

Abstract
The regulation of cell mass (cell growth) is often tightly coupled to the cell division cycle (cell proliferation). Ribosome biogenesis and the control of rDNA transcription through RNA polymerase I are known to be critical determinants of cell growth. Here we show that granulocytic cells deficient in the c-MYC antagonist MAD1 display increased cell volume, rDNA transcription and protein synthesis. MAD1 repressed and c-MYC activated rDNA transcription in nuclear run-on assays. Repression of rDNA transcription by MAD1 was associated with its ability to interact directly with the promoter of upstream binding factor (UBF), an rDNA regulatory factor. Conversely, c-MYC activated transcription from the UBF promoter. Using siRNA, UBF was shown to be required for c-MYC-induced rDNA transcription. These data demonstrate that MAD1 and c-MYC reciprocally regulate rDNA transcription, providing a mechanism for coordination of ribosome biogenesis and cell growth under conditions of sustained growth inhibition such as granulocyte differentiation.
Keywords: growth, MYC, MAD, RNA polymerase I, UBF
Introduction
In mammals, cell growth is tightly coupled to the cell division cycle of rapidly proliferating cells (Pardee, 1989). However, in other contexts, such as cell hypertrophy or during the reduction in cell mass associated with differentiation, these processes become uncoupled. Cell growth is a complex process involving the synthesis of macromolecules such as rRNA and protein as well as the generation of energy through anabolic pathways. A central component of cell growth is protein synthesis, necessitating the generation of functional ribosomes (Peculis, 2002). The requirement for regulation of ribosome biogenesis is evident in studies showing that mutations in genes encoding key components of the ribosome-biosynthetic pathway result in both reduced cell growth and size (Jorgensen et al, 2002). Despite the biological importance of cell growth in normal development and in diseases such as cancer, the molecules that coordinate these processes in mammalian systems remain poorly defined.
Ribosome biogenesis and subsequent increased protein synthesis in response to growth stimuli require the coordinated synthesis of numerous molecular components of the functional ribosome by all three RNA polymerases (reviewed in Larminie et al, 1998). RNA polymerase I transcribes multiple copies of the gene that encodes the 45S ribosomal RNA precursor of the 18S, 5.8S and 28S rRNA within the nucleoli (rDNA transcription). RNA polymerase III transcribes the 5S component of rRNA and various tRNA molecules that are required for translation. RNA polymerase II transcribes mRNAs that encode for a large number of ribosomal subunit proteins. Finally, post-translational mechanisms, such as phosphorylation of the ribosomal subunit protein S6 by S6 kinase, also regulate the function of the ribosome (Dufner and Thomas, 1999). Normal development requires mechanisms that acutely respond to the demand for growth as well as mechanisms for the sustained regulation of growth in processes such as differentiation. During the process of differentiation, many cell types undergo an arrest in the G1 phase of the cell cycle. Arrest of the cell division cycle is often accompanied by a reduction in cell growth and ribosome biogenesis. Indeed, some cell types such as granulocytes reduce their size during differentiation, suggesting that growth is being inhibited disproportionately to division during the final cell cycles prior to terminal differentiation. Elucidating the molecular mechanisms that control this tightly orchestrated process and how it is linked to ribosome biogenesis is an important question to be addressed.
The MAX network of transcription factors is comprised of a group of bHLH-Zip proteins that form heterodimers with the bHLH-Zip protein MAX. These proteins include the MYC family of transcriptional activators (c, N and L-MYC) and the MAD family of transcriptional repressors. Both the MYC and MAD families of proteins have been implicated in the regulation of cell growth in addition to their established role in cell division. MYC in both Drosophila and mammalian cells promotes cell growth and protein synthesis (Iritani and Eisenman, 1999; Johnston et al, 1999; Schuhmacher et al, 1999). Consistent with this, characterization of transcriptional targets of c-MYC has revealed a number of genes involved in the promotion of various aspects of cell growth (Coller et al, 2000; Guo et al, 2000; Boon et al, 2001). In contrast, the MYC antagonist MAD1 reduces cell size when overexpressed and reduces expression of a number of molecules implicated in certain aspects of ribosome biogenesis and protein translation (Iritani et al, 2002).
Recent studies have specifically implicated MYC in the regulation of ribosome biogenesis. First, MYC can regulate expression of ribosomal subunit proteins that are transcribed by RNA polymerase II (Guo et al, 2000; Kim et al, 2000; Shiio et al, 2002). Second, MYC can directly regulate gene transcription mediated by RNA polymerase III via interaction with TFIIIB (Gomez-Roman et al, 2003). Third, MYC regulates a series of proteins important in nucleolar function and rRNA processing that are transcribed by RNA polymerase II (Greasley et al, 2000; Zeller et al, 2001; Schlosser et al, 2003). While these mechanisms link proteins of the MAX network to ribosome biogenesis, we demonstrate here that MAD1 and c-MYC can regulate the final key component and major rate-limiting step of ribosome biogenesis: transcription of the rDNA genes. MAD1 and c-MYC were able to directly regulate the expression of upstream binding factor (UBF), an HMG-box protein whose expression facilitates rDNA transcription in vivo. siRNA was used to block UBF accumulation and this prevented c-MYC from fully activating rDNA transcription. These data indicate that members of the MAX network of transcription factors regulate rDNA transcription, providing a mechanism for the coordination of ribosome biogenesis and cell growth during granulocyte differentiation.
Results
MAD1 negatively regulates cell growth and rDNA transcription during granulocyte differentiation
Granulocytic cells deficient in MAD1 undergo extra cell divisions before terminally differentiating (Foley et al, 1998); thus, we sought to determine if MAD1 also regulates cell growth during granulocyte differentiation. We examined whole bone marrow from Mad1-null and wild-type mice for differences in cell volume and observed an increased volume only in cells of the myeloid fraction from Mad1-null mice (Figure 1A). To determine the subpopulation of granulocytic cells contributing to this increase in cell volume, we isolated both the immature and mature granulocytic cell fractions and found that the immature granulocytes from Mad1-null animals displayed a statistically significant greater volume than wild-type cells (Figure 1B). We also looked at the total RNA content of the immature fraction, gating on G0/G1 cells to exclude any cell cycle differences, and found an increase in total RNA in cells from Mad1-null mice (Figure 1C). Over 80% of total cellular RNA is rRNA (Paule, 1998), suggesting that Mad1-null cells may have elevated levels of rRNA. As rRNA is transcribed within the nucleolus and the rate of rRNA synthesis is related to nucleolar size (Derenzini et al, 2000), we determined the size of nucleoli in granulocytic cells from Mad1-null and wild-type mice. Indeed, cells from Mad1-null mice displayed larger nucleoli (2.3-fold) compared to wild-type cells (Figure 1D).
Figure 1.
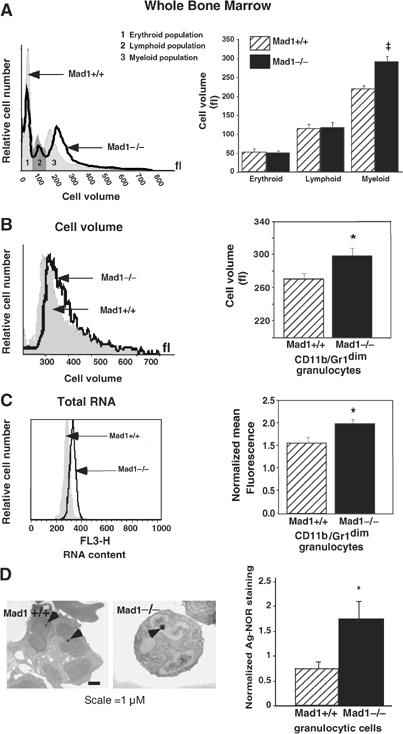
MAD1 regulates cell growth during granulocyte differentiation. (A) Representative cell volume profiles (left) and pooled data (right) of whole bone marrow cells from wild-type (WT; Mad1+/+) and Mad1-null (Mad1−/−) mice were determined using a Sysmex CDA500 system. Results are the mean±s.e.m. from four WT and seven Mad1−/− mice. ‡P=0.002 compared to cells from WT mice. (B) Immature granulocytes (CD11b/Gr-1dim staining) were purified from WT and Mad1−/− mouse bone marrow using FACS as described (Walkley et al, 2002). Representative cell volume profiles (left) and pooled data (right) of granulocytes (n=4 for each genotype) are shown. *P<0.05 compared to cells from WT mice. (C) Total RNA content of immature granulocytes in the G0/G1 phase of the cell cycle. Cell populations were purified as in (B), stained for RNA and DNA content using acridine orange and gated on G0/G1. Representative RNA staining profiles (left) and pooled data (right) of granulocytes (n=4 WT, 6 Mad1−/−) are shown. *P<0.05 compared to WT mice. (D) Size of granulocytic nucleoli from WT and Mad1−/− mice as demonstrated by Ag-NOR staining. Arrowheads point to nucleolar regions of granulocytes (left). Total area of Ag-NOR-stained nucleoli was calculated (right) (n=10 WT, 20 Mad1−/−). *P<0.05 compared to WT mice.
To further investigate the possibility that loss of MAD1 leads to a higher rate of rRNA synthesis, we looked at a major rate-limiting step in rRNA accumulation: transcription of the 45S rRNA precursor by RNA polymerase I (Paule, 1998). To determine if rates of rDNA transcription were altered in granulocytes from Mad1-null mice, we used an ex vivo culture system where primary granulocytes in whole bone marrow were stimulated with stem cell factor (SCF) and G-CSF. Cells from Mad1-null mice had higher rates of synthesis of total RNA (34% increase) and rDNA transcription (2.5-fold) as measured by incorporation of [3H]uridine into RNA and nuclear run-on analyses of the 45S gene (loading of RNA polymerase I onto the 45S rDNA gene), respectively (Figure 2A and B). This was associated with an increase in cell volume (10% increase) and rate of total protein synthesis (2.4-fold) as determined by biosynthetic labeling with [35S]methionine (Figure 2C and D). Significantly, cell proliferation rates remained the same for both wild-type and Mad1-null cells (Figure 2E). To rule out differences in ploidy, DNA content was measured and found to be equivalent for wild-type and Mad1-null cells (data not shown). These data demonstrate that, under these culture conditions, the MAX network contributes to the regulation of growth and rDNA transcription in granulocytes independent of cell proliferation rate.
Figure 2.
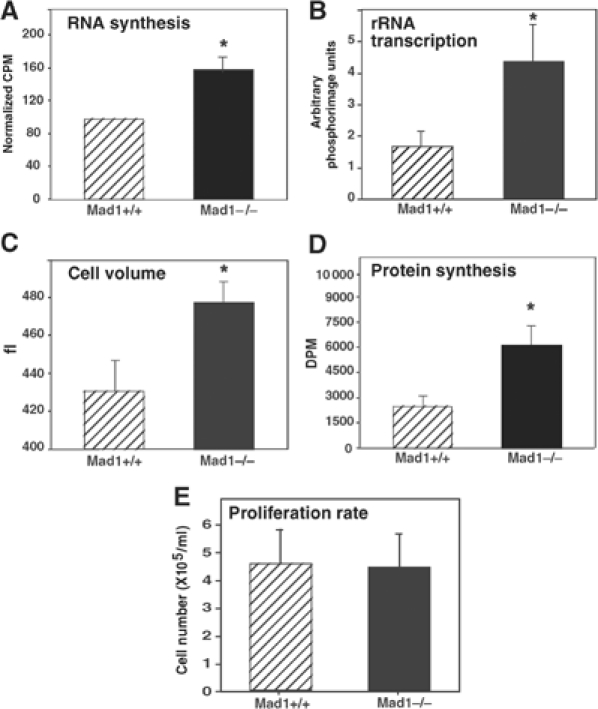
MAD1 regulates RNA synthesis, rDNA transcription and protein synthesis in granulocytes. Cultures of primary granulocytes from wild-type (WT; Mad1+/+) and Mad1-null (Mad1−/−) mice were stimulated with SCF and G-CSF. Cells were analyzed after 5 days of culture and equal cell numbers were used for all assays. (A) RNA synthesis as measured by incorporation of [3H]uridine into total cellular RNA (n=4 for each genotype). *P<0.05 compared to WT mice. (B) rDNA transcription was measured by nuclear run-on analysis of the 45S rRNA precursor (n=4 WT, 4 Mad1−/−). *P<0.05 compared to WT mice. (C) Cell volume of cultured granulocytes (n=8 WT, 7 Mad1−/−). *P<0.05 compared to WT mice. (D) Protein synthesis as measured by incorporation of [35S]methionine into total cellular protein (n=4 WT, 5 Mad1−/−). *P<0.05 compared to WT mice. (E) Proliferation rate as determined by granulocyte concentration 5 days after wild-type and Mad1−/− cultures were seeded with 1 × 105 cells/10 ml of culture medium (n=7 WT, 7 Mad1−/−).
MAD1 and c-MYC can regulate rDNA transcription in various cell types
The above studies demonstrated that loss of MAD1 correlated with increased rDNA transcription in granulocytes. We therefore examined if enforced expression of MAD1 could regulate rDNA transcription as measured by nuclear run-on analyses of the 45S gene. The assays were performed in NIH3T3 cells: a well-characterized system for the study of rDNA transcription that allows for retroviral-mediated expression of potential regulatory proteins. Indeed, NIH3T3 cells infected with a retroviral vector expressing MAD1 displayed 40% lower transcription of the 45S gene compared to control cells (P<0.001) (Figure 3A).
Figure 3.
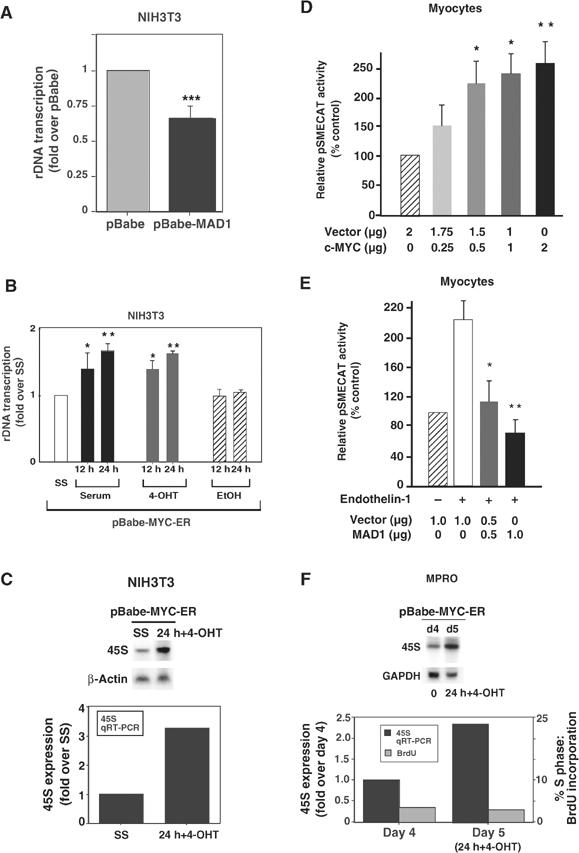
Regulation of rDNA transcription by MAD1 and c-MYC. (A) Proliferating cultures of NIH3T3 cells were infected with pBabe and pBabe-MAD1 expression vectors. Cells were then analyzed for endogenous rDNA transcription by nuclear run-on assay. Results are the mean±s.e.m. of four independent experiments. ***P<0.001 compared to pBabe cells. (B) Cultures of pBabe-MYC-ER NIH3T3 cells were incubated in DMEM containing 0.5% serum for 24 h (SS) and then stimulated with 10% serum (▪), 4-OHT (200 nM) ( ) or vehicle (EtOH) (
) or vehicle (EtOH) ( ) for 12 and 24 h before being analyzed for endogenous rDNA transcription by run-on assays (n=5; *P<0.05 and **P<0.01 compared to SS cells); or (C) 24 h for expression of the 45S rRNA precursor by Northern blot (β-actin control) and qRT-PCR (normalized to β-2-microglobulin expression). (D) Unstimulated (nonhypertrophic) cultures of primary neonatal cardiomyocytes were transfected with pSMECAT, pCMV-β-Gal (see Supplementary Figure 1) and an increasing amount of a c-MYC expression vector. After 24 h, cells were assayed for CAT activity (n=5). *P<0.05 and **P<0.01 compared to cells transfected with the empty expression vector (% control). (E) Cultures of primary neonatal cardiomyocytes were transfected with pSMECAT, pCMV-β-Gal (see Supplementary Figure 1) and an increasing amount of an MAD1 expression vector and then stimulated with the hypertrophic agent endothelin-1 (10−7 M). After 24 h, cells were assayed for CAT activity (n=5). *P<0.05 and **P<0.01 compared to hypertrophic cells transfected with the empty expression vector (% control). (F) Cultures of pBabe-MYC-ER MPRO granulocytic cells were induced to differentiate (for 4 days) and then stimulated with 4-OHT (200 nM) for 24 h before being assayed for 45S RNA expression by Northern blot (GAPDH control) and qRT-PCR (normalized to GAPDH expression). At 0 and 24 h, BrdU incorporation was assayed to determine the % of cells in S phase.
) for 12 and 24 h before being analyzed for endogenous rDNA transcription by run-on assays (n=5; *P<0.05 and **P<0.01 compared to SS cells); or (C) 24 h for expression of the 45S rRNA precursor by Northern blot (β-actin control) and qRT-PCR (normalized to β-2-microglobulin expression). (D) Unstimulated (nonhypertrophic) cultures of primary neonatal cardiomyocytes were transfected with pSMECAT, pCMV-β-Gal (see Supplementary Figure 1) and an increasing amount of a c-MYC expression vector. After 24 h, cells were assayed for CAT activity (n=5). *P<0.05 and **P<0.01 compared to cells transfected with the empty expression vector (% control). (E) Cultures of primary neonatal cardiomyocytes were transfected with pSMECAT, pCMV-β-Gal (see Supplementary Figure 1) and an increasing amount of an MAD1 expression vector and then stimulated with the hypertrophic agent endothelin-1 (10−7 M). After 24 h, cells were assayed for CAT activity (n=5). *P<0.05 and **P<0.01 compared to hypertrophic cells transfected with the empty expression vector (% control). (F) Cultures of pBabe-MYC-ER MPRO granulocytic cells were induced to differentiate (for 4 days) and then stimulated with 4-OHT (200 nM) for 24 h before being assayed for 45S RNA expression by Northern blot (GAPDH control) and qRT-PCR (normalized to GAPDH expression). At 0 and 24 h, BrdU incorporation was assayed to determine the % of cells in S phase.
While MAD:MAX complexes bind DNA and repress transcription, MYC:MAX complexes bind the same canonical sites and typically activate transcription (Eisenman, 2001). Given this, we sought to determine if c-MYC could activate rDNA transcription. NIH3T3 cells were infected with a retroviral vector expressing the c-MYC-ER fusion protein, an inducible form of c-MYC that can be rapidly activated in cells by the addition of 4-OH-tamoxifen (4-OHT) (Eilers et al, 1991; Littlewood et al, 1995). Cells expressing c-MYC-ER were cultured in media containing low serum, c-MYC was activated by the addition of 4-OHT and the rate of rDNA transcription was analyzed. Following induction of c-MYC activity, nuclear run-on analyses of the 45S gene demonstrated a significant increase (70%) in rDNA transcription as compared to control serum starved cells (P<0.01) that approximated the fold induction observed with serum (Figure 3B). Since the number of nuclei assayed per time point were normalized for DNA content before measurement of RNA polymerase I transcription, elevated gene copy number could not account for the increased rDNA transcription. In addition, Northern and quantitative real-time PCR (qRT-PCR) analyses of 45S rRNA precursor expression levels in these cells demonstrated a 2- to 3-fold increase in c-MYC-induced 45S expression when normalized to control transcripts (Figure 3C).
To exclude the possibility that the ability of MAD1 to repress and c-MYC to activate rDNA transcription was an indirect consequence of the ability of these proteins to regulate cell proliferation, we examined the effect of expression of MAD1 or c-MYC on rDNA transcription in primary cultures of terminally differentiated neonatal cardiomyocytes. These cells were transfected with an rDNA reporter gene, pSMECAT (Hannan et al, 1996b), allowing us to examine nonproliferative growth (hypertrophic growth) that is dependent on increased rates of rDNA transcription (Brandenburger et al, 2001). c-MYC conferred a dose-dependent activation of rDNA transcription (up to 2.5-fold) in unstimulated cardiomyocytes (Figure 3D), consistent with studies demonstrating that conditional cardiomyocyte-specific overexpression of c-MYC stimulates hypertrophic growth (Xiao et al, 2001). Conversely, MAD1 significantly inhibited rDNA transcription to below basal levels in myocytes undergoing hypertrophy in response to the growth stimulant endothelin-1 (Luyken et al, 1996) (Figure 3E). Under the same conditions, c-MYC and MAD1 had no effect on a cotransfected control reporter gene, pCMV-β-Gal (Supplementary Figure 1). We also assayed the effect of enforced c-MYC expression on endogenous rDNA transcription rates in differentiated murine MPRO granulocytes, a model system for in vitro differentiation. MPRO cells infected with the c-MYC-ER-expressing retrovirus were induced to differentiate for 4 days, then c-MYC was activated by addition of 4-OHT for 24 h before expression of the 45S rRNA precursor was examined. Northern blot and qRT-PCR analyses demonstrated a 2.3-fold increase in expression of the 45S precursor, while cell cycle analysis showed that the cells remain arrested with approximately 3% of cells in S phase (Figure 3F). After c-MYC induction, cells also remained morphologically differentiated (data not shown). Together these findings indicate that the effects of c-MYC and MAD1 on rDNA transcription occur independently of cell division and cell type.
The rDNA transcription factor UBF is regulated by c-MYC and MAD1
Transcription from rDNA genes is tightly regulated during changing cellular states such as the switch from quiescence to proliferation (Grummt et al, 1976; Paule, 1998), during cellular differentiation (Larson et al, 1993; Cavanaugh et al, 1995) or from basal growth to hypertrophy (Hannan et al, 1996a). The transcription factor UBF is an important regulator of rDNA transcription and, like proteins of the MAX network, expression of UBF is tightly regulated in many systems in response to changing cellular growth requirements (Larson et al, 1993; Cavanaugh et al, 1995). For example, differentiation of L6 myoblasts correlates with a simultaneous decrease in UBF expression and rDNA transcription (Larson et al, 1993). Conversely, serum refeeding of serum-deprived NIH3T3 fibroblasts leads to a rapid accumulation of UBF mRNA and protein (Supplementary Figure 2), which precedes the activation of rDNA transcription (Glibetic et al, 1995). We therefore examined expression of UBF during differentiation of the human HL-60 and murine MPRO granulocytic cell lines and correlated this with expression of c-MYC and MAD1. As previously described, expression of c-MYC is reduced and MAD1 is induced during granulocyte differentiation (Figure 4A). In parallel to the induction of MAD1, UBF protein (Figure 4A) and mRNA (Figure 4B) were significantly reduced during granulocyte differentiation. Strikingly, in HL-60 cells, many transcripts including β-actin (Figure 4B) reduced on differentiation. However, UBF mRNA is more tightly regulated, being absolved by day 1, consistent with the effects on protein levels.
Figure 4.
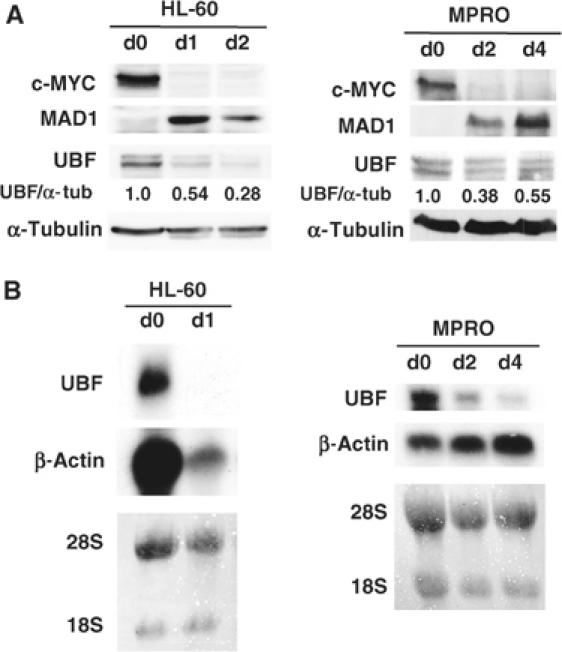
Regulation of c-MYC, MAD1 and UBF during granulocyte differentiation. (A) Human (HL-60) and mouse (MPRO) granulocytic cell lines were induced to differentiate with ATRA (for 1 and 2 days) and AGN194024 (for 2 and 4 days) respectively as compared to undifferentiated cells (day 0). Protein from cells was analyzed by Western blot for c-MYC, MAD1 and UBF expression. α-Tubulin was used as a protein loading control and ratios of UBF to α-tubulin were calculated. (B) Cells were differentiated as indicated in (A) and UBF mRNA levels were analyzed by Northern blot. Lanes are equally loaded with total RNA as demonstrated by β-actin expression and/or stained for 18S and 28S rRNA. d0=day 0, d1=day 1, d2=day 2 and d4=day 4.
UBF regulates rDNA transcription and binds to multiple sites across the rDNA gene including the proximal promoter (Moss and Stefanovsky, 2002; O'Sullivan et al, 2002). We therefore performed chromatin immunoprecipitation (ChIP) assays on undifferentiated and differentiated MPRO cells to determine if binding of UBF to the rDNA gene was likewise reduced during granulocyte differentiation. Sites within three regions of the rDNA gene, all of which bind varying amounts of UBF, were examined: the 5′-enhancer sequence (ENH), the proximal rDNA promoter (upstream control element, UCE) and the externally transcribed spacer (ETS) (Figure 5A). As a negative control, we also examined UBF binding to the promoter of Lactoferrin, a gene that is transcriptionally activated in mature granulocytes. Binding of UBF to the rDNA gene was enriched, specifically at the ENH and UCE sites, in proliferating cells (day 0) whereas following differentiation (day 4) there was a marked reduction in UBF binding (Figure 5B and C) that correlated with a reduction in 45S precursor transcript (Figure 5D). Comparatively no UBF binding was demonstrated at the Lactoferrin promoter (Figure 5C). These results demonstrate that, as expression of c-MYC decreases and MAD1 increases, both the amount of UBF bound to the rDNA gene and the rate of rDNA transcription correlate with UBF expression during granulocyte differentiation.
Figure 5.
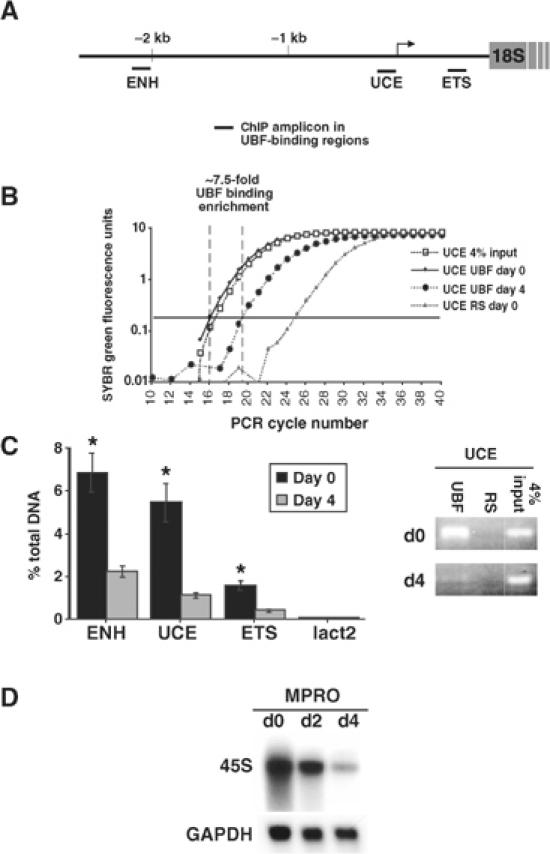
Regulation of UBF binding to the rDNA gene during granulocyte differentiation. MPRO cells were differentiated for 4 days and UBF binding to the rDNA gene was determined by ChIP assays using qRT-PCR. (A) Three regions of the murine rDNA gene were examined: the 5′ enhancer region (ENH); the upstream control element (UCE); and the 5′ external transcribed spacer (ETS). (B) Representative qRT-PCR amplification curves as displayed by ABI Prism 7000 are shown for the UCE amplicon. RS: control ChIP with preimmune rabbit sera. The arbitrary amplification threshold is depicted as the horizontal bar running across the graph. (C) Data presented as % of DNA site immunoprecipitated with anti-UBF compared to that present in total input DNA (based on 4% input; calculated as described in Frank et al, 2001). A site within the Lactoferrin promoter (lact2) was used as a negative control. Results are the mean±s.e.m. from three independent experiments. *P<0.05 compared to day-4 ChIPs. The representative ethidium bromide gel shows the amount of UCE product present after 19 cycles (within the exponential phase) of PCR of ChIP samples. (D) Cells were assayed for 45S rRNA expression by Northern blot (GAPDH control).
The above data suggest regulation of UBF gene expression by c-MYC and MAD1, and consistent with this, activation of the c-MYC-ER fusion protein in quiescent NIH3T3 fibroblasts was sufficient to induce the expression of UBF mRNA (2.7-fold) and protein (2- to 3-fold) (Figure 6A). Induction of UBF protein was quantitatively similar to that obtained by stimulating NIH3T3 cells with serum (Supplementary Figure 2). As well, similar effects were observed in granulocytes, as induction of c-MYC in differentiated MPROs led to an approximate four-fold increase in UBF mRNA (data not shown). Conversely, enforced expression of MAD1 in proliferating fibroblasts reduced the expression of UBF (Figure 6B).
Figure 6.
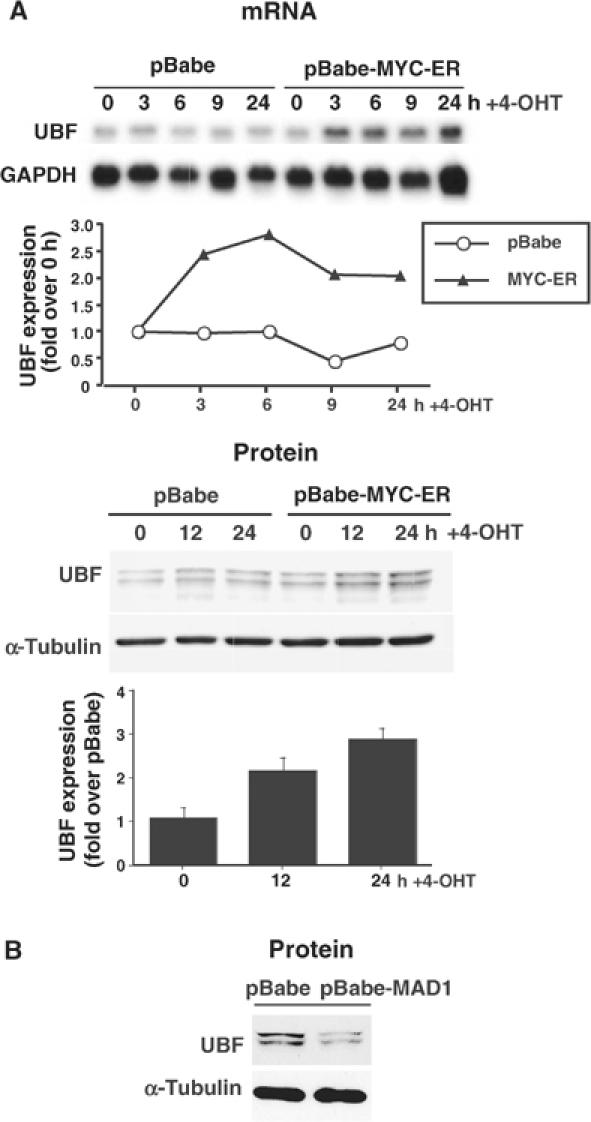
Proteins of the MAX network can regulate the expression of UBF. (A) Cultures of pBabe and pBabe-MYC-ER NIH3T3 cells were incubated in DMEM containing low serum and then stimulated with 4-OHT for 3–24 h before being analyzed for UBF mRNA expression by Northern blot (GAPDH control). UBF expression at each time point was quantitated by qRT-PCR analysis (normalized to GAPDH expression). The same cells were also analyzed by Western blot for UBF protein expression (α-tubulin control) and UBF levels were quantitated from at least four independent Western blots/experiments and represented as fold increase in MYC-ER cells over pBabe cells for each time point. (B) Proliferating NIH3T3 cells were infected with the pBabe or pBabe-MAD1 retrovirus, selected and analyzed by Western blot for expression of UBF (α-tubulin control).
Interestingly, a significant increase in the level of UBF mRNA (2.5-fold) was observed within 3 h of c-MYC activation, suggesting direct regulation of the UBF gene by the MYC and MAD1 family (Figure 6A). To determine if c-MYC and MAD1 were able to interact directly with the UBF promoter during granulocyte differentiation, we again performed ChIP assays in MPRO cells. PCR primer sets were designed to amplify a select set of potential c-MYC/MAD1-binding sites (E boxes) spanning the UBF promoter (Figure 7A). ChIP results show that c-MYC and MAD1 binding of the UBF promoter was enriched in undifferentiated (day 0) and differentiated (day 4) granulocytes, respectively (Figure 7B). Enrichment of binding of both c-MYC and MAD1 was statistically significant at a more distal E box (UBF-2) and a canonical E box (UBF-5) immediately upstream of the transcriptional start site (Figure 7B). In contrast, no significant changes in binding were detected at the non-E-box-containing site in the promoter region of the Lactoferrin gene (lact1) (Figure 7B). Consistent with a role for c-MYC and MAD1 in modulating expression of UBF in vivo, ChIP analyses for acetylated histone H3 demonstrated reduced acetylation at the UBF promoter, particularly at the E-box sites surrounding the transcriptional start (UBF-5 and -7), in differentiated granulocytes (Figure 7B). Furthermore, c-MYC and MAD1 regulated expression of a UBF promoter reporter construct in terminally differentiated and proliferating cells respectively, and this regulation was abrogated by deletion of the distal E boxes (Supplementary Figure 3). Together, these data demonstrate that UBF is a direct gene target of c-MYC/MAD1 in granulocytes.
Figure 7.
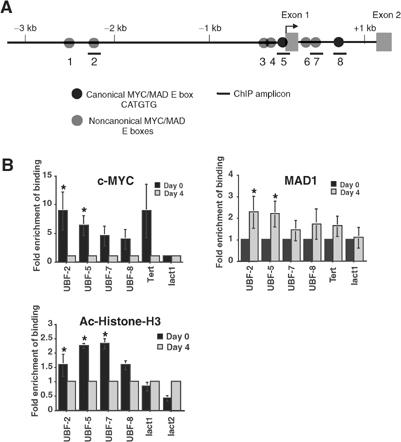
Association of MAD1 and c-MYC with the UBF promoter during granulocyte differentiation. MPRO cells were differentiated and ChIP analyses were performed using antibodies to c-MYC, MAD1 and acetylated histone H3 (Ac-Histone-H3). Binding of immunoprecipitated proteins to the UBF promoter was assessed by qRT-PCR. (A) Schematic representation of the murine UBF gene, with MYC/MAD E-box sites indicated. (B) Quantitation of c-MYC, MAD1 and Ac-Histone-H3 binding to the UBF promoter before (day 0) and 4 days after differentiation. Data are expressed as fold enrichment relative to day 4 (c-MYC and Ac-Histone-H3) or day 0 (MAD1). The lact1 site is a negative control for all ChIPs. The Tert and lact2 sites are positive controls for c-MYC/MAD1 and Ac-Histone-H3 (active at day 4) ChIPs, respectively. Results are the mean±s.e.m. from a minimum of three independent experiments. *P<0.05 compared to day-4 ChIPs (c-MYC and Ac-Histone-H3) or day-0 ChIPs (MAD1).
Is UBF required for c-MYC induction of rDNA transcription?
To determine if the induction of UBF expression by c-MYC was required for the ability of c-MYC to activate rDNA transcription, we used small interfering RNA (siRNA) to block UBF expression. A transfected siRNA oligo to UBF, siUBF-B, was able to efficiently reduce endogenous levels of both UBF isoforms by over 95% in NIH3T3 cells (Figure 8A). In contrast, siRNA to the enhanced green fluorescent protein (siEGFP) had no effect on UBF levels (Figure 8A). The dose of the transfected siUBF-B was titrated to determine a concentration that would block c-MYC-induced UBF expression without affecting basal levels of UBF in low serum (inset, Figure 8B). Inhibition of UBF expression using this concentration of siUBF-B significantly attenuated (72% reduction after 12 h) the ability of c-MYC to activate rDNA transcription as measured by nuclear run-on assays (Figure 8B). In parallel experiments, siRNA to EGFP had no effect on rDNA transcription induced by c-MYC. Thus c-MYC regulates rDNA transcription in part by modulation of the cellular levels of the rDNA transcription factor UBF. Conversely, to examine the role of UBF in the ability of MAD1 to repress transcription of the pSMECAT rDNA reporter gene, we enforced expression of UBF in cells cotransfected with MAD1 and pSMECAT. Interestingly and consistent with the findings for c-MYC, UBF expression was able to reverse MAD1-mediated repression of rDNA transcription in both proliferating fibroblasts and in postmitotic cardiomyocytes (Supplementary Figure 4).
Figure 8.
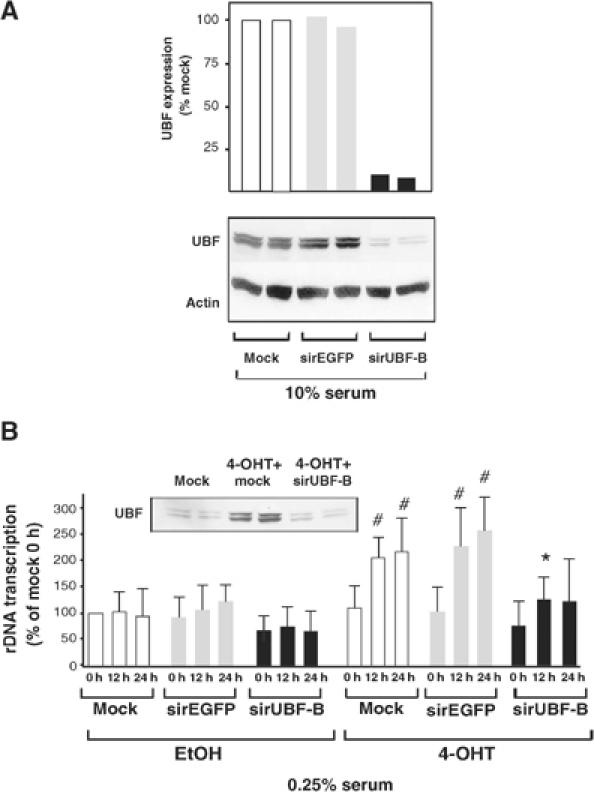
UBF is required for activation of rDNA transcription by c-MYC. Cultures of pBabe-MYC-ER NIH3T3 cells were transfected with 25 nM siRNA oligos to the 5′ region of the EGFP (sirEGFP) or UBF (sirUBF-B) mRNA and were incubated in (A) DMEM containing 10% serum for 24 h and analyzed for expression of UBF by Western blot; or (B) DMEM containing 0.25% serum for 24 h (designated 0 h) before cells were stimulated with either vehicle (EtOH) or 4-OHT for 12 and 24 h, then analyzed for expression of UBF by Western blot or endogenous rRNA synthesis as described above. Inset Western shows UBF expression in 0.25% serum following c-MYC activation and siUBF-B transfection. Note: UBF levels in siUBF-B transfected lane are equivalent to baseline levels found prior to c-MYC activation. All Western blots show UBF expression from two independent transfection experiments. Data are expressed as % of mock transfected unstimulated (A) or EtOH-stimulated 0 h (B) cells. Mock: transfection without siRNA. (n=4); #P<0.05 compared to EtOH-stimulated mock transfection; *P<0.05 compared to 4-OHT-stimulated sirEGFP transfection.
Discussion
Members of the MAX network of proteins regulate rDNA transcription
We have demonstrated that during granulocyte differentiation loss of the MYC antagonist MAD1 results in larger immature granulocytes with higher total RNA content and larger nucleoli. Nuclear run-on assays in primary granulocytes and fibroblast-derived cell lines demonstrated that MAD1 was able to repress, and c-MYC activate, rDNA transcription. As rRNA synthesis is coupled to cell division in proliferating cells, it was possible that the effects on rDNA transcription were secondary to those on the cell cycle. However, this is unlikely as MAD1 and c-MYC regulated rDNA transcription in postmitotic cardiomyocytes and differentiated granulocytes. When considered with the recent description of MYC as a regulator of expression of genes required for ribosome assembly (Kim et al, 2000) and as a regulator of RNA polymerase III-mediated transcription of tRNA and 5S rRNA transcripts (Gomez-Roman et al, 2003), our demonstration of the ability of the MAX network to regulate rDNA transcription provides a mechanism for the coordinate regulation of all aspects of ribosome biogenesis. This suggests that MAX-network proteins are central controllers of cell growth. Further, since the MAX-network proteins are established regulators of cell proliferation, these factors are likely to be key players in regulating the complex balance between cell proliferation and growth that is required for mammalian development during differentiation and the determination of organ size.
Mechanism of rDNA transcription regulation by c-MYC and MAD-1
Efficient transcription from the mammalian ribosomal gene promoter requires a multiprotein complex including UBF and Rrn3 (Moss and Stefanovsky, 2002). In addition to Rrn3, regulation of UBF activity appears to be one of the primary mechanisms by which rDNA transcription rates are regulated under changing growth conditions (reviewed in Hannan et al, 1998). Interestingly, despite the relative abundance of UBF, it appears to be limiting for rDNA transcription, and variations in UBF concentration and DNA binding (Figure 5C and D) correlate with both changes in rDNA transcription rates and growth in a variety of systems (Hannan et al, 1996b). UBF protein levels are regulated at the level of transcription in a manner very similar to primary response genes such as SRF and c-MYC. We examined a possible link between the ability of proteins of the MAX network to regulate rDNA transcription and the expression of UBF, and found that c-MYC and MAD1 directly regulate UBF gene expression. Moreover, UBF expression was required for c-MYC to activate rDNA transcription, as reduction in UBF protein using siRNA attenuated activation of rDNA transcription by c-MYC. Therefore, in a context where rDNA transcription is repressed over days, such as granulocyte differentiation, c-MYC and MAD1 become important regulators of rDNA transcription and this is achieved, at least in part, via the modulation of UBF levels.
The precise mechanism by which changes in UBF levels regulate rDNA transcription is yet to be resolved. Through DNA binding of its HMG1 boxes, UBF has been shown to induce a chromatin-like structure termed the enhancersome in which approximately 140 bp of DNA is looped into a single turn (Moss and Stefanovsky, 2002 and references therein). In addition to enabling SL-1 recruitment, this structure may allow UBF to displace histone H1 and thus compete with the repressive effects of chromatin (Kermekchiev et al, 1997; Moss and Stefanovsky, 2002). Surprisingly, UBF binding in vivo is not restricted to the core promoter but binds to multiple sites distributed across the entire transcribed rDNA repeat including the intergenic spacers (Moss and Stefanovsky, 2002; O'Sullivan et al, 2002). These findings suggest that UBF may have a more generalized structural role over the rDNA repeat in addition to its recognized role in stable transcription complex formation at the promoter. Given the high number of UBF-binding sites in the rDNA repeat, it is plausible that alterations in UBF content, along with post-translational modifications, may regulate the loading of UBF onto the rDNA locus. Consistent with this, we demonstrate that the reduction in UBF expression observed during granulocyte differentiation correlated with a reduction in the amount of UBF associated with the rRNA genes at all sites assayed. Thus, like chromatin-remodeling proteins, UBF may have several roles including gene activation, initiation complex formation and transcriptional enhancement depending on where it is bound throughout the rRNA gene.
While reduction of UBF accumulation by RNAi significantly attenuated the ability of c-MYC to activate rDNA transcription, it did not completely reduce rDNA transcription to uninduced levels. This suggests that there may be additional mechanisms by which proteins of the MAX network regulate rDNA transcription. For example, members of the RB family of pocket proteins can associate with the rDNA promoter and repress transcription (Cavanaugh et al, 1995; Voit et al, 1997; Ciarmatori et al, 2001). As MAX-network proteins have been documented to regulate the activity of pocket proteins (Vlach et al, 1996; Queva et al, 1999), this may provide an additional mechanism of regulation. Indeed, granulocytic cell lines deficient in MAD1 display reduced accumulation of the pocket protein p130 during differentiation (McArthur et al, 2002). Also, under some conditions, c-MYC protein has been detected in the nucleolus, raising the possibility of a more direct mechanism of rDNA transcription activation (Arabi et al, 2003). Interestingly, Gomez-Roman et al (2003) observed that c-MYC was able to induce transcription of the 5SrRNA gene within 3 h and this rapid induction was associated with an interaction between c-MYC and TFIIIB. It is possible that such a direct mechanism may function in addition to or distinct from mechanisms operating primarily during differentiation.
Regulation of cell growth during differentiation
Proteins of the MAX network display consistent and characteristic changes of expression during cellular differentiation in many cell types. As both MYC and MAD proteins have been demonstrated to regulate cell growth, proteins of the MAX network are attractive candidates to mediate the reduction in growth that accompanies many forms of differentiation, such as that of granulocytes as described in this study. The process of differentiation is also characterized by the acquisition of specialized cellular phenotypes. In some instances, differentiated cells are capable of growth, most notably cardiac and skeletal muscle. Intriguingly, MYC has also been implicated in growth control in this context (Pollack et al, 1994; Xiao et al, 2001). As such, proteins of the MAX network may primarily function as growth regulators, tempting one to speculate that their ability to regulate cell growth may be necessary for their effects on differentiation.
Finally, the ability of the MAX-network proteins to regulate rDNA transcription and cell growth may have implications for the role of these proteins in cancer. The identification of mechanisms by which MYC can regulate ribosome biogenesis may allow direct testing of the requirement of cell growth control for the oncogenic properties of MYC. Thus, the link between the MAX network and growth suggests that cancers with dysregulated expression of MYC or loss of function of MAD proteins may display sensitivity to cancer therapies directed at protein synthesis.
Materials and methods
Mice
The Mad1−/− (Foley et al, 1998) mice have been described previously. All mice used were aged 8–10 weeks. Mice had been backcrossed four generations to the C57/BL6J background from the hybrid C57/BL6J 129/SV background.
Isolation of cell populations and cell analyses
Granulocyte populations were isolated from adult mouse whole bone marrow as described (Walkley et al, 2002). Differentiation of HL-60 and MPRO cells was induced by stimulation with 10−5 M All-trans retinoic acid and 10−7 M of the retinoid agonist AGN 194204, respectively (McArthur et al, 2002). Cell volume was determined using a Sysmex CDA 500 system. RNA content was determined by staining with acridine orange (Molecular Probes) (Darzynkiewicz, 1994). Nucleoli were visualized using transmission electron microscopy by staining for Ag-Nor proteins (Ploton et al, 1984).
5-Bromo-2-deoxyuridine (BrdU) incorporation and analysis were carried out as described (Walkley et al, 2002) with a 30 min at 37°C BrdU incorporation time prior to fixation.
Biosynthetic labeling
Granulocytes for biosynthetic labeling and nuclear run-on analyses were obtained by culturing adult mouse whole bone marrow cells for 5 days in DMEM supplemented with 20% FCS, SCF and rhG-CSF as described (Walkley et al, 2002).
35S-methionine labeling was performed by incubating cultured granulocytes for 30 min at 37°C in methionine-free DMEM followed by the addition of 50 μCi/ml trans-35S-labeled methionine for 60 min at 37°C. After termination of labeling, cells were lysed and protein was TCA precipitated before incorporated 35S was measured by scintillation counting.
RNA synthesis was analyzed by suspending cells in DMEM (10% FCS) containing 50 Ci of [5,6-3H]uridine and incubating for 5 min at 37°C followed by 10 min in media without the radiolabel. After washing the cells, RNA was isolated and 3H incorporation was measured by scintillation counting.
Nuclear run-on transcription
Isolation of de novo-synthesized RNA from isolated nuclei was carried out as described (Hannan et al, 1999) using equal numbers of nuclei for each time point (DNA concentration was also determined). RNA amounts were then quantitated and normalized for recovery using a 3H-labeled riboprobe. Transcription from the 45S rDNA gene in the presence of 5 mg/ml α-amanitin (inhibits RNA polymerases II and III) was measured by hybridization of in vitro-synthesized 32P-labeled run-on transcripts to immobilized plasmids containing the mouse 45S gene as described (Hannan et al, 1996a). Radioactive hybrids were detected and quantitated using Molecular Dynamics PhosphorImager.
Northern analysis
Northern analysis was performed on 15–20 μg of total RNA extracted from NIH3T3, MPRO and HL-60 cells using Trizol reagent (Invitrogen). Following electrophoresis, the RNA was transferred to nylon membranes that were stained for total RNA with 0.5 M sodium acetate (pH 5.2) and 0.04% methylene blue dye, and the images were scanned. Membranes were probed using cDNA probes to a fragment (+1 to + 153 relative to the start of transcription) of the externally transcribed spacer region of the mouse 45S gene, UBF, β-actin or GAPDH.
cDNA synthesis and qRT-PCR analysis of cDNA/RNA expression
Cells were lysed in Trizol reagent (Invitrogen) and RNA was prepared using standard procedures. cDNA was prepared using random primers and Superscript II reverse transcriptase (Invitrogen) as described by the manufacturer. qRT-PCR was performed using the SYBR Green® dye detection method (Applied Biosystems). Primers were designed using Primer Express 2® software (Applied Biosystems) and their sequences are listed in Supplementary Table 2.
Western analysis
Immunoblotting was performed on 50–100 μg of total protein extracts from NIH3T3, MPRO and HL-60 cells. Following SDS–PAGE, proteins were transferred to PVDF membranes and analyzed using the antisera listed below and enhanced chemiluminescence (ECL) detection. The following antibodies were used: rabbit anti-c-MYC (Santa Cruz, sc-764), anti-MAD1 (Santa Cruz, sc-222), anti-UBF1/2 (Brandenburger et al, 2001) and mouse anti-α-tubulin (Sigma). Quantitation of UBF (compared to α-tubulin) was determined using ImageJ 1.29 software (http://rsb.info.nih.gov/ij/download.html).
Chromatin immunoprecipitation analysis
ChIP assays were carried out as described with modifications (Walkley et al, 2004). Undifferentiated and differentiated MPROs were fixed in 0.8% formaldehyde and ChIP assays were performed in triplicate using 1.5 × 106 cells per IP (including no-antibody control). For c-Myc/Mad1/acetylated histone H3 ChIPs, 5 μg of antibody was used for each IP (anti-c-MYC (sc-764), anti-MAD1 (sc-222) and anti-acetyl-histone H3 (Upstate #06-599)); for UBF ChIPs, 6 μl of either anti-UBF1/2 (Brandenburger et al, 2001) or preimmune rabbit sera control was used for each IP. qRT-PCR was carried out in triplicate using the SYBR Green® dye detection method (Applied Biosystems). Calculations for the percent of total DNA bound were performed as described (Frank et al, 2001) and are shown as such or as fold change in percent of total DNA bound. Primers were designed using Primer Express 2® software (Applied Biosystems) and their sequences are listed in Supplementary Table 1.
Cell culture, retroviral gene transfer and transfection
pBabe-MYC-ER retroviruses were used to infect NIH3T3 cells (Littlewood et al, 1995). For the siRNA experiments, oligo duplexes (25 nM) were transfected into pBabe-MYC-ER or pBabe NIH3T3 cells using Lipofectamine (Invitrogen) according to the manufacturer's recommendations. Following transfection, the medium was replaced with DMEM containing 0.25–0.5% FCS to induce cell cycle arrest. After 24 h, cells were stimulated with 4-OHT for 12 or 24 h before being harvested for Western or nuclear run-on analysis. Cardiomyocytes were isolated and transfected with the following: the rDNA reporter construct, pSMECAT (0.5 μg); a control reporter, pCMV-β-Gal (0.5 μg); and the indicated amounts of MAD1 and c-MYC expression constructs as described (Brandenburger et al, 2001). After transfection with MAD1, hypertrophy was initiated (Brandenburger et al, 2001). Cells were harvested and cell lysates prepared to assay for CAT or β-Gal activity as described (Hannan et al, 1999).
RNA interference
RNAs were chemically synthesized by Proligo. Sequences from the open reading frame of mouse UBF and EGFP were selected to obtain 21-nt sense and antisense strand oligos with symmetric 2-nt 3′ overhangs of identical sequence. The oligo RNA sequences against UBF1 and EGFP are provided in Supplementary Table 3.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Tables
Acknowledgments
This work was supported by a Special Fellowship of the Leukemia and Lymphoma Society (GAM), a National Health and Medical Research Council of Australia (NHMRC) Fellowship (RDH) and by grants from the NHMRC (GAM, RBP and RDH) and the National Heart Foundation of Australia (RDH). CRW is a recipient of an Australian Postgraduate Award. We thank Robert Ramsay, Ricky Johnstone and Helena Richardson for their critical reading of the manuscript and Ben Williams for assistance with cell culture.
References
- Arabi A, Rustum C, Hallberg E, Wright AP (2003) Accumulation of c-Myc and proteasomes at the nucleoli of cells containing elevated c-Myc protein levels. J Cell Sci 116: 1707–1717 [DOI] [PubMed] [Google Scholar]
- Boon K, Caron HN, van Asperen R, Valentijn L, Hermus MC, van Sluis P, Roobeek I, Weis I, Voute PA, Schwab M, Versteeg R (2001) N-myc enhances the expression of a large set of genes functioning in ribosome biogenesis and protein synthesis. EMBO J 20: 1383–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandenburger Y, Jenkins A, Autelitano DJ, Hannan RD (2001) Increased expression of UBF is a critical determinant for rRNA synthesis and hypertrophic growth of cardiac myocytes. FASEB J 15: 2051–2053 [DOI] [PubMed] [Google Scholar]
- Cavanaugh AH, Hempel WM, Taylor LJ, Rogalsky V, Todorov G, Rothblum LI (1995) Activity of RNA polymerase I transcription factor UBF blocked by Rb gene product. Nature 374: 177–180 [DOI] [PubMed] [Google Scholar]
- Ciarmatori S, Scott PH, Sutcliffe JE, McLees A, Alzuherri HM, Dannenberg JH, te Riele H, Grummt I, Voit R, White RJ (2001) Overlapping functions of the pRb family in the regulation of rRNA synthesis. Mol Cell Biol 21: 5806–5814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coller HA, Grandori C, Tamayo P, Colbert T, Lander ES, Eisenman RN, Golub TR (2000) Expression analysis with oligonucleotide microarrays reveals that MYC regulates genes involved in growth, cell cycle, signaling, and adhesion. Proc Natl Acad Sci USA 97: 3260–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzynkiewicz Z (1994) Simultaneous analysis of cellular RNA and DNA content. Methods Cell Biol 41: 401–420 [DOI] [PubMed] [Google Scholar]
- Derenzini M, Trere D, Pession A, Govoni M, Sirri V, Chieco P (2000) Nucleolar size indicates the rapidity of cell proliferation in cancer tissues. J Pathol 191: 181–186 [DOI] [PubMed] [Google Scholar]
- Dufner A, Thomas G (1999) Ribosomal S6 kinase signaling and the control of translation. Exp Cell Res 253: 100–109 [DOI] [PubMed] [Google Scholar]
- Eilers M, Schirm S, Bishop JM (1991) The MYC protein activates transcription of the alpha-prothymosin gene. EMBO J 10: 133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenman RN (2001) Deconstructing myc. Genes Dev 15: 2023–2030 [DOI] [PubMed] [Google Scholar]
- Foley KP, McArthur GA, Queva C, Hurlin PJ, Soriano P, Eisenman RN (1998) Targeted disruption of the MYC antagonist MAD1 inhibits cell cycle exit during granulocyte differentiation. EMBO J 17: 774–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B (2001) Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev 15: 2069–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glibetic M, Taylor L, Larson D, Hannan R, Sells B, Rothblum L (1995) The RNA polymerase I transcription factor UBF is the product of a primary response gene. J Biol Chem 270: 4209–4212 [DOI] [PubMed] [Google Scholar]
- Gomez-Roman N, Grandori C, Eisenman RN, White RJ (2003) Direct activation of RNA polymerase III transcription by c-Myc. Nature 421: 290–294 [DOI] [PubMed] [Google Scholar]
- Greasley PJ, Bonnard C, Amati B (2000) Myc induces the nucleolin and BN51 genes: possible implications in ribosome biogenesis. Nucleic Acids Res 28: 446–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grummt I, Smith VA, Grummt F (1976) Amino acid starvation affects the initiation frequency of nucleolar RNA polymerase. Cell 7: 439–445 [DOI] [PubMed] [Google Scholar]
- Guo QM, Malek RL, Kim S, Chiao C, He M, Ruffy M, Sanka K, Lee NH, Dang CV, Liu ET (2000) Identification of c-Myc responsive genes using rat cDNA microarray. Cancer Res 60: 5922–5928 [PubMed] [Google Scholar]
- Hannan KM, Hannan RD, Rothblum LI (1998) Transcription by RNA polymerase I. Front Biosci 3: d376–d398 [DOI] [PubMed] [Google Scholar]
- Hannan R, Stefanovsky V, Arino T, Rothblum L, Moss T (1999) Cellular regulation of ribosomal DNA transcription: both rat and Xenopus UBF1 stimulate rDNA transcription in 3T3 fibroblasts. Nucleic Acids Res 27: 1205–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan RD, Luyken J, Rothblum LI (1996a) Regulation of ribosomal DNA transcription during contraction-induced hypertrophy of neonatal cardiomyocytes. J Biol Chem 271: 3213–3220 [DOI] [PubMed] [Google Scholar]
- Hannan RD, Stefanovsky V, Taylor L, Moss T, Rothblum LI (1996b) Overexpression of the transcription factor UBF1 is sufficient to increase ribosomal DNA transcription in neonatal cardiomyocytes: implications for cardiac hypertrophy. Proc Natl Acad Sci USA 93: 8750–8755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iritani BM, Delrow J, Grandori C, Gomez I, Klacking M, Carlos LS, Eisenman RN (2002) Modulation of T-lymphocyte development, growth and cell size by the Myc antagonist and transcriptional repressor Mad1. EMBO J 21: 4820–4830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iritani BM, Eisenman RN (1999) c-Myc enhances protein synthesis and cell size during B lymphocyte development. Proc Natl Acad Sci USA 96: 13180–13185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston LA, Prober DA, Edgar BA, Eisenman RN, Gallant P (1999) Drosophila myc regulates cellular growth during development. Cell 98: 779–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P, Nishikawa JL, Breitkreutz BJ, Tyers M (2002) Systematic identification of pathways that couple cell growth and division in yeast. Science 297: 395–400 [DOI] [PubMed] [Google Scholar]
- Kermekchiev M, Workman JL, Pikaard CS (1997) Nucleosome binding by the polymerase I transactivator upstream binding factor displaces linker histone H1. Mol Cell Biol 17: 5833–5842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Li Q, Dang CV, Lee LA (2000) Induction of ribosomal genes and hepatocyte hypertrophy by adenovirus-mediated expression of c-Myc in vivo. Proc Natl Acad Sci USA 97: 11198–11202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larminie CG, Alzuherri HM, Cairns CA, McLees A, White RJ (1998) Transcription by RNA polymerases I and III: a potential link between cell growth, protein synthesis and the retinoblastoma protein. J Mol Med 76: 94–103 [DOI] [PubMed] [Google Scholar]
- Larson DE, Xie W, Glibetic M, O'Mahony D, Sells BH, Rothblum LI (1993) Coordinated decreases in rRNA gene transcription factors and rRNA synthesis during muscle cell differentiation. Proc Natl Acad Sci USA 90: 7933–7936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI (1995) A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res 23: 1686–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luyken J, Hannan RD, Cheung JY, Rothblum LI (1996) Regulation of rDNA transcription during endothelin-1-induced hypertrophy of neonatal cardiomyocytes. Hyperphosphorylation of upstream binding factor, an rDNA transcription factor. Circ Res 78: 354–361 [DOI] [PubMed] [Google Scholar]
- McArthur GA, Foley KP, Fero ML, Walkley CR, Deans AJ, Roberts JM, Eisenman RN (2002) MAD1 and p27(KIP1) cooperate to promote terminal differentiation of granulocytes and to inhibit Myc expression and cyclin E-CDK2 activity. Mol Cell Biol 22: 3014–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss T, Stefanovsky VY (2002) At the center of eukaryotic life. Cell 109: 545–548 [DOI] [PubMed] [Google Scholar]
- O'Sullivan AC, Sullivan GJ, McStay B (2002) UBF binding in vivo is not restricted to regulatory sequences within the vertebrate ribosomal DNA repeat. Mol Cell Biol 22: 657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee AB (1989) G1 events and regulation of cell proliferation. Science 246: 603–608 [DOI] [PubMed] [Google Scholar]
- Paule MR (1998) Transcription of Ribosomal Genes by Eukaryotic RNA Polymerase I, 1st edn. Austin, TX: Landes Bioscience [Google Scholar]
- Peculis BA (2002) Ribosome biogenesis: ribosomal RNA synthesis as a package deal. Curr Biol 12: R623–R624 [DOI] [PubMed] [Google Scholar]
- Ploton D, Menager M, Adnet JJ (1984) Simultaneous high resolution localization of Ag-NOR proteins and nucleoproteins in interphasic and mitotic nuclei. Histochem J 16: 897–906 [DOI] [PubMed] [Google Scholar]
- Pollack PS, Houser SR, Budjak R, Goldman B (1994) c-myc gene expression is localized to the myocyte following hemodynamic overload in vivo. J Cell Biochem 54: 78–84 [DOI] [PubMed] [Google Scholar]
- Queva C, McArthur GA, Ramos LS, Eisenman RN (1999) Dwarfism and dysregulated proliferation in mice overexpressing the MYC antagonist MAD1. Cell Growth Differ 10: 785–796 [PubMed] [Google Scholar]
- Schlosser I, Holzel M, Murnseer M, Burtscher H, Weidle UH, Eick D (2003) A role for c-Myc in the regulation of ribosomal RNA processing. Nucleic Acids Res 31: 6148–6156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuhmacher M, Staege MS, Pajic A, Polack A, Weidle UH, Bornkamm GW, Eick D, Kohlhuber F (1999) Control of cell growth by c-Myc in the absence of cell division. Curr Biol 9: 1255–1258 [DOI] [PubMed] [Google Scholar]
- Shiio Y, Donohoe S, Yi EC, Goodlett DR, Aebersold R, Eisenman RN (2002) Quantitative proteomic analysis of Myc oncoprotein function. EMBO J 21: 5088–5096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlach J, Hennecke S, Alevizopoulos K, Conti D, Amati B (1996) Growth arrest by the cyclin-dependent kinase inhibitor p27Kip1 is abrogated by c-Myc. EMBO J 15: 6595–6604 [PMC free article] [PubMed] [Google Scholar]
- Voit R, Schafer K, Grummt I (1997) Mechanism of repression of RNA polymerase I transcription by the retinoblastoma protein. Mol Cell Biol 17: 4230–4237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkley CR, Purton LE, Snelling HJ, Yuan YD, Nakajima H, Pierre Chambon P, Chandraratna RAS, McArthur GA (2004) Identification of the molecular requirements for an RAR alpha-mediated cell cycle arrest during granulocytic differentiation. Blood 103: 1286–1295 [DOI] [PubMed] [Google Scholar]
- Walkley CR, Yuan YD, Chandraratna RA, McArthur GA (2002) Retinoic acid receptor antagonism in vivo expands the numbers of precursor cells during granulopoiesis. Leukemia 16: 1763–1772 [DOI] [PubMed] [Google Scholar]
- Xiao G, Mao S, Baumgarten G, Serrano J, Jordan MC, Roos KP, Fishbein MC, MacLellan WR (2001) Inducible activation of c-Myc in adult myocardium in vivo provokes cardiac myocyte hypertrophy and reactivation of DNA synthesis. Circ Res 89: 1122–1129 [DOI] [PubMed] [Google Scholar]
- Zeller KI, Haggerty TJ, Barrett JF, Guo Q, Wonsey DR, Dang CV (2001) Characterization of nucleophosmin (B23) as a Myc target by scanning chromatin immunoprecipitation. J Biol Chem 276: 48285–48291 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Tables