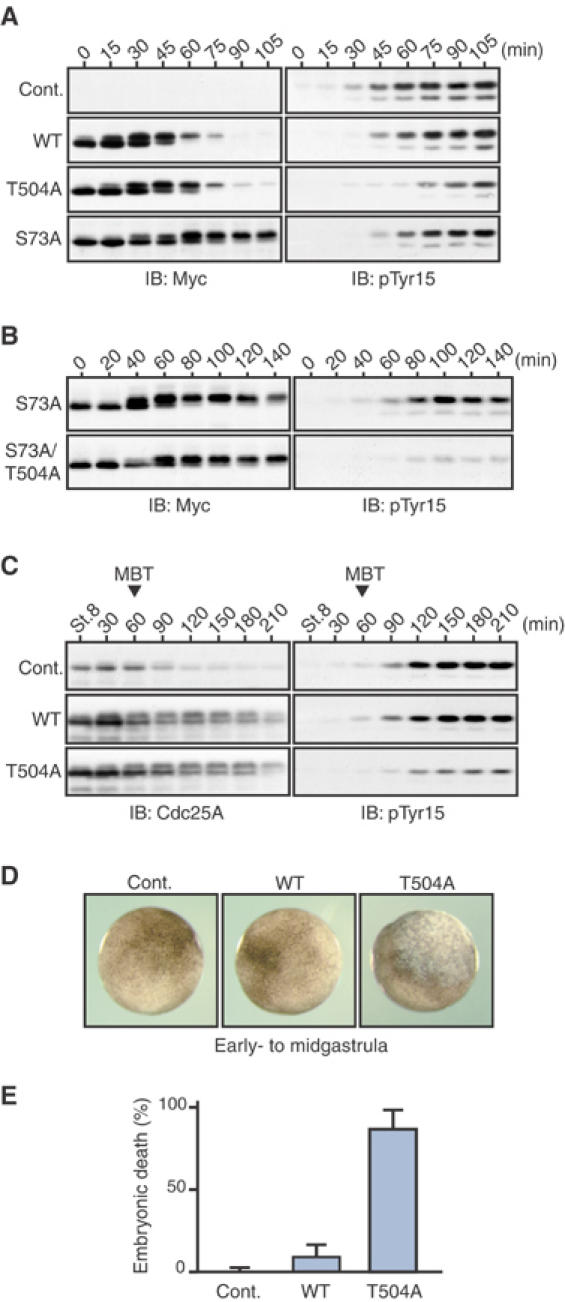
Requirement of Cdc25A phosphorylation on Thr504 for the DNA replication checkpoint. (A, B) Activated eggs were injected with 1 ng of mRNA encoding Myc-tagged wild-type Cdc25A or indicated Myc-tagged Cdc25A mutants, reinjected 2.5 h later with 2 ng of Δ60-Chk1 mRNA, and then analyzed by immunoblotting using anti-Myc or anti-Cdk1 phospho-Tyr15 antibodies. (C–E) One-cell embryos were uninjected (Cont.) or injected with 1 ng of mRNA encoding either wild-type Cdc25A or T504A Cdc25A, cultured, and analyzed for immunoblotting (after stage 8 with anti-Cdc25A or anti-Cdk1 phospho-Tyr15 antibodies; C), for the external morphology (D) and for the percentage embryonic death at stage 11 (E).
We also tested the requirement of Chk1-mediated phosphorylation of Cdc25A Thr504 for the physiological DNA replication at the MBT. When expressed ectopically (four-fold over endogenous levels) in early embryos, wild-type Cdc25A had no appreciable effect on Cdk1 Tyr15 phosphorylation (or cell cycle elongation; Shimuta et al, 2002) at the MBT, but the T504A mutant did have a significant inhibitory effect on it (although this mutant, like wild-type Cdc25A, became degraded at the MBT) (Figure 3C). Consequently, most of the embryos expressing the T504A mutant but not the wild-type Cdc25A died at the early- to midgastrula stages with apoptotic phenotypes (Figures 3D and E). Consistent with our previous results (Shimuta et al, 2002), the expression of the stable S73A mutant had no appreciable effect on Cdk1 Tyr15 phosphorylation at the MBT (although it affected cell viability at late gastrula stages) (not shown). Thus, these results indicate that Chk1-mediated Thr504 phosphorylation, rather than degradation itself, of Cdc25A is essential for the DNA replication checkpoint at the MBT. We obtained essentially similar results with the aphidicolin-induced replication checkpoint in early embryos (data not shown). Taken together, these results strongly suggest that at the DNA replication checkpoint Chk1 inhibits the Cdk Tyr15 phosphatase Cdc25A by phosphorylating Thr504, thereby contributing largely to Cdk1 Tyr15 phosphorylation and cell cycle arrest.
Inhibition of the Cdc25A/Cdk–cyclin interactions by Thr504 phosphorylation
Because Chk1 was suggested to inhibit Cdc25A by phosphorylating Thr504, we first tested whether this phosphorylation could directly inhibit the phosphatase activity of Cdc25A. To do this in vitro, we incubated purified Cdc25A protein first with Δ60-Chk1 (to phosphorylate it on Thr504; see Figure 4A) and then with Tyr15-phospholylated Cdk1–cyclin A1 (which served as substrate for Cdc25A). Interestingly, wild-type Cdc25A preincubated with Δ60-Chk1 dephosphorylated Cdk1 Tyr15 ∼1.5-fold less efficiently than that preincubated with kinase-dead Δ60-Chk1 (DA) (Figure 4A). However, even the T504A mutant showed a very similar decrease in phosphatase activity toward Cdk1 Tyr15 after incubation with Δ60-Chk1. A phosphatase-dead Cys428 → Ser Cdc25A mutant (C428S) could not dephosphorylate Cdk1 Tyr15 at all. Thus, although Chk1 could directly and partially inhibit the phosphatase activity of Cdc25A, this inhibition was not likely to be due to Thr504 phosphorylation.
We then asked whether Chk1 phosphorylation of Thr504 could inhibit Cdc25A from physically interacting with Cdk–cyclin complexes. For this, we expressed in eggs GST-fused Cdc25A (wild type or T504A) together with or without Δ60-Chk1 and then examined its interaction with endogenous cyclins A1, B1, or E1 by GST pulldown assays. (In this experiment, both the wild-type and T504A forms of Cdc25A had, in fact, S73A and C428S mutations to avoid Δ60-Chk1-induced degradation and to form a stable complex with Cdk–cyclin complexes (Xu and Burke, 1996), respectively.) In the absence of Δ60-Chk1 expression, wild-type Cdc25A and the T504A mutant bound to Cdk1–cyclin A1 (in Xenopus eggs, cyclin A1 is bound only to Cdk1), Cdk1–cyclin B1, or Cdk2–cyclin E1 with equal efficiency (Figure 4B, left panel). In the presence of Δ60-Chk1 expression, however, wild-type Cdc25A bound to all the Cdk–cyclin complexes significantly less efficiently than the T504A mutant. In fact, when the binding was normalized to the input of cyclins (the levels of cyclins A1 and B1 considerably increased after Δ60-Chk1 expression, probably due to Chk1-induced G2 arrest; Figure 4B, left panel), only the binding of wild-type Cdc25A showed a significant decrease after expression of Δ60-Chk1 (Figure 4B, right panel). To confirm these results in more refined conditions, we performed GST pulldown assays after mixing (and incubating) two different types of egg extracts, one containing GST-Cdc25A (with or without Δ60-Chk1) and the other containing Myc-tagged cyclins A1, B1, or E1. Even in this case, in the presence of Δ60-Chk1, wild-type Cdc25A bound to (ectopic) Cdk1–cyclin A1 or Cdk1–cyclin B1 significantly less efficiently than the T504A mutant (Figure 4C). (For unknown reasons, ectopic cyclin E1 could not interact with GST-Cdc25A in egg extracts; not shown.) Thus, these results strongly suggest that Chk1 phosphorylation of Thr504 inhibits Cdc25A from interacting with various Cdk–cyclin complexes.
From these results, Cdc25A might interact with the various Cdk–cyclin complexes via the C-terminal tail containing Thr504, with Chk1 phosphorylation of Thr504 inhibiting the interactions. To test this possibility, first we made a C-terminal 23-amino-acid (499–521) truncated Cdc25A mutant and tested for its interactions with Cdk–cyclin complexes in eggs. This mutant (Cdc25A-ΔC23), unlike wild-type Cdc25A, could not appreciably bind to Cdk1–cyclin A1, Cdk1–cyclin B1, or Cdk2–cyclin E1 (Figure 4D), implicating the C-terminal tail (containing Thr504) in interactions with the various Cdk–cyclin complexes. We then introduced Ala mutations into the three C-terminal basic residues (Arg499, Lys501, and Arg503) of full-length Cdc25A (because these C-terminal basic residues were located adjacent to Thr504 and well conserved in other Cdc25 family members; see Figure 5A). This C-terminal mutant, Cdc25A-3A, showed a significantly reduced binding to all the Cdk–cyclin complexes, particularly Cdk2–cyclin E1 (Figure 4E). Thus, the three C-terminal basic residues were important for Cdc25A to interact with the various Cdk–cyclin complexes, supporting the idea that Chk1 phosphorylation of the adjacent Thr504 residue acts to inhibit the C-terminal interactions.
Figure 5.
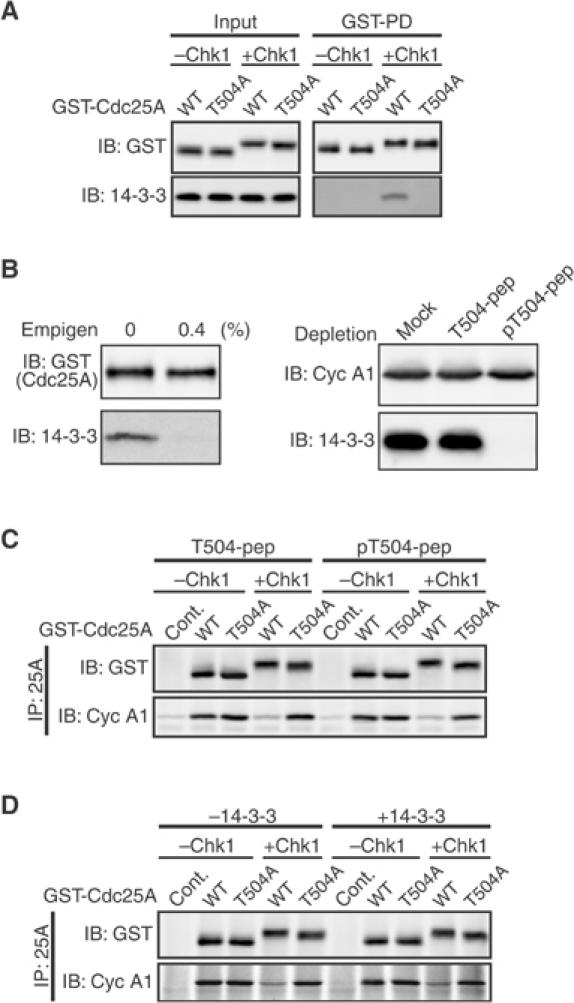
No requirement of 14-3-3 binding for the inhibitory effect of Thr504 phosphorylation. (A) Activated eggs expressing GST-Cdc25A proteins (together with or without Δ60-Chk1) as in Figure 4B were subjected to GST pulldown assays and analyzed by immunoblotting using anti-GST antibody or anti-human 14-3-3β antibody (which can recognize all 14-3-3 isoforms). (B) Chk1-phosphorylated GST-Cdc25A protein (wild type) was GST-pulled down from activated eggs, washed with or without 0.4% Empigen, and analyzed by immunoblotting as in (A) (left). Extracts from the eggs expressing GST-cyclin A1 (together with Xe-Wee1B; see Figure 4C legend) were treated with the indicated peptide-coupled beads and immunoblotted for cyclin A1 or 14-3-3 protein (right). See Materials and methods for details. (C) GST-Cdc25A proteins (phosphorylated or not by Chk1) and egg extracts (equivalent to 10 eggs) were both prepared as in (B) (GST-Cdc25A proteins being eluted from glutathione beads), mixed together, and incubated for 1 h at 4°C. GST-Cdc25A proteins were then immunoprecipitated with anti-Cdc25A antibody (IP) and analyzed by immunoblotting using anti-GST or anti-cyclin A1 antibodies. As control of GST-Cdc25A proteins (wild type or T504A), GST alone was used (Cont.). (D) GST-Cdc25A proteins prepared as in (C) were first incubated or not with 0.1 μg of recombinant Xenopus 14-3-3ɛ protein in 30 μl of an egg extraction buffer for 15 min at 4°C and then with (GST-)Cdk1–cyclin A1 complexes (purified from 10 eggs) for 1 h. (Xenopus 14-3-3ɛ protein can bind to phosphorylated Thr504 of Cdc25A; see Materials and methods.) The mixture was then analyzed as in (C). In (A–D), all the GST-Cdc25A constructs had both S73A and C428S mutations.
No requirement of 14-3-3 binding for the inhibitory effect of Thr504 phosphorylation
A recent study shows that 14-3-3 proteins bind to human Cdc25A at the Chk1-phosphorylated Thr507 residue (equivalent to Thr504 of Xenopus Cdc25A), suggesting a role for this binding in negatively regulating Cdc25A (Chen et al, 2003). We tested whether 14-3-3 binding could indeed mediate the inhibitory effect of Thr504 phosphorylation that we observed here. Initially, we confirmed that endogenous 14-3-3 proteins can bind to Chk1-phosphorylated Thr504 of Xenopus Cdc25A in eggs (Figure 5A). We then attempted to determine whether purified Chk1-phosphorylated Cdc25A (stripped of associated 14-3-3 proteins) could bind to Cdk1–cyclin A1 in egg extracts depleted of endogenous 14-3-3 proteins. For this, Chk1-phosphorylated (GST-)Cdc25A protein was isolated from eggs, and was washed with the zwitterionic detergent Empigen (Lee et al, 2001) to strip more than 96% of the associated 14-3-3 proteins (Figure 5B, left). On the other hand, the egg extracts (containing ectopic cyclin A1) were treated with short phospho-Thr504-containing peptides (coupled to agarose beads) (pT504-pep) to remove centrifugally more than 99% of the endogenous 14-3-3 proteins that would bind to phospho-Thr504; mock treatment with beads alone or control treatment with unphosphorylated Thr504 peptide beads (T504-pep) could not deplete 14-3-3 proteins at all (Figure 5B, right). (Cyclin A1 could not be depleted by any treatment.) When incubated with control egg extracts treated with T504-pep, purified Chk1-phosphorylated wild-type Cdc25A bound to Cdk1–cyclin A1 much less efficiently than Chk1-phosphorylated T504A or unphosphorylated wild-type or T504A Cdc25A proteins (Figure 5C, left), similar to the result with intact eggs (Figure 4B). However, essentially the same result was obtained even with egg extracts treated with pT504-pep (Figure 5C, right), suggesting that 14-3-3 binding was not required for the inhibition of the Cdc25A/Cdk1–cyclin A1 interaction by Chk1-mediated Thr504 phosphorylation. To confirm this unexpected result more directly, we performed the binding assays in vitro in the presence or absence of recombinant 14-3-3 proteins. Interestingly, even in the absence of 14-3-3 proteins, purified Chk1-phosphorylated wild-type Cdc25A (treated with Empigen) bound to purified Cdk1–cyclin A1 (having no associated 14-3-3 proteins) substantially less efficiently than the T504A mutant; addition of 14-3-3 proteins did not appreciably affect the binding (Figure 5D). This in vitro result was essentially the same as that obtained with Empigen-untreated Cdc25A proteins (not shown). Taken together, these results suggest strongly that the inhibitory effect of Thr504 phosphorylation does not require 14-3-3 binding.
Chk1 regulation of other Cdc25 family members by C-terminal phosphorylation
We noticed that all known Cdc25 family members from yeast to humans have, in their C-terminal tail, a serine or threonine residue (in the motif Arg/Lys-X-X-Ser/Thr) that seems to correspond to Thr504 of Xenopus Cdc25A; some examples are shown in Figure 6A. To examine whether Chk1 could also regulate other Cdc25 family members by their C-terminal phosphorylation, we first tested whether Ser549 of human Cdc25B, Thr533 of Xenopus Cdc25C, and Ser457 of Drosophila String (a Cdc25 homolog; Edgar and O'Farrell, 1989), each corresponding to Thr504 of Xenopus Cdc25A, could be phosphorylated by Chk1 or Chk2 in vitro. These sites (in GST fusion peptides) were all phosphorylated, albeit to somewhat different degrees, by Δ60-Chk1, but not by Chk2, which could phosphorylate control Xenopus Cdc25C S287 efficiently (Figure 6B). Thus, as for Cdc25A (Figure 2B), Chk1 but not Chk2 was able to phosphorylate Cdc25B, Cdc25C, and String on their C-terminal residue.
Figure 6.
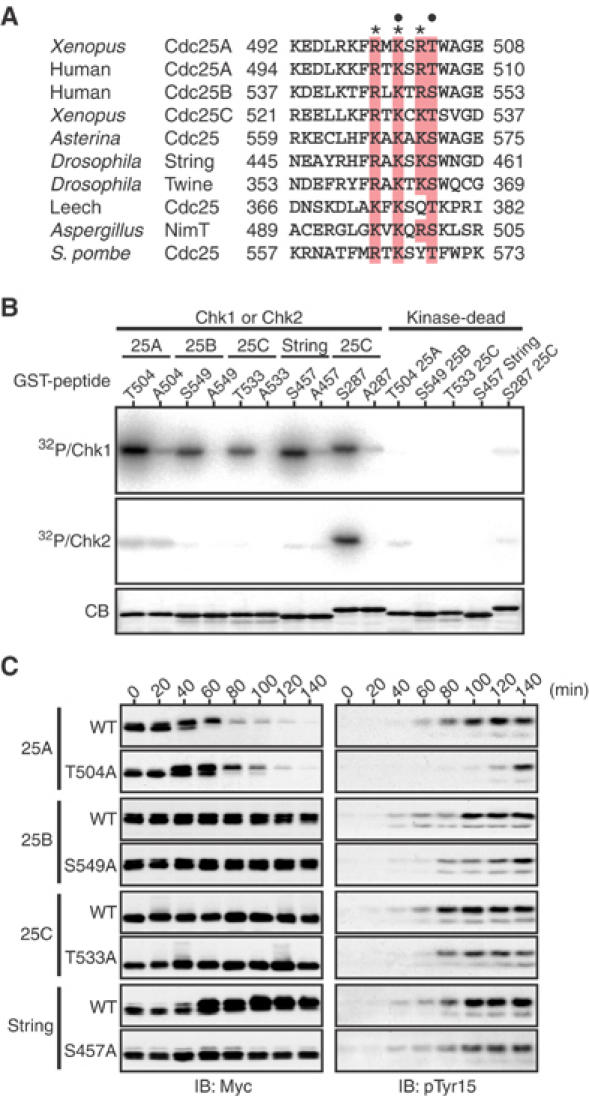
Chk1 regulation of other Cdc25 family members by C-terminal phosphorylation. (A) Alignment of the C-terminal tails of Cdc25 family members. A conserved consensus Chk1/Chk2 phosphorylation motif (R/K-X-X-S/T) and three other conserved basic residues are dotted and asterisked, respectively. (B) GST-Cdc25 C-terminal peptide fusion proteins (wild-type and Ser/Thr → Ala mutant forms of Cdc25A, 25B, 25C, or String) were phosphorylated by Δ60-Chk1 or Chk2 in vitro and analyzed as in Figure 2B. (C) Activated eggs were injected with 1 ng of mRNA encoding indicated forms of (Myc-tagged) Cdc25 phosphatases, reinjected 2.5 h later with 2 ng of Δ60-Chk1 mRNA, and then analyzed as in Figure 3A.
To determine whether these phosphatases could be functionally inhibited by their C-terminal phosphorylation by Chk1, we tested the effect of ectopic expression of S549A Cdc25B, T533A Cdc25C, or S457A String on Chk1-induced cell cycle arrest, as performed earlier for T504A Cdc25A (Figure 3A). When first analyzed for their expression in eggs, neither the wild-type form nor the C-terminal Ala mutant form of Cdc25B or Cdc25C showed an appreciable mobility shift or degradation after Δ60-Chk1 expression (Figure 6C, left). On the other hand, wild-type String, but not its C-terminal Ala mutant, showed a prominent mobility shift and, for unknown reasons, a significant increase in levels after Δ60-Chk1 expression. With all these phosphatases, however, the C-terminal Ala mutant, particularly that of Cdc25B or String, inhibited Chk1-induced Cdk1 Tyr15 phosphorylation more strongly than the wild-type form, if not comparably to T504A Cdc25A (Figure 6C, right). Thus, under these experimental conditions, C-terminal phosphorylation of the respective phosphatases was required, more or less, for Chk1-induced cell cycle arrest. These results suggest that like Xenopus Cdc25A, human Cdc25B, Xenopus Cdc25C, and Drosophila String all can be functionally inhibited, at least in part, by their C-terminal phosphorylation by Chk1.
Regulation of various Cdc25/Cdk–cyclin interactions by C-terminal phosphorylation
Finally, we tested whether Chk1-mediated C-terminal phosphorylation could inhibit interactions of the various Cdc25 phosphatases with their Cdk–cyclin substrates, as performed earlier for Cdc25A (Figure 4B). For this, we initially determined with which Cdk–cyclin complexes Cdc25B, Cdc25C, and String could interact. When expressed in eggs, both Cdc25B and String interacted not only with Cdk1–cyclin A1 and Cdk1–cyclin B1 but also with Cdk2–cyclin E1 (despite their proposed function at the G2/M transition; Edgar and O'Farrell, 1989; Lammer et al, 1998); Cdc25C interacted only with Cdk1–cyclin B1 (consistent with it being an M-phase phosphatase) (Figure 7A). We then compared the interactions of the wild-type and C-terminal Ala mutant forms of the respective phosphatases with their Cdk–cyclin substrates after expression of Δ60-Chk1. Compared to their C-terminal Ala mutants, wild-type Cdc25B and String bound significantly less efficiently to both Cdk1–cyclin A1 and Cdk2–cyclin E1 and, somewhat less efficiently, to Cdk1–cyclin B1, in the presence of Δ60-Chk1; wild-type Cdc25C also bound to Cdk1–cyclin B1 somewhat less efficiently than its C-terminal Ala mutant (Figure 7B). Thus, Chk1-mediated C-terminal phosphorylation apparently could inhibit Cdc25B, Cdc25C, and String from interacting with their Cdk–cyclin substrates, although, in these cases, the inhibitory effect on binding to Cdk1–cyclin B1 was somewhat weaker than that observed with Cdc25A (cf. Figure 4B).
Figure 7.
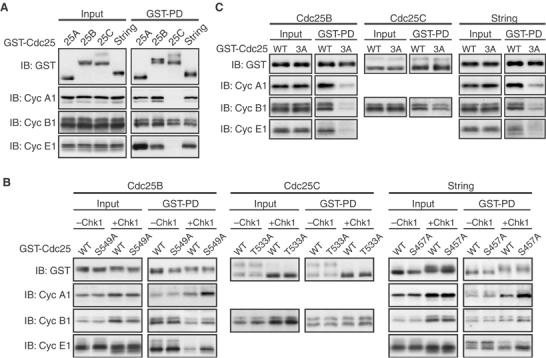
Regulation of various Cdc25/Cdk–cyclin interactions by C-terminal phosphorylation. (A, C) Activated eggs were injected with 2 ng of mRNA encoding indicated forms of GST-Cdc25 phosphatases, cultured for 2.5 h, and then subjected to GST pulldown assays as in Figure 4B. (B) Activated eggs were injected with 2 ng of mRNA encoding indicated forms of GST-Cdc25 phosphatases, incubated for 2.5 h, reinjected or not with 2 ng of Δ60-Chk1 mRNA, and then 1.5 h later subjected to GST pulldown assays. In (A–C), all the GST-Cdc25 constructs were phosphatase-dead forms (like the C428S mutant of Cdc25A).
Like Xenopus Cdc25A, human Cdc25B, Xenopus Cdc25C, and Drosophila String all had three basic residues N-terminally adjacent to their C-terminal Chk1 phosphorylation site (Figure 6A). Therefore, we also tested the requirement of the three basic residues of the respective phosphatases for interactions with their Cdk–cyclin substrates, by mutating the three basic residues to Ala (3A). When expressed in eggs, Cdc25B-3A and String-3A bound to Cdk1–cyclin A1, Cdk1–cyclin B1, and Cdk2–cyclin E1 significantly less efficiently than their wild-type forms; Cdc25C-3A bound to Cdk1–cyclin B1 slightly less efficiently than the wild-type form (Figure 7C). Thus, as in Xenopus Cdc25A (Figure 4E), the three basic residues were required, more or less, for the respective phosphatases to bind efficiently to their Cdk–cyclin substrates. These results, together with the conservation of the relevant C-terminal residues in virtually all Cdc25 family members (Figure 6C), may suggest that Chk1 phosphorylation of the C-terminal site inhibits all Cdc25 phosphatases from C-terminally interacting with their Cdk–cyclin substrates.
Discussion
A very recent paper reports that, during an unperturbed cell cycle, Chk1 phosphorylates human Cdc25A on a C-terminal 14-3-3 binding site (Thr507) and, thereby, may inhibit its interaction with Cdk1–cyclin B1 but not with Cdk1/2–cyclin A or Cdk2–cyclin E (Chen et al, 2003). With a quite different approach, here we independently show that Chk1 phosphorylates Xenopus Cdc25A on Thr504 (equivalent to Thr507 in human Cdc25A) and inhibits it from interacting not only with Cdk1–cyclin B1 but also with Cdk1–cyclin A1 and Cdk2–cyclin E1. Unexpectedly, however, this inhibition does not require 14-3-3 binding to Chk1-phosphorylated Thr504. Moreover, and importantly, Chk1 can phosphorylate all other examined Cdc25 family members on their C-terminal site and inhibit them from interacting with their Cdk–cyclin substrates. Thus, Chk1 seems to affect cell cycle progression through all stages by inhibiting interactions between various Cdc25 phosphatases and their Cdk–cyclin substrates.
Cdc25A is unstable during interphase of the normal cell cycle due to the basal activity of Chk1 (Mailand et al, 2000; Molinari et al, 2000; Zhao et al, 2002; Sørensen et al, 2003), which is probably important to maintain genomic integrity (Bartek and Lukas, 2003; Donzelli and Draetta, 2003). Similarly, accelerated degradation of Cdc25A by activated Chk1 or Chk2 has been thought to be central to the S and G2 cell cycle checkpoints induced by various genotoxic stresses (see Bartek and Lukas, 2003; Donzelli and Draetta, 2003). However, a recent study shows that a stable mutant of human Cdc25A cannot overcome the S-phase checkpoint induced by various types of stress (Goloudina et al, 2003). Strikingly, in our study, Chk1-induced Cdc25A degradation per se is not essential for the Chk1-induced cell cycle arrest or the DNA replication checkpoint in Xenopus embryos; rather, Chk1 inhibition of the Cdc25A's interactions with various Cdk–cyclin complexes is essential for those events (Figures 3 and 4). It seems, therefore, that inhibition of interactions with Cdk–cyclin substrates, rather than destabilization, is a primary mechanism by which Chk1 inhibits Cdc25A. If so, the previously observed checkpoint bypass by overexpressed human Cdc25A (Mailand et al, 2000, 2002; Molinari et al, 2000; Falck et al, 2001) might be due to an escape of the overexpressed Cdc25A from the presently uncovered inhibition mechanism rather than simply from degradation. Consistent with this possibility, very strong expression of wild-type Cdc25A, like moderate expression of the T504A mutant, can overcome the DNA replication checkpoint and induce apoptosis in early embryos (data not shown, but see Figure 3E).
Previous studies raised the notion that different Cdc25 family members may be differently inhibited by Chk1 or Chk2. Thus, while vertebrate Cdc25A becomes targeted for degradation by Chk1 or Chk2 as already mentioned (see also Figure 1), vertebrate Cdc25C and the fission yeast Cdc25 are either excluded from the nucleus (Peng et al, 1997; Sanchez et al, 1997; Kumagai and Dunphy, 1999; Lopez-Girona et al, 1999) or are catalytically inactivated after phosphorylation by Chk1 or Chk2 (Blasina et al, 1999; Furnari et al, 1999). Human Cdc25B seems to be sequestrated in response to UV irradiation (Bulavin et al, 2001), but the role of Chk1 or Chk2 in this regulation is not known. In addition, Drosophila String (Cdc25) undergoes a Grapes (Chk1)-dependent degradation at the MBT (Edgar and O'Farrell, 1989; Sibon et al, 1997), but this degradation is not likely to be a direct effect of Chk1-mediated phosphorylation (see Figure 6C), unlike the case with Xenopus Cdc25A (Shimuta et al, 2002). However, we show here that all known Cdc25 family members from yeast to humans have a C-terminally located serine or threonine residue that seemingly corresponds to Thr504 of Xenopus Cdc25A, and that phosphorylation of the C-terminal residue by Chk1 can more or less inhibit human Cdc25B, Xenopus Cdc25C, and Drosophila String from functionally interacting with their Cdk–cyclin substrates (Figures 6 and 7). Thus, it seems very likely that Chk1 inhibits virtually all Cdc25 family members by a common mechanism, or by interfering with their interactions with their Cdk–cyclin substrates (Figure 8). Clearly, this type of inhibition is a major mechanism by which Chk1 inhibits Cdc25A, as discussed above. It then remains to be determined how largely this type of inhibition, as compared with other types of inhibition, contributes to Chk1 inhibition of other Cdc25 phosphatases.
Figure 8.
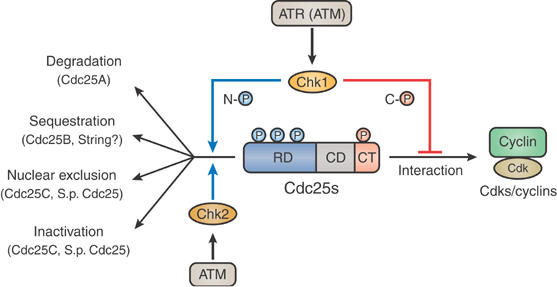
Model for the regulation of Cdc25 phosphatases by Chk1 and Chk2. In response to genotoxic stress, both Chk1 and Chk2 phosphorylate various Cdc25 phosphatases on multiple N-terminal sites (N-℗) and inhibit them by various mechanisms; however, only Chk1 phosphorylates the various phosphatases on a C-terminal site (C-℗) and inhibits them from C-terminally interacting with their Cdk–cyclin substrates. Chk1 also functions during an unperturbed cell cycle. See text for details.
Reportedly, the N-terminus of human Cdc25A binds to Cdk complexes containing cyclins A and E but not cyclin B (Saha et al, 1997), allowing the recent proposal that the Chk1-inhibited C-terminal interaction observed with human Cdc25A may be specific to Cdk1–cyclin B1 (Chen et al, 2003). Obviously, however, the C-terminal tail of Xenopus Cdc25A (containing Thr504) is absolutely required for this phosphatase to interact not only with Cdk1–cyclin B1 but also with Cdk1–cyclin A1 and Cdk2–cyclin E1 (Figure 4D); other Cdc25 phosphatases also require their C-terminal tail to interact with their Cdk–cyclin substrates (see Figure 7C). Specifically, three conserved basic residues adjacent to the C-terminal Chk1 phosphorylation site, as well as two other conserved C-terminal basic residues (in Cdc25A (Chen et al, 2003) and Cdc25B (Wilborn et al, 2001)), are required, more or less, for the various phosphatases to interact with their Cdk–cyclin substrates (Figures 4E and 7C). Therefore, it seems that Chk1 phosphorylation of the adjacent C-terminal site acts to inhibit the C-terminal interactions of the various phosphatases. In the case of (human) Cdc25A, this inhibition has been proposed to involve the binding of 14-3-3 proteins to the C-terminal Chk1-phosphorylated site (Chen et al, 2003). In our study, however, this inhibition does not require 14-3-3 binding, as evidenced by experiments involving extensive depletion of 14-3-3 proteins from both Chk1-phosphorylated Cdc25A and egg extracts (Figure 5). Upon binding to Cdc25A, therefore, 14-3-3 proteins might have some other role(s) such as prevention of the Chk1-phosphorylated site from dephosphorylation.
Another important point that emerged from the present study is that Chk2 seems to be strikingly different from Chk1 in regulating Cdc25 phosphatases. Chk1 and Chk2 share many common substrates and have partly overlapping roles (Zhou and Elledge, 2000; Bartek and Lukas, 2003). Specifically, these two kinases, although structurally very different (Xu et al, 2002; Katsuragi and Sagata, 2004), phosphorylate the common substrates on largely overlapping sites (Rhind and Russell, 2000; Bartek and Lukas, 2003; see also Figure 1B). Strikingly, however, Chk2 cannot phosphorylate Xenopus Cdc25A (as well as human Cdc25A; Chen et al, 2003) or any other examined Cdc25 family members on their C-terminal Chk1 phosphorylation site (Figures 2B and 6B). Thus, Chk2 seems to have a more limited role in regulating Cdc25 phosphatases than Chk1. This may explain, at least in part, why the IR-induced DNA damage checkpoint absolutely requires Chk1, in addition to Chk2, and why only Chk1 is required during the normal cell cycle (Liu et al, 2000; Zhao et al, 2002; Sørensen et al, 2003). Altogether, it seems that only Chk1, but not Chk2, inhibits virtually all Cdc25 phosphatases by a novel common mechanism during both perturbed and unperturbed cell cycles (Figure 8).
Materials and methods
cDNAs and in vitro transcription
cDNAs encoding Xenopus Chk1, Chk2, Δ60-Chk1, their kinase-dead (or dominant-negative) mutants, Cdc25A, or Cdc25C were described (Oe et al, 2001; Shimuta et al, 2002). A cDNA encoding human Cdc25B was a gift from H Okayama. A cDNA encoding Drosophila String was isolated from a cDNA library of Drosophila embryos (a gift from E Nitasaka). cDNAs encoding Xenopus cyclins A1 or B1 were donated by T Hunt. A cDNA encoding Xenopus 14-3-3ɛ was isolated by PCR of the oocyte cDNA library. All the cDNA constructs were subcloned into either the N-terminally Myc-tagged or GST-tagged pT7-G(UKII+) transcription vectors (Okamoto et al, 2002). In vitro mutagenesis and transcription of the cDNAs were performed as described (Shimuta et al, 2002). The cDNA encoding Xenopus 14-3-3ɛ was also subcloned into the pGEX-3X vector to bacterially produce GST-14-3-3ɛ proteins.
GST fusion peptides
See Supplementary data for GST fusion peptides used in Figures 1B, 2B and 6B.
Antibodies and immunoblotting
Anti-Xenopus phospho-Thr504 peptide antibody was raised in rabbits against the phosphorylated peptides (KFRMKSRpTWAGERSK) and affinity-purified by standard methods. Routinely, proteins equivalent to one egg or embryo were analyzed by immunoblotting (Shimuta et al, 2002), using the above-described anti-phospho-Thr504 antibody, anti-Xenopus Cdc25A antibody (Shimuta et al, 2002), anti-Myc antibody (A-14, Santa Cruz), anti-GST antibody (Z-5 or B-14, Santa Cruz), anti-Xenopus cyclin A1 or B1 antibodies (a gift from J Maller), anti-Xenopus cyclin E1 antibody (a gift from T Kishimoto), anti-Cdk1 phospho-Tyr15 antibody (♯911, Cell Signaling), anti-human Chk1 phospho-Ser345 antibody (♯2341, Cell Signaling), or anti-human 14-3-3β antibody (H8, Santa Cruz).
In vitro kinase and phosphatase assays
For in vitro Chk1/Chk2 kinase assays, GST-fused Cdc25 peptides were incubated with [γ-32P]ATP and either Δ60-Chk1 protein or Chk2 protein (immunoprecipitated with anti-Myc antibody from eggs overexpressing them) and analyzed essentially as described (Shimuta et al, 2002). In vitro Cdc25A phosphatase assays (in Figure 4A) were performed under strict conditions, involving sequential kinase–phosphatase reactions (see Supplementary data for details).
Eggs and embryos
Unfertilized eggs and embryos were prepared, cultured, staged, and microinjected as described (Shimuta et al, 2002). Routinely, the unfertilized eggs were used after artificial activation with the calcium ionophore A23187 (Shimuta et al, 2002). In some experiments, early blastula embryos at stage 7 were treated with aphidicolin (100 μg/ml) or caffeine (10 mM).
Egg extracts
Activated eggs expressing ectopic proteins were homogenized in 5–10 μl/egg of an extraction buffer (20 mM sodium phosphate (pH 8.0), 80 mM β-glycerophosphate, 0.2% Tween 20, 1 mM EDTA, 1 mM dithiothreitol (DTT), 2 μM pepstatin A, 10 μg/ml aprotinin, and 0.2 mM PMSF) in the presence of phosphatase inhibitors (10 μM okadaic acid, 1 μM microcystin LR, and 3 μM tautomycin) at 4°C. The homogenates were centrifuged briefly, and the resulting supernatants served as egg extracts.
GST pulldown assays
GST fusion proteins were pulled down from egg extracts using glutathione-Sepharose beads (Amersham). Co-pulled-down proteins (routinely equivalent to 10 eggs) were then analyzed by immunoblotting using appropriate antibodies. When necessary for further use, GST fusion proteins were eluted from the beads with 10 mM glutathione in the egg extraction buffer.
Depletion of 14-3-3 proteins
Chk1-phosphorylated (GST-)Cdc25A protein was GST-pulled down from egg extracts, washed with 0.4% Empigen BB to remove associated 14-3-3 proteins (Lee et al, 2001), and eluted for further use. To deplete endogenous 14-3-3 proteins (that would bind to Chk1-phosphorylated Thr504 of Cdc25A) from egg extracts, 10 μg of phospho-Thr504-containing peptides (residues 497–511), covalently linked to SulfoLink coupling gel (Pierce), was incubated with 300 μl of egg extracts (equivalent to 60 eggs) for 30 min at 4°C, and centrifuged briefly; the supernatant was treated again with fresh peptide beads. By this treatment, more than 99% of both endogenous 14-3-3 proteins (that could react with anti-human 14-3-3β antibody; see Results) and ectopically expressed Xenopus 14-3-3ɛ (not shown) could be depleted from the extracts.
Supplementary Material
Supplementary data
Acknowledgments
We thank Drs J Maller, T Kishimoto, T Hunt, H Okayama, and E Nitasaka for reagents, and K Gotoh for typing the manuscript. This work was supported by the scientific grants from the Ministry of Education, Science and Culture of Japan and the CREST Research Project of Japan Science and Technology Agency to NS.
References
- Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15: 2177–2196 [DOI] [PubMed] [Google Scholar]
- Barlow C, Hirotsune S, Paylor R, Liyanage M, Eckhaus M, Collins F, Shiloh Y, Crawley JN, Ried T, Tagle D, Wynshaw-Boris A (1996) ATM-deficient mice: a paradigm of ataxia telangiectasia. Cell 86: 159–171 [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas J (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3: 421–429 [DOI] [PubMed] [Google Scholar]
- Blasina A, Weyer IV, Laus MC, Luyten WHML, Parker AE, McGowan CH (1999) A human homologue of the checkpoint kinase Cds1 directly inhibits Cdc25 phosphatase. Curr Biol 9: 1–10 [DOI] [PubMed] [Google Scholar]
- Bulavin DV, Higashimoto Y, Popoff IJ, Gaarde A, Basrur V, Potapova O, Appella E, Fornace AJ (2001) Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 411: 102–107 [DOI] [PubMed] [Google Scholar]
- Busino L, Donzelli M, Chiesa M, Guardavaccaro D, Ganoth D, Dorrello NV, Hershko A, Pagano M, Draetta GF (2003) Degradation of Cdc25A by β-TrCP during S phase and in response to DNA damage. Nature 426: 87–91 [DOI] [PubMed] [Google Scholar]
- Chen MS, Ryan CE, Piwnica-Worms H (2003) Chk1 kinase negatively regulates mitotic function of Cdc25A phosphatase through 14-3-3 binding. Mol Cell Biol 23: 7488–7497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzelli M, Draetta GF (2003) Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep 4: 671–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar BA, O'Farrell PH (1989) Genetic control of cell division patterns in the Drosophila embryo. Cell 57: 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falck J, Mailand N, Syljuåsen RG, Bartek J, Lukas J (2001) The ATM–Chk2–Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 410: 842–847 [DOI] [PubMed] [Google Scholar]
- Furnari B, Blasina A, Boddy MN, McGowan CH, Russell P (1999) Cdc25 inhibited in vivo and in vitro by checkpoint kinases Cds1 and Chk1. Mol Biol Cell 10: 833–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goloudina A, Yamaguchi H, Chervyakova DB, Appella E, Fornace AJ, Bulavin DV (2003) Regulation of human Cdc25A stability by serine 75 phosphorylation is not sufficient to activate a S-phase checkpoint. Cell Cycle 2: 473–478 [PubMed] [Google Scholar]
- Guo Z, Kumagai A, Wang SX, Dunphy WG (2000) Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev 14: 2745–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Weinert TA (1989) Checkpoints: controls that ensure the order of cell cycle events. Science 246: 629–634 [DOI] [PubMed] [Google Scholar]
- Hoffmann I, Draetta G, Karsenti E (1994) Activation of the phosphatase activity of human cdc25A by a cdk2–cyclin E dependent phosphorylation at the G1/S transition. EMBO J 13: 4302–4310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinno S, Suto K, Nagata A, Igarashi M, Kanaoka Y, Nojima H, Okayama H (1994) Cdc25A is a novel phosphatase functioning early in the cell cycle. EMBO J 13: 1549–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuragi Y, Sagata N (2004) Regulation of Chk1 kinase by autoinhitition and ATR-mediated phosphorylation. Mol Biol Cell 15: 1680–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Dunphy WG (1999) Binding of 14-3-3 proteins and nuclear export control the intracellular localization of the mitotic inducer Cdc25. Genes Dev 13: 1067–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai A, Guo Z, Emami KH, Wang SX, Dunphy WG (1998) The Xenopus Chk1 protein kinase mediates a caffeine-sensitive pathway of checkpoint control in cell-free extracts. J Cell Biol 142: 1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammer C, Wagerer S, Saffrich R, Mertens D, Ansorge W, Hoffmann I (1998) The cdc25B phosphatase is essential for the G2/M phase transition in human cells. J Cell Sci 111: 2445–2453 [DOI] [PubMed] [Google Scholar]
- Lee J, Kumagai A, Dunphy WG (2001) Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol Biol Cell 12: 551–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, Donehower LA, Elledge SJ (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev 14: 1448–1459 [PMC free article] [PubMed] [Google Scholar]
- Lopez-Girona A, Furnari B, Mondesert O, Russell P (1999) Nuclear localization of Cdc25 is regulated by DNA damage and a 14-3-3 protein. Nature 397: 172–175 [DOI] [PubMed] [Google Scholar]
- Mailand N, Falck J, Lukas C, Syljuåsen RG, Welcker M, Bartek J, Lukas J (2000) Rapid destruction of human Cdc25A in response to DNA damage. Science 288: 1425–1429 [DOI] [PubMed] [Google Scholar]
- Mailand N, Podtelejnikov AV, Groth A, Mann M, Bartek J, Lukas J (2002) Regulation of G2/M events by Cdc25A through phosphorylation-dependent modulation of its stability. EMBO J 21: 5911–5920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Huang M, Elledge SJ (1998) Linkage of ATM to cell cycle regulation by the Chk2 protein kinase. Science 282: 1893–1897 [DOI] [PubMed] [Google Scholar]
- Molinari M, Mercurio C, Dominguez J, Goubin F, Draetta GF (2000) Human Cdc25A inactivation in response to S phase inhibition and its role in preventing premature mitosis. EMBO Rep 1: 71–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurse P (1997) Checkpoint pathways come of age. Cell 91: 865–867 [DOI] [PubMed] [Google Scholar]
- Oe T, Nakajo N, Katsuragi Y, Okazaki K, Sagata N (2001) Cytoplasmic occurrence of the Chk1/Cdc25 pathway and regulation of Chk1 in Xenopus oocytes. Dev Biol 229: 250–261 [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nakajo N, Sagata N (2002) The existence of two distinct Wee1 isoforms in Xenopus: implications for the developmental regulation of the cell cycle. EMBO J 21: 2472–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica-Worms H (1997) Mitotic and G2 checkpoint control: regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 277: 1501–1505 [DOI] [PubMed] [Google Scholar]
- Rhind N, Russell P (2000) Chk1 and Cds1: linchpins of the DNA damage and replication checkpoint pathways. J Cell Sci 113: 3889–3896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha P, Eichbaum Q, Silberman ED, Mayer BJ, Dutta A (1997) p21CIP1 and Cdc25A: competition between an inhibitor and an activator of cyclin-dependent kinases. Mol Cell Biol 17: 4338–4345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ (1997) Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 277: 1497–1501 [DOI] [PubMed] [Google Scholar]
- Sarkaria JN, Busby EC, Tibbetts RS, Roos P, Taya Y, Karnitz LM, Abraham RT (1999) Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res 59: 4375–4382 [PubMed] [Google Scholar]
- Shiloh Y (2001) ATM and ATR: networking cellular responses to DNA damage. Curr Opin Genet Dev 11: 71–77 [DOI] [PubMed] [Google Scholar]
- Shimuta K, Nakajo N, Uto K, Hayano Y, Okazaki K, Sagata N (2002) Chk1 is activated transiently and targets Cdc25A for degradation at the Xenopus midblastula transition. EMBO J 21: 3694–3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sibon OCM, Stevenson VA, Theurkauf WE (1997) DNA-replication checkpoint control at the Drosophila midblastula transition. Nature 388: 93–97 [DOI] [PubMed] [Google Scholar]
- Sørensen CS, Syljuåsen RG, Falck J, Schroeder T, Rönnstrand L, Khanna KK, Zhou BB, Bartek J, Lukas J (2003) Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 3: 247–258 [DOI] [PubMed] [Google Scholar]
- Takizawa CG, Morgan DO (2000) Control of mitosis by changes in the subcellular location of cyclin-B1–Cdk1 and Cdc25C. Curr Opin Cell Biol 12: 658–665 [DOI] [PubMed] [Google Scholar]
- Walworth NC (2000) Cell-cycle checkpoint kinases: checking in on the cell cycle. Curr Opin Cell Biol 12: 697–704 [DOI] [PubMed] [Google Scholar]
- Wilborn M, Free S, Ban A, Rudolph J (2001) The C-terminal tail of the dual-specificity Cdc25B phosphatase mediates modular substrate recognition. Biochemistry 40: 14200–14206 [DOI] [PubMed] [Google Scholar]
- Xu X, Burke SP (1996) Roles of active site residues and the NH2-terminal domain in the catalysis and substrate binding of human Cdc25. J Biol Chem 271: 5118–5124 [DOI] [PubMed] [Google Scholar]
- Xu X, Tsvetkov LM, Stern DF (2002) Chk2 activation and phosphorylation-dependent oligomerization. Mol Cell Biol 22: 4419–4432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Watkins JL, Piwnica-Worms H (2002) Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc Natl Acad Sci USA 99: 14795–14800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BB, Elledge SJ (2000) The DNA damage response: putting checkpoints in perspective. Nature 408: 433–439 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data