

Abstract
Kekkon proteins negatively regulate the epidermal growth factor receptor (EGFR) during oogenesis in Drosophila. Their structural relative in mammals, LRIG1, is a transmembrane protein whose inactivation in rodents promotes skin hyperplasia, suggesting involvement in EGFR regulation. We report upregulation of LRIG1 transcript and protein upon EGF stimulation, and physical association of the encoded protein with the four EGFR orthologs of mammals. Upregulation of LRIG1 is followed by enhanced ubiquitylation and degradation of EGFR. The underlying mechanism involves recruitment of c-Cbl, an E3 ubiquitin ligase that simultaneously ubiquitylates EGFR and LRIG1 and sorts them for degradation. We conclude that LRIG1 evolved in mammals as a feedback negative attenuator of signaling by receptor tyrosine kinases.
Keywords: cancer, growth factor, signal transduction, tyrosine kinase, ubiquitin ligase
Introduction
Cell fate determination, as well as rapid responses to extracellular cues, is critically mediated by soluble growth factors and their transmembrane receptors. Once activated by growth factors, the receptors simultaneously launch both ‘positive signals', which lead to cell stimulation, and ‘negative signals', which regulate the amplitude and duration of these positive signals (reviewed in Dikic and Giordano, 2003). A delicate balance between positive and negative signals is critical for normal cell homeostasis, and its disturbance is often implicated in disease development. An example is provided by the ErbB family of receptor tyrosine kinases (RTKs) and their ligands, soluble factors of the epidermal growth factor (EGF) family (Yarden and Sliwkowski, 2001). The prevalence of positive signals, promoted by autocrine loops, overexpressed or mutated ErbB receptors, as well as unleashed downstream effectors, leads to excessive cell proliferation and often associates with human cancer.
Unlike positive signals, which are relatively well understood, the nature and mediators of signal desensitization are only beginning to be unraveled. A major source of information on negative ErbB signals arises from developmental genetics of invertebrate organisms like Caenorhabditis elegans and Drosophila. A major mediator of definitive negative signals, Sli-1/c-Cbl, was discovered in both worms and mammals. In C. elegans, loss of Sli-1 leads to excessive vulva formation, and naturally occurring aberrant forms of c-Cbl are oncogenic in mammals (Thien et al, 2001). Studies in mammalian cells identified c-Cbl as an E3 ubiquitin ligase, which tags ligand-activated ErbB-1 molecules with ubiquitin, thereby promoting their sorting to degradation in lysosomes (reviewed in Shtiegman and Yarden, 2003). In insects, a group of negative regulators undergo transcriptional up-regulation following activation of the EGF-receptor (reviewed in Shilo, 2003). An example is kekkon-1, which encodes a transmembrane protein that physically binds to and inhibits EGF receptor molecules (Ghiglione et al, 1999). The six leucine-rich repeats (LRRs) of Kekkon-1 are necessary for recognition of EGFR, and for consequent inhibition of activation by growth factors (Ghiglione et al, 2003; Alvarado et al, 2004). The multiple Kekkon proteins of insects have no clear orthologs in mammals (MacLaren et al, 2004). However, the three mammalian LRIG genes share domain organization with Kekkons (Suzuki et al, 1996; Nilsson et al, 2001; Guo et al, 2004; Holmlund et al, 2004). The extracellular regions of both the murine Lrig1/Lig-1 (Suzuki et al, 1996) and the human LRIG1 (Nilsson et al, 2001) share 15 LRRs followed by three immunoglobulin (Ig) domains. Interestingly, disruption of the LRIG1 gene in mice resulted in fertile animals that developed skin defects (Suzuki et al, 2002).
The structural similarity of LRIG1 to Kekkons predicts that LRIG1 will interact with and restrict ErbB signaling in mammals, but no relevant data have been reported. Consistent with negative signaling, LRIG1 is downregulated in tumors of renal cell carcinoma (Thomasson et al, 2004). The present study addressed the possible physical and functional interactions between human LRIG1 and ErbB receptors. We report direct ErbB–LRIG1 interactions, which inhibit ErbB signaling through a mechanism that involves enhancement of receptor ubiquitylation and accelerated intracellular degradation.
Results
LRIG1 mRNA is induced by EGF and the encoded protein physically associates with all the four ErbBs
The large family of transmembrane proteins containing both LRRs and Ig domains includes Kekkons of insects and LRIGs of mammals, but the evolutionary relatedness of the two groups remains unclear. To address this question, we analyzed cDNA and genomic databases of both vertebrates and invertebrates. Interestingly, whereas six orthologous Kekkons were identified in insects (MacLaren et al, 2004), no clear orthologs could be identified in species like Homo sapiens. On the other hand, our analyses detected clear LRIG orthologs in insects and in nematodes, in addition to the previously reported orthologs in Fugu and Ciona (Guo et al, 2004). The phylogram shown in Figure 1A presents the predicted relationships within the extended LRIG family, including two orthologs in nematodes, a single representative in Drosophila, an ortholog in Ciona intestinalis and the three human forms. This analysis excludes the possibility that Drosophila Kekkon and human LRIG1 are orthologs. Rather, the most parsimonious explanation is that they are distant family relatives, which were already distinct in a shared arthropod–vertebrate–nematode (bilaterian) ancestor.
Figure 1.
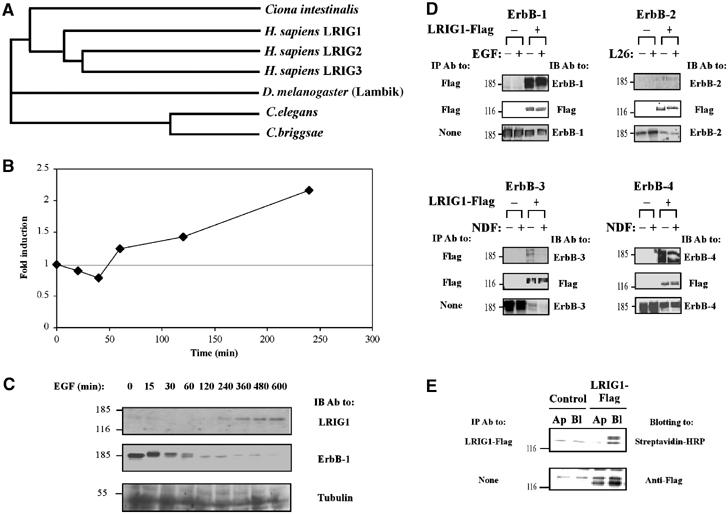
LRIG1 mRNA is induced by EGF and the encoded protein physically associates with the EGF receptor and other ErbB proteins. (A) LRIG proteins form a distinct, conserved LRR-Ig subfamily in metazoans. LRIG orthologs assembled from predicted proteomes of completed genomes were used for multi-alignment using ClustalW, gap trimming and tree construction. The following proteins were assembled: human LRIG1 (gene accession AB018349), LRIG2 (AY358288) and LRIG3 (AY505340); Ciona intestinalis gene 77.18.1; D. melanogaster gene CG8434 (Lambik); C. briggsae predicted protein P6018; and C. elegans predicted protein T21D12.9. (B) Subconfluent HeLa cells (∼1.7 × 106 cells/plate) were serum starved for 24 h, followed by treatment with EGF (20 ng/ml) for the indicated time intervals. Cells were then extracted, and total RNA prepared, followed by reverse transcription with random hexamer primers. Real-time PCR was carried out with primers specific to LRIG1. The level of gene expression was quantified in comparison to a standard curve created by serial dilutions of the template cDNA. The experiment was repeated thrice. (C) HeLa cells were treated with EGF for the indicated intervals as in (B). Whole-cell extracts were resolved by electrophoresis and analyzed by IB with the indicated antibodies. (D) HEK-293T cells co-expressing a Flag-tagged LRIG1 and the indicated ErbB proteins were incubated for 15 min at 37°C with the respective ligands, or with a mAb to ErbB-2 (L26), prior to cell lysis. Cleared cell extracts were subjected to IP and IB, as indicated. (E) Filter-grown MDCK cells stably expressing a Flag peptide-tagged LRIG1 were labeled on the apical (Ap) or the basolateral (Bl) surface with biotin. Whole-cell extracts were prepared and subjected to blotting either directly (lower panel) or after IP. For control we used the parental, untransfected MDCK cells.
Ligand-dependent feedback loops characterize the known negative regulators of RTKs in insects (Shilo, 2003). Accordingly, transcription from the Kek1 gene is regulated by EGFR signaling during oogenesis and in processes leading to development of the wing and eye (Ghiglione et al, 1999). To test the possibility that LRIG1, like Kekkon1, acts in a feedback loop upon EGF stimulation, we examined the effect of EGF on LRIG1 mRNA in HeLa cells, by using real-time PCR (Figure 1B) and DNA arrays (data not shown). Quantification of LRIG1 transcripts in both tests revealed a two- to four-fold induction over a period of a few hours, consistent with the time course of induction of other feedback regulators. As expected, by using western blotting, we observed upregulation of the respective protein 2 h following EGF treatment, concurrent with gradual disappearance of the respective receptor, ErbB-1 (Figure 1C).
It is interesting to note that negative feedback regulators, like Argos and Kekkon-1, physically interact with the Drosophila EGFR following their induction (Schweitzer et al, 1995; Ghiglione et al, 1999). Hence, we examined co-immunoprecipitation of LRIG1 with each of the four ErbB proteins. When transiently expressed in HEK-293T cells together with individual ErbBs, a Flag-tagged LRIG1 underwent co-immunoprecipitation with all four ErbBs, including ErbB-2 and ErbB-3, which exhibited relatively weak interactions with LRIG1 (Figure 1D). These analyses revealed two potentially important features. First, LRIG1–ErbB interactions existed even in the absence of stimulatory ligands (i.e., EGF and the Neu differentiation factor (NDF)) or antibodies (in the case of ErbB-2). Second, ectopic expression of LRIG1 was associated with a significant reduction in the level of expression of each ErbB protein, suggesting linkage between LRIG1 induction and receptor stability, a possibility we address below. This effect was specific to ErbB proteins; examination of the fibroblast growth factor receptor and the low-affinity nerve growth factor receptor detected no downregulation by LRIG1 (data not shown).
In insects, both EGFR (Sapir et al, 1998) and Kekkon-1 (Ghiglione et al, 2003) localize to the apical surface of wing imaginal discs, in contrast to all four ErbB proteins, which localize to the basolateral side of epithelial sheets in mammals (Shelly et al, 2003). To test the possible asymmetric distribution of LRIG1, we expressed it in polar Madin–Darby canine kidney (MDCK) cells, which underwent surface labeling with biotin from either the basolateral or the apical side. This experiment indicated that LRIG1 was almost exclusively targeted to the basolateral surface (Figure 1E). In conclusion, the results presented in Figure 1 envisage the following scenario: LRIG1 expression is induced upon receptor activation, and the newly synthesized protein localizes to the basolateral surface of epithelial cells, where it physically interacts with all members of the ErbB family.
LRIG1–ErbB recognition involves both ectodomains
The extracellular LRRs of Kekkon-1 are necessary for recognition of the ectodomain of the Drosophila EGFR (Ghiglione et al, 2003; Alvarado et al, 2004). To examine possible involvement of the cytoplasmic domain of ErbB-1/EGFR in the recognition of LRIG1, we precipitated LRIG1 from extracts of HEK-293T cells expressing either the wild-type (WT) receptor or a truncation mutant (Figure 2A). This analysis revealed that LRIG1 interacts not only with WT receptors, but also with a mutant lacking the whole intracellular domain (mutant denoted VRK; Shelly et al, 2003). Consistent with the inference that cytoplasmic motifs of ErbB molecules are not essential for LRIG1 binding, engineered ectodomains of all four ErbBs fused to the Fc portion of IgG (denoted IgB-1 through 4; Chen et al, 1996) specifically co-precipitated with LRIG1 when co-expressed (Figure 2B). To identify the ErbB-binding portion of LRIG1, we deleted the LRRs (ΔLRR), the Ig-like domain (ΔIg), or both (ΔLRRIg; see Figure 2C). By co-expressing ErbB-1 and individual LRIG1 mutants and testing co-immunoprecipitation, we concluded that the LRRs and the Ig domains are each sufficient for receptor binding, such that ErbB-1 recognition is abolished only when both are deleted (Figure 2D).
Figure 2.
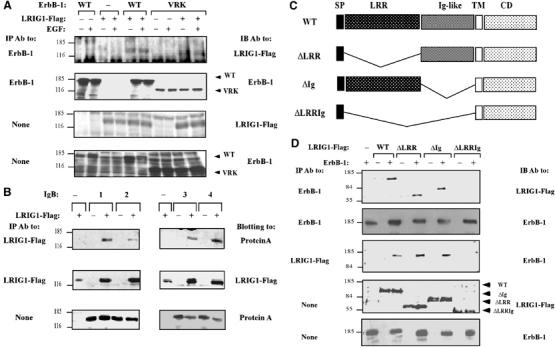
Recognition between LRIG1 and ErbB-1 occurs via the respective ectodomains. (A) HEK-293T cells co-expressing a Flag peptide-tagged LRIG1 and WT-ErbB-1, or a mutant lacking the whole cytoplasmic domain (VRK), were analyzed. Monoclonal antibodies to either Flag or the ectodomain of ErbB-1 were used to probe immunoprecipitates or whole-cell extracts. (B) Whole extracts derived from HEK-293T cells co-expressing LRIG1-Flag and the indicated Fc–ErbB fusion proteins (IgBs) were analyzed by using the indicated antibodies. (C) Schematic diagram depicting the domain structure of LRIG1, including a signal peptide (SP), a LRR, three Ig-like domains, a transmembrane domain (TM) and a cytoplasmic domain (CD). Also shown are deletion mutants. (D) HEK-293T cells co-expressing ErbB-1 and the indicated forms of LRIG1-Flag were extracted and analyzed either directly or following IP with the indicated antibodies.
LRIG1 enhances ligand-induced receptor ubiquitylation and degradation
As ectopic expression of LRIG1 was associated with partial disappearance of all ErbB molecules (Figure 1D), we concentrated on one receptor, ErbB-1, whose activation-dependent degradation is relatively well understood (reviewed in Shtiegman and Yarden, 2003). This process involves recruitment of an E3 ubiquitin ligase, c-Cbl, which sorts receptor molecules to degradation by tagging them with ubiquitin. Consistent with previous reports, only faint labeling of ErbB-1 with a peptide-tagged ubiquitin was noted in resting cells, but receptor ubiquitylation was enhanced upon short incubation with EGF (10 min; Figure 3A). As expected, an ectopically expressed c-Cbl significantly enhanced ubiquitylation and accelerated degradation of ErbB-1. Surprisingly, introduction of LRIG1 exerted remarkable effects, similar to those of c-Cbl, namely: enhanced ligand-induced ubiquitylation and an associated acceleration of receptor degradation (Figure 3A). In line with cooperation between LRIG1 and c-Cbl, we observed maximal ligand-induced degradation of ErbB-1 in cells co-expressing c-Cbl and LRIG-1.
Figure 3.
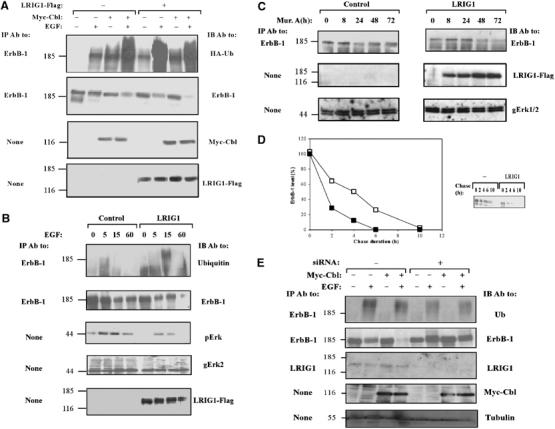
LRIG1 enhances ubiquitylation and degradation of ErbB-1. (A) HEK-293T cells co-expressing ErbB-1 and HA-tagged ubiquitin, along with Myc-Cbl and LRIG1-Flag, as indicated, were stimulated with EGF (100 ng/ml) for 10 min. Cell extracts were analyzed using the indicated antibodies. (B) HEK-293 cells stably expressing pIND-LRIG1-Flag, or a control vector, were transfected with a plasmid encoding ErbB-1, and 24 h later they were treated with Muristerone A (0.1 μM) for another 24 h. Thereafter, cultures were treated at 37°C with EGF (100 ng/ml) for the indicated intervals. Cell extracts were analyzed either directly (None), or after IP, and the immunoprecipitates were analyzed by using the indicated antibodies, including antibodies to Erk2 (gErk2) and the doubly phosphorylated form of Erks (pErk). (C) HEK-293 cells stably expressing the ecdysone receptor were stably transfected with a plasmid expressing LRIG1-Flag from the ecdysone-inducible promoter (pIND-LRIG1-Flag). For control we used an empty pIND plasmid. Cells were incubated at 37°C without or with Muristerone A (2 μM) for the indicated time intervals. Thereafter, cell extracts were prepared and the endogenous ErbB-1 immunoprecipitated. Immunoprecipitates and whole-cell extracts were analyzed with antibodies to ErbB-1 or to the Erk1 and Erk2 proteins. (D) CHO cells expressing ErbB-1, either alone (open symbols) or together with LRIG1-Flag (closed symbols), were metabolically labeled for 16 h with 35S-labeled amino acids. Thereafter, cells were chased at 37°C in fresh medium containing EGF (100 ng/ml). An autoradiogram of the immunoprecipitated ErbB-1 is shown, along with the respective quantification of the ErbB-1 signals. (E) HeLa cells were transfected with siRNA-encoding plasmids and the indicated vectors, together with a pBabe-puro vector for Puromycin selection. At 24 h post transfection, cells were re-plated in the presence of 2 μg/ml Puromycin. After 48 h, cells were stimulated with EGF (100 ng/ml) for 10 min and cell extracts analyzed either directly or following IP with the indicated antibodies.
To emulate ligand-induced upregulation of LRIG1, we established a HEK-293 cell system that expresses LRIG1 from an ecdysone-inducible promoter. Following selection of inducible clones, we transiently expressed ErbB-1 in this cell system, and analyzed its ubiquitylation by utilizing anti-ubiquitin antibodies. Using this experimental system, we found that, upon induction with the respective insect hormone (Muristerone A), the newly synthesized LRIG1 molecules enhanced EGF-induced ubiquitylation and degradation of ErbB-1 (Figure 3B). In addition, antibodies to the doubly phosphorylated form of the mitogen-activated protein kinase (MAPK/Erk) revealed that downstream signaling to MAPK/Erk was inhibited upon LRIG1 induction. Further, analysis of the weakly expressed endogenous EGF receptors of HEK-293 cells showed that inducible expression of LRIG1 led to a time-dependent decrease in endogenous receptor levels (Figure 3C).
To quantitatively analyze the effect of LRIG1 on receptor fate, we metabolically labeled Chinese hamster ovary (CHO) cells and followed the kinetics of degradation of a transiently expressed ErbB-1 (Figure 3D). In the absence of EGF and an ectopic LRIG1, ErbB-1 displayed the normal half-life of approximately 8 h (data not shown), which was shortened to approximately 4 h upon stimulation with EGF. Interestingly, ectopic LRIG1 shortened the half-life of ErbB-1 even in the absence of EGF (data not shown). Nevertheless, the respective half-life determined in EGF-stimulated, LRIG1-expressing cells was as short as 2 h, in line with the assertion that LRIG1 can accelerate receptor degradation. To firmly establish this conclusion, we used RNA interference (Brummelkamp et al, 2002) to knock down expression of endogenous LRIG1. Preliminary tests selected several sequences of LRIG1, which reduced protein expression by 50–80% when expressed as siRNAs. When one of the siRNA constructs was introduced to HeLa cells or human keratinocytes (HaCat) along with c-Cbl, the cells partly lost their ability to ubiquitylate and downregulate ErbB-1 in response to treatment with EGF (Figure 3E and data not shown). Collectively, these results suggest that the feedback loop involving upregulation of LRIG1 expression leads to the formation of receptor–LRIG1 complexes, which sort ligand-activated receptors to intracellular degradation, thereby restricting growth factor signaling.
c-Cbl interacts with and ubiquitylates LRIG1
In view of LRIG1's structure, which predicts no conventional E3 ubiquitin ligase function, and the additive effect of EGF, c-Cbl and LRIG1 on receptor degradation (Figure 3A), we hypothesized that LRIG1 can enhance recruitment of c-Cbl to ErbB-1. To test the involvement of c-Cbl, we utilized a well-characterized dominant-negative mutant, namely 70Z-Cbl, which negates EGF-induced receptor inactivation (Thien et al, 2001). When co-expressed with LRIG1, 70Z-Cbl stabilized the receptor and reduced EGF-induced receptor ubiquitylation (Figure 4A), implying that LRIG1-mediated ubiquitylation and destabilization of ErbB-1 involve a functional c-Cbl. To test the possible interactions between LRIG1 and c-Cbl, we prepared two cytoplasmic truncation mutants of LRIG1 (Figure 4B) and tested their physical interactions with c-Cbl in HEK-293T cells (Figure 4C). As full-length LRIG1 underwent co-immunoprecipitation with c-Cbl, but deletion of a portion of the cytoplasmic domain of LRIG1 abolished co-immunoprecipitation, we concluded that c-Cbl binds, either directly or indirectly, to LRIG1 sequences distal to the transmembrane region. To corroborate this conclusion, we established an in vitro assay utilizing bacterially expressed portions of the cytoplasmic domain of LRIG1, fused to glutathione-S-transferase (GST; see Figure 4B), in combination with HA-tagged c-Cbl derived from mammalian cells. As predicted by analyses performed with living cells, a full cytoplasmic portion of LRIG1, as well as an amino-terminal portion, recognized c-Cbl, but the carboxyl-terminal portion of LRIG1 displayed no c-Cbl binding (data not shown). A series of internal deletion mutants mapped the Cbl docking site to a segment encompassing amino acids 900–939 of LRIG1 (see Figure 4B and D). Likewise, by employing truncation mutants of c-Cbl, we concluded that the amino-terminal half of the molecule is involved in binding to LRIG1 (data not shown).
Figure 4.
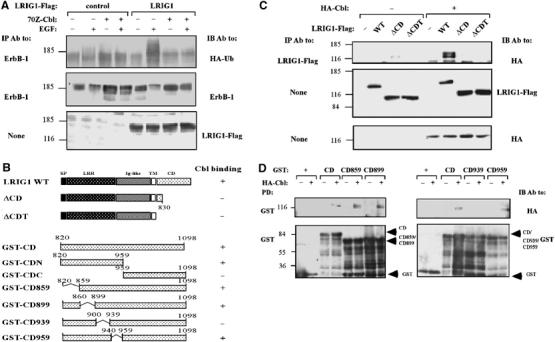
The juxtamembrane domain of LRIG1 interacts with c-Cbl. (A) HEK-293T cells co-expressing ErbB-1 and HA-tagged ubiquitin, along with a dominant-negative form of c-Cbl, 70Z-Cbl and LRIG1-Flag, as indicated, were stimulated with EGF (100 ng/ml) for 10 min. Cell extracts were analyzed using the indicated antibodies. (B) Schematic diagrams of WT-LRIG1 and mutants lacking portions of the cytoplasmic domain. Also shown are diagrams of GST–LRIG1 fusion proteins containing either the full cytoplasmic domain (GST-CD), or the indicated regions. Binding of c-Cbl to individual forms of LRIG1 is indicated in the right column. (C) HEK-293T cells co-expressing HA-c-Cbl, or a control vector, and the indicated forms of LRIG1-Flag were extracted and co-immunoprecipitation analyzed with the indicated antibodies. (D) The indicated glutathione immobilized GST-LRIG1 proteins (1 μg; see (B)) were incubated with extracts derived from HEK-293T cells transfected with a control or a HA-c-Cbl plasmid. Proteins pulled down (PD) were washed extensively, resolved by electrophoresis and detected using the indicated antibodies. The lower panels show the various GST fusion proteins (arrowheads).
Recruitment of c-Cbl to multimolecular complexes, such as the ErbB-1 signaling particle, results in degradation of not only ErbB-1, but also several components, including Grb2 and Shc (Ettenberg et al, 2001). Hence, ubiquitylation of LRIG1 and subsequent degradation may occur subsequent to physical association with c-Cbl and ErbB-1. To test this prediction, we followed the rate of LRIG1 degradation in cells treated with a protein synthesis inhibitor. The data presented in Figure 5A indicate that co-expression of ErbB-1 or treatment with EGF accelerates degradation of LRIG1, in line with inducible recruitment of an E3 ligase. As a proteasome inhibitor blocked LRIG1 degradation (Figure 5A) and LRIG1 mutants that are unable to bind c-Cbl (ΔCDT) or ErbB-1 (ΔLRRIg) underwent only limited degradation under similar conditions (data not shown), we inferred that EGF-mediated recruitment of c-Cbl to LRIG1 sorts the latter to degradation in the 26S proteasome. In line with this scenario, an ubiquitylated LRIG1 was detectable under basal conditions, and the extent of poly-ubiquitylation was increased upon overexpression of ErbB-1 (Figure 5B). As expected, a truncation mutant of LRIG1 lacking most of the cytoplasmic portion (ΔCD; see Figure 4B) underwent only faint ubiquitylation, overexpression of ErbB-1 significantly enhanced degradation of WT LRIG1, and further enhancement was observed upon stimulation with EGF. Moreover, the dominant-negative form of c-Cbl, 70Z-Cbl, completely abolished ubiquitylation of LRIG1 in ErbB-1-expressing HEK-293T cells stimulated with EGF (Figure 5C), in line with the possibility that c-Cbl is responsible for ErbB-1-mediated ubiquitylation of LRIG1. Conceivably, by recruiting c-Cbl to the vicinity of ErbB-1, both LRIG1 and ErbB-1 undergo ubiquitylation and subsequent degradation, but how EGF stimulates Cbl-mediated degradation of LRIG1 is addressed by another set of experiments (see below).
Figure 5.
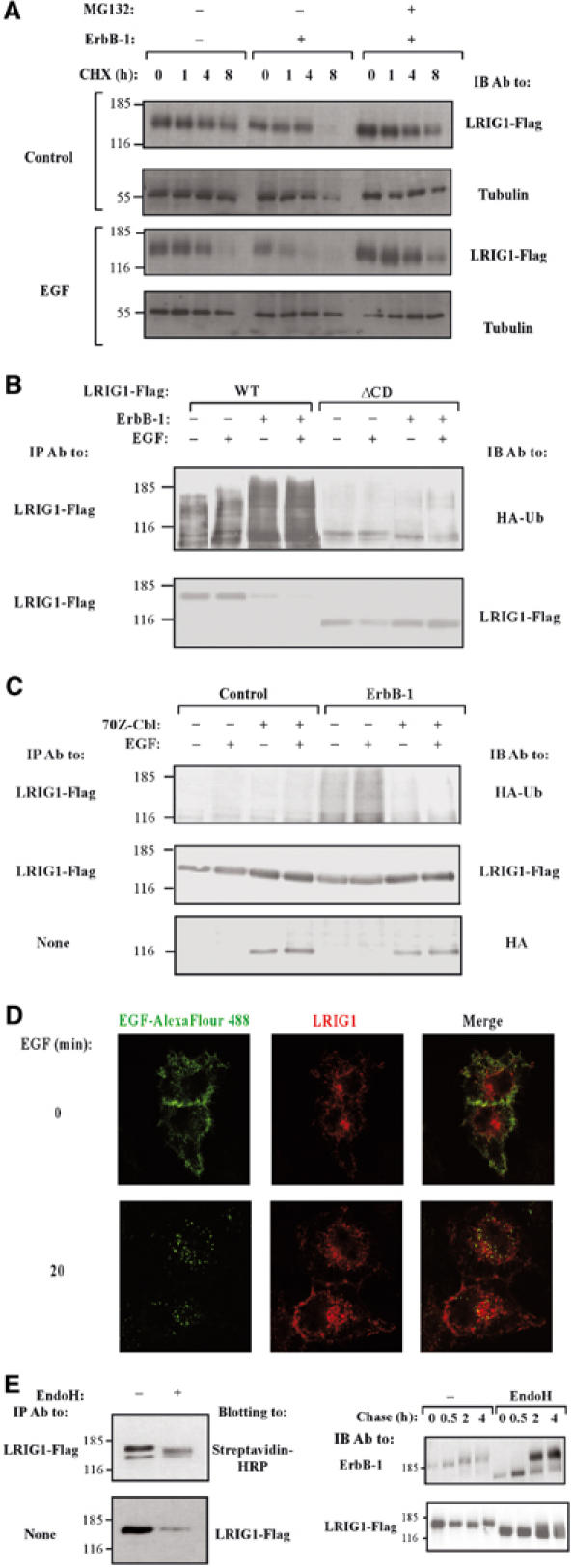
Subcellular localization of LRIG1, ubiquitylation by c-Cbl and proteasomal degradation. (A) CHO cells transiently expressing LRIG1, either alone or together with ErbB-1, were treated with cycloheximide (CHX, 10 μg/ml) for the indicated intervals in the absence or presence of EGF (100 ng/ml) and MG132 (10 μM). Cells were then extracted, and analyzed directly with the indicated antibodies. (B) HEK-293T cells co-expressing the indicated forms of LRIG1-Flag and HA-ubiquitin, in the absence or presence of ErbB-1, were stimulated with EGF (100 ng/ml) for 10 min. Cells were extracted using SDS (1%) containing buffer, and analyzed with the indicated antibodies. (C) HEK-293T cells co-expressing LRIG1-Flag and HA-tagged ubiquitin, along with ErbB-1 and the dominant-negative form of c-Cbl, 70Z-Cbl, as indicated, were stimulated with EGF (100 ng/ml) for 10 min. Cell extracts were analyzed using the indicated antibodies. (D) HeLa cells co-expressing ectopic ErbB-1 and Flag-tagged LRIG1 were pre-incubated for 40 min at 4°C with an EGF conjugated to Alexa Fluor 488. Thereafter, cells were either fixed (0 min), or incubated for 20 min at 37°C. This was followed by fixation, permeabilization, staining with an anti-Flag antibody and detection using a Cy3-conjugated secondary antibody. Confocal micrographs reflecting the distribution of fluorescent EGF and Flag-LRIG1 are shown, along with merge panels depicting both signals. (E) Left panels: The surface of LRIG1-expressing CHO cells was labeled with biotin, followed by IP of LRIG1 and treatment with EndoH (New England Biolabs, Beverly, MA, USA), as indicated. Right panels: CHO cells transiently co-expressing ErbB-1 or LRIG1 were subjected to a 20 min-long metabolic labeling with [35S]methionine (pulse), followed by a variable length chase. Thereafter, ErbB-1 and LRIG1 immunoprecipitates were untreated or treated with EndoH, resolved by electrophoresis and detected using autoradiograpy.
Subcellular localization of LRIG1
In line with previous studies of green fluorescent proteins fused to LRIG1 (Nilsson et al, 2003) and LRIG2 (Holmlund et al, 2004), our immunofluorescence analyses localized LRIG1-Flag molecules to the cell surface, as well as to intracellular structures, which were identified as both trans-Golgi vesicles and early endosomes (Figure 5D and data not shown). The surface localization of LRIG1 corresponded to the plasma membrane, filopodia and cell-to-cell boundaries, similar to the distribution of an ErbB-1-bound fluorescent analog of EGF (EGF-Alexa Fluor 488; Figure 5D). This analog enabled us to track the rapid formation of ErbB-1-containing endosomes. In most cases, the vesicles formed contained no LRIG1, suggesting segregation of ligand–receptor complexes from LRIG1 at the entry to the endocytic pathway. Likewise, upon short stimulation with EGF, we observed translocation of c-Cbl to endosomes, in line with previous reports (Levkowitz et al, 1998; de Melker et al, 2001), but Cbl-containing endosomes only rarely included LRIG1 molecules (data not shown). Notably, we were unable to detect the previously reported transient association of c-Cbl with the plasma membrane (de Melker et al, 2001), which leaves open the possibility that a small fraction of LRIG1 transiently associates with c-Cbl and ErbB-1 at the membrane, but LRIG1 molecules are left behind (or undergo degradation) upon internalization of Cbl–receptor complexes.
The duality of intracellular and membranal LRIG1 led us to study the phenomenon. Consistent with surface localization, LRIG1 molecules were labeled with a membrane-impermeable analog of biotin (Figures 1E and 5E). As resistance to endoglycosidase H (EndoH) is usually acquired upon translocation of EndoH-sensitive high-mannose precursors from the ER to the Golgi apparatus, we treated the biotinylated form of LRIG1 with EndoH (Figure 5E; left panels). This experiment revealed that the majority of surface-associated LRIG1 remains sensitive to EndoH. Pulse-chase metabolic labeling confirmed rapid acquisition of EndoH resistance by ErbB-1 molecules (Figure 5E; right panels). In contrast, LRIG1 underwent only limited glycosylation following exit from the ER and it remained largely EndoH-sensitive (Figure 5E), in line with the prevalence of cytoplasmic and perinuclear staining.
LRIG1-induced receptor ubiquitylation is independent of the Cbl docking site of ErbB-1, but it involves tyrosine phosphorylation of c-Cbl
The major mechanism that enables c-Cbl to bind with, and subsequently ubiquitylate ErbB-1, entails c-Cbl binding to phosphorylated tyrosine-1045 of ErbB-1 (Levkowitz et al, 1999). To address the mechanism underlying LRIG1-induced receptor ubiquitylation, we utilized Y1045F-ErbB-1, a molecule whose Cbl docking site was mutated to a phenylalanine. Unlike WT-ErbB-1, the mutant underwent only minimal EGF- and Cbl-dependent ubiquitylation (Figure 6A). Nevertheless, Y1045F-ErbB-1 displayed increased ubiquitylation, as well as enhanced EGF-induced degradation, in the presence of ectopic LRIG1, indicating a mechanism independent of the direct Cbl docking on ErbB-1. This conclusion was verified by utilizing Y1045F-ErbB-1 in combination with two LRIG1 mutants. These are ΔLRRIg-LRIG1, which binds c-Cbl but cannot bind ErbB-1 (Figure 2D), and ΔCDT-LRIG1, which binds ErbB-1 but cannot recruit c-Cbl (Figure 4C). As expected, neither mutant of LRIG1 could mimic the effect of the parental molecule on ubiquitylation and degradation of Y1045F-ErbB-1 (Figure 6B and C).
Figure 6.
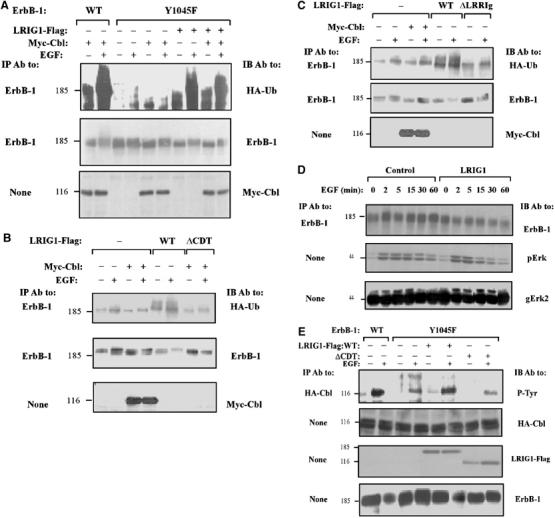
LRIG1-induced ubiquitylation of ErbB-1 is independent of direct docking of c-Cbl at ErbB-1. (A) HEK-293T cells co-expressing ErbB-1, either WT or Y1045F, along with HA-tagged ubiquitin, without or with Myc-Cbl and LRIG1-Flag, were stimulated with EGF (100 ng/ml) for 10 min. Cell extracts were analyzed using the indicated antibodies. (B, C) HEK-293T cells co-expressing Y1045F-ErbB-1 and HA-tagged ubiquitin, along with Myc-Cbl and the indicated forms of LRIG1, were stimulated with EGF (100 ng/ml) for 10 min and cell extracts analyzed using the indicated antibodies. (D) CHO cells expressing Y1045F-ErbB-1, either alone or with LRIG1-Flag, were stimulated with EGF (100 ng/ml) for the indicated time intervals. Cell extracts were analyzed either directly, or after immunoprecipitation with antibodies to ErbB-1, Erk2, and the phosphorylated form of Erk proteins (pErk). (E) CHO cells co-expressing ErbB-1, either WT or Y1045F, along with HA-Cbl and either WT or ΔCDT-LRIG1-Flag, were stimulated with EGF (100 ng/ml) for 10 min. Tyrosine phosphorylation of c-Cbl was analyzed using the indicated antibodies.
Consistent with the notion that ligand-induced receptor degradation is a major signal-attenuating process, Y1045F-ErbB-1 is endowed with enhanced signaling and delayed desensitization (Waterman et al, 2002). Indeed, when expressed in CHO cells and stimulated with EGF, Y1045F-ErbB-1 underwent very inefficient degradation, and downstream signaling to MAPK was relatively steady. In contrast, ectopic expression of LRIG1 restored both ligand-induced degradation of Y1045F-ErbB-1 and down-regulation of MAPK signals emanating from mutant, as well as WT-ErbB-1 molecules (Figure 6D and data not shown). In conclusion, by recruiting c-Cbl to ErbB-1 via a mechanism independent of the Cbl docking site at tyrosine 1045, LRIG1 enhances desensitization of ErbB signaling.
It is important to note that physical engagement of c-Cbl is not sufficient for receptor ubiquitylation; to sort RTKs for degradation, Cbl must first undergo trans-phosphorylation (Levkowitz et al, 1999). Hence, the ability of LRIG1 to recruit c-Cbl to ErbB-1 might be reflected not only by receptor and LRIG1 ubiquitylation, but also by tyrosine phosphorylation of c-Cbl. Consistent with this prediction, EGF-induced tyrosine phosphorylation of c-Cbl in cells expressing WT-ErbB-1 was stronger than in cells expressing Y1045F-ErbB-1, but ectopic expression of LRIG1 enhanced Cbl modification in both cell types (Figure 6E and data not shown). ΔCDT-LRIG1, a form unable to recruit c-Cbl, did not elevate Cbl phosphorylation beyond the control level, which is probably mediated by Grb2 or other adaptors (Waterman et al, 2002). Conceivably, when LRIG1 brings c-Cbl molecules to the vicinity of ErbB-1, no ubiquitylation occurs unless the tyrosine kinase is stimulated by a ligand, and c-Cbl undergoes phosphorylation/activation. This model explains the stimulatory effect of EGF on degradation of LRIG1 (Figure 5A) and LRIG1-mediated ubiquitylation/degradation of ErbB-1, although both LRIG1–ErbB-1 (Figure 1D) and LRIG1–Cbl interactions (Figure 4) appear ligand-independent.
LRIG1 negates EGF-induced signals
The unveiled ability of LRIG1 to bind with and downregulate expression of ErbB proteins predicts suppression of ErbB signals. This possibility was addressed by analyzing both mitogenic and apoptotic signals emanating from ligand-activated ErbB-1 molecules of HEK-293 and A431 cells, respectively. When HEK-293 cells expressing LRIG1 from an ecdysone-inducible promoter (Ecr-293 cells) were stimulated with EGF, they underwent enhanced proliferation, which was suppressed upon induction of LRIG1 expression (Figure 7A). The inhibitory action of LRIG1 was verified by analyzing transcription from the c-fos promoter, a shared target for many growth factors (Figure 7B). EGF-induced stimulation of COS-7 cells expressing a c-fos reporter gene elevated transcription by 10-fold. WT-LRIG1, unlike its truncation mutants which are unable to bind c-Cbl or ErbB-1, inhibited the c-fos signal, suggesting that LRIG1 downregulates mitogenic signaling downstream of ErbB-1. To address apoptotic signals, we employed the well-characterized system of human A431 squamous carcinoma cells, which highly overexpress ErbB-1 due to gene amplification. In response to EGF, the growth of these cells is inhibited and they undergo programmed cell death (Gulli et al, 1996). To test the effect of LRIG1, we prepared a line expressing the ecotrophic receptor for retroviruses (denoted A431R), and then established by retroviral infection several clones that ectopically express LRIG1. When compared to the parental cells, LRIG1-expressing clones displayed normal growth in culture, but increased the ability to escape the inhibitory effect of EGF, as reflected by the number of cell colonies (Figure 7C). Consistent with these observations, a representative LRIG1-overexpressing clone exhibited reduced expression of ErbB-1, and an associated reduction in EGF-induced active MAPK/Erk (Figure 7D). Furthermore, by using a Ras-binding domain (RBD) of Raf1 in the form of a GST fusion protein (de Rooij and Bos, 1997), we found that the initial rate of EGF-induced Ras activation was not affected by LRIG1, but the ectopically expressed protein desensitized Ras at later time points (Figure 7E). In conclusion, the results obtained in engineered derivatives of HEK-293 and A431 cells are consistent with a model that attributes to LRIG1 a negative regulatory function, which involves physical association with growth factor receptor molecules, recruitment of an E3 ubiquitin ligase and consequent degradation of active receptors.
Figure 7.
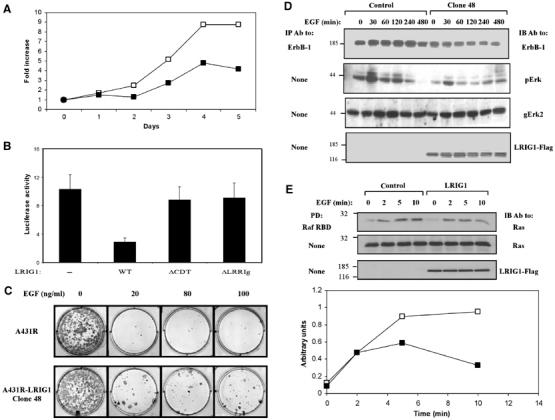
LRIG1 negates EGF-induced signals. (A) HEK-293 cells expressing the ecdysone receptor were stably transfected with a plasmid expressing LRIG1-Flag from an ecdysone-inducible promoter (pIND-LRIG1-Flag). For control, we used an empty pIND plasmid. Parental cells (open symbols) and a mixture of stable clones expressing Flag-tagged LRIG1 (closed symbols) were plated in 96-well plates (5000 cells/well) in the presence of Muristerone A (2 μM) and EGF (20 ng/ml). Cell proliferation was monitored for 5 days by using MTT. (B) COS-7 cells co-expressing a fos-luciferase reporter gene, and the indicated forms of LRIG1-Flag were treated without or with EGF (20 ng/ml) for 12 h. The luminescence signal was determined and presented as the mean±s.d. of six cultures, relative to unstimulated cells. (C) A clone of A431R cells stably expressing a Flag-tagged LRIG1 (clone 48) was plated at a density of 200 cells/cm2, in the presence of the indicated concentrations of EGF. At 14 days after plating, cells were fixed using methanol, stained with Giemsa, and photographed. The parental A431R cells were used as control. (D) A431R and A431R-LRIG1 (clone 48) cells were stimulated in the presence of EGF (50 ng/ml) for the indicated time intervals. Thereafter, cell extracts were analyzed with the indicated antibodies. (E) A431R and A431R-LRIG1 (clone 48) cells were untreated (open symbols) or treated with EGF (20 ng/ml; closed symbols) for 2–10 min and cell extracts subjected to a Raf1 RBD pull-down assay. Immunoblotting with anti-Ras antibodies is shown, along with quantification of Ras signals.
Discussion
Our study establishes for the first time a role for LRIG1, an LRR-Ig protein, as a feedback negative regulator of RTKs in mammalian cells. LRIG1 belongs to a distinct family, which is widely represented in both vertebrates and invertebrates (Figure 1A). Single LRIG orthologs exist in the nematodes C. elegans and C. briggsae. Likewise, a single clear ortholog exists in each sequenced insect genome, namely: bee-Apis mellifera, mosquito-Anopholes gambiae, Drosophila melanogaster and D. pseudoobscura. Moreover, a single LRIG homolog appears in the urochordate Ciona intestinalis, indicating that it is a ‘pre-duplication' member of the chordate LRIG family (Guo et al, 2004). The six Kekkon genes have no clear orthologs in vertebrates or in nematodes, and they are distantly related to the LRIG proteins. Hence, it can be concluded that Kekkons and the LRIG ortholog in D. melanogaster have co-existed in insects as separate genes over extensive evolutionary time. Nevertheless, the findings reported in this study and in other studies (Ghiglione et al, 1999, 2003; Alvarado et al, 2004) imply that both LRIG1 and Kekkons evolved as negative regulators of RTKs, although the mechanisms underlying their actions appear distinct: inhibition of receptor activation in the case of Kekkons (Ghiglione et al, 1999, 2003), and accelerated receptor degradation in the case of LRIG1.
By identifying Cbl-mediated receptor degradation as a mechanism that underlies negative signaling by LRIG1, our study links LRIG1 to a growing list of proteins that help desensitize RTK signaling. Negative regulators of RTK signaling may be divided into two groups (Dikic and Giordano, 2003). The immediate early regulators, such as Ras-GAP, Cbl and the cytoplasmic tyrosine kinase Ack/Ark, are ubiquitously expressed proteins whose function requires no de novo protein synthesis. The delayed negative regulators, such as Sprouty (reviewed in Guy et al, 2003) and RALT (Fiorini et al, 2002), are transcriptionally induced upon receptor stimulation and, therefore, they reach their peak of activity only 2–3 h after stimulation. LRIG1 belongs to the latter group of delayed regulators (see Figure 1B and C). Although both Kekkon-1 and LRIG1 physically interact with all four members of the ErbB family (Ghiglione et al, 2003; Figure 1D), their mechanisms of action differ. Conceivably, the shared LRRs and Ig-like domains of LRIG1 and Kekkon-1 enable recognition of extracellular motifs common to all ErbB proteins (Figure 2B), but the divergent cytoplasmic domain of LRIG1 evolved an ability to recruit c-Cbl (Figure 4D).
In light of the finding that LRIG1 acts within a framework of a negative feedback loop, it is interesting to consider the common attributes of the currently known delayed negative regulators. These proteins not only share time courses of transcriptional induction by RTKs but also physically associate with activated receptors. Thus, the newly synthesized Argos and LRIG1 bind to the extracellular region of the EGF receptor (Schweitzer et al, 1995; and Figure 1D), whereas RALT (Fiorini et al, 2002) and Sprouty proteins (Guy et al, 2003) bind either directly or indirectly to the cytoplasmic domains of RTKs. Interestingly, like LRIG1 (Figure 5C), RALT (Fiorini et al, 2002) and Sprouty proteins (Guy et al, 2003) undergo ubiquitylation and subsequent degradation, suggesting similar modes of post-translational control.
The following mechanism of LRIG1 modulation of RTKs emerges from our studies. In resting cells, LRIG1 and other delayed negative regulators are only weakly expressed, but upon RTK stimulation, the LRIG1 gene undergoes transcriptional activation. A few hours from ErbB activation, LRIG1 level reaches a high steady state, which probably enables physical attachment to all available ErbB receptors. Like RALT, but unlike Cbl, LRIG1 binds to both ligand-activated and nonactivated receptors. Likewise, the juxtamembrane domain of LRIG1 constitutively recruits a third component, c-Cbl, whose E3 ubiquitin ligase remains sedentary unless phosphorylated on a specific tyrosine residue (Levkowitz et al, 1999; Thien et al, 2001). Upon ligand binding and activation of c-Cbl, all components of the putative complex undergo ubiquitylation, but LRIG1 dissociates and undergoes proteasomal degradation once a Cbl–receptor complex is internalized. An important feature of the proposed model is the ability of LRIG1 to sort for degradation receptors, which are otherwise uncoupled to the Cbl-mediated pathway of endocytosis and degradation. This feature has been demonstrated with Y1045F-ErbB-1 (Figure 6A), and it implies that LRIG1 evolved as a surrogate mechanism that enables Cbl recruitment and subsequent receptor inactivation.
Along with signaling and evolutionary aspects, our results may bear relevance to pathologies in which RTKs are involved. For example, ErbB proteins have been implicated in human cancer of epithelial and glial origins, due to frequent amplification and aberrations of the corresponding genes, as well as occurrence of autocrine loops (reviewed in Yarden and Sliwkowski, 2001). Hence, it will be interesting to test the possibility that oncogenic ErbB versions, such as EGFRvIII, escape regulation by LRIG1. Consistent with antagonistic interactions in cancer, a recent survey of renal cell carcinomas discovered reciprocal relationships between an overexpressed ErbB-1 and a downregulated LRIG1 (Thomasson et al, 2004). The psoriasiform epidermal hyperplasia of Lrig1/Lig-1-defective mice, as well as inverse relationships between LRIG1 levels and proliferative ability of psoriatic human keratinocytes (Suzuki et al, 2002), are additional manifestations of the inhibitory potential of LRIG1. Nevertheless, further research, and more surveys of clinical specimens, will be necessary to address the proposition that LRIG1 can suppress oncogenic signals by means of recruiting an E3 ubiquitin ligase to active growth factor receptors.
Materials and methods
Reagents and antibodies
Lipofectamin was supplied by Gibco BRL (Grand Island, NY, USA). MG132, Puromycin and G418 were from Calbiochem (San Diego, CA, USA). Murine monoclonal antibody (mAb) SG565 to ErbB-1 was generated in our laboratory and used for immunoprecipitation (IP). For immunoblot (IB) analysis of ErbB-1, we used an mAb from Alexis Biochemicals (Lausen, Switzerland). Polyclonal antibody A940 to LRIG1 was generated in rabbits that were immunized with human LRIG1 cytoplasmic domain fused to GST. Anti-Flag-agarose beads for IP were from Sigma (St Louis, MO, USA). Hemagglutinin (HA) rat mAb was purchased from Roche Molecular Biochemicals (Mannheim, Germany). Antibodies to GST were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Peroxidase-conjugated and fluorescently labeled antibodies were purchased from Jackson ImmunoResearch (West Grove, PA, USA). Peroxidase-conjugated protein A was from ICN (Costa Mesa, CA, USA).
Construction of expression vectors
Full-length human LRIG1 cDNA was amplified by using mRNA from 487GM glioma cells. The QuickChange mutagenesis kit (Stratagene, Cedar Creek, UK) and PCR-based strategies were used to generate mutants. The LRIG1 cytoplasmic domain was cloned into pGEX-2TK for expression of GST fusion proteins in bacteria.
Cell culture and transfection
Plasmid transfection to subconfluent cell cultures was performed using the calcium phosphate method or Lipofectamine, and cells were harvested 48 h later. The total amount of DNA in each transfection was normalized with the respective empty vector. Clones of MDCK cells were selected and cultured as described (Shelly et al, 2003). EcR-293 cells (HEK-239 cells constitutively expressing subunits of the ecdysone receptor) were transfected with pIND-Flag-LRIG1, and stable clones cultured in medium supplemented with G418 (700 μg/ml) and Zeocin (200 μg/ml). A431R cells stably expressing an ecotrophic virus receptor were infected with a pBabe-LRIG1 retrovirus produced in Pheonix cells (obtained from G Nolan, Stanford University).
Small-interference RNA
Three LRIG1-specific 19 nucleotide sequences corresponding to nucleotides 605–623, 1170–1188 and 1494–1512 were designed and inserted into the pSUPER vector as described (Brummelkamp et al, 2002). These constructs were referred to as LR-605, LR-1170 and LR-1494, respectively. HeLa cells were transfected with the relevant siRNA vector together with pBabe-puro for selection using the calcium phosphate method. At 24 h post transfection, cells were re-plated in the presence of 2 μg/ml Puromycin.
Analysis of transfected cells and Raf1 RBD assays
Transfected cells were extracted and analyzed essentially as described (Levkowitz et al, 1999). The RBD of Raf1, in the form of a GST fusion protein, was isolated and used as described previously (de Rooij and Bos, 1997).
Cell surface biotinylation
For biotinylation, cells were washed three times with ice-cold phosphate-buffered saline (PBS) and then incubated for 60 min at 4°C with N-hydroxysuccinimide-biotin (biotin-X-NHS, 0.5 mg/ml; Calbiochem) dissolved in borate buffer (10 mM boric acid, 150 mM NaCl (pH 8.0)). Coupling of biotin was blocked by cell rinsing with a solution of 15 mM glycine in PBS.
Immunofluorescence
Cells were fixed for 15 min in 3% paraformaldehyde and permeabilized for 10 min with Triton X-100. For staining, coverslips were incubated for an hour at room temperature with primary antibodies. After extensive washing, the coverslips were incubated for 40 min with Cy2- or Cy3-conjugated secondary antibodies. For EGF uptake, prior to fixation, transfected cells were washed in PBS and incubated at 4°C for 40 min with buffer containing EGF (2 μg/ml) conjugated to Alexa Fluor 488 (Molecular Probes, Leiden, The Netherlands). After binding, cells were transferred to 37°C for 20 min, washed in PBS and fixed. After staining with primary and fluorescently labeled secondary antibodies, coverslips were mounted in moviol, and immunofluorescence was analyzed using a Nikon Eclipse TE300 confocal microscope.
Cell survival assay
A431R cells were plated in 96-well plates at a density of 500 cells/well, in the presence of increasing concentrations of EGF. Cell survival was assayed 9 days after plating, by adding 3-(4,5-dimethylthiazol-z-yl)-2,5-diphenyl tetrazolium bromide (MTT; final concentration 0.05 mg/ml) and incubating for 2 h. Signals were quantified by determining the optical density at 540–630 nm after lysis of the cells in acidic isopropanol. Likewise, stable clones of Ecr-293 were plated in 96-well plates at a density of 5000 cells/well in the presence of EGF (20 ng/ml). Cellular growth was followed over a period of 5 days.
Acknowledgments
We thank Drs Wallace Langdon, Gary Nolan, Reuven Agami and Dirk Bohmann for cells and plasmids, and members of our group for useful insights. Our laboratory is supported by research grants from Minerva, the Prostate Cancer Foundation, the J&R Center for Scientific Research, the National Cancer Institute (grant CA72981) and the Willner Family Center for Vascular Biology. Y Yarden is the incumbent of the Harold and Zelda Goldenberg Professorial Chair.
References
- Alvarado D, Rice AH, Duffy JB (2004) Bipartite inhibition of Drosophila epidermal growth factor receptor by the extracellular and transmembrane domains of Kekkon 1. Genetics 167: 187–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) Stable suppression of tumorigenicity by virus-mediated RNA interference. Cancer Cell 2: 243–247 [DOI] [PubMed] [Google Scholar]
- Chen X, Levkowitz G, Tzahar E, Karunagaran D, Lavi S, Ben Baruch N, Leitner O, Ratzkin BJ, Bacus SS, Yarden Y (1996) An immunological approach reveals biological differences between the two NDF/heregulin receptors, ErbB-3 and ErbB-4. J Biol Chem 271: 7620–7629 [PubMed] [Google Scholar]
- de Melker AA, van der Horst G, Calafat J, Jansen H, Borst J (2001) c-Cbl ubiquitinates the EGF receptor at the plasma membrane and remains receptor associated throughout the endocytic route. J Cell Sci 114: 2167–2178 [DOI] [PubMed] [Google Scholar]
- de Rooij J, Bos JL (1997) Minimal Ras-binding domain of Raf1 can be used as an activation-specific probe for Ras. Oncogene 14: 623–625 [DOI] [PubMed] [Google Scholar]
- Dikic I, Giordano S (2003) Negative receptor signalling. Curr Opin Cell Biol 15: 128–135 [DOI] [PubMed] [Google Scholar]
- Ettenberg SA, Magnifico A, Cuello M, Nau MM, Rubinstein YR, Yarden Y, Weissman AM, Lipkowitz S (2001) Cbl-b-dependent coordinated degradation of the epidermal growth factor receptor signaling complex. J Biol Chem 276: 27677–27684 [DOI] [PubMed] [Google Scholar]
- Fiorini M, Ballaro C, Sala G, Falcone G, Alema S, Segatto O (2002) Expression of RALT, a feedback inhibitor of ErbB receptors, is subjected to an integrated transcriptional and post-translational control. Oncogene 21: 6530–6539 [DOI] [PubMed] [Google Scholar]
- Ghiglione C, Amundadottir L, Andresdottir M, Bilder D, Diamonti JA, Noselli S, Perrimon N, Carraway IK (2003) Mechanism of inhibition of the Drosophila and mammalian EGF receptors by the transmembrane protein Kekkon 1. Development 130: 4483–4493 [DOI] [PubMed] [Google Scholar]
- Ghiglione C, Carraway KL, Amundadottir LT, Boswell RE, Perrimon N, Duffy JB (1999) The transmembrane molecule kekkon 1 acts in a feedback loop to negatively regulate the activity of the Drosophila EGF receptor during oogenesis. Cell 96: 847–856 [DOI] [PubMed] [Google Scholar]
- Gulli LF, Palmer KC, Chen YQ, Reddy KB (1996) Epidermal growth factor-induced apoptosis in A431 cells can be reversed by reducing the tyrosine kinase activity. Cell Growth Differ 7: 173–178 [PubMed] [Google Scholar]
- Guo D, Holmlund C, Henriksson R, Hedman H (2004) The LRIG gene family has three vertebrate paralogs widely expressed in human and mouse tissues, and a homolg in Ascidiacea. Genomics 84: 157–165 [DOI] [PubMed] [Google Scholar]
- Guy GR, Wong ES, Yusoff P, Chandramouli S, Lo TL, Lim J, Fong CW (2003) Sprouty: how does the branch manager work? J Cell Sci 116: 3061–3068 [DOI] [PubMed] [Google Scholar]
- Holmlund C, Nilsson J, Guo D, Starefeldt A, Golovleva I, Henriksson R, Hedman H (2004) Characterization and tissue-specific expression of human LRIG2. Gene 332: 35–43 [DOI] [PubMed] [Google Scholar]
- Levkowitz G, Waterman H, Ettenberg SA, Katz M, Tsygankov AY, Alroy I, Lavi S, Iwai K, Reiss Y, Ciechanover A, Lipkowitz S, Yarden Y (1999) Ubiquitin ligase activity and tyrosine phosphorylation underlie suppression of growth factor signaling by c-Cbl/Sli-1. Mol Cell 4: 1029–1040 [DOI] [PubMed] [Google Scholar]
- Levkowitz G, Waterman H, Zamir E, Kam Z, Oved S, Langdon WY, Beguinot L, Geiger B, Yarden Y (1998) c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev 12: 3663–3674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLaren CM, Evans TA, Alvarado D, Duffy JB (2004) Comparative analysis of the Kekkon molecules, related members of the LIG superfamily. Dev Genes Evol (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Nilsson J, Starefeldt A, Henriksson R, Hedman H (2003) LRIG1 protein in human cells and tissues. Cell Tissue Res 312: 65–71 [DOI] [PubMed] [Google Scholar]
- Nilsson J, Vallbo C, Guo D, Golovleva I, Hallberg B, Henriksson R, Hedman H (2001) Cloning, characterization, and expression of human LIG1. Biochem Biophys Res Commun 284: 1155–1161 [DOI] [PubMed] [Google Scholar]
- Sapir A, Schweitzer R, Shilo BZ (1998) Sequential activation of the EGF receptor pathway during Drosophila oogenesis establishes the dorsoventral axis. Development 125: 191–200 [DOI] [PubMed] [Google Scholar]
- Schweitzer R, Howes R, Smith R, Shilo BZ, Freeman M (1995) Inhibition of Drosophila EGF receptor activation by the secreted protein Argos. Nature 376: 699–702 [DOI] [PubMed] [Google Scholar]
- Shelly M, Mosesson Y, Citri A, Lavi S, Zwang Y, Melamed-Book N, Aroeti B, Yarden Y (2003) Polar expression of ErbB-2/HER2 in epithelia: bimodal regulation by Lin-7. Dev Cell 5: 475–486 [DOI] [PubMed] [Google Scholar]
- Shilo BZ (2003) Signaling by the Drosophila epidermal growth factor receptor pathway during development. Exp Cell Res 284: 140–149 [DOI] [PubMed] [Google Scholar]
- Shtiegman K, Yarden Y (2003) The role of ubiquitylation in signaling by growth factors: implications to cancer. Semin Cancer Biol 13: 29–40 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Miura H, Tanemura A, Kobayashi K, Kondoh G, Sano S, Ozawa K, Inui S, Nakata A, Takagi T, Tohyama M, Yoshikawa K, Itami S (2002) Targeted disruption of LIG-1 gene results in psoriasiform epidermal hyperplasia. FEBS Lett 521: 67–71 [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Sato N, Tohyama M, Wanaka A, Takagi T (1996) cDNA cloning of a novel membrane glycoprotein that is expressed specifically in glial cells in the mouse brain. LIG-1, a protein with leucine-rich repeats and immunoglobulin-like domains. J Biol Chem 271: 22522–22527 [DOI] [PubMed] [Google Scholar]
- Thien CB, Walker F, Langdon WY (2001) RING finger mutations that abolish c-Cbl-directed polyubiquitination and downregulation of the EGF receptor are insufficient for cell transformation. Mol Cell 7: 355–365 [DOI] [PubMed] [Google Scholar]
- Thomasson M, Hedman H, Guo D, Ljungberg B, Henriksson R (2004) LRIG1 and epidermal growth factor receptor in renal cell carcinoma: a quantitative RT–PCR and immunohistochemical analysis. Br J Cancer 89: 1285–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman H, Katz M, Rubin C, Shtiegman K, Lavi S, Elson A, Jovin T, Yarden Y (2002) A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J 21: 303–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden Y, Sliwkowski MX (2001) Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2: 127–137 [DOI] [PubMed] [Google Scholar]