

Abstract
Protein expression depends significantly on the stability, translation efficiency and localization of mRNA. These qualities are largely dictated by the RNA-binding proteins associated with an mRNA. Here, we report a method to visualize and localize RNA–protein interactions in living mammalian cells. Using this method, we found that the fragile X mental retardation protein (FMRP) isoform 18 and the human zipcode-binding protein 1 ortholog IMP1, an RNA transport factor, were present on common mRNAs. These interactions occurred predominantly in the cytoplasm, in granular structures. In addition, FMRP and IMP1 interacted independently of RNA. Tethering of FMRP to an mRNA caused IMP1 to be recruited to the same mRNA and resulted in granule formation. The intimate association of FMRP and IMP1 suggests a link between mRNA transport and translational repression in mammalian cells.
Keywords: fragile X syndrome; microscopy, fluorescence; RNA-binding proteins; RNA transport; translation, genetic
Introduction
From synthesis to destruction, mRNAs are associated with an array of proteins. Proteins control the efficiency of transcription, processing, nuclear export, translation, localization and degradation of mRNA. The importance of regulation at the level of mRNA has become increasingly apparent with the discovery of disease causing defects in these processes (Conne et al, 2000; Calkhoven et al, 2002). For instance, the absence of an RNA-binding protein, the fragile X mental retardation protein (FMRP), results in the most frequently inherited cause of mental retardation in humans (Turner et al, 1996). FMRP contains two hnRNP K homology (KH) domains and an RGG box. The RGG box binds strongly to G quartet-containing RNA sequences, including that encoding the RGG box itself (Darnell et al, 2001; Schaeffer et al, 2001). As FMRP contains both a nuclear localization sequence (NLS) and a nuclear export sequence (NES) (Eberhart et al, 1996), it has the potential to interact with RNA in both subcellular compartments. Because FMRP is clearly visible in the dendrites of neurons (Feng et al, 1997), early hypotheses proposed that it may play a role in mRNA localization. However, recent reports have demonstrated that FMRP can repress the translation of associated mRNAs in vitro (Laggerbauer et al, 2001; Li et al, 2001). Given that in the absence of FMRP some of its mRNA targets can show changes in abundance (Miyashiro et al, 2003), increased or decreased association with polysomes (Brown et al, 2001; Zalfa et al, 2003), and altered subcellular localization (Miyashiro et al, 2003), it is likely that FMRP plays many roles in cells.
The subcellular localization of mRNAs is important for many cellular functions. Some mRNAs are transported to specific regions so that the synthesis of their protein products is confined (Palacios and St Johnston, 2001). In fibroblasts and neurons, the mRNA for β-actin is actively transported to regions of polarized cell growth (Lawrence and Singer, 1986; Bassell et al, 1998). This requires a 54-nucleotide (nt)-sequence element (or zipcode) located in the transcript's 3′ untranslated region (3′UTR) and its cognate protein, called the zipcode-binding protein 1 (ZBP1) in chicken cells (Kislauskis et al, 1994; Ross et al, 1997) and insulin-like growth factor II mRNA-binding protein 1 (IMP1) in humans (Nielsen et al, 1999). If the process of β-actin mRNA localization is disrupted, directionality and persistence of cell movement are significantly reduced (Shestakova et al, 2001). ZBP1/IMP1 contains two RNA recognition motifs (RRMs) and four KH domains, which bind the β-actin zipcode (Farina et al, 2003). Like many other RNA-binding proteins involved in mRNA transport, ZBP1/IMP1 is found in granular structures, which mostly coincide with β-actin mRNA (Zhang et al, 2001; Farina et al, 2003). Analysis of β-actin mRNA transport granules in neurons has shown that they contain ZBP1 (Zhang et al, 2001) and translational components (Bassell et al, 1998). Granule formation and RNA binding both require intact KH domains, a feature conserved between chicken ZBP1 (Farina et al, 2003) and human IMP1 (Nielsen et al, 2002).
One of the major problems in studying RNA–protein interactions is the inability to reconcile in vitro findings with observations of functions in vivo. This is particularly important for studies of interactions that take place during mRNA transport. The observation of RNA-binding protein movements does not always allow accurate assessment of where they contact mRNA. For example, GFP-Staufen cannot be observed to move to the posterior with oskar mRNA in Drosophila oocytes, possibly because most GFP-Staufen particles do not contain oskar mRNA, which is expressed at much lower levels than the fusion protein (Palacios and St Johnston, 2002). We wished to investigate possible RNA–protein interactions between candidate RNA-binding proteins, such as FMRP and IMP1, and mRNAs that are localized in vivo. To accomplish this, a system to visualize RNA–protein interactions was constructed. Using this system, we observed, for the first time, the association of RNA-binding proteins with specific RNA sequences in their native location in living cells.
Results
A method to visualize RNA–protein interactions in living cells
We have constructed a genetically encoded reporter of RNA–protein interactions. In this system, a portion of the Venus fluorescent protein (Nagai et al, 2002) is tethered to an mRNA of interest by the well-characterized bacteriophage MS2 coat protein–RNA operator interaction (Lim and Peabody, 1994) (Figure 1A). The complementary portion of Venus is fused to an RNA-binding protein of interest. If the RNA-binding protein is able to associate with the RNA sequence of interest, it will bring the two portions of Venus in close proximity to form a fluorescent complex (Figure 1B) while simultaneously identifying their site of interaction within the cell. The use of Venus, rather than yellow fluorescent protein, has advantages in that it folds faster and fluoresces more brightly (Nagai et al, 2002). The complementation of yellow fluorescent protein fragments in mammalian cells has previously been described as bimolecular fluorescence complementation (BiFC) (Hu et al, 2002); our RNA-bridged method represents trimolecular fluorescence complementation (TriFC).
Figure 1.
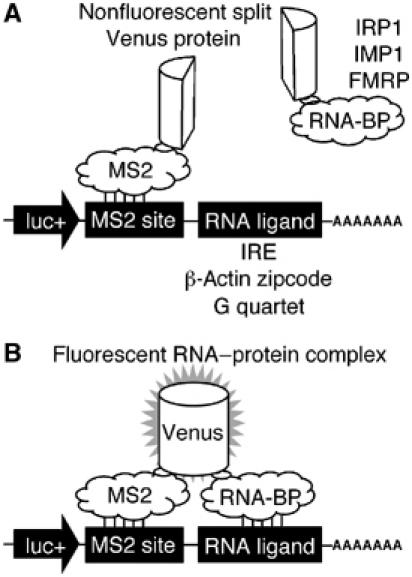
A TriFC method to study RNA–protein interactions in living cells. A portion of the Venus fluorescent protein is attached to a reporter mRNA by the bacteriophage MS2 coat protein–RNA interaction. The complementing portion of Venus is fused to an RNA-binding protein (A). If the RNA-binding protein interacts with a sequence of interest within the reporter mRNA, the two portions of Venus are brought into close proximity to form a fluorescent product (B).
To test our system, we incorporated the β-actin zipcode RNA sequence and the IMP1 coding sequence into TriFC plasmid vectors (Supplementary Figures S1, S2 and S3) and cotransfected them into COS-7 cells. We used COS-7 cells for this study because they have been shown to contain components of the β-actin mRNA localization machinery, and zipcode-containing mRNAs show significant directed motility when expressed in these cells (Fusco et al, 2003). They express low levels of FMRP and FXR2 but no detectable FXR1, as judged by Western analysis (data not shown). When the β-actin zipcode RNA and IMP1 were expressed in the TriFC system, a bright fluorescent signal was detected. The distribution of fluorescence was predominantly cytoplasmic, and particulates with large granules were visible throughout the cytoplasm (Figure 2A). COS-7 cells are asymmetric, and increased fluorescence was often observed at peripheral locations.
Figure 2.
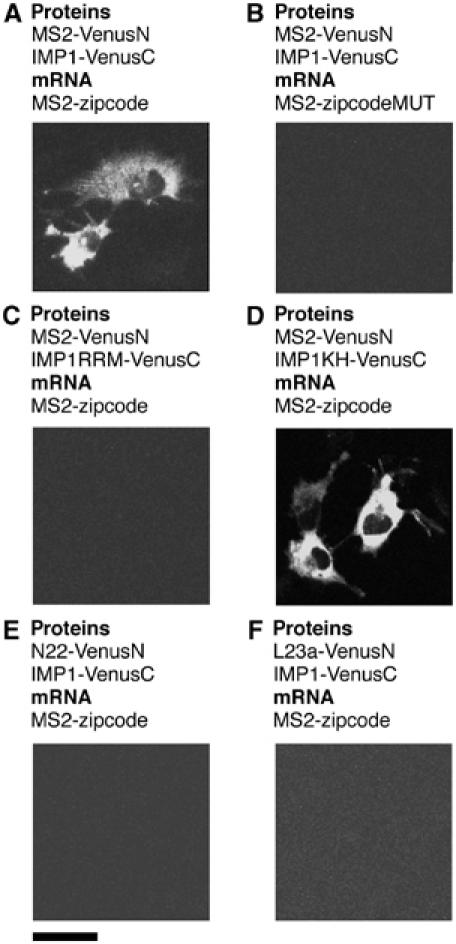
Detection of specific IMP1–zipcode interactions in living cells. Cells were transfected with plasmids expressing protein fusions to either the N- or C-terminal complementing portions of Venus (VenusN or VenusC, respectively) and reporter mRNAs as indicated. Scale bar, 20 μm.
To confirm the specificity of the signal, we expressed the MS2 and IMP1 fusion proteins in the absence of reporter mRNA and could not detect a fluorescent signal (Supplementary Figure S4A). If the zipcode sequence was removed from the reporter, mRNA fluorescence could no longer be detected (Supplementary Figure S4B). Likewise, the MS2 operator sequence was also necessary for fluorescence complementation (Supplementary Figure S4C). Mutation of a core element within the 54 nt chicken β-actin zipcode has previously been observed to prevent the peripheral localization of zipcode-containing mRNAs and abolish ZBP1 binding (Ross et al, 1997). If the corresponding changes were incorporated into the human β-actin zipcode used in this study, no interaction with IMP1 could be detected by TriFC (Figure 2B). This demonstrates that the interactions between IMP1 and the β-actin zipcode observed by TriFC were biologically relevant.
The RNA-binding activity of IMP1 has been mapped to the four tandem KH domains in its C-terminal section (Nielsen et al, 2002). We divided the coding sequence of IMP1 into two portions: one encompassing the N-terminal RRM domains and the other containing the four KH domains. In TriFC analysis, we observed that fluorescence complementation could only occur when the β-actin zipcode RNA was tested against the KH domain-containing portion of IMP1 (Figure 2C and D). This confirms previous in vitro work (Nielsen et al, 2002) and further validates the reliability of TriFC for the analysis of RNA–protein interactions.
When reporter mRNAs were expressed on separate plasmids, one RNA incorporating the MS2 operator sequence and the other incorporating the zipcode sequence, and coexpressed along with the MS2 and IMP1 fusion proteins, fluorescence was not observed (Supplementary Figure S4D). Therefore, both RNA elements had to be present on a single transcript for a signal to be detected. This indicates that the association of mRNAs bearing complementing portions of Venus fluorescent protein with similar complexes during processing, export or translation was not sufficient to give a response in the TriFC system. When we replaced the MS2 or IMP1 proteins with sequences from proteins N and L23a, respectively, no fluorescence was observed in either case (Figure 2E and F). Both N and L23a are strong RNA-binding protein, which do not play a role in β-actin mRNA localization. This further demonstrates that the interaction observed between IMP1 and the β-actin zipcode could be specifically detected using TriFC.
To extend the applicability of this approach, we looked at the well-studied interaction between the iron response element (IRE) stem–loop RNA structure and the iron regulatory protein (IRP1). In cells expressing IRE and IRP1 sequences in the TriFC system, we detected a cytoplasmic signal that was not granular as for the zipcode–IMP1 interaction (Figure 3A). This diffuse cytoplasmic fluorescence would be expected of nonlocalized mRNAs. This signal could not be detected in the absence of reporter mRNA (Figure 3D), if the IRE was absent from the reporter transcript (Figure 3C) or if a critical G in the loop of the IRE sequence was mutated to an A (Figure 3B). This single base change decreases the affinity for IRP1 by more than 100-fold in vitro (Ke et al, 1998), and as shown here eliminates fluorescence complementation in vivo.
Figure 3.
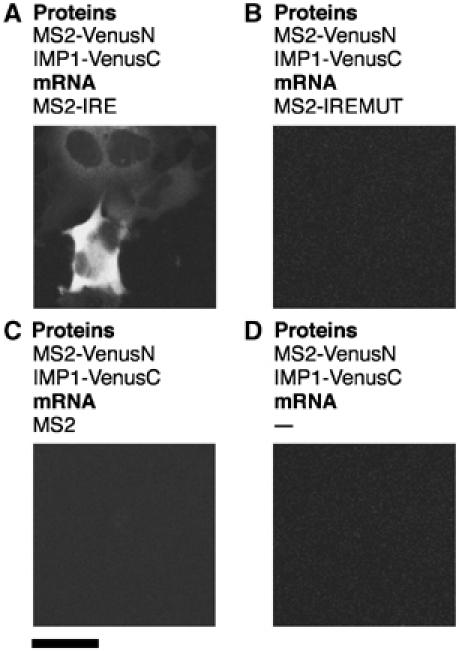
Detection of specific IRP1–IRE interactions in living cells. Cells were transfected with plasmids expressing the MS2 coat protein fusion to the N-terminal complementing portion of Venus protein (MS2-VenusN) and IRP1 fused to the C-terminal complementing portion of Venus (IRP1-VenusC), and reporter mRNAs as indicated. Scale bar, 20 μm.
Previous reports have demonstrated specific binding of FMRP to the 3′UTR of FMR1 mRNA (Brown et al, 1998; Li et al, 2001), although a recent study did not support this, demonstrating binding of FMRP to G quartet sequences in vitro (Schaeffer et al, 2001). These included a 101-base purine-rich sequence containing 12 GG doublets encoding the RGG box in the FMR1 mRNA (Schaeffer et al, 2001). The identification of two distinct FMRP attachment sites on FMR1 mRNA is not necessarily contradictory, as FMRP contains multiple, different RNA-binding domains and has affinity for distinct RNA homopolymers (Siomi et al, 1993). We examined the interaction between FMRP and the 3′UTR of FMR1 mRNA or G quartet RNA sequences. We chose the G quartet motif from the semaphorin 3F 3′UTR as it demonstrated strong binding to FMRP in vitro (Darnell et al, 2001) and showed an altered polysome association in fragile X cells (Brown et al, 2001), indicating that this would be a key interaction in vivo. This smaller 37-base motif contains five GG doublets. We isolated the FMR1 isoform 18 cDNA from a rat hippocampal library and used this as a source of the FMRP ORF and FMR1 3′UTR sequences. This isoform retains the NLS, NES, RRM and RGG domains but lacks several regions of unknown function.
Punctate fluorescence was detected predominantly in the cytoplasm when the FMRP–G quartet interaction was examined by TriFC (Figure 4A). If the semaphorin 3F sequences were removed (Figure 4C) or if two GG doublets predicted to be Hoogsteen bonded in the G quartet structure were substituted with C's, no fluorescence could be detected (Figure 4D). Interestingly, if one Hoogsteen-bonded G was replaced by a U, faint but significant fluorescence could be observed (Figure 4B). Here we also show that FMRP associates with its own mRNA's 3′UTR in living cells. The distribution of FMRP–FMR1 3′UTR interactions was indistinguishable from those of FMRP and the semaphorin 3F G quartet sequence (Figure 4E), indicating that FMRP may exhibit multiple attachment sites on its own mRNA. There are two conserved blocks of four and five GG doublets in FMR1 3′UTR that may form G quartets. However, whether they or other conserved blocks in the large, highly conserved 3′UTR are responsible for association with FMRP remains to be determined.
Figure 4.
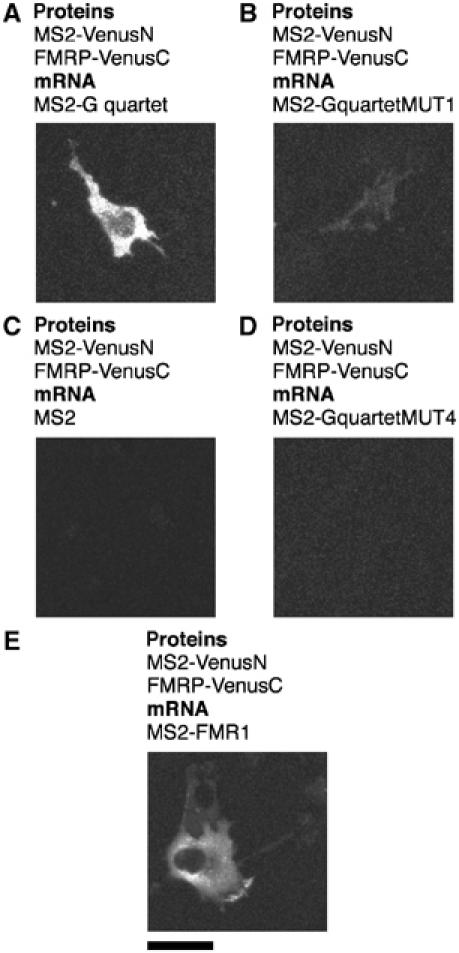
Detection of specific FMRP–RNA interactions in living cells. Cells were transfected with plasmids expressing the MS2 coat protein fusion to the N-terminal complementing portion of Venus protein (MS2-VenusN) and FMRP isoform 18 fused to the C-terminal complementing portion of Venus (VenusC), and reporter mRNAs containing the MS2 operator and additional sequences as indicated. Scale bar, 20 μm.
Common proteins associate with the β-actin zipcode-, FMR1 3′UTR- and G quartet-containing RNAs
Previous research has shown that both IMP1 (Zhang et al, 2001) and FMRP (De Diego Otero et al, 2002) form particles and are transported to peripheral processes of neurons. Given that the distributions of FMRP–FMR1 3′UTR (Figure 4E) and IMP1–β-actin zipcode interactions (Figure 2A) were very similar, we hypothesized that the two mRNAs might bind common regulatory factors. In addition to IMP1 and FMRP, we used the polypyrimidine tract-binding protein 1 (PTB1) and the human homolog of Drosophila staufen (hStau1). These were good candidates as PTB binds IGF-II RNA along with human IMP1 (Nielsen et al, 1999) and it associates with localized RNA in Xenopus oocytes (Cote et al, 1999). Staufen is a well-established mRNA localization factor in Drosophila (St Johnston et al, 1991), and in mammalian neurons hStau1 associates with RNA and travels to the distal cytoplasm of neuronal dendrites (Kohrmann et al, 1999).
When tested by TriFC, all four test proteins bound to the β-actin zipcode, the FMR1 3′UTR and the semaphorin 3F G quartet sequences (Figure 5). FMRP, IMP1 and hStau1 all interacted with RNA predominantly in the cytoplasm. In contrast, PTB interactions occurred only in the nucleoplasm (Figure 5). These interactions were specific as none of the tested proteins interacted with a reporter mRNA containing only the MS2 operator in its 3′UTR (Figure 5) or a mutated β-actin zipcode sequence (Supplementary Figure S5). The mRNA of c-myc had previously been shown to be localized to the perinuclear cytoplasm (Veyrune et al, 1996), and could be used to test whether any of these proteins were general mRNA localization factors. FMRP, IMP1 and PTB did not interact with a reporter containing the c-myc 3′UTR (Figure 5). However, cytoplasmic fluorescence that was often enriched around the nuclei of transfected cells was observed for hStau1 and the c-myc 3′UTR in TriFC analysis (Figure 5). This indicates that hStau1 is a general factor involved in the transport of mRNAs to several locations in the cells. This hypothesis is supported by the finding that Staufen is required for both anterior and posterior mRNA localization in Drosophila oocytes (St Johnston et al, 1991). We consistently found that approximately 80% of COS-7 cells fluoresced when transfected with TriFC plasmids expressing the β-actin zipcode sequence and IMP1. These corresponded to the transfected cells in the population as determined by coexpression of a cotransfected mRFP1 fusion protein (data not shown). This was observed for all positive associations detected by TriFC, indicating that the association of these molecules was not cell state dependent in this context.
Figure 5.

IMP1, FMRP, hStau and PTB associate with multiple mRNA sequences in vivo. Cells were transfected with plasmids expressing the MS2 coat protein fused to the N-terminal complementing portion of Venus, an RNA-binding protein (IMP1, FMRP, hStau or PTB) fused to the C-terminal complementing portion of Venus, and a reporter mRNA containing an MS2 coat protein recognition site and RNA sequence of interest as indicated. Scale bar, 20 μm.
Since the interaction of FMRP with the β-actin zipcode and that of IMP1 with the FMR1 3′UTR had not previously been tested, we also examined these findings with immunoprecipitation experiments. Cells were transfected with plasmids designed to express the MS2 coat protein fused to enhanced green fluorescent protein (EGFP), a reporter mRNA incorporating the β-actin zipcode or FMR1 3′UTR sequences, and either FLAG-tagged IMP1 or red fluorescent protein-tagged FMRP. Immunoprecipitation from lysates with anti-GFP antibodies followed by immunoblotting showed a specific association between IMP1 and the β-actin zipcode or FMR1 3′UTR RNA sequences, as well as FMRP and the β-actin zipcode or FMR1 3′UTR RNA sequences (Figure 6). FMRP and IMP1 could not be detected in immmunoprecipitations from cells expressing reporter mRNAs without β-actin zipcode or FMR1 3′UTR RNA sequences, confirming the results obtained using the new TriFC method. Only a small proportion (∼5%) of the total tagged IMP1 or FMRP was present in the precipitated complex. This may indicate that these proteins are bound to other mRNAs or involved in other processes in the cell; however, only the location of the interacting fraction is specifically observed by TriFC.
Figure 6.

IMP1 and FMRP associate with zipcode and FMR1 3′UTR RNA sequences. Immunoprecipitation confirms the association of IMP1 and FMRP with β-actin zipcode- and FMR1 3′UTR-containing mRNAs. An EGFP fusion to the MS2 coat protein allowed the immunoprecipitation of reporter mRNAs containing an MS2 coat protein recognition site and an RNA sequence of interest. Immunoprecipitations were performed using anti-GFP antibodies. FMRP and IMP1 were coexpressed as mRFP1 and FLAG-tagged fusion proteins, respectively. The theoretical molecular weights of FMRP-mRFP1 and FLAG-IMP1 are 91 and 66 kDa, respectively. The efficiency of immunoprecipitation was shown by immunoblotting with anti-GFP antibodies (lower panel). The theoretical molecular weight of MS2-EGFP is 40 kDa. The association of FMRP and IMP1 with reporter mRNAs was determined by immunoblotting with anti-FLAG and anti-FMRP antibodies, respectively.
IMP1 and FMRP associate independently of RNA
Because both IMP1 and FMRP associated with β-actin zipcode, G quartet and FMR1 3′UTR sequences in the cytoplasm, we wanted to determine if these two proteins interacted by BiFC analysis directly or indirectly via a bridging molecule. Expression of FMRP and IMP1 fused to complementing portions of the Venus fluorescent protein yielded a strong fluorescent signal that was particularly granular in appearance (Figure 7). As negative controls, IRP1 and L23a did not show detectable fluorescence when tested against IMP1 or FMRP (Figure 7). As a positive control, we confirmed the homodimerization of FMRP that has been observed previously in vitro (Laggerbauer et al, 2001). In addition to FMRP self-association, we also detected strong fluorescence when IMP1 was fused to complementing portions of the Venus fluorescent protein (Figure 7). Homodimerization has previously been observed for the Xenopus homolog of IMP1, VgRBP (Git and Standart, 2002), but had not been reported for chicken or mammalian IMP1 family proteins.
Figure 7.

FMRP and IMP1 interact in the absence of reporter mRNA. Expression of protein fusions to complementing portions of Venus demonstrated homomeric and heteromeric interactions between FMRP and IMP1. The unrelated IRP1 and L23a RNA-binding proteins do not interact with either FMRP or IMP1.
To determine whether the interaction between IMP1 and FMRP was RNA dependent, we performed immunoprecipitation experiments in the presence of RNase A or an RNase inhibitor, in parallel to the results shown in Figure 7. VenusC-tagged FMRP could effectively pull down IMP1, in a manner that was independent of RNA (Figure 8A). Likewise, tagged FMRP co-immunoprecipitated with IMP1 in the presence of RNase A (Figure 8B). This indicates that IMP1 and FMRP interact with each other without a bridging RNA.
Figure 8.

FMRP and IMP1 associate via protein–protein interactions. (A) Cells were transfected with plasmids expressing FMRP fused to VenusC and FLAG-tagged IMP1. FMRP-VenusC was immunoprecipitated with an anti-GFP antibody. To test for nonspecific immunoprecipitation, an irrelevant antibody, anti-COX IV, was used in a control reaction. Immunoblotting with anti-GFP antibodies was used to confirm the immunoprecipitation of FMRP-VenusC (theoretical molecular weight of 93 kDa). The association of FLAG-IMP1 (theoretical molecular weight of 66 kDa) was determined by immunoblotting with anti-FLAG antibodies. A reaction in which RNase A was used in place of RNasin determined the RNA-independent nature of the interaction. (B) Cells were transfected with plasmids expressing an IMP1-Venus fusion protein and mRFP1 tagged FMRP (both of theoretical molecular weight 91 kDa). Immunoprecipitations were performed as in (A). The association of FMRP with IMP1 was determined by immunoblotting with anti-FMRP antibodies. In a control reaction, lysates expressing only FMRP-mRFP1 were subjected to immunoprecipitation.
IMP1 and FMRP form a complex on mRNAs and promote granule formation
Given that IMP1 and FMRP form a complex, we hypothesized that the binding of one could recruit the other to an mRNA. To test this, we fused either IMP1 or FMRP to the MS2 coat protein. The MS2 coat protein folds as an obligate dimer; therefore, one or the other of these fusions could be expressed alongside the components of the TriFC to provide a fraction of reporter mRNAs carrying MS2 dimers with both a portion of Venus and either IMP1 or FMRP attached (Figure 9A). When FMRP was tethered to a reporter mRNA, IMP1 was efficiency recruited, as judged by fluorescent complex formation (Figure 9B). Similarly, tethered IMP1 could recruit FMRP to a common RNA–protein complex (Figure 9B). Fluorescence was punctate in the cytoplasm, indicating that the association of IMP1/FMRP is sufficient to sequester mRNAs into granules. This was specific, as tethering an unrelated protein, human placental alkaline phosphatase (hPLAP), to mRNA did not result in detectible fluorescence (Figure 9B).
Figure 9.
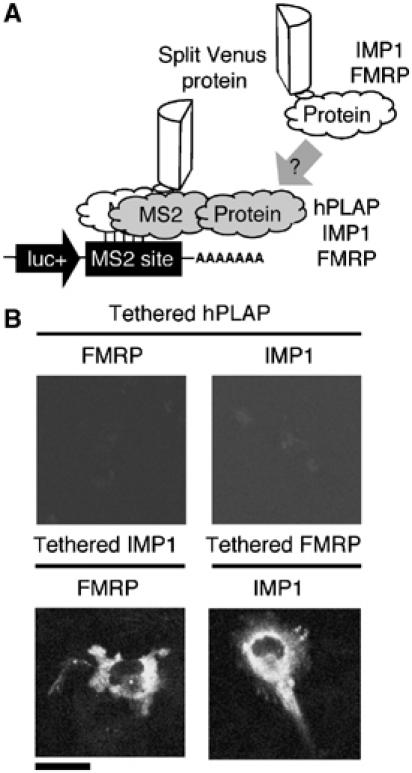
Tethered IMP1 recruits FMRP to an mRNA, and tethered FMRP can recruit IMP1 to an mRNA. (A) Schematic representation of how TriFC can be used to analyze the recruitment of specific protein to an mRNP complex. (B) Cells were transfected with plasmids expressing reporter mRNA containing the MS2 coat protein recognition site and the MS2 coat protein fused to the N-terminal complementing portion of Venus. A soluble form of hPLAP, IMP1 or FMRP was tethered to the reporter mRNA by fusion to the MS2 coat protein sequence. Recruitment of IMP1 or FMRP to reporter mRNAs was determined by coexpression of fusions to the C-terminal portion of Venus.
Discussion
The TriFC method described here enables simple detection and localization of RNA–protein interactions within a living cell. As RNAs and proteins can be expressed in their native location, both the physical and biological properties of these complexes can be investigated. The nature of TriFC necessitates the expression of its components in a transgenic manner; however, the sensitivity of the Venus fluorescent protein (Rekas et al, 2002) should allow the detection of most interactions when components are expressed at low, physiologically relevant levels. The ability to visualize the sites of RNA–protein association removes the uncertainty involved in indirect methods of analysis. In addition, the ability to study RNA–protein interactions directly in living cells enables interactions that are likely to be biologically relevant to be sorted from those that are not, a limitation of in vitro assays. In this case, although FMRP binding to the FMR1 3′UTR had been reported (Brown et al, 1998), Schaeffer et al (2001) argued recently that the more avid binding to a G quartet sequence in the coding region would be the only biologically relevant interaction in vivo. Here we show that contrary to that proposal, FMRP is found associated with the FMR1 3′UTR in living cells, indicating that this is a significant binding site. Therefore, FMRP may exhibit multiple attachment sites within its own mRNA.
The observation of RNA–protein interactions by TriFC in vivo has one significant limitation, as do all in vivo systems. Because complexes form in an extremely heterogeneous natural environment, endogenous proteins or RNAs could bring TriFC components into close proximity. This enables fluorescence to be detected in cases when TriFC components do not interact directly, but associate via a bridging RNA or protein. This can be resolved by analysis of purified components in vitro or by RNAi knock down of candidate bridging molecules. It is important to note that this potential also represents a major strength of TriFC. Heterologous in vivo systems or in vitro analysis of purified components would not allow these interactions to be detected. It is noteworthy that if RNA-binding proteins form part of the final mRNP, they would be expected to be important in its regulation (de Moor and Richter, 2001). The extent to which this occurs in vivo is not currently understood, due to a lack of methods that enable this kind of analysis. TriFC can be used to address the questions that existing methods for studying RNA–proteins interactions are not well suited to answer.
Using TriFC analysis, we have demonstrated that the β-actin zipcode, FMR1 3′UTR and the semaphorin 3F G quartet RNA sequences associate with a similar set of RNA-binding proteins. Two of these RNA-binding proteins, FMRP and IMP1, associate with each other via protein–protein interactions. The binding of one of these two proteins to an mRNA is sufficient to recruit the other and form granules. This indicates that FMRP and IMP1 show multiple modes of attachment to mRNAs, either by directly binding RNA or by interacting with other RNA-bound proteins. The extent to which endogeneous FMRP and IMP1 interact in various tissues and physiological states remains to be determined. Future studies of complex formation and colocalization in tissues with specific antibodies will be needed to address these issues. The ability of FMRP to attach to mRNAs in distinct ways may help explain the many roles that FMRP plays in vivo (Antar and Bassell, 2003).
The Drosophila FMRP homolog (dFMR1) has recently been found associated with Argonaute-2 (Ago-2), a component of the RNA-induced silencing complex (RISC) (Caudy et al, 2002; Ishizuka et al, 2002). The RISC is a large RNA–protein complex involved in the sequence-specific post-transcriptional silencing of gene expression via small interfering RNAs (siRNAs) or micro-RNAs (miRNAs) (Carthew, 2002). These new findings of an interaction between dFMR1 and the RISC could present a new way for FMRP to associate with its targets, via hydrogen bonding of small RNA templates. Interestingly, the untranslated BC1 RNA was found in FMRP immunoprecipitates from mouse brains (Zalfa et al, 2003). BC1 was found to mediate the association of FMRP with MAP1B mRNA by direct base-pairing (Zalfa et al, 2003), indicating that, at least in one case in mammals, small RNAs can modify the activity of FMRP. New data presented in our article show that a protein with a well-defined role in RNA transport (IMP1/ZBP1) can interact with a protein with a known role in gene silencing (FMRP). The association of ZBP1 and the β-actin zipcode RNA sequence can occur with high affinity using purified components (Farina et al, 2003). This argues that the binding of ZBP1/IMP1 to mRNAs does not require siRNAs or miRNAs. In addition, we have shown that the association of IMP1 and FMRP is RNA independent. Hence it seems unlikely that the association of IMP1 and FMRP on β-actin zipcode-containing mRNAs requires additional small RNAs. This does not rule out the possibility that FMRP-associated RISC components could play a role in the mechanistic aspects of translational silencing of IMP1/FMRP-bound mRNAs. The precise mechanisms used by FMRP to modify the expression of its target mRNAs are unclear and require further study.
ZBP1/IMP1 has a well-established role in mRNA transport in fibroblasts and neurons. It has been proposed that localized mRNAs are translationally silenced during transport. In neurons, mRNAs contained within transport granules are not competent for translation (Krichevsky and Kosik, 2001). Indeed given that ribosomes are likely to form a matrix in the cell cytoplasm (Rocha et al, 2000), association with polysomes may prevent efficient mRNA transport. Experimental support for this idea has been obtained in neurons, where ferritin mRNA, which is normally confined to the cell body, can efficiently migrate into dendrites following treatment with translational inhibitors (Lu et al, 1998). Our finding that IMP1 forms a complex with FMRP provides a possible link between mRNA transport factors and a protein that could function as a translational repressor in some circumstances. We propose a model, in which FMRP inhibits the translation of IMP1-associated mRNAs to facilitate efficient transport of mRNPs (Figure 10). It is possible that FMRP inhibits the translation of localized mRNAs, such as β-actin, only during transport so that translation is delayed but the net amount of protein synthesized is not lowered. A role for FMRP in β-actin mRNA regulation is supported by the observation that the dendritic spines of fragile X patients are dysmorphic (Rudelli et al, 1985), resembling filopodia. The transition from immature filopodia to mature spines is accompanied by the deposition of high local concentrations of β-actin (Zhang and Benson, 2002). Loss of translational repression could impair the efficiency of β-actin mRNA transport, impairing β-actin protein deposition at synaptic sites. Some FMRP-bound mRNAs show reduced dendritic localization in neurons harvested from Fmr1 knockout mice (Miyashiro et al, 2003). It is possible that a role of FMRP in the life cycle of these mRNAs is to recruit IMP1 by protein–protein interactions. This could then enable the formation of transport granules, and subsequent dendritic mRNA localization.
Figure 10.
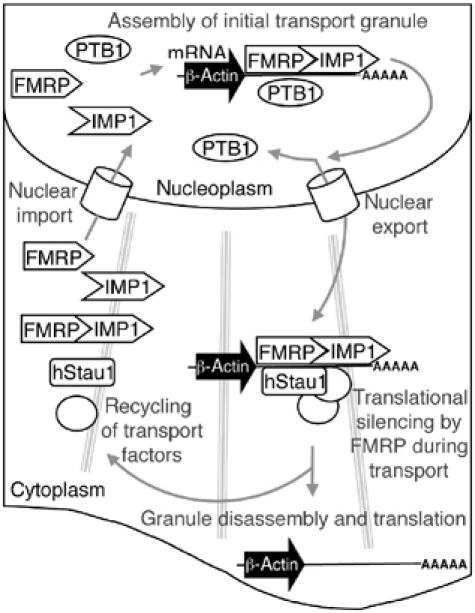
Model of the mRNA localization process involving IMP1 and FMRP. The localization process begins in the nucleus, with IMP1 and FMRP associating with the RNA at the transcription site. In the example shown, the target is β-actin mRNA. IMP1 and FMRP may bind sequentially, or attach as a preformed complex. The binding of PTB in the nucleus may in some way facilitate the granule assembly or nuclear export processes. The hStau1 protein could attach in the nucleus or the cytoplasm along with other general mRNA localization components, such as molecular motors. The granule then travels to its final location at the cell periphery. While in transit, attached FMRP silences the translation of transported mRNAs. Once anchored at its final location, the granule complex is disassembled. Removal of FMRP leaves mRNAs competent for translation. The free transport factors then return to take part in further rounds of mRNA localization.
Another RNA-binding protein, PTB1, also associated with semaphorin 3F 3′UTR G quartet, the FMR1 3′UTR and the β-actin zipcode. These interactions were distinct from those with IMP1, FMRP and hStau1, as they were confined to the nucleoplasm. A number of nuclear-cytoplasmic shuttling RNA-binding proteins, including PTB1, have been implicated in mRNA localization (Wilkinson and Shyu, 2001), but a direct role for PTB1 in peripheral mRNA localization is unlikely given the exclusively nuclear sites of interactions demonstrated here. It is notable that in yeast an exclusively nuclear protein has been implicated in ASH1 mRNA localization (Long et al, 2001) and subsets of mRNAs are differentially associated with the nuclear export machinery (Hieronymus and Silver, 2003). A pathway for localized mRNAs that begins in the nucleus of mammalian cells is supported by our findings.
Human Stau1 was associated with the c-myc 3′UTR. This association was observed in a distribution dissimilar to the sites of hStau1–β-actin zipcode, hStau1–FMR1 3′UTR and hStau1–G quartet interactions. As has previously been observed for reporter mRNAs containing the c-myc 3′UTR (Veyrune et al, 1996), fluorescence for the hStau1–c-myc 3′UTR interaction was enriched in the perinuclear cytoplasm. The association of hStau1 with mRNAs that are transported to distinct locations is at first surprising. However, Staufen is required for both anterior and posterior mRNA localization in Drosophila oocytes (St Johnston et al, 1991). These observations suggest that Staufen has a general role in mRNA transport. This is supported by the observation that rat Staufen appears to be responsible for bulk RNA transport to neuronal dendrites (Tang et al, 2001). Recently, 2–5% of nonlocalized mRNA molecules were observed to display directed, microtubule-dependent movements (Fusco et al, 2003), further indicating that a general mRNA transport machinery exists.
We show that all four IMP1, FMRP, PTB and hStau proteins are present on three different mRNAs. Since it is well established that these proteins do not share sequence specificity in their RNA-binding characteristics, we conclude that a sequence-specific RNA-binding protein most likely binds first to the RNA and provides a platform where the other proteins can subsequently attach. In the case of β-actin zipcode RNA, this role would be performed by IMP1/ZBP1. This is supported by three lines of evidence. Firstly, it has been shown that ZBP1, the IMP1 ortholog, binds to β-actin mRNA immediately as it is transcribed in the nucleus (Oleynikov and Singer, 2003). Secondly, the interaction of ZBP1 and the β-actin zipcode RNA sequence can occur with high affinity using purified components (Farina et al, 2003). Thirdly, we have shown that FMRP can associate with IMP1 by protein–protein interactions, providing a mode for subsequent attachment of RNA-binding proteins in a way that is independent of direct RNA binding. The association of these proteins on mRNAs may be conserved through evolution. In Xenopus oocytes, the IMP1 homolog VgRBP and the PTB homolog colocalize with Vg1 mRNA (Cote et al, 1999; Kroll et al, 2002). In addition, the PTB homolog Smooth is found in the bicoid mRNA localization complex from Drosophila oocytes. As this localization event requires Staufen (Arn et al, 2003), these two proteins may also associate on bicoid mRNA. These observations raise the possibility that these RNA-binding proteins often act as sets to regulate the life cycles of mRNA. The TriFC system can facilitate the rapid testing of RNA–protein interactions in vivo and places these events in their correct subcellular location. Understanding how these arrays of interactions influence mRNA expression remains a major challenge for future work.
Materials and methods
Plasmid expression vectors
All expression vectors were based on pRL-null (Promega, NCBI accession number AF025844), and expressed proteins and mRNAs of interest from the SV40 promoter, subcloned from pGL3-Control (Promega, U47296). Reporter mRNAs contain a human optimized firefly luciferase (luc+, from pGL3-Control) followed by an MS2 coat protein recognition site (AAACAAGAGGATTACCCTTGT). The parental vector contained a small cloning cassette (5′-NheI, NotI, NgoMIV, XhoI, EcoRV-3′) 3′ to the MS2 coat protein recognition site. RNA sequences of interest were synthesized as oligonucleotides or by PCR and inserted into the cloning cassette. Sequences used in this study were as follows: the human β-actin zipcode (nt 1202–1260, NM_001101) and a mutated version with 10 nt substitutions according to Ross et al (1997); an IRE from the rabbit ferritin 5′UTR (nt 33–60, X14578) and a mutated version corresponding to the HL1 variant from Ke et al (1998); a G quartet sequence from the human semaphorin 3F 3′UTR (nt 2536–2572, NM_004186), a mutated version with one G changed to a T (G2549T), and a mutated version where two pairs of Hoogsteen-bonded G's were replaced with C's (G2548C, G2549C, G2552C, G2553C); the full-length c-myc 3′UTR (nt 1741–2056, K02276); and the full-length rat FMR1 3′UTR (nt 1927–4189, AY240947).
Fusion proteins were joined by a linker encoding DVLE and the initiator M of the C-terminal fusion partner was replaced by H in all plasmids. Protein sequences used were as follows: the MS2 bacteriophage coat protein (amino acids (aa) 1–129, V29I/dIFG variant of 721932A) (Lim and Peabody, 1994); the EGFP (Clontech, aa 1–239, AAB02572); the Venus fluorescent protein (Nagai et al, 2002) (EYFP with mutations F46L/F64L/M153T/V163A/S175G, aa 1–154 or 155–239); monomeric red fluorescent protein (mRFP1, aa 1–225, AAM54544) (Campbell et al, 2002); the human IMP1 (full length: aa 1–577; RRM domain-containing segment: aa 1–196; KH domain-containing segment: aa 197–577; NP_006537); rat ribosomal protein L23a (aa 1–156, CAA46336); the rat polypyrimidine tract-binding protein (PTB1, aa 1–530, CAA43202); human staufen protein (hStau1, aa 1–496, NP_059348); rabbit IRP1 (aa 1–889, AAA31255); the RNA-binding peptide from bacteriophage lambda N protein (aa 1–22, AAA56728); FMRP isoform 18 (aa 1–581, AAP15341); and a soluble form of hPLAP (aa 24–506, AAA51706). A plasmid that expresses FLAG-tagged IMP1 was generated by subcloning the full-length coding region into pFLAG-CMV2 (Sigma). All fusion proteins were tested for expression by transfection and Western blotting (Supplementary Figure S6).
Cell culture
COS-7 cells were grown at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum (FBS), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. Cells were transfected using FuGENE6 (Roche). Plasmids for expression of RNA reporters were transfected at 4 × the molarity of fusion protein expression plasmids.
Microscopy
Cells for microscopy were plated on poly-D-lysine-coated 35 mm tissue culture dishes with number 0 coverslip bottoms (MatTek). Images of live cells were acquired using a Ziess 510 Axiovert 200M inverted confocal microscope with a Plan-Neofluor × 40/1.3 oil DIC objective 48 h after transfection. The 514 nm line of an argon laser was used in conjunction with a 530 nm long-pass emission filter and Zeiss LSM 510 Imaging Software. Images are representative of the majority of transfected cells, observed in at least three independent experiments. In order to confirm that the combinations of plasmids that did not fluoresce did enter the cells, cotransfection experiments were performed with an L23a-mRFP1 fusion protein expression plasmid. This also showed that transfection efficiencies were comparable for different TriFC experiments.
Immunoprecipitation
Cells were transfected in 100 mm diameter dishes (BD Falcon). After 48 h, cells were washed with PBS and extracted in 550 μl lysis buffer containing 50 mM Tris–HCl (pH 8.0), 150 mM KCl, 1 mM EGTA, 5 mM MgCl2, 1 mM DTT, 0.25% Nonidet P-40, 0.05% deoxycholic acid, 40 U ml−1 RNasin (Promega) and 1 × EDTA-free complete protease inhibitor cocktail (Roche) for 20 min at 4°C. To determine if the FMRP–IMP1 interaction was RNA dependent, a lysis buffer that contained 10 μg ml−1 RNase A instead of RNasin was used. Cell lysates were collected by scraping and vigorous mixing, and cell debris was removed by centrifugation at 16 000 g for 1 min. The clarified lysate (500 μl) was incubated for 15 min with 3 μg anti-GFP (1 814 460, Roche) or anti-COX IV (20E8, Molecular Probes) monoclonal antibodies, and 0.9 μg antibody affinity capture ligand (Upstate). This mixture was applied to a prewetted spin column containing IP capture resin (Upstate) and washed four times with 500 μl of lysis buffer by centrifugation at 1500 g. Immune complexes were collected in 60 μl 1 × elution buffer (Upstate) by centrifugation at 500 g. Aliquots of 20 μl were used for Western blotting.
SDS–PAGE and Western blotting
Cell samples were denatured in loading buffer (final concentration: 50 mM Tris, 4% SDS, 12% glycerol, 2% 2-mercaptoethanol, 0.01% Coomassie brilliant blue) for 10 min at 94°C and separated on 12.5% Tris-glycine PAGE gels. Gels were then electrotransfered onto 0.2 μm nitrocellulose (100 V, 1 h) in transfer buffer (25 mM Tris–HCl (pH 8.3), 192 mM glycine, 20% methanol) and then blocked with 2% (w/v) fat-free milk powder in TBS (5 mM Tris–HCl (pH 7.4), 20 mM NaCl) and 0.1% Tween 20. Anti-FMRP (1C3, Chemicon), anti-FLAG (M2, Sigma) or anti-GFP (1 814 460, Roche) monoclonal antibodies were diluted 1:5000 in TBS, 0.1% fat-free milk powder and 0.1% Tween 20 and used for immunodetection by overnight incubation. After three 10 min washes in TBS and 0.1% Tween 20, horseradish peroxidase-conjugated goat anti-mouse IgG (1:10 000, Bio-Rad) was used as a secondary antibody. The secondary antibody binding was carried out for 1 h at room temperature, followed by three 10 min washes in TBS. Antibody-bound proteins were visualized using a chemiluminescent substrate.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Acknowledgments
We thank Jan Christiansen, Luc DesGroseillers, Thomas Hughes, Annie Kao, Tom Kerppola, Atsushi Miyawaki, David Peabody, Roger Tsien and Marvin Wickens for generously providing plasmid constructs, Andrew McNaughton for assistance with confocal microscopy, Annie Chien and Nattanan Panjaworayan for providing data prior to publication and John Cutfield and Richard MacKnight for critical reading of the manuscript. Oliver Rackham was supported by a University of Otago postgraduate scholarship. This work was supported by Marsden Fund of NZ and Health Research Council grants to Chris M Brown and Warren Tate.
References
- Antar LN, Bassell GJ (2003) Sunrise at the synapse: the FMRP mRNP shaping the synaptic interface. Neuron 37: 555–558 [DOI] [PubMed] [Google Scholar]
- Arn EA, Cha BJ, Theurkauf WE, Macdonald PM (2003) Recognition of a bicoid mRNA localization signal by a protein complex containing Swallow, Nod, and RNA binding proteins. Dev Cell 4: 41–51 [DOI] [PubMed] [Google Scholar]
- Bassell GJ, Zhang H, Byrd AL, Femino AM, Singer RH, Taneja KL, Lifshitz LM, Herman IM, Kosik KS (1998) Sorting of beta-actin mRNA and protein to neurites and growth cones in culture. J Neurosci 18: 251–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown V, Jin P, Ceman S, Darnell JC, O'Donnell WT, Tenenbaum SA, Jin X, Feng Y, Wilkinson KD, Keene JD, Darnell RB, Warren ST (2001) Microarray identification of FMRP-associated brain mRNAs and altered mRNA translational profiles in fragile X syndrome. Cell 107: 477–487 [DOI] [PubMed] [Google Scholar]
- Brown V, Small K, Lakkis L, Feng Y, Gunter C, Wilkinson KD, Warren ST (1998) Purified recombinant Fmrp exhibits selective RNA binding as an intrinsic property of the fragile X mental retardation protein. J Biol Chem 273: 15521–15527 [DOI] [PubMed] [Google Scholar]
- Calkhoven CF, Muller C, Leutz A (2002) Translational control of gene expression and disease. Trends Mol Med 8: 577–583 [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY (2002) A monomeric red fluorescent protein. Proc Natl Acad Sci USA 99: 7877–7882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carthew RW (2002) RNA interference: the fragile X syndrome connection. Curr Biol 12: R852–R854 [DOI] [PubMed] [Google Scholar]
- Caudy AA, Myers M, Hannon GJ, Hammond SM (2002) Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev 16: 2491–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conne B, Stutz A, Vassalli JD (2000) The 3′ untranslated region of messenger RNA: a molecular ‘hotspot' for pathology. Nat Med 6: 637–641 [DOI] [PubMed] [Google Scholar]
- Cote CA, Gautreau D, Denegre JM, Kress TL, Terry NA, Mowry KL (1999) A Xenopus protein related to hnRNP I has a role in cytoplasmic RNA localization. Mol Cell 4: 431–437 [DOI] [PubMed] [Google Scholar]
- Darnell JC, Jensen KB, Jin P, Brown V, Warren ST, Darnell RB (2001) Fragile X mental retardation protein targets G quartet mRNAs important for neuronal function. Cell 107: 489–499 [DOI] [PubMed] [Google Scholar]
- De Diego Otero Y, Severijnen LA, van Cappellen G, Schrier M, Oostra B, Willemsen R (2002) Transport of fragile X mental retardation protein via granules in neurites of PC12 cells. Mol Cell Biol 22: 8332–8341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Moor CH, Richter JD (2001) Translational control in vertebrate development. Int Rev Cytol 203: 567–608 [DOI] [PubMed] [Google Scholar]
- Eberhart DE, Malter HE, Feng Y, Warren ST (1996) The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum Mol Genet 5: 1083–1091 [DOI] [PubMed] [Google Scholar]
- Farina KL, Huttelmaier S, Musunuru K, Darnell R, Singer RH (2003) Two ZBP1 KH domains facilitate beta-actin mRNA localization, granule formation, and cytoskeletal attachment. J Cell Biol 160: 77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST, Hersch SM (1997) Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci 17: 1539–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco D, Accornero N, Lavoie B, Shenoy SM, Blanchard JM, Singer RH, Bertrand E (2003) Single mRNA molecules demonstrate probabilistic movement in living mammalian cells. Curr Biol 13: 161–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Git A, Standart N (2002) The KH domains of Xenopus Vg1RBP mediate RNA binding and self-association. RNA 8: 1319–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieronymus H, Silver PA (2003) Genome-wide analysis of RNA–protein interactions illustrates specificity of the mRNA export machinery. Nat Genet 33: 155–161 [DOI] [PubMed] [Google Scholar]
- Hu CD, Chinenov Y, Kerppola TK (2002) Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell 9: 789–798 [DOI] [PubMed] [Google Scholar]
- Ishizuka A, Siomi MC, Siomi H (2002) A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev 16: 2497–2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y, Wu J, Leibold EA, Walden WE, Theil EC (1998) Loops and bulge/loops in iron-responsive element isoforms influence iron regulatory protein binding. Fine-tuning of mRNA regulation? J Biol Chem 273: 23637–23640 [DOI] [PubMed] [Google Scholar]
- Kislauskis EH, Zhu X, Singer RH (1994) Sequences responsible for intracellular localization of beta-actin messenger RNA also affect cell phenotype. J Cell Biol 127: 441–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohrmann M, Luo M, Kaether C, DesGroseillers L, Dotti CG, Kiebler MA (1999) Microtubule-dependent recruitment of Staufen-green fluorescent protein into large RNA-containing granules and subsequent dendritic transport in living hippocampal neurons. Mol Biol Cell 10: 2945–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krichevsky AM, Kosik KS (2001) Neuronal RNA granules: a link between RNA localization and stimulation-dependent translation. Neuron 32: 683–696 [DOI] [PubMed] [Google Scholar]
- Kroll TT, Zhao WM, Jiang C, Huber PW (2002) A homolog of FBP2/KSRP binds to localized mRNAs in Xenopus oocytes. Development 129: 5609–5619 [DOI] [PubMed] [Google Scholar]
- Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A, Fischer U (2001) Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum Mol Genet 10: 329–338 [DOI] [PubMed] [Google Scholar]
- Lawrence JB, Singer RH (1986) Intracellular localization of messenger RNAs for cytoskeletal proteins. Cell 45: 407–415 [DOI] [PubMed] [Google Scholar]
- Li Z, Zhang Y, Ku L, Wilkinson KD, Warren ST, Feng Y (2001) The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res 29: 2276–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim F, Peabody DS (1994) Mutations that increase the affinity of a translational repressor for RNA. Nucleic Acids Res 22: 3748–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long RM, Gu W, Meng X, Gonsalvez G, Singer RH, Chartrand P (2001) An exclusively nuclear RNA-binding protein affects asymmetric localization of ASH1 mRNA and Ash1p in yeast. J Cell Biol 153: 307–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, McLaren RS, Winters CA, Ralston E (1998) Ribosome association contributes to restricting mRNAs to the cell body of hippocampal neurons. Mol Cell Neurosci 12: 363–375 [DOI] [PubMed] [Google Scholar]
- Miyashiro KY, Beckel-Mitchener A, Purk TP, Becker KG, Barret T, Liu L, Carbonetto S, Weiler IJ, Greenough WT, Eberwine J (2003) RNA cargoes associating with FMRP reveal deficits in cellular functioning in Fmr1 null mice. Neuron 37: 417–431 [DOI] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A (2002) A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol 20: 87–90 [DOI] [PubMed] [Google Scholar]
- Nielsen FC, Nielsen J, Kristensen MA, Koch G, Christiansen J (2002) Cytoplasmic trafficking of IGF-II mRNA-binding protein by conserved KH domains. J Cell Sci 115: 2087–2097 [DOI] [PubMed] [Google Scholar]
- Nielsen J, Christiansen J, Lykke-Andersen J, Johnsen AH, Wewer UM, Nielsen FC (1999) A family of insulin-like growth factor II mRNA-binding proteins represses translation in late development. Mol Cell Biol 19: 1262–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleynikov Y, Singer RH (2003) Real-time visualization of ZBP1 association with beta-actin mRNA during transcription and localization. Curr Biol 13: 199–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios IM, St Johnston D (2001) Getting the message across: the intracellular localization of mRNAs in higher eukaryotes. Annu Rev Cell Dev Biol 17: 569–614 [DOI] [PubMed] [Google Scholar]
- Palacios IM, St Johnston D (2002) Kinesin light chain-independent function of the kinesin heavy chain in cytoplasmic streaming and posterior localisation in the Drosophila oocyte. Development 129: 5473–5485 [DOI] [PubMed] [Google Scholar]
- Rekas A, Alattia JR, Nagai T, Miyawaki A, Ikura M (2002) Crystal structure of venus, a yellow fluorescent protein with improved maturation and reduced environmental sensitivity. J Biol Chem 277: 50573–50578 [DOI] [PubMed] [Google Scholar]
- Rocha EP, Guerdoux-Jamet P, Moszer I, Viari A, Danchin A (2000) Implication of gene distribution in the bacterial chromosome for the bacterial cell factory. J Biotechnol 78: 209–219 [DOI] [PubMed] [Google Scholar]
- Ross AF, Oleynikov Y, Kislauskis EH, Taneja KL, Singer RH (1997) Characterization of a beta-actin mRNA zipcode-binding protein. Mol Cell Biol 17: 2158–2165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudelli RD, Brown WT, Wisniewski K, Jenkins EC, Laure-Kamionowska M, Connell F, Wisniewski HM (1985) Adult fragile X syndrome. Clinico-neuropathologic findings. Acta Neuropathol (Berl) 67: 289–295 [DOI] [PubMed] [Google Scholar]
- Schaeffer C, Bardoni B, Mandel JL, Ehresmann B, Ehresmann C, Moine H (2001) The fragile X mental retardation protein binds specifically to its mRNA via a purine quartet motif. EMBO J 20: 4803–4813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shestakova EA, Singer RH, Condeelis J (2001) The physiological significance of beta -actin mRNA localization in determining cell polarity and directional motility. Proc Natl Acad Sci USA 98: 7045–7050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi H, Siomi MC, Nussbaum RL, Dreyfuss G (1993) The protein product of the fragile X gene, FMR1, has characteristics of an RNA-binding protein. Cell 74: 291–298 [DOI] [PubMed] [Google Scholar]
- St Johnston D, Beuchle D, Nusslein-Volhard C (1991) Staufen, a gene required to localize maternal RNAs in the Drosophila egg. Cell 66: 51–63 [DOI] [PubMed] [Google Scholar]
- Tang SJ, Meulemans D, Vazquez L, Colaco N, Schuman E (2001) A role for a rat homolog of staufen in the transport of RNA to neuronal dendrites. Neuron 32: 463–475 [DOI] [PubMed] [Google Scholar]
- Turner G, Webb T, Wake S, Robinson H (1996) Prevalence of fragile X syndrome. Am J Med Genet 64: 196–197 [DOI] [PubMed] [Google Scholar]
- Veyrune JL, Campbell GP, Wiseman J, Blanchard JM, Hesketh JE (1996) A localisation signal in the 3′ untranslated region of c-myc mRNA targets c-myc mRNA and beta-globin reporter sequences to the perinuclear cytoplasm and cytoskeletal-bound polysomes. J Cell Sci 109 (Part 6): 1185–1194 [DOI] [PubMed] [Google Scholar]
- Wilkinson MF, Shyu AB (2001) Multifunctional regulatory proteins that control gene expression in both the nucleus and the cytoplasm. BioEssays 23: 775–787 [DOI] [PubMed] [Google Scholar]
- Zalfa F, Giorgi M, Primerano B, Moro A, Di Penta A, Reis S, Oostra B, Bagni C (2003) The fragile X syndrome protein FMRP associates with BC1 RNA and regulates the translation of specific mRNAs at synapses. Cell 112: 317–327 [DOI] [PubMed] [Google Scholar]
- Zhang HL, Eom T, Oleynikov Y, Shenoy SM, Liebelt DA, Dictenberg JB, Singer RH, Bassell GJ (2001) Neurotrophin-induced transport of a beta-actin mRNP complex increases beta-actin levels and stimulates growth cone motility. Neuron 31: 261–275 [DOI] [PubMed] [Google Scholar]
- Zhang W, Benson DL (2002) Developmentally regulated changes in cellular compartmentation and synaptic distribution of actin in hippocampal neurons. J Neurosci Res 69: 427–436 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6