

Abstract
We describe a high-temperature (75°C) transition in the Mu integration complex that causes efficient and true reversal of the integration reaction. A second reversal pathway, first described as ‘foldback' reversal for the HIV integrase, was also observed upon disassembly/reassembly of the Mu complex at normal temperatures. Both true and foldback reversal severed only one or the other of the two integrated Mu ends, and each exhibited distinct metal ion specificities. Our results directly implicate an altered transposase configuration in the Mu strand transfer complex that inhibits reversal, thereby regulating the directionality of transposition.
Keywords: directionality of transposition, disintegration, hairpin formation, target conformation, transposase
Introduction
Transposable elements are self-propagating units that have been referred to as selfish DNA (Doolittle and Sapienza, 1980; Orgel and Crick, 1980). They drive genome evolution in many ways, accumulating to constitute nearly half the genome in humans and up to 90% in some plants (Kazazian, 2004). Strategies for the genetic success of these elements must involve mechanisms that maintain the forward momentum and prevent reversal of integration, among others (Craig et al, 2002).
Phage Mu provides a paradigm for the study of transposition (see Mizuuchi, 1997; Chaconas and Harshey, 2002). The transposition chemistry of Mu is similar to that of the DNA of retroviral elements like HIV (Engelman et al, 1991). These elements are also related in the structure of the catalytic domains of their integrases, and in the sequence patterns at their DNA termini (Dyda et al, 1994; Bujacz et al, 1995; Rice and Mizuuchi, 1995; Lee and Harshey, 2003). During Mu transposition from supercoiled DNA, the transposase (MuA) nicks the left (L) and right (R) ends to liberate free 3′OHs (Figure 1A). This requires Escherichia coli HU protein and Mg2+, and is followed by MuB-dependent strand transfer (ST) of the cleaved ends to phosphodiester bonds placed 5 bp apart on opposite strands of a supercoiled target. When this nucleoprotein complex (type II) is deproteinized, the 5 bp across the donor–target joint are disrupted, giving a ‘θ' appearance to the product. Although the active form of MuA in the transpososome is a tetramer, only two subunits perform successive cleavage and ST (Namgoong and Harshey, 1998; Williams et al, 1999). Under altered reaction conditions (addition of 10–15% DMSO), the need for DNA supercoiling and for HU and MuB proteins is bypassed, allowing transposition to occur from a pair of oligonucleotides derived from the R end, which encode two (R1–R2) of the three MuA binding sites at this end (Figure 1B; Savilahti et al, 1995).
Figure 1.
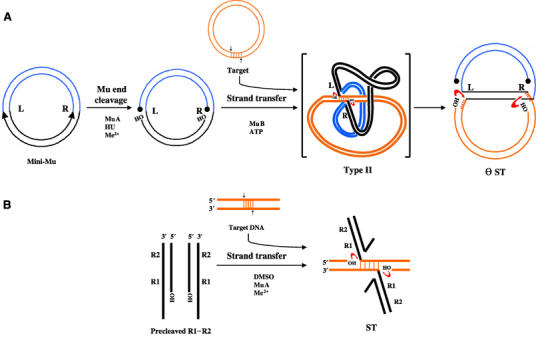
Mu DNA transposition. (A) On supercoiled plasmid substrates (supercoils not shown), in the presence of HU and divalent metal ions (Me2+), MuA nicks L and R ends. Cleaved ends attack target DNA, whose capture is promoted by MuB. The 5 bp joint in the fused donor–target type II complex is disrupted upon deproteinization, revealing the θ ST intermediate, which retains supercoiling in the Mu domain. (B) Precleaved oligonucleotide substrates containing R1 and R2 subsites within the R end can perform ST under DMSO conditions. In (A) and (B), free 3′OHs in the ST intermediates can potentially reverse integration by attacking the transposon–target joint as shown (red arrows). Circular knobs, 5′ phosphate groups; black, Mu DNA; blue, flanking non-Mu DNA; orange, target DNA.
A chemical reversal of integration is called disintegration, and would involve an attack of the adjacent 3′OHs of target DNA in the ST intermediate, on phosphodiester bonds joining the transposon to the target (Figure 1, red arrows). Such a reaction was first described for HIV-1 integrase (Chow et al, 1992), and subsequently found in other systems (Jonsson et al, 1993; Polard et al, 1996; Beall and Rio, 1998; Melek and Gellert, 2000). Disintegration is not normally observed in these systems, and special substrates or altered pH and metal ion concentrations were used in order to do so. We now report the discovery of three reaction conditions that efficiently reverse Mu integration on plasmid as well as oligonucleotide substrates. Two reversal pathways could be distinguished—true reversal and foldback reversal. Our results suggest that most disintegration reactions described to date used the foldback mechanism, and that true reversal may require rearranging the transposase active site through mechanisms of the kind induced by high temperatures and/or base-pair mismatches in the target as reported here for Mu. A novel aspect of our results is that they suggest a mechanism for regulating the directionality of transposition.
Results
Disassembly/reassembly of the transpososome promotes disintegration of θ ST intermediates via the foldback pathway
Deproteinized θ ST products (see Figure 1A) retain supercoiling in the Mu sequence of the donor, and resolve into a series of topoisomers upon electrophoresis (Figure 2A, lane 1). Upon reincubation of θ ST with MuA and HU under normal conditions for 2 h, a novel product (D) was seen (lane 2). Its formation depended on both MuA and HU (lanes 3 and 4), but not MuB (not shown). The reaction was specific to Mg2+ (see Table I). MuA(E392A), a catalytically inactive variant (Kim et al, 1995), was inactive in the formation of D (lane 5). DNA supercoiling in the Mu domain was required, and the reassembled complex had a tetrameric form of MuA, similar to that found in complexes prior to disassembly (Supplementary Figure 1; Lavoie et al, 1991).
Figure 2.
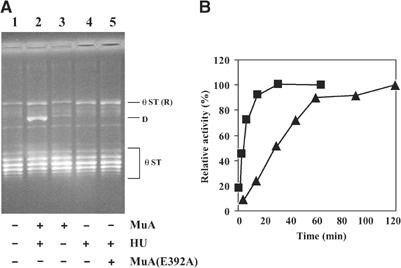
Formation of a novel product upon disassembly/reassembly of MuA on θ ST intermediates. (A) θ STs were incubated with indicated proteins for 2 h at 30°C prior to agarose gel electrophoresis. (R), relaxed form; D, novel product. (B) Kinetics of formation of θ ST (▪) and D (▴). Relative activity refers to percentage of the final product.
Table 1.
Metal ion specificities for disintegration
| Disintegration pathway | Treatment | Activity (relative %) |
||||||
|---|---|---|---|---|---|---|---|---|
| Mg2+ | Ca2+ | Mn2+ | Co2+ | Zn2+ | No metal | |||
| (1) θ ST | FB | (a) Directly in ions | 100 | <2 | <2 | <2 | <2 | <2 |
| (b) Preassembly in Mg2+ | 100 | <2 | 9 | 220 | 76 | <2 | ||
| (2) Half-target substrate | FB | (a) Directly in ions | 100 | <2 | <2 | 15 | <2 | <2 |
| (b) Preassembly in Mg2+ | 100 | <2 | 550 | 126 | 400 | <2 | ||
| (c) Assembly | 100 | N | N | 30 | N | 115 | ||
| (3) Full-target substrate | ? | (a) Directly in ions | 100 | <2 | 5 | 20 | <2 | <2 |
| (b) Preassembly in Mg2+ | # | # | # | # | # | # | ||
| (c) Assembly | 100 | N | N | 35 | N | 170 | ||
| (4) Type II | T | Preassembly in Mg2+ | 100 | 10 | 56 | 240 | 220 | <2 |
| (5) Type IIo | T | Preassembly in Mg2+ | 100 | 62 | 53 | 44 | 54 | <2 |
| FB, foldback; T, true reversal; #, complex did not survive gel filtration; N, no detectable complex. Activity is relative to that obtained with Mg2+. | ||||||||
The kinetics of formation of D is shown in Figure 2B. While the ST reaction was 80% complete by 10 min, formation of D was slower, taking nearly an hour to reach similar levels. Approximately 20% of the input substrate was converted to D at the final time point (see Figure 2A, lane 2). Disassembly and reassembly of MuA was essential, as continued incubation of the original type II complex for 2 h did not give rise to D (not shown).
The D band was purified and examined by electron microscopy (Figure 3A). All molecules observed (>100) were exclusively circles with tails. Length measurements (on >20 molecules) showed that the circles corresponded to the mini-Mu donor (4.1 kb) and the tail to the target plasmid (2.8 kb). Thus, either one or the other end of Mu had likely severed in these molecules. Restriction analysis showed an equal mixture of products disintegrated at the left or the right end (Supplementary Figure 2).
Figure 3.
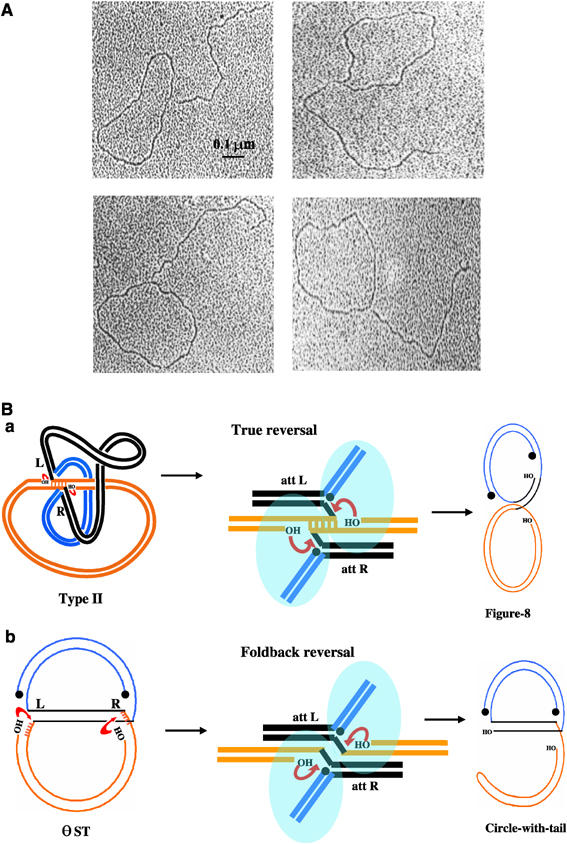
Characterization of the novel product D. (A) Electron micrographs of D (see text). (B) Two target site configurations within a Mu transpososome (a and b, central panel) would lead to two different single-ended disintegration products. Blue ovals represent active MuA subunits, which work in trans, that is, the subunit bound to one end catalyzes reaction at the other end. Other symbols are as in Figure 1.
Upon reassembly of the transposase on θ ST, the target could have assumed two possible configurations (Figure 3B, central panel). In the true reversal pathway (a), the target base pairs are hydrogen bonded as in the original type II complex. Single-ended disintegration would give figure-8 products where the donor is still linked to the target at one Mu end. If the target DNA had flipped to assume a non-hydrogen-bonded ‘foldback' configuration (b), single-ended disintegration would yield circle-with-tail products. In either pathway, disintegration could occur using one of two sources as nucleophiles—target hydroxyls (as shown) or water. In the foldback pathway, if the nucleophiles were target hydroxyls, the tail would end in a hairpin; if water, the tail would be open. Two-dimensional agarose gel analysis revealed that D was predominantly a hairpin product (Supplementary Figure 3).
In summary, disintegration of either one or the other end of Mu occurs fairly efficiently upon disassembly followed by reassembly of the transpososome on plasmid θ ST substrates. This reaction requires a catalytically active form of MuA, HU protein, Mg2+ ions and supercoiling within the Mu domain. Product structure suggests that disintegration occurs by a foldback pathway that is chemically but not structurally/mechanistically analogous to a true reversal of integration; the major product ends in a hairpin and is likely the result of transesterification involving target hydroxyls.
Disintegration of half-target oligonucleotide ST substrates also occurs via the foldback pathway
To characterize this reaction further, oligonucleotide substrates similar to those described earlier were employed (Chow and Brown, 1994; Mazumder et al, 1994). The half-target oligomer was designed to resemble a ST joint at the right end (Figure 4A), the expectation being that MuA would bind each half-substrate and reconstitute the full-target site (Figure 4B). The target sequence immediately flanking Mu was 5′-ACCTG; this sequence will be mismatched if a full target is reconstituted by base pairing.
Figure 4.
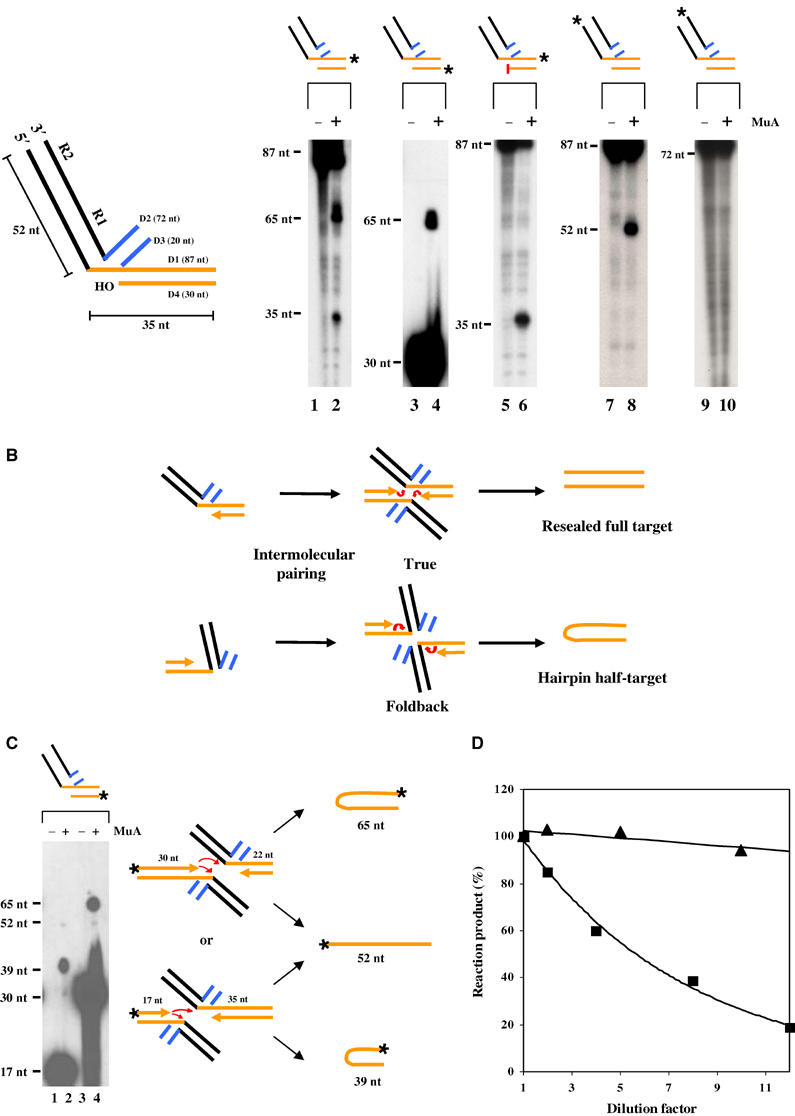
Disintegration of half-target ST substrates. (A) Substrates contain R1–R2 sites from the Mu right end (black), a non-Mu flank (D2/D3; blue) and a half-target flank (D1/D4; orange). Radiolabeled (*) substrates were incubated with (+) or without (−) MuA for 2 h at 30°C (lanes 1–10). The red bar denotes a dideoxy 3′ end. (B) Two different initial target conformations (upper and lower) will lead to different product outputs after intermolecular pairing. The red arrows are as in Figure 1. (C) Disintegration from a mixture of substrates with different half-target lengths. Lanes 1 and 2, label on the shorter target substrate; lanes 3 and 4, label on the longer target substrate. The 17 and 30 nt species are unreacted substrates. See text for details. (D) Effect of dilution on the yield of disintegration products from θ (▴) and oligonucleotide (▪) ST substrates. In each reaction, the same amount of substrate was diluted to different extents in the reaction buffer, while the concentration of MuA was kept constant (see text). Reactions were fractionated in denaturing polyacrylamide gels, and analyzed by phosphorimaging.
Under DMSO reaction conditions, incubation with MuA of the half-target substrate labeled at the 3′ end of the 87 nt D1 oligonucleotide gave 65 and 35 nt products (ratio ∼2:1) on denaturing gels (lane 2). The 65 nt species could arise from use of target hydroxyls as nucleophiles, either reconstituting a full target via true reversal or joining the top and bottom strands of the target flank via foldback reversal to produce a hairpin product as shown (Figure 4B); the 35 nt species could arise from hydrolysis of the Mu–target junction. When the radiolabel was on the 5′ end of the 30 nt D4, it again gave a 65 nt band, confirming that target hydroxyls had participated in ST (lane 4). Decreasing the length of the 5 nt single-stranded flanking target region to 4 nt by increasing the length of D4 gave a 66 nt product, reconfirming this inference; however, any further increase in the length of D4 abolished the reaction (data not shown). Incorporation of a dideoxy nucleotide at the 3′ end of D4 abrogated production of the 65 nt species as expected, while retaining the 35 nt species (lane 6); the latter must therefore arise from hydrolysis. When D1 was labeled at the 5′ end, a 52 nt species was produced, consistent with cleavage at the Mu–target junction (lane 8). When D2 was labeled at the 3′ end, no product was observed (lane 10), showing that this strand did not participate in the reaction. Similar results were seen upon labeling the 3′ end of D3 (not shown). Finally, DNA sequencing confirmed that the 65 nt species came from joining the target region of D1 with D4.
Because the target is reconstituted from two identical halves in the experiment above, the 65 nt species could have arisen from either pathway (Figure 4B). To distinguish between the two possibilities, mixing experiments were carried out between substrates with different target lengths. The substrate in Figure 4A was mixed with a similar substrate but shorter by 13 nt on both strands of the target. In separate mixing experiments, the radiolabel was either on the shorter substrate (Figure 4C, lanes 1 and 2) or on the longer one (Figure 4C, lanes 3 and 4), as indicated in the schematic. Disintegration by the foldback pathway will give a 39 nt hairpin in the first case and a 65 nt hairpin in the second case. This is what was observed (lanes 2 and 4, respectively). Disintegration by the true reversal pathway will give a 52 nt product in both cases. Only miniscule amounts (<5%) of such a product could be seen upon overexposure of the gel. These experiments confirmed that the major product (>95%) was a hairpin, irrespective of whether the target sequence was capable of base pairing or not (not shown).
Formation of the hairpin was dependent on the presence of Mg2+ and was not generally supported by other metal ions (see below; Table I). Thus, disintegration from both plasmid θ substrates (Figures 2 and 3) and oligonucleotide ST substrates (Figure 4) occurs by the foldback pathway under these conditions, and shows a similar metal ion specificity.
The forward integration reaction of Mu occurs in trans, that is, transposase bound to one Mu end catalyzes reaction at the other end (Aldaz et al, 1996; Savilahti and Mizuuchi, 1996; Namgoong and Harshey, 1998). The disintegration reaction on oligonucleotide substrates is therefore expected to be intermolecular. To test this, the reaction mixture was serially diluted over a 12-fold range. Each reaction contained the same amount of substrate (2 pmol), but was diluted differently in the reaction buffer as indicated (Figure 4D). The MuA concentration was kept constant at 0.3 μM, equivalent to approximately 1–2 MuA monomers per binding site in the undiluted sample. The effect of dilution was striking on oligonucleotide substrates, where the reaction dropped to 50% at six-fold dilution and to 20% at 12-fold dilution (Figure 4D). Assuming synapsis between two MuA-bound molecules to be second order, the rate of the intermolecular reaction should decrease as the square of the dilution factor. The data tend toward this pattern. In contrast, the reaction on plasmid θ substrates, expected to be intramolecular since the two ends are on the same plasmid molecule, was not significantly affected. The trans activity of MuA subunits was also confirmed in experiments where wild-type MuA and the catalytically inactive variant MuA(E392A) were preloaded separately on half-target substrates and then mixed; reaction occurred on the half-target substrate bound by MuA(E392A) (not shown).
We conclude that MuA catalyzes disintegration by the foldback pathway on the half-target substrates. While target 3′OHs are the major source of the nucleophile, hydrolysis of the Mu–target junction also occurs. Like the forward integration reaction, disintegration also occurs in trans and likely uses the same active site.
Mismatches within the 5 bp target integration site enhance disintegration on full-target ST substrates
We next wished to test the disintegration behavior of a full-target site, which resembles a single-ended integrant as shown in Figure 5. Such substrates have been used in retroviral systems (Chow et al, 1992; Jonsson et al, 1993; Mazumder et al, 1994). The single nick in the target could still allow the DNA to swivel and assume either one of the two conformations (Figure 3B; we will refer to the flipped target conformation as ‘quasi-foldback' in this case). Nucleophilic attack by the single target hydroxyl on the Mu–target junction in either conformation would generate a 52 nt full-length target (Figure 5, right). This reaction is not expected to be efficient, since an intermolecular complex would have to accommodate two full-target sites, resulting perhaps in overcrowding and steric clashes. Indeed, the reaction was not efficient (lane 2). However, introduction of mismatches into the 5 bp target flank improved activity dramatically (lanes 3–14). A centrally placed mismatch showed the highest stimulation (lane 10), its activity being greater than that of a substrate where all five base pairs were mismatched (compare lanes 4 and 10). Mismatches on either side of the central base pair did not behave in a symmetrical fashion (compare lane 6 with 14, and lane 8 with 12), and may represent sequence context effects. As with the half-target substrate, disintegration with a full-target substrate also showed a fairly stringent requirement for Mg2+ (see Table I).
Figure 5.
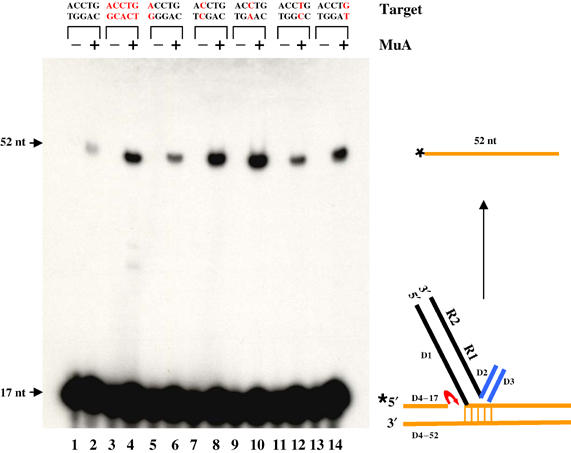
Disintegration of full-target substrates. ST substrates carrying mismatches (red bases) within the 5 bp target flank were labeled (*) as indicated, incubated with (+) or without (−) MuA, and analyzed as described in Figure 4.
In summary, base unpairing within the target flank is necessary to stimulate disintegration on full-target substrates. Given the intermolecular nature of this reaction, two full-target DNAs must be accommodated within the complex, which is apparently not stable (see Table I). The substrate design does not permit a definitive conclusion with regard to the conformation of the target in the complex, but based on the finding that the metal ion specificity for this reaction is similar to that for foldback reversal and different from true reversal (see next section), it is likely that base unpairing promotes a quasi-foldback target configuration.
High temperatures promote disintegration within type II complexes by the true reversal pathway
Results presented in Figures 2, 3, 4 and 5 suggest that the Mu transposase is inherently capable of reversing integration, yet does not normally do so. That the predominant configuration of the target under these conditions is a foldback arrangement suggests that the potentially reactive components (target hydroxyls, Mu–target junctions) are not correctly juxtaposed for reversal within the ST active site, and that their organization must be perturbed in order to do so. Could we test this conclusion by altering DNA conformation by other means? We tried changing pH and temperature conditions. The temperature reactions are shown in Figure 6A. Type II ST complexes were incubated at increasing temperatures in normal reaction buffer for 5 min, and deproteinized prior to electrophoresis. At very high temperatures (75°C), a novel product D* (similar in mobility to D in Figure 2) was obtained. The reaction showed a sharp temperature optimum. Approximately 50% of the input substrate was converted to D*, and the reaction reached a plateau around 5 min at this temperature (Figure 6B). Addition of EDTA abolished the reaction, showing that Mg2+ was required (data not shown). All metal ions tested functioned to varying degrees: Co2+ and Zn2+ were the most efficient, taking the reaction to near completion (see below; Table I).
Figure 6.
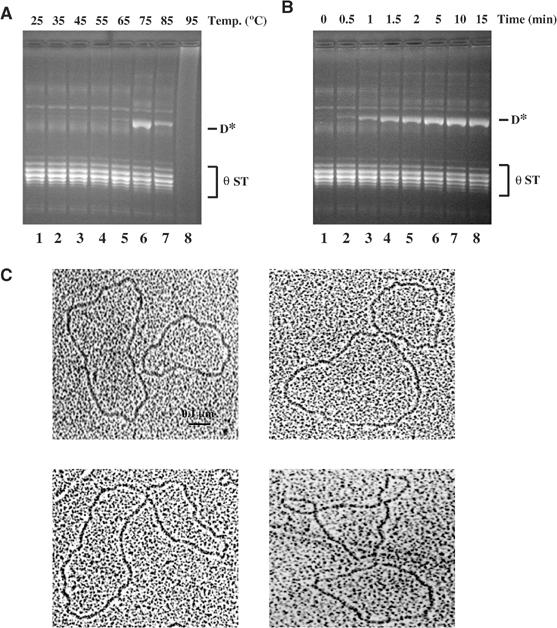
True reversal at high temperatures. (A) Type II complexes formed at 30°C were incubated at the indicated temperatures for 5 min, followed by addition of SDS prior to electrophoresis in 1% agarose gels. (B) Time course of the reaction at 75°C. (C) Electron micrographs of the novel product D* obtained in (A).
The D* band was excised and examined by electron microscopy. It was composed exclusively of two circles linked at a single point (Figure 6C; >100 molecules examined). The sizes of the circles were consistent with one being the mini-Mu donor (4.1 kb) and the other the target (2.8 kb) plasmid. This is the expected configuration for single-ended disintegration by the true reversal pathway (see Figure 3Ba). Restriction digestion of this product showed that it contained an equal mixture of molecules disintegrated at the left or right end (Supplementary Figure 4). Incubation of the θ complexes at alkaline pH (8–11) for 10 min at 30°C also gave the D* band, but the reaction was inefficient (<5% that at high temperature; not shown).
In summary, incubation of type II ST complexes at 75°C for 5 min results in efficient disintegration of one or the other Mu end by true reversal. This reaction shows broad metal ion specificity, and is most efficient in the presence of Co2+ and Zn2+ ions.
Tm of target DNA influences the temperature range of disintegration on oligonucleotide substrates but does not change the temperature optimum
The sharp temperature optimum for the high-temperature disintegration of type II complexes is striking (Figure 6A). At these temperatures, changes in either DNA and/or protein could influence disintegration. A test for the importance of DNA would be to alter the base composition (and hence Tm) of target DNA and determine if the optimum shifts in a predictable manner. To do so, we first tested whether this reaction could be reproduced on oligonucleotide substrates.
Integration reactions were performed using precleaved donor substrates and an end-labeled 76 bp target carrying a central mismatch, in order to direct all integrations to this site (Figure 7A). Mu ends attack target positions flanking the central mismatch as shown (Yanagihara and Mizuuchi, 2002). We will refer to the resulting ST complexes as type IIO, to denote that they were formed on oligonucleotides. As in type II complexes, the initial transposon–target joint is expected to be preserved in type IIO complexes as well. The reactions were electrophoresed on native 1.5% agarose gels to separate complexes from unreacted target. Gel slices containing these complexes were excised and equilibrated on ice in the reaction buffer (see Materials and methods), incubated at various temperatures for 5 min to induce disintegration, followed by DNA extraction and fractionation on denaturing polyacrylamide gels (Figure 7B).
Figure 7.
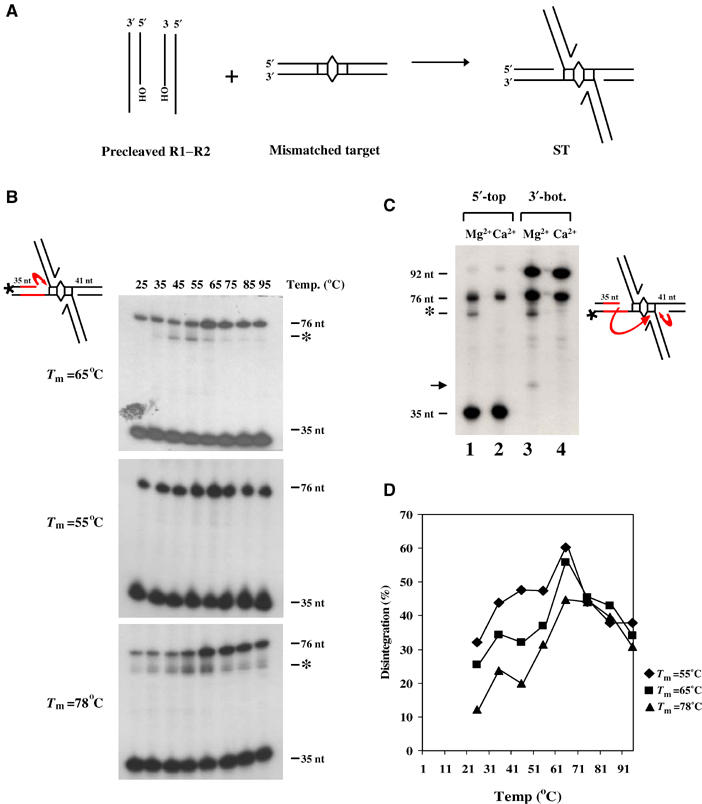
High-temperature disintegration of oligonucleotide substrates. (A) Directed integration to a site with a mismatched base pair. (B) Three targets, differing in the sequence of 20 bp flanking one side of the mismatch (red lines), were labeled at the indicated 5′ end. After integration, complexes were separated from free target on agarose gels, subjected to in-gel disintegration at the indicated temperatures, and electrophoresed on denaturing polyacrylamide gels. *, unexpected product. (C) Substrate with Tm=65°C was labeled either as in (B) (lanes 1 and 2) or at the 3′ end of the bottom strand as shown (lanes 3 and 4). Reactions were at 55°C as described in (B), except that lanes 2 and 4 contained Ca2+. See text for details. (D) Graphical representation of reactions in (B). ‘% disintegration' refers to % of input substrate converted to the 76 nt product.
Two products were observed—a 35 nt species, which comes from the unreacted target flank, and a 76 nt species, which is the result of disintegration by true reversal (Figure 7B, top panel). The efficiency of disintegration was similar to that on plasmid substrates. However, unlike the sharp optimum observed with type II complexes, disintegration of type IIO complexes occurred over a wider temperature range (and even at room temperature!), with an optimum at 65°C. As with type II complexes, these reactions also exhibited broad metal ion specificity (see below; Table I).
We next synthesized two sets of oligonucleotide targets in which 20 residues flanking one side of the mismatch on the labeled strand (indicated in red; all Tm's refer to the Tm of the 35-nt region) were either AT-rich (Tm=55°C) or GC-rich (Tm=78°C), the expectation being that, if thermal motion of DNA was responsible for realigning the reactive DNA components, the temperature optimum should shift to lower or higher values, respectively. We observed that the AT-rich substrate was indeed more proficient in disintegration at lower temperatures, as judged by a higher percentage of products at these temperatures (Figure 7B, middle panel). On the other hand, the GC-rich substrate was more proficient in disintegration at higher temperatures (Figure 7B, bottom panel).
The data in Figure 7B are shown graphically in Figure 7D. Strikingly, all three substrates showed an optimum of 65°C. A second common peak around 35°C was also seen. The behavior of the three substrates converged around 70°C. A common component of all three complexes is the MuA protein. Thus, the data suggest that while the Tm of the target DNA may influence the reaction, temperature-induced transitions in protein structure are likely primarily responsible for promoting disintegration.
Reactions in the top and bottom panels in Figure 7B also yielded an unexpected product (*), characterized further in Figure 7C using the substrate with Tm=65°C. In lanes 1 and 2 the radio label was at the 5′ end of the top strand as in Figure 7B, while in lanes 3 and 4 it was at the 3′ end of the bottom strand as indicated on the right. Reactions were incubated at 55°C (the optimum for * formation; see Figure 7B) in the presence of either Mg2+ (lanes 1 and 3) or Ca2+ (lanes 2 and 4). In lanes 3 and 4, the 92 nt species is the undisintegrated bottom strand with the attached Mu strand (equivalent to the unreacted 35 nt species in lane 1), while the 76 nt species comes from disintegration on the bottom strand (short red arrow on the right). Ca2+ ions supported formation of the 76 nt product, but not the * product (lanes 2 and 4). The 41 nt product in lane 3 (black arrow) likely comes from hydrolysis at the Mu–target joint on the bottom strand. Similar data were obtained for the substrate with Tm=78°C (not shown).
Since the * product was observed when either the top or the bottom strand was labeled, it might represent a hairpin product resulting from an attack of the top strand hydroxyl on the bottom target strand. Maxam–Gilbert sequencing of the * product, however, disproved this possibility. Rather, this product represented an attack of the target hydroxyl on the opposite transposon–target junction (long red arrow on the left). This suggests that this is a product from the foldback pathway, and therefore the target must have rearranged during gel electrophoresis in this small fraction of type IIO complexes (see Discussion). That Ca2+ does not support its formation is consistent with the stringent metal ion specificity of the foldback reaction discussed below.
The two disintegration pathways manifest distinct metal ion specificities
Divalent metal ions are required for Mu transpososome assembly, as well as for transposition (see Chaconas and Harshey, 2002): either Mg2+ or Mn2+ ions function equally well. Ca2+ supports assembly and not Mu end cleavage (Mizuuchi et al, 1992), but promotes ST of cleaved ends on oligonucleotide substrates (Savilahti et al, 1995). Co2+ and Zn2+ can cleave and ST, but cannot support assembly (Wang et al, 1996). We initially noticed that the foldback reaction on both plasmid and half-target substrates was not supported by Mn2+ (see Table I, rows 1a and 2a). Among other metals, only Co2+ showed some activity (15%) on half-target substrates (Table I, row 2a). Since Co2+ and Zn2+ do not support assembly, we preassembled both plasmid and half-target complexes in Mg2+, depleted the Mg2+ by EDTA treatment followed by gel filtration, then retested the different metals for disintegration. We now observed that, except for Ca2+, all the other three metal ions promoted disintegration to varying degrees, in some cases significantly better than with Mg2+ (Table I, rows 1b and 2b). To test if the failure of these metals to function when added directly to the reaction was due to failure of complex assembly, the substrates were assayed for assembly by electrophoresis through agarose gels; indeed, assembly was only detected with Co2+, consistent with levels of disintegration observed when Co2+ was added directly (Table I, compare rows 2a and 2c). The similar behavior of half- and full-target complexes (Table I, compare rows 2a and 2c with 3a and 3c), suggests that the reaction observed on the latter substrates also follows the foldback pathway. Full-target complexes assembled in Mg2+ were unstable and did not survive gel filtration (Table I, row 3b). Interestingly, both the half- and full-target complexes were seen to assemble in the absence of metals (Table I, rows 2c and 3c), an observation consistent with a requirement for metal ions only to create or maintain a single-stranded character within fully base-paired Mu ends prior to cleavage (Lee and Harshey, 2003). Thus, with the exception of Mg2+, all other metal ions tested interfere with the assembly of foldback complexes.
In contrast, disintegration by true reversal was supported by all metal ions on both kinds of substrates (Table I, rows 4 and 5). ST complexes formed with Mg2+ were depleted for metal by gel filtration (Table I, row 4) or by electrophoresis through agarose gels (Table I, row 5) prior to testing. Plasmid ST complexes generated with Mn2+ could also be disintegrated with Mn2+ (not shown).
In summary, the two reversal pathways show distinct metal ion specificities for assembly and for phosphoryl transfer. Ca2+ ions support disintegration by true reversal but not foldback reversal (see also Figure 7C) while, with the exception of Mg2+, most metal ions inhibit assembly of complexes that catalyze disintegration by the foldback pathway.
Discussion
The integration or ST step of Mu transposition should at best go to 50% completion at equilibrium, because the reversal of integration is an isoenergetic reaction (see Mizuuchi, 1997). Yet integration does go to completion. One strategy to drive the reaction forward would be to promote rapid dissociation of the complex after ST. This does not happen with the Mu complex, which performs additional roles following transposition (see Chaconas and Harshey, 2002). The results presented here suggest that Mu employs an alternative strategy for controlling the directionality of transposition. They are consistent with the conclusion that a conformational change ‘locks' the active site upon ST. This can be overcome by perturbing protein/DNA structure to reverse integration.
Two disintegration pathways: regulatory implications for transposition
We have reported two types of disintegration events. In the first ‘true reversal', a target 3′OH attacks the junction phosphodiester proximal to it, repairing the broken strand in the target and releasing the Mu end. In the second ‘pseudo' or ‘foldback reversal', the 3′OH attacks the junction phosphodiester distal to it (and on the opposite strand) to release the Mu end, but forms a hairpin on the target. On plasmid substrates, both reactions disintegrate only one of the two Mu ends. We find different metal ion specificities for the two types of reactions. We argue below that the majority of disintegrations reported to date in the literature are of the foldback type, also referred to as ‘macroscopic reversal' of integration (Gerton et al, 1999).
A novel aspect of the reversal of Mu integration reported here is that it implicates an altered active site anatomy in blocking reversal. Thus, only when type II products were stripped of proteins and MuA reassembled on them did the reaction reverse (Figures 2 and 3). The structure of the products obtained here was consistent with a switch in the original orientation of the target to assume a foldback configuration (Figure 3Bb and Supplementary Figure 3). Similar results were obtained with half-target oligonucleotide substrates (Figure 4). Full-target substrates required base-pair mismatches within the 5 bp target flank for efficient disintegration (Figure 5). We infer that this reaction also occurred by the foldback pathway because of its metal ion specificity (discussed below); base unpairing may serve to create a swivel at the single nick in these substrates and hence a quasi-foldback arrangement of the target joint.
The experiments above suggested that the normally unfavorable geometry of the ST active site could be rearranged to promote reversal. This inference was further tested by changing protein/DNA conformation with increasing temperatures (Figure 6). Very high temperatures (75°C on plasmid substrates, 65°C on oligonucleotide substrates) were seen to reverse integration efficiently (50–100% of substrate converted, depending on the metal ion; Figures 6 and 7 and Table I). The target joints in these complexes would not have had the opportunity to rearrange; indeed, product structure was consistent with true reversal (see Figure 3Ba). Differences in the temperature range and optimum for true reversal using plasmid versus oligonucleotide substrates (compare Figures 6 and 7) could be due to the single base-pair mismatch in the oligonucleotide target. On the latter substrates, true reversal is observed even at room temperatures.
Disintegration is likely performed by the same active site residues used for integration (Figure 2 and data not shown). Studies with the related HIV integrase have reached a similar conclusion (Gerton et al, 1999). Like integration, disintegration also occurs in trans (Figure 4D and data not shown). However, unlike integration, which is strongly coupled at both ends (Namgoong and Harshey, 1998; Williams et al, 1999), disintegration occurred only at one or the other Mu end (Figures 3 and 6 and Supplementary Figures 2 and 4). Given the high efficiency of disintegration (20% substrate conversion by foldback, 50–100% conversion by true reversal), independent events at each end should still have been detectable as a double-ended disintegration event that regenerates the starting plasmid substrates. The absence of such products not only suggests a loss of coordination but a negative cooperativity between the active sites. This feature of the reaction was not revealed in prior experiments due to the nature of the substrates employed.
While both DNA and protein must contribute to the active site environment, the high-temperature results would appear to favor protein conformation as the primary determinant of the negative regulation. If a change in DNA conformation alone could overcome the barrier to disintegration, and if high temperatures promote this event via DNA melting, one would not expect a narrow optimum of 75°C, as observed with type II complexes (Figure 6A). Mu insertions occur nearly randomly in a target (Mizuuchi and Mizuuchi, 1993; Haapa-Paananen et al, 2002), and one would expect a broader DNA melting profile for the collective integration sites. Although disintegration was observed over a wider temperature range when the Tm of the target DNA flank was manipulated, the temperature optimum remained constant (Figure 7D), suggesting a change in protein rather than DNA structure at the high temperature. In the foldback reaction on the other hand, the flipped target likely repositions the reacting DNA moieties in the active site. We suggest that the minor foldback product observed during high-temperature reversal on oligonucleotide substrates (Figure 7), is the result of target rearrangement during gel electrophoresis of the complex.
High temperatures (65–75°C) are known to reverse topoisomerase II-linked DNA breaks induced by various drugs (Hsiang and Liu, 1989; Bromberg and Osheroff, 2001). That these enzymes cannot cleave DNA at suboptimal (high or low) temperatures but can religate enzyme-associated cleaved molecules at these temperatures has served as a basis for a DNA religation assay. Like the reversal of Mu integration, cleaved complexes show a rapid rate of reversal as well as a narrow range of the transition temperature. Heat-induced transitions have been shown to occur within the enzyme and to be dependent on Mg2+ and DNA (see Bromberg and Osheroff, 2001, and references therein).
Metal ion specificity of disintegration: plasticity of the transposase active site
The Mu transposase belongs to a superfamily of integrases with a ‘DDE' motif that uses a single active site to catalyze the hydrolysis and joining of transposon ends to target (see Craig et al, 2002). DDE residues coordinate divalent metal ions, which facilitate the phosphoryl transfer reaction chemistry. In all systems studied, Mg2+ and Mn2+ support both cleavage and ST, although Mg2+ is likely the biologically relevant cation. In Mu, integration is normally strictly dependent on Mg2+ or Mn2+. However, preassembled substrates can be cleaved by Zn2+ and Co2+ (Wang et al, 1996), and precleaved substrates can carry out ST in the presence of Ca2+ (Savilahti et al, 1995). Since a single active site carries out both cleavage and ST, the differential metal ion selectivity in cleavage versus ST must reflect either conformational differences between the active sites during the two steps or differences in the way the two distinct nucleophiles are activated. Metal ions are also needed for assembly of the Mu transpososome, but this requirement is substantially reduced on prenicked substrates (Savilahti et al, 1995) or substrates with mismatched Mu termini (Lee and Harshey, 2003), implying that on fully base-paired substrates metal ions assist in the DNA opening observed around Mu ends (Wang et al, 1996; Kobryn et al, 2002).
The metal ion specificities for disintegration by the two pathways reported here (Table I) suggest that there must be conformational differences in their active sites. A clear finding is that Ca2+ ions support true but not foldback reversal, and that the foldback reaction has a near-stringent requirement for Mg2+. The latter observation was traced to an inhibitory action of most of these metal ions on complex assembly, which occurred even in the absence of metal on ST substrates (see Table I). Once assembled, however, Mn2+, Co2+ and Zn2+ were all proficient in foldback disintegration. Co2+ and Zn2+ were consistently superior in both pathways.
Since both disintegration pathways use the same target nucleophile, the differences in metal ion specificity are likely due to the different target site conformation within the active sites. That true but not foldback reversal is supported by both Mg2+ and Ca2+ highlights the similarity between the forward and reverse steps of ST, since precleaved ends ST in the presence of Ca2+ (Savilahti et al, 1995). The ability of a single active site to accommodate different sets of components to catalyze phosphoryl transfer speaks to its remarkable plasticity (see Baker and Mizuuchi, 2002).
General implications of the disintegration reaction
The disintegration reaction was first reported for HIV-1 integrase using full-target substrates like those in Figure 5 (Chow et al, 1992). Use of half-target substrates showed predominantly a foldback pathway for disintegration by HIV-1 integrase (Mazumder et al, 1994). Using specialized short ‘crossbone' substrates, HIV and MLV integrases also showed predominantly foldback activity, although some apparently true reversal was reported (Chow and Brown, 1994). However, there was no way of distinguishing true from quasi-foldback reversal in these experiments, as inferred here with the Mu substrates. A reaction equivalent to disintegration has been reported for IS911 transposase (Polard et al, 1996), P element transposase (Beall and Rio, 1998) and the RAG1/2 proteins involved in immunoglobulin rearrangements (Melek and Gellert, 2000). As observed for the Mu foldback reaction, hairpin formation as well as hydrolysis of the transposon–target junction contributes to disintegration in many of these systems. Based on the experiments reported here for Mu, we argue that true reversal might only occur during conformational transitions in the nucleoprotein complex as observed here at high temperatures.
DNA mismatches were recently shown to efficiently target Mu (Yanagihara and Mizuuchi, 2002) and RAG-mediated transposition (Tsai et al, 2003), and suppress the use of other target sites. Speculation that target site selection by these transposases may be intimately linked to mutagenic and metabolic processes that transiently present favorable DNA structures to the transposition machinery should be tempered with our findings that Mu integrations at such sites are highly favored toward disintegration (Figure 7).
A common theme to emerge from in vitro studies of transposition is that regulatory mechanisms exist to counter reversal of integration. While the basic mechanism may be a shared regulatory feature of most transposable elements, the molecular details are likely to be varied. Understanding structural transitions within the active site by comparison of the structure of transpososomes arrested at various stages of integration/disintegration will provide a fruitful area for future investigation. It should be possible from these studies to manipulate the transposase to stimulate disintegration, with obvious applications for intervention in the integration of elements such as HIV (Craigie, 2002).
Materials and methods
Proteins and DNA substrates
Mu A, B and E. coli HU were purified as described (Namgoong and Harshey, 1998). pZW140 (Wang et al, 1996) was the mini-Mu donor, and Litmus 28 (New England Biolabs or NEB) the plasmid target. Oligonucleotides were from Integrated DNA Technologies, Coralville, IA, and 3′ dideoxy substrates from Sigma-Genosys. Radiolabeling at the 5′ end used [γ-32p]ATP and T4 polynucleotide kinase, and at the 3′ end [α-32P]cordycepin phosphate and terminal nucleotidyl transferase (NEB). Precleaved substrates comprising the following two oligonucleotides were annealed as described top strand, 5′-AAGTTTTCGCATTTATCGTGAAACGCTTTCGCGTT TTTCGTGCGCCGCTTCA-3′; bottom strand, 5′-GATCACTCATTGAAGCGGCGCACGAAAAACGCGAA AGCG TTTCACGATAAATGCGAAAACTT-3′. Terminal dinucleotide on cleaved strand is underlined. Other oligonucleotides were as follows:
D1, 5′-AAGTTTTCGCATTTATCGTGAAACGCTTTCGCGTT TTTCGTGCGCCGC TTCAACCTGTGTCTAGCTGTAC TACGTCAGTCCGATACT-3′; D2, 5′-GTAGCAGACTGATCACTCATTGAAGCGGCGCACGA AAAACGCGA AAGCGTTTCACGATAAATGCGAAAACTT-3′; D3, 5′-ATGAGTGATCAGTCTGCTAC-3′; D4, 5′-AGTATCGGACTGACGTAGTA CAGCTAGACA-3′; D4–17, 5′-TAACTGTCTCGCGCATA-3′, D4–52, 5′-AGTATCGGACTGACGTAGTACAGCTAGACACAGG TTATGCGCGAGACAGTTA-3′. Sequence of the target fragment with a single mismatch was as in Yanagihara and Mizuuchi (2002). The sequence of the low-Tm (55°C) target was as follows: top strand, 5′-CGTTCATTAGCACAAAAAAAAAAAAAAAAAAAAAA ACGGCACATAAGATCAGAAGTTAACTAGCACTAGT ACTTGC-3′; bottom strand, 5′-GCAAGTACTAGTGC TAGTTAACTTCTGATCTTATG TGCGGTTTTTTTTTTTTTTTTTTTTTTTGTG CTAATGAACG-3′. The underlined 20 nt sequences were those replaced in the Tm=65°C target. For the high-Tm (78°C) target, the underlined region was GGI repeats in the top strand and C's in the bottom strand.
ST on plasmid substrates
Reactions (20 μl, 1 × ) contained 1 μg each of mini-Mu donor and target plasmids, 0.2 μg HU, 0.5 μg MuA, 0.1 μg MuB in 25 mM Tris–HCl, pH 8.0, 140 mM KCl, 10 mM MgCl2 and 2 mM ATP. Incubation was at 30°C for 1 h, followed by heparin addition (0.05 mg/ml final) to remove loosely bound protein, prior to electrophoresis on 1% TAE agarose gels.
Purification of θ ST products
A 100 × ST reaction was electrophoresed in a 1% agarose gel, and the DNA was visualized by ethidium bromide staining. Type II complex band was excised, and DNA was electroeluted in 20 ml of 1 × TAE buffer. The eluate was concentrated 50-fold by speed-vacuum centrifugation, and DNA was purified on Qiagen QIAquick PCR purification spin columns.
Disintegration of θ ST and type II complexes
Reactions (20 μl) containing 0.5 μg of purified θ ST DNA, 0.5 μg MuA, 0.2 μg HU in 25 mM Tris–HCl, pH 8.0, 140 mM KCl and 10 mM MgCl2 were incubated at 30°C for 2 h. SDS was added to a final concentration of 0.1% before electrophoresis in 1% agarose gels.
High-temperature disintegration of type II complexes was performed in the same buffer for 5 min.
Disintegration of half- and full-target oligonucleotide substrates
Reactions (20 μl) containing 2 pmol of oligo-substrate, 6 pmol MuA, 25 mM Hepes (K+), pH 7.6, 140 mM KCl, 10 mM MgCl2 and 10% DMSO were incubated at 30°C for 2 h. Following extraction with phenol/chloroform, DNA was precipitated, resuspended in 10 μl of Maxam and Gilbert gel loading buffer, and fractionated on 12% urea–PAGE.
High-temperature in-gel disintegration of type IIO complexes
ST reactions (20 μl) containing 4 pmol of precleaved Mu DNA, 4 pmol of labeled target, 12 pmol MuA in 25 mM Hepes (K+), pH 7.6, 140 mM KCl, 10 mM MgCl2, 10% glycerol and 15% DMSO were first incubated at 30°C for 1 h. Following heparin addition, reactions were electrophoresed on 1.5% agarose gels in 1 × TBE buffer at 4.5 V/cm for 2.5 h. ST complex band was located by exposure to an X-ray film at 4°C for an hour. Appropriate gel slices (∼0.5 cm3) were excised and equilibrated on ice for 30 min in 500 μl of 25 mM Hepes (K+) (pH 7.6) and 10 mM MgCl2. After incubation at various temperatures for 5 min to induce disintegration, DNA was purified using Bio-Rad Freeze ‘N Squeeze spin columns, extracted with phenol/chloroform, precipitated and fractionated on 12% urea–PAGE.
Disintegrations in different metal ions
Concentrations of metal ions were as follows: 10 mM CaCl2, 2 mM MnCl2 and 1 mM CoCl2 or ZnCl2. These were added directly to the reaction or after preassembly of MuA in 10 mM MgCl2. The latter reactions were incubated at room temperature for 10 min, stopped by addition of 20 mM EDTA and heparin, and purified over Bio-Gel P-100 spin columns; the various metal ions were added back, followed by incubation at 30°C for 1 h (oligo-substrates) or 2 h (θ ST), or 5 min at high temperatures (type II and type IIO complexes).
Reactions monitoring transpososome assembly were electrophoresed on 1.5% agarose gels in 1 × TBE buffer at 4.5 V/cm for 2.5 h. Gels were dried and exposed to film.
Quantitation and DNA sequencing
DNA band intensities were quantified using Bio-Rad Multianalyst software. Sequencing was by the Maxam–Gilbert procedure (Ausubel, 1994).
Electron microscopy
D and D* products were excised from gels and purified as described above for θ STs. DNA was spread on to grids by the cytochrome c–ammonium acetate method (Coggins, 1987).
Supplementary Material
Supplementary Figures
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Acknowledgments
We thank A Vetcher and S Levine at UT Dallas, and J Mendenhall at UT Austin for help with electron microscopy. We thank M Jayaram for helpful comments on the manuscript. This work was supported by a grant from the National Institutes of Health (GM33247). Partial support was provided by the Robert Welch Foundation (F-1351).
References
- Aldaz H, Schuster E, Baker TA (1996) The interwoven architecture of the Mu transposase couples DNA synapsis to catalysis. Cell 85: 257–269 [DOI] [PubMed] [Google Scholar]
- Ausubel FM (1994) Current Protocols in Molecular Biology. New York: John Wiley & Sons [Google Scholar]
- Baker TA, Mizuuchi K (2002) Chemical mechanisms for mobilizing DNA. In Mobile DNA II, Craig NL, Craigie R, Gellert M, Lambowitz AM (eds) pp 12–23. Washington, DC: ASM Press [Google Scholar]
- Beall EL, Rio DC (1998) Transposase makes critical contacts with, and is stimulated by, single-stranded DNA at the P element termini in vitro. EMBO J 17: 2122–2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg KD, Osheroff N (2001) DNA cleavage and religation by human topoisomerase II alpha at high temperature. Biochemistry 40: 8410–8418 [DOI] [PubMed] [Google Scholar]
- Bujacz G, Jaskolski M, Alexandratos J, Wlodawer A, Merkel G, Katz RA, Skalka AM (1995) High-resolution structure of the catalytic domain of avian sarcoma virus integrase. J Mol Biol 253: 333–346 [DOI] [PubMed] [Google Scholar]
- Chaconas G, Harshey RM (2002) Transpsition of phage Mu DNA. In Mobile DNA II, Craig NL, Craigie R, Gellert M, Lambowitz AM (eds) pp 384–402. Washington, DC: ASM Press [Google Scholar]
- Chow SA, Brown PO (1994) Juxtaposition of two viral DNA ends in a bimolecular disintegration reaction mediated by multimers of human immunodeficiency virus type 1 or murine leukemia virus integrase. J Virol 68: 7869–7878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow SA, Vincent KA, Ellison V, Brown PO (1992) Reversal of integration and DNA splicing mediated by integrase of human immunodeficiency virus. Science 255: 723–726 [DOI] [PubMed] [Google Scholar]
- Coggins LW (1987) Preparation of nucleic acids for electron microscopy. In Electron Microscopy in Molecular Biology: A Practical Approach, Sommerville J, Scheer U (eds) pp 1–29. Oxford, Washington, DC: IRL Press [Google Scholar]
- Craig NL, Craigie R, Gellert M, Lambowitz AM (2002) Mobile DNA II. Washington, DC: ASM Press [Google Scholar]
- Craigie R (2002) Retroviral DNA integration. In Mobile DNA II, Craig NL, Craigie R, Gellert M, Lambowitz AM (eds) pp 613–630. Washington, DC: ASM Press [Google Scholar]
- Doolittle WF, Sapienza C (1980) Selfish genes, the phenotype paradigm and genome evolution. Nature 284: 601–603 [DOI] [PubMed] [Google Scholar]
- Dyda F, Hickman AB, Jenkins TM, Engelman A, Craigie R, Davies DR (1994) Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases. Science 266: 1981–1986 [DOI] [PubMed] [Google Scholar]
- Engelman A, Mizuuchi K, Craigie R (1991) HIV-1 DNA integration: mechanism of viral DNA cleavage and DNA strand transfer. Cell 67: 1211–1221 [DOI] [PubMed] [Google Scholar]
- Gerton JL, Herschlag D, Brown PO (1999) Stereospecificity of reactions catalyzed by HIV-1 integrase. J Biol Chem 274: 33480–33487 [DOI] [PubMed] [Google Scholar]
- Haapa-Paananen S, Rita H, Savilahti H (2002) DNA transposition of bacteriophage Mu. A quantitative analysis of target site selection in vitro. J Biol Chem 277: 2843–2851 [DOI] [PubMed] [Google Scholar]
- Hsiang YH, Liu LF (1989) Evidence for the reversibility of cellular DNA lesion induced by mammalian topoisomerase II poisons. J Biol Chem 264: 9713–9715 [PubMed] [Google Scholar]
- Jonsson CB, Donzella GA, Roth MJ (1993) Characterization of the forward and reverse integration reactions of the Moloney murine leukemia virus integrase protein purified from Escherichia coli. J Biol Chem 268: 1462–1469 [PubMed] [Google Scholar]
- Kazazian HH Jr (2004) Mobile elements: drivers of genome evolution. Science 303: 1626–1632 [DOI] [PubMed] [Google Scholar]
- Kim K, Namgoong SY, Jayaram M, Harshey RM (1995) Step-arrest mutants of phage Mu transposase: implications in nucleoprotein assembly, DNA cleavage and strand transfer. J Biol Chem 270: 1472–1479 [DOI] [PubMed] [Google Scholar]
- Kobryn K, Watson MA, Allison RG, Chaconas G (2002) The Mu three-site synapse: a strained assembly platform in which delivery of the L1 transposase binding site triggers catalytic commitment. Mol Cell 10: 659–669 [DOI] [PubMed] [Google Scholar]
- Lavoie BD, Chan BS, Allison RG, Chaconas G (1991) Structural aspects of a higher order nucleoprotein complex: induction of an altered DNA structure at the Mu–host junction of the Mu type 1 transpososome. EMBO J 10: 3051–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Harshey RM (2003) The conserved CA/TG motif at Mu termini: T specifies stable transpososome assembly. J Mol Biol 330: 261–275 [DOI] [PubMed] [Google Scholar]
- Mazumder A, Engelman A, Craigie R, Fesen M, Pommier Y (1994) Intermolecular disintegration and intramolecular strand transfer activities of wild-type and mutant HIV-1 integrase. Nucleic Acids Res 22: 1037–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melek M, Gellert M (2000) RAG1/2-mediated resolution of transposition intermediates: two pathways and possible consequences. Cell 101: 625–633 [DOI] [PubMed] [Google Scholar]
- Mizuuchi K (1997) Polynucleotidyl transfer reactions in site-specific DNA recombination. Genes Cells 2: 1–12 [DOI] [PubMed] [Google Scholar]
- Mizuuchi M, Baker TA, Mizuuchi K (1992) Assembly of the active form of the transposase–Mu DNA complex: a critical control point in Mu transposition. Cell 70: 303–311 [DOI] [PubMed] [Google Scholar]
- Mizuuchi M, Mizuuchi K (1993) Target site selection in transposition of phage Mu. Cold Spring Harb Symp Quant Biol 58: 515–523 [DOI] [PubMed] [Google Scholar]
- Namgoong SY, Harshey RM (1998) The same two monomers within a MuA tetramer provide the DDE domains for the strand cleavage and strand transfer steps of transposition. EMBO J 17: 3775–3785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgel LE, Crick FH (1980) Selfish DNA: the ultimate parasite. Nature 284: 604–607 [DOI] [PubMed] [Google Scholar]
- Polard P, Ton-Hoang B, Haren L, Betermier M, Walczak R, Chandler M (1996) IS911-mediated transpositional recombination in vitro. J Mol Biol 264: 68–81 [DOI] [PubMed] [Google Scholar]
- Rice P, Mizuuchi K (1995) Structure of the bacteriophage Mu transposase core: a common structural motif for DNA transposition and retroviral integration. Cell 82: 209–220 [DOI] [PubMed] [Google Scholar]
- Savilahti H, Mizuuchi K (1996) Mu transpositional recombination: donor DNA cleavage and strand transfer in trans by the Mu transposase. Cell 85: 271–280 [DOI] [PubMed] [Google Scholar]
- Savilahti H, Rice PA, Mizuuchi K (1995) The phage Mu transpososome core: DNA requirements for assembly and function. EMBO J 14: 4893–4903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CL, Chatterji M, Schatz DG (2003) DNA mismatches and GC-rich motifs target transposition by the RAG1/RAG2 transposase. Nucleic Acids Res 31: 6180–6190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Namgoong SY, Zhang X, Harshey RM (1996) Kinetic and structural probing of the precleavage synaptic complex (type 0) formed during phage Mu transposition. Action of metal ions and reagents specific to single-stranded DNA. J Biol Chem 271: 9619–9626 [DOI] [PubMed] [Google Scholar]
- Williams TL, Jackson EL, Carritte A, Baker TA (1999) Organization and dynamics of the Mu transpososome: recombination by communication between two active sites. Genes Dev 13: 2725–2737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagihara K, Mizuuchi K (2002) Mismatch-targeted transposition of Mu: a new strategy to map genetic polymorphism. Proc Natl Acad Sci USA 99: 11317–11321 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4