

Abstract
Chronic activation of the phosphoinositide 3-kinase (PI3K)/PTEN signal transduction pathway contributes to metastatic cell growth, but up to now effectors mediating this response are poorly defined. By simulating chronic activation of PI3K signaling experimentally, combined with three-dimensional (3D) culture conditions and gene expression profiling, we aimed to identify novel effectors that contribute to malignant cell growth. Using this approach we identified and validated PKN3, a barely characterized protein kinase C-related molecule, as a novel effector mediating malignant cell growth downstream of activated PI3K. PKN3 is required for invasive prostate cell growth as assessed by 3D cell culture assays and in an orthotopic mouse tumor model by inducible expression of short hairpin RNA (shRNA). We demonstrate that PKN3 is regulated by PI3K at both the expression level and the catalytic activity level. Therefore, PKN3 might represent a preferred target for therapeutic intervention in cancers that lack tumor suppressor PTEN function or depend on chronic activation of PI3K.
Keywords: metastasis, orthotopic prostate tumor model, phosphoinositide 3-kinase, PTEN, 3D cell culture
Introduction
The phosphoinositide 3-kinase (PI3K)/PTEN signaling pathway regulates a diverse set of cellular responses including growth, development, survival, motility, adhesion, immune cell function and glucose transport (Katso et al, 2001; Roymans and Slegers, 2001). Class IA PI3K molecules are heterodimers consisting of a regulatory subunit, p85, and a catalytic subunit, p110, whereby several different isoforms exist for each subunit (Wymann and Pirola, 1998). PI3K is only transiently activated after growth factor stimulation of normal cells, and is rapidly turned off through tumor suppressor PTEN function. PTEN negatively regulates PI3K signaling by dephosphorylating its second messenger phospholipid products, thereby ensuring that activation of the pathway occurs in a transient and controlled fashion (Vazquez and Sellers, 2000). PTEN is one of the most frequently inactivated tumor suppressor genes in human cancer (Trotman and Pandolfi, 2003), and its loss results in chronic activation of the PI3K pathway and correlates with increased metastatic behavior (Wang et al, 2003). Sustained PI3K activation can also be achieved by activated forms of PI3K itself and correlates with increased invasiveness or growth in semi-solid matrices (Jimenez et al, 1998; Klippel et al, 1998; Kobayashi et al, 1999).
Conventional gene expression analysis using microarrays to identify cancer therapy targets is hampered by the chromosomal instability of tumor cells and typically results in a large number of differentially expressed genes with uncertain disease relevance. Instead of comparing disease end points, we investigated the molecular changes induced after simulating PI3K hyperactivation experimentally, which allowed to compare pairs of otherwise isogenic cells under normal growth conditions in subsequent gene profiling studies (Kaufmann et al, 2004). This approach was combined with three-dimensional (3D) culture conditions using basement membrane-containing extracellular matrix, which provides for a more relevant system to recapitulate tumor cell growth and metastasis than standard 2D culture conditions using plastic culture surfaces (Bissell et al, 2003). In fact, controlled modulation of PI3K signaling has little phenotypic consequences on 2D cell growth (Stolarov et al, 2001), but dramatically affects the growth phenotype under 3D culture conditions (Kaufmann et al, 2004).
A number of kinases and other signaling molecules, which mediate PI3K-regulated events, represent candidate cancer therapy targets (Luo et al, 2003). However, most of these proteins act rather upstream in the PI3K signaling cascade and also regulate multiple functions in normal cells (Wymann et al, 2003). Therefore, their inhibition is likely to cause side effects. Here, we describe the identification, validation and biochemical characterization of a novel kinase molecule, which mediates invasive prostate cancer cell growth further downstream of a hyperactive PI3K pathway in cell-based assays and a mouse tumor model.
Results
PKN3 mediates growth on basement membrane downstream of PI3K
Sustained activation of PI3K signaling is required for growth of invasive PTEN−/− PC-3 prostate cancer cells on basement membrane matrix, whereas modulation of MEK-MAP kinase signaling has little effect in this cell type (Kaufmann et al, 2004; Supplementary Figure 1). By expression profiling, we identified genes that are PI3K-dependently expressed in cells grown on basement membrane-containing matrigel (Kaufmann et al, 2004). One of these genes encoded a kinase with homology to the protein kinase C superfamily: PKN3, also known as PKNβ (Manning et al, 2002; Mukai, 2003), has not yet been linked to the PI3K/PTEN pathway or to invasive growth.
After verifying its PI3K-dependent expression (Supplementary Figure 2A), we tested whether PKN3 expression is required for PI3K-induced cell growth under 3D culture conditions. PC-3 cells were treated with three different PKN3-specific antisense molecules, the so-called GeneBlocs (GBs) (Sternberger et al, 2002), and subsequently seeded on matrigel (Figure 1A). GBs specific for inhibiting expression of PTEN or p110β, an isoform of the catalytic subunit of PI3K, served as negative or positive controls. In PC-3 cells, p110β appears to represent the predominant isoform for mediating PI3K signaling as compared to p110α (Supplementary Figure 3). Parallel samples were analyzed to confirm specific reduction of PKN3 mRNA expression (not shown; see reduction of protein expression in Figure 1B). Inhibition of PKN3 expression interfered with growth on matrigel to the same extent as inhibition of p110β (Figure 1A), which was previously shown to be required for growth of PC-3 cells on basement membrane matrix (Czauderna et al, 2003b; Supplementary Figure 3). PTEN GB treatment had no effect on the PTEN−/− PC-3 cells in the matrigel assay, similar to a mismatch control antisense molecule (mm) of PKN3 GB3. Three GB molecules targeting distinct sites in the PKN3 mRNA all inhibited PC-3 cell growth on matrigel, whereas their corresponding four-nucleotide mismatch controls did not (Supplementary Figure 4A). To verify the GB-mediated PKN3 protein knockdown, we raised antibodies against the PKN3 kinase domain. PKN3-specific GBs that interfered with PC-3 cell growth on matrigel also efficiently reduced PKN3 protein levels, whereas none of the control antisense molecules had any effect (Figure 1B). Importantly, the PKN3 GBs did not interfere with expression of the kinase molecules PKN1 or PKN2 (also known as PRK1 and PRK2), which are close homologs of PKN3 (Mukai, 2003). Both were shown to be regulated by PI3K (Flynn et al, 2000) and implicated in prostate cancer (Metzger et al, 2003). Furthermore, PKN1 mRNA expression was also PI3K-dependent in PC-3 cells grown on matrigel (Supplementary Figure 2B). In contrast to PKN3, however, neither PKN1 nor PKN2 protein knockdown interfered with the ability of PC-3 to grow on matrigel (Supplementary Figures 4B and C). These experiments suggest that PKN3, but not PKN1 or PKN2, is required for PC-3 growth on matrigel.
Figure 1.
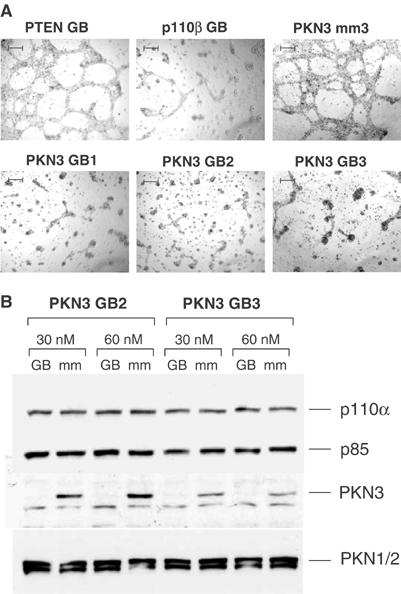
PKN3 mediates growth on basement membrane matrix in PTEN−/− prostate cancer cells. (A) PC-3 cells transfected with 30 nM of the indicated GB antisense or mismatch molecules (mm) were grown for 48 h on matrigel; size bars: 200 μm. The experiment was reproduced by independent transfections. (B) PC-3 cells were grown for 48 h in the presence of the indicated sets of PKN3 GB or mm control. Protein extracts were analyzed by immunoblotting using PKN3-specific antiserum. The filter was reprobed using antibodies against PKN1 and PKN2. The PI3K subunits p110α and p85 served as loading controls.
PKN3 is expressed in a PI3K-dependent manner and is upregulated in patient prostate tumor samples
To investigate whether sustained PI3K signaling is not only required but may even be sufficient for increased expression of PKN3, we generated stably transfected pools of a normal human breast epithelial cell line (MCF-10A) expressing an inducible version of a constitutively active PI3K, M-p110*-ER, from promoters with different strengths. The activity of M-p110*-ER can be stimulated by the addition of 4-hydroxytamoxifen (4-OHT), which results in chronic activation of the PI3K pathway (Klippel et al, 1998). Both stable M-p110*-ER pools exhibited increased basal PI3K signaling compared to the control cell population, as indicated by the elevated phosphorylation of the downstream effector p70S6 kinase (p70S6K) (Figure 2A, lanes 1, 5 and 9). In cells with lower M-p110*-ER levels PI3K signaling was further enhanced by 4-OHT treatment (lanes 5 and 6), whereas it was almost maximal without 4-OHT in cells with high M-p110*-ER levels (lanes 9 and 10). Endogenous PKN3 protein expression was strongly induced in M-p110*-ER cells compared to control cells, and the induction level correlated with the respective level of PI3K signaling (compare lanes 1 and 2, 5 and 6, and 9 and 10); the identity of the PKN3 signal was confirmed by the disappearance of PKN3 in GB-treated samples (lanes 13 and 14). In MCF-10A cells, increased PKN3 expression was also dependent on chronic activation of the PI3K pathway, as indicated in samples treated with LY294002 (LY), a small-molecule inhibitor of PI3K (lanes 7 and 11). Rapamycin, which inhibits mTOR-p70S6K signaling, also abrogated PKN3 expression in MCF-10A cells (lanes 8 and 12), indicating that this PI3K effector pathway contributes to PKN3 induction in these cells. PKN1/2 expression was regulated by PI3K to some extent, but was not as readily induced by M-p110*-ER as PKN3. Taken together, this indicates that hyperactivation of the PI3K pathway is not only required but can also be sufficient for enhanced PKN3 expression.
Figure 2.
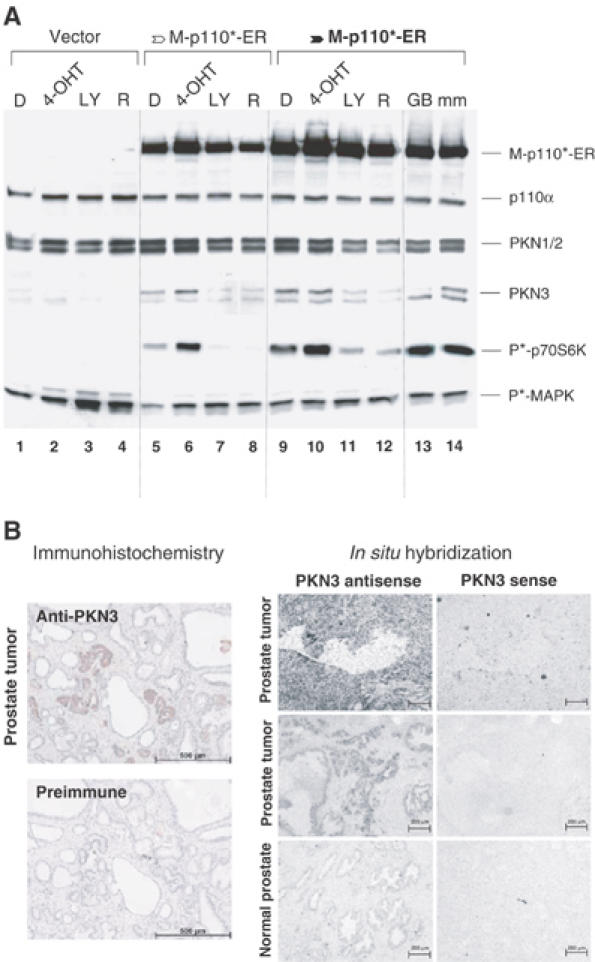
PKN3 is PI3K-dependently expressed and upregulated in prostate tumor samples. (A) Stable MCF-10A cells that direct expression of a 4-OHT-inducible version of an activated PI3K, M-p110*-ER, from a weaker (open arrow) or a stronger promoter (filled arrow, bold) were analyzed compared to vector-transfected cells. The cells were incubated in serum-free medium with or without DMSO (D), 200 nM 4-OHT, 10 μM LY or 20 nM rapamycin (R) overnight. Parallel samples were treated with 30 nM PKN3 GB3 or mismatch (mm) control to confirm the identity of the PKN3 signal. Cell extracts were analyzed using the indicated antibodies. Inhibition of PI3K signaling was confirmed by dephosphorylation of p70S6K at T389 (P*-p70S6K); MAP kinase phosphorylation at T202/Y204 (P*-MAPK) served as control. (B) Immunohistochemical analysis of two adjacent human prostate tumor tissue sections was performed using anti-PKN3 antiserum or pre-immune serum (left panels). The samples were hematoxylin counterstained to monitor the glandular tissue structure; size bars: 500 μm. Normal and tumor prostate tissue samples were compared by in situ hybridization using a PKN3-specific antisense probe; the specificity of the signal was confirmed by hybridizing adjacent tissue sections with the sense probe (right panels); size bars: 200 μm.
Since loss of PTEN function and the concomitant activation of PI3K signaling have been correlated with the development of metastatic prostate cancer, we wanted to test whether PKN3 could be detected in tissue samples of patient prostate tumors. Immunohistochemical analysis of adjacent tissue sections was carried out using PKN3 preimmune or immune serum. Only the immune serum-treated samples exhibited regions with positive staining (Figure 2B, left). Close inspection revealed that most of the tumor cells in the sample stained positive for PKN3 while the surrounding nontumorigenic tissue was negative. These results were corroborated by in situ hybridization studies, where prostate tumor tissues showed elevated staining only with a PKN3-antisense probe compared to normal prostate tissue (Figure 2B, right).
Induced inhibition of PKN3 expression interferes with formation of lymph node metastasis in an orthotopic mouse prostate tumor model
For long-term loss of function studies to validate candidate targets in mouse tumor model systems, we recently established a vector-derived expression system for inducible shRNA molecules (Czauderna et al, 2003b). Doxycyline (Dox) treatment induces U6tetO promoter-controlled shRNA expression by inactivating the tetracycline repressor (TetR). We isolated stable PC-3 cell populations expressing two different shRNAs targeting PKN3. Dox-induced shRNA expression resulted in efficient knockdown of PKN3 protein levels after 48 h (Figure 3A). Pools of stable cells directing induced inhibition of p110α or expressing an unrelated control shRNA were analyzed in parallel. Respective cell populations were seeded on matrigel and photographed at different time points. As expected from experiments shown above (Figure 1), cells with shRNA-mediated knockdown of PKN3 expression exhibited impaired growth on extracellular matrix (Figure 3B). As described earlier, inhibition of p110β, but not of p110α, interferes with matrigel growth of PC-3 cells (Supplementary Figure 3; Czauderna et al, 2003b), whereas p110α represents the predominant PI3K subunit in other cell types (Figure 5A). Accordingly, induced inhibition of p110α had no effect in PC-3 cells in the experiment shown here and served as a control for Dox treatment and shRNA induction. This result confirmed that vector-derived inducible expression of PKN3 shRNA in these stable cell populations was indeed functional.
Figure 3.
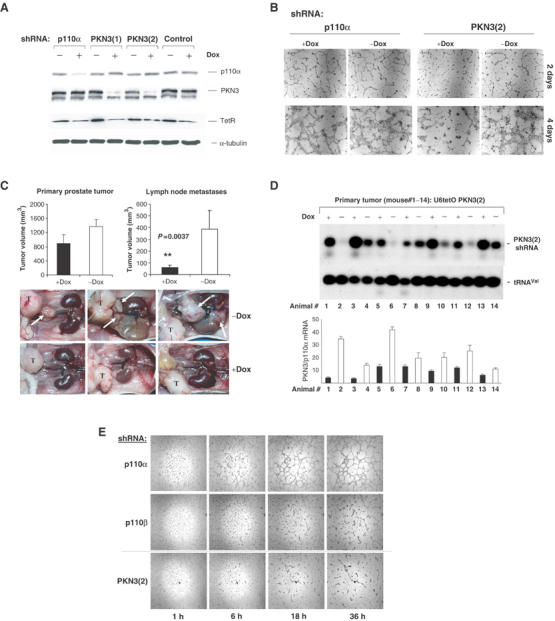
Inducible knockdown of PKN3 expression inhibits metastasis in an orthotopic mouse prostate tumor model. (A) PC-3 cells stably transfected with vectors that direct Dox-dependent expression of two different PKN3 shRNAs were analyzed in parallel to p110α or an unrelated control shRNA. Cells were grown±100 ng/ml Dox, and extracts were analyzed by immunoblotting. Expression of the TetR was confirmed using an anti-TetR antibody; α-tubulin served as loading control. (B) PKN3(2) shRNA and p110α shRNA cells analyzed in (A) were seeded on matrigel and photographed after 2 and 4 days; size bars: 200 μm. (C) PC-3 cells with inducible PKN3(2) shRNA were transplanted intraprostatically into nude mice. One group of animals received Dox (black bars), the second group was mock-treated (white bars); each group consisted of eight animals. After 56 days, the mice were killed and evaluated for primary tumor and lymph node metastases development (top). Each bar represents the mean tumor volume±s.e. The reduction in lymph node metastases formation in the Dox-treated group is statistically significant according to the Mann–Whitney test. Representative in situ pictures of three animals of each group are shown (bottom). The primary prostate tumor is labeled with ‘T' and the position of lumbar and renal lymph node metastases is indicated by white arrows. (D) RNA samples extracted from PC-3 prostate tumors of seven animals of each group were analyzed by Northern blotting for induction of PKN3 shRNA; tRNAVal served as loading control (top). PKN3 mRNA levels after shRNA induction were quantified by Taqman analysis relative to p110α mRNA (bottom); the results are mean of triplicates±s.e. Black bars indicate the results for Dox-treated animals, white bars for the control group. (E) PC-3 cell populations stably expressing shRNA specific for p110α (negative control), p110β (positive control) and PKN3 were analyzed by time-lapse-video microscopy on matrigel. Pictures taken at the indicated times after seeding are shown at × 2.5 magnification.
Figure 5.
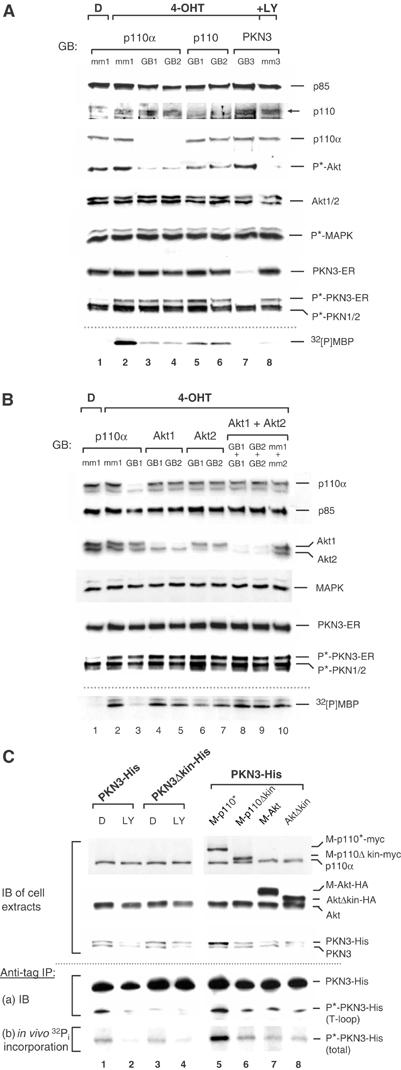
PI3K regulates PKN3 activity. (A) HeLa cells stably expressing PKN3-ER were grown in the presence of 40 nM of the indicated GBs or their mismatch (mm) controls in regular growth medium for 48 h. PKN3-ER activity was induced with 200 nM 4-OHT in DMSO (D) 12 h before cell lysis; 10 μM LY was added to a control sample. Cell extracts were analyzed by probing with the indicated antibodies. Specific inhibition of PI3K signaling was confirmed by testing for phospho(S473)-Akt (P*-Akt) and phospho(T202/Y204)-MAP kinase (P*-MAPK) levels. Anti-ER precipitates were subjected to in vitro kinase activity using MBP as substrate; radiolabeled MBP (32[P]MBP) was detected by autoradiography (bottom). (B) PKN3-ER cells were treated with GBs and analyzed as in (A). For combination of two GBs, each individual GB was employed at 30 nM. (C) The indicated His-tagged PKN3 derivatives were transiently coexpressed in COS-7 cells in the presence of empty vector (lanes 1–4) or expression vectors for activated PI3K (M-p110*-myc), activated Akt (M-Akt-HA) or their respective inactive versions (M-p110Δkin-myc; AktΔkin-HA) (lanes 5–8). The cells were depleted in phosphate-free medium and metabolically labeled with 32Pi±10 μM LY or DMSO (D) as indicated. Duplicate samples were kept in phosphate-free medium without label, and protein expression was detected by immunoblotting (IB) of the extracts (upper part). In vivo phosphorylation of PKN3-His was detected by anti-His IP followed by (a) IB and (b) autoradiography of the labeled samples (lower part).
Next, stable PKN3 shRNA PC-3 cells were transplanted intraprostatically into nude mice (Stephenson et al, 1992). The animals were split into two groups, one group was treated with Dox to induce shRNA expression and the second group was mock-treated. After 56 days the mice were killed and analyzed for primary tumor formation and lymph node metastasis. The Dox-treated group of mice showed a small effect on primary tumor growth; however, metastases formation was strongly inhibited (Figure 3C). Similar results were obtained in a separate experiment with a cell population expressing the second PKN3 shRNA; also, Dox treatment of mice transplanted with untransfected PC-3 cells did neither affect tumor nor metastases formation (not shown).
Northern-blot analysis of RNA extracted from the primary tumors showed that samples obtained from Dox-treated mice exhibited on average increased expression of PKN3 shRNA compared to samples from the control group (Figure 3D, top panel). In fact, overall there was an inverse correlation between the level of shRNA induction and the amount of PKN3 mRNA detected in these samples: high shRNA levels in tumors resulted in reduced levels of PKN3 mRNA (Figure 3D, lower panel). Leakiness in shRNA expression as well as limited knockdown in PKN3 mRNA in certain samples may have been caused by loss of TetR expression after 56-day implantation or by contaminating PKN3 signals from mouse cells in the respective human tumor cell material. Nevertheless, the data strongly suggest that the decreased PKN3 levels in the primary tumors were indeed the cause for the reduced metastases formation. This result correlates with the results obtained from the matrigel growth assay and indicates that PKN3 may be an important mediator of invasive signaling.
The finding that PKN3 supports metastasis formation rather than tumor growth suggests that PKN3 might modulate cell adhesion and/or migration. shRNA-mediated reduction in PKN3 expression appeared to affect PC-3 cell morphology and the structure of the tubulin network, indicating cytoskeletal rearrangements (Supplementary Figure 5). To analyze whether these alterations affect migration and motility, PC-3 cell populations expressing shRNA specific for p110α, p110β and PKN3 were analyzed by time-lapse-video microscopy on matrigel, since changes caused by reduced PKN3 levels are more evident under 3D growth conditions (Figure 3E). Equal plating of the cells was observed 1 h after seeding. Subsequent time points indicated that cells expressing p110β- or PKN3-specific shRNAs were impaired in forming network-like structures on matrigel, which correlated with reduced cell numbers compared to the p110α shRNA control. The entire video clips for the p110α shRNA control and the PKN3 shRNA cells can be viewed in the Supplementary section. The data indicate that motility and cell–cell contacts might influence PC-3 cell growth on matrigel.
PKN3 is an AGC-type kinase and its catalytic activity is regulated by PI3K
After validating PKN3 as a downstream effector of activated PI3K by independent gene silencing approaches, we wanted to characterize its catalytic activity to investigate whether it could serve as a target for therapeutic intervention in cancer. We transiently expressed various recombinant PKN3 derivatives in HeLa cells (Figure 4A, top), and immunoblotting of cell extracts with PKN3-specific antiserum revealed approximately equal expression of the different proteins (upper blot). The cell extracts were analyzed in parallel using an antibody that recognizes the activation loop phosphorylation (T-loop) site in the catalytic domain of PKN1 or PKN2 (middle blot). Phosphorylation of this conserved threonine residue, which is present in PKN3 at position 718, is required for activation of AGC-type kinases (Parekh et al, 2000). Endogenous PKN1/2 and all recombinant PKN3 molecules were detected by this phospho-specific antibody, except for the fragment carrying a threonine to alanine substitution at position 718 (TA718), indicating that wild-type (wt) PKN3 is phosphorylated at this residue. The PKN3 derivatives were precipitated from the lysate via their Myc tag and after a series of stringent washes subjected to an in vitro protein kinase assay using myelin basic protein (MBP) as a substrate in the presence of radiolabeled ATP. The fragment comprising just the kinase domain exhibited catalytic activity, which was specific, since fragments with mutations in the catalytic center (lysine to arginine, KR588) or the T-loop phosphorylation site (TA718) had no detectable activity (lower panel). The full-length molecule, however, had substantially increased kinase activity compared to the catalytic domain fragment. ΔN, the derivative lacking the N-terminal region, appeared to be inactive despite the fact that it overlaps the kinase domain fragment, which was active by itself, and was phosphorylated at T718. This suggests that the middle region of PKN3 imposes a negative regulatory function on the catalytic domain that is relieved in the presence of the N-terminus in the full-length protein. Our results were confirmed with HA-tagged versions of PKN3 (not shown).
Figure 4.
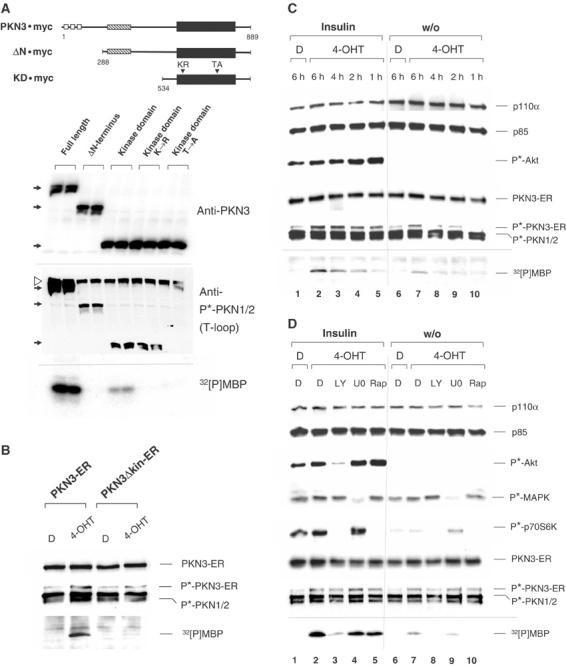
Characterization of PKN3 catalytic activity. (A) Full-length or truncated PKN3 versions were modified with the Myc epitope at the C-terminus and transiently expressed in HeLa cells. Schematic structures of the molecules are shown (top): Presumed functional regions (Mukai, 2003) are represented by white (ACC-finger domains), hatched (C2-like domain) and black (kinase domain) boxes. The fragment lacking the first 287 amino acids from the N-terminus is labeled ΔN. A protein fragment spanning the kinase domain at the C-terminus is labeled KD. KR588 (KR) and TA718 (TA) denote point mutations in the ATP-binding site (lysine to arginine or glutamic acid (KE588) at position 588 behaved identically) and in the activation loop (T-loop) phosphorylation site (threonine to alanine, position 718) of the catalytic domain. The first and last amino acids of each fragment are indicated. Cell extracts were analyzed using anti-PKN3 antiserum or anti-phospho(T-loop)-PKN1/2 antibody (P*-PKN1/2) as indicated. The position of wt and truncated PKN3 molecules is indicated by black arrows at the left of each filter; the position of endogenous phosphorylated PKN1/2 molecules is shown by a white arrowhead. Anti-Myc precipitates were tested for kinase activity in vitro using MBP as a substrate; radiolabeled MBP (32[P]MBP) was detected by autoradiography (bottom). (B) HeLa cells stably expressing a 4-OHT-regulatable version of PKN3, PKN3-ER, and its inactive version, PKN3Δkin-ER (carrying mutation KE588), were stimulated for 30 min with 200 nM 4-OHT in DMSO (D). Cell extracts were immunoblotted and tested for in vitro kinase activity after precipitation with anti-ER antibody. (C) Serum-starved cells were stimulated in a time course with or without insulin (10 μg/ml)±4-OHT. Activation of the PI3K pathway was confirmed with anti-phospho(S473)-Akt antibody (P*-Akt). (D) Quiescent cells were stimulated for 6 h as in (C) ±10 μM LY, 10 μM U0126 (U0) or 20 nM rapamycin (Rap). Inhibitor treatment was monitored by P*-Akt, phospho(T202/Y204)-MAP kinase (P*-MAPK) and phospho(T389)-p70S6K (P*-p70S6K) levels.
Having established a biochemical assay for PKN3 enzymatic activity we wanted to test whether its catalytic function is regulated by PI3K as shown for other AGC-type kinases (Parekh et al, 2000). In order to investigate the requirements for PKN3 activation without using overexpression, we established stable HeLa cell pools expressing an inducible version of PKN3, PKN3-ER, and its kinase-deficient version, PKN3Δkin-ER. The PKN3-ER fusion protein had no detectable kinase activity in the absence of 4-OHT, also increased phosphorylation at the T-loop site (T718) was 4-OHT dependent (Figure 4B). PKN3Δkin-ER had no activity confirming specificity of the 4-OHT-induced response. Consistent with transient transfection experiments (Supplementary Figure 6), phosphorylation at T718 in the inactive PKN3Δkin-ER was weaker in this inducible system than in the active kinase molecule. Next, HeLa PKN3-ER cells were starved in serum-free medium overnight and subsequently stimulated in the presence or absence of 4-OHT with or without insulin for the indicated times (Figure 4C). Insulin treatment strongly activated the PI3K pathway in HeLa cells, as indicated by increased phosphorylation of the downstream effectors Akt and p70S6K, but had little or no effect on the MEK-MAP kinase pathway (Figure 4D). PKN3-ER kinase activity as well as T-loop phosphorylation were substantially increased in the presence of 4-OHT plus insulin (Figure 4C, lanes 1–5) compared to 4-OHT treatment alone (lanes 6–10). In contrast to the immediate Akt activation, PKN3-ER activity increased steadily during 6 h insulin treatment, suggesting that PKN3 is not a direct PI3K effector and requires activation of additional components within the signaling cascade, which then mediate PKN3 stimulation further downstream. Similar data were obtained with serum stimulation (not shown).
To investigate the contribution of various signaling pathways to PKN3 activation, 6 h insulin stimulation was carried out in the absence or presence of LY, U0126 or Rapamycin. Successful inhibitor treatment was monitored by reduced phosphorylation of Akt, MAP kinase or p70S6K (Figure 4D). Inhibition of PI3K strongly interfered with the insulin-mediated increase in PKN3 activity and also blocked PKN3 basal activity (lanes 3 and 8), whereas inhibition of MEK-MAP kinase signaling had no substantial effect (lanes 4 and 9). Rapamycin did not affect the insulin-mediated increase in PKN3 activity, but it reduced the basal activity (lanes 5 and 10), suggesting that mTOR-p70S6K signaling downstream of PI3K can contribute to PKN3 stimulation to a certain extent. This set of experiments suggests that activation of PI3K can regulate PKN3 activity.
PKN3 catalytic activity depends on PI3K, but not on Akt
We wanted to investigate the contribution of the PI3K pathway for PKN3 activation in more detail without having to rely on pharmacological inhibitors, such as LY. Therefore, we inhibited p110α and p110β expression in the stable HeLa PKN3-ER cell system, to interfere with PI3K activity. Two different GBs for each subunit were employed and each one caused a specific reduction in p110α or p110β protein levels (Figure 5A); specific reduction in mRNA levels was also more than 90% for each subunit (not shown). In both cases PI3K signaling was inhibited, as indicated by the reduction in phospho-Akt levels (Figure 5A, compare lanes 2 to 6), although inhibition of p110α expression had a more pronounced effect than inhibition of p110β expression. This is in contrast to the result obtained for PC-3 cells, where p110β was the predominant subunit compared to p110α, although the identical GBs had been used (see Supplementary Figure 3). This indicates that p110α represents the predominant isoform in this particular type of HeLa cells (please note: different from the HeLa line used by Czauderna et al, 2003a), whereas p110β is the predominant species in PC-3 cells. In other cell types, we have observed equal contributions of both subunits to PI3K signaling (not shown). The inhibitory effect of both p110α GBs on Akt phosphorylation was comparable to that of LY treatment; total Akt protein levels and MAP kinase phosphorylation were not affected. The level of inhibition of PI3K signaling correlated with the reduction in 4-OHT-dependent PKN3-ER activity: Similar to the effect on phospho-Akt levels, inhibition of p110α expression had a more pronounced effect on PKN3-ER activity than inhibition of p110β expression in regular growth medium (lanes 2–6). GB-mediated inhibition of PKN3-ER expression abrogated the detection of kinase activity, confirming the specificity of the kinase reaction. Even though PKN3 T-loop phosphorylation was required for PKN3 activation (Figure 4A, Supplementary Figure 6), it was not sufficient for PKN3 activation, as indicated in the p110α GB- or LY-treated samples (Figure 5A, lanes 3, 4 and 8).
Since inhibition of p110α expression reduced Akt phosphorylation and PKN3 activation to a comparable extent, we next analyzed the contribution of Akt to PI3K-dependent PKN3 activation. Two different GB molecules that specifically inhibit expression of either Akt1 or Akt2 were identified and tested for their ability to interfere with PKN3-ER activity, single and in combination; Akt3 protein expression was not detected in this HeLa cell line (not shown). Akt1 GBs specifically reduced Akt1, Akt2 GBs reduced Akt2 protein levels and the combination of both types substantially reduced the expression of all detectable Akt species, whereas MAP kinase levels were not affected (Figure 5B, lanes 4–9). Compared to the p110α GB control, which inhibited 4-OHT-dependent PKN3-ER kinase activation, none of the Akt GBs was able to reproducibly inhibit PKN3 activity, even when Akt1 and Akt2 were simultaneously reduced. This indicates that Akt does not appear to mediate PI3K-dependent activation of PKN3 catalytic activity in this cellular context.
The experiments shown in Figure 4 and Supplementary Figure 6 suggested that phosphorylation of PKN3 within the T-loop is required, but not sufficient for its catalytic activity. Therefore, it is likely that other PI3K-mediated signals, such as additional phosphorylation events, are necessary as well. To investigate whether hyperactivation of PI3K or Akt, as they occur in PTEN-deficient cancers, can contribute to increased PKN3 phosphorylation, we monitored PI3K-dependent phosphorylation of PKN3 in vivo. COS-7 cells transfected with recombinant PKN3-His or PKN3Δkin-His were metabolically labeled with orthophosphate in the presence or absence of LY. In parallel, PKN3-His was coexpressed with a constitutively active PI3K (M-p110*) or its kinase-defective control (M-p110Δkin), as well as with a constitutively active Akt (M-Akt) or the inactive control (AktΔkin) (Klippel et al, 1996; Kulik et al, 1997). Unlabeled duplicate samples were analyzed by immunoblotting (Figure 5C, upper part); the radiolabeled samples were analyzed by immunoprecipitation, followed by blotting and autoradiography (lower part). In agreement with the experiments shown above, the immunoblot analysis revealed that PKN3 is PI3K-dependently expressed also in this system, as indicated by the LY-treated samples (lanes 1–4) and the fact that coexpression of M-p110* increased the amount of PKN3-His above basal levels (lane 5 compared to lanes 1, 3, 6 and 8). By contrast, M-Akt (lane 7) was not able to elevate PKN3-His expression above basal levels. Radiolabeled cell extracts from 10-cm plates were precipitated with limiting amounts of antibody in order to compensate for the different expression levels. Immunoblotting confirmed approximately equal loading (Figure 5C, lower panels). In COS-7 cells, T-loop phosphorylation was strongly dependent on PI3K and again on the presence of an active PKN3. Importantly, coexpression of M-p110*, but not of M-Akt, substantially enhanced the total incorporation of radiolabeled phosphate into PKN3-His above basal levels (lane 5), indicating that hyperactivation of PI3K is sufficient to stimulate increased phosphorylation of this effector protein. Taken together, this shows that activation of PI3K signaling is required and can also be sufficient for increased PKN3 expression and phosphorylation.
PKN3 is an effector of various signal transduction pathways involved in transformation
Our data indicated that PKN3 mediates malignant growth of cells with an activated PI3K pathway, such as PTEN-defective tumor cells. To investigate its role in malignant growth regulated by additional signaling pathways, we analyzed human breast epithelial cells that can be inducibly transformed by activating expression of an oncogenic form of Ras, RasV12. Pools of stably transfected MCF-10A cells were mock-stimulated or stimulated for RasV12 expression in the presence or absence of LY or the MEK inhibitor U0126. RasV12 induction was functional, as shown by enhanced phosphorylation of the downstream effector MAP kinase, and resulted in increased expression of PKN3 (Figure 6A, compare lanes 1 and 5). The increase in endogenous PKN3 expression was dependent on PI3K (lanes 5–7), in agreement with data shown above. Interestingly, Ras-mediated upregulation of PKN3 was also blocked in the presence of U0126, which blocked MAP kinase phosphorylation (lane 8).
Figure 6.
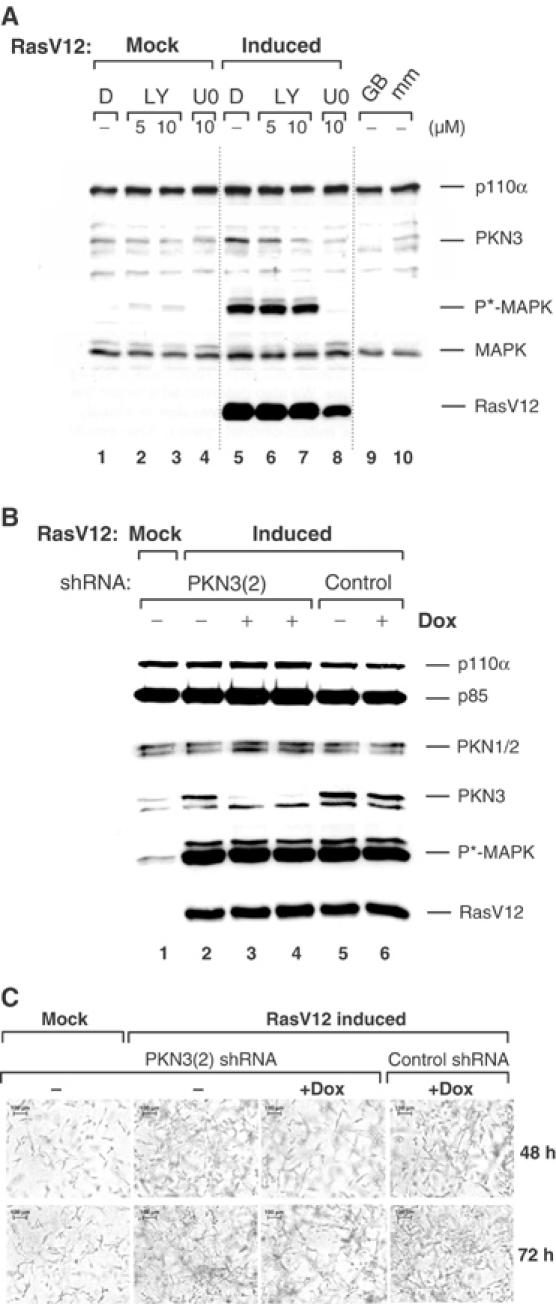
Upregulation of PKN3 in mammary epithelial cells expressing oncogenic Ras. (A) MCF-10A cells stably transfected with an inducible RasV12 were induced or mock-treated±LY or U0126 (U0) in DMSO (D) as indicated. Pathway activation/inhibition was monitored via phospho(T202/Y204)-MAP kinase (P*-MAPK) levels. The identity of the PKN3 protein band was confirmed by analysis of extracts from cells treated with PKN3 GB3 (GB) or its mismatch (mm) (lanes 9 and 10). (B) Double-stable MCF-10A cells expressing inducible RasV12 plus Dox-inducible PKN3(2) or control shRNAs were grown in duplicate samples±0.5 μg/ml (lane 3) or 1 μg/ml Dox (lanes 4 and 6) for 48 h. One set of samples was analyzed by immunoblotting (B), the second set was grown in 3D cultures (C). (C) Cells were embedded into collagen gels in 24 wells±Dox with or without RasV12 induction. Photographs were taken at the indicated times; size bars: 100 μm.
To test whether inhibition of RasV12-induced PKN3 expression can interfere with growth of these cells in 3D cultures, we isolated double stable populations, after transfection of an inducible PKN3 shRNA or a control shRNA vector into the MCF10A RasV12 background. As shown above, induction of RasV12 strongly enhanced MAP kinase phosphorylation as well as PKN3 expression, but did not affect PKN1/2 expression (Figure 6B, lanes 1 and 2). Dox-mediated PKN3 shRNA induction caused downregulation of PKN3 protein expression without reducing PKN1/2 levels (lanes 3 and 4). Parallel samples were embedded into collagen gels (Grunert et al, 2003) and grown for the indicated times in the presence or absence of RasV12 induction with or without Dox treatment (Figure 6C). RasV12 strongly stimulated MCF10A cell growth in the collagen matrix, and Dox-mediated inhibition of PKN3 expression partially, but reproducibly, reversed this effect. In contrast, Dox treatment of cells expressing a control shRNA continued to grow like RasV12-transformed cells. This result shows that enhanced expression of PKN3 in cells transformed by oncogenic Ras can contribute to their invasive potential. Depending on the cellular context, PKN3 can be regulated by various signal transduction pathways that mediate cell growth and transformation.
Discussion
We show that PKN3, an AGC-type kinase, can contribute to malignant growth of cells with increased PI3K activation: Inhibition of PKN3 expression using different types of gene silencing tools interfered with growth of prostate and breast epithelial cells in 3D assay systems (Figures 1, 3 and 6). Pools of stably transfected PC-3 cells were impaired in forming metastases in an orthotopic mouse tumor model after induction of PKN3 shRNA expression, although the effect on primary tumor formation was relatively small (Figure 3C). This suggests that PKN3 may primarily be required for metastatic growth of prostate cancer cells. In agreement with this, reduced levels of PKN3 appeared to cause cytoskeletal rearrangements and decreased cell motility (Supplementary Figure 5, Figure 3E). This resulted in reduced formation of network-like structures on extracellular matrix, which can be viewed by time-lapse-video microscopy (Supplementary section).
Reducing PI3K levels or its activity in cells with a chronically active pathway interferes predominantly with growth in 3D cultures, but not with growth on plastic (Kaufmann et al, 2004). This is consistent with results demonstrating that restoring PTEN expression in PTEN−/− cells to physiological levels reduced PI3K activation without affecting growth on plastic, but it induced changes in cell morphology and attachment (Stolarov et al, 2001). shRNA-directed inhibition of PI3K also strongly interfered with metastases formation in mice after orthotopic transplantation of stably transfected PC-3 cells (Czauderna et al, 2003b). Therefore, sustained activation of PI3K signaling may in certain cell types primarily affect cell functions that contribute to invasiveness, such as loss of cell–cell contacts, reduced adherence, increased motility or anchorage independence (Di Cristofano et al, 1998; Klippel et al, 1998; Kobayashi et al, 1999), and some of these functions might be mediated by PKN3.
A number of studies have demonstrated that growth in 3D cultures using extracellular matrix components represents a better indicator for the malignant potential of tumor cells than 2D culture conditions (Weaver et al, 1997; Sahai and Marshall, 2003; Wolf et al, 2003). Cells with increased malignant potential have a growth advantage on matrigel matrix consisting of basement membrane components (Petersen et al, 1992; Muthuswamy et al, 2001). Even signaling events exhibit striking differences when 3D and 2D growth conditions were compared (Wang et al, 1998; Cukierman et al, 2001). We would have failed to detect the PI3K dependence of PKN3 expression in RNA samples obtained from cells grown in regular 2D plastic cultures. Similarly, EPSTI-1, which regulates epithelial–stromal cell interactions in breast tumors, was only identified in cells grown in 3D cultures (Gudjonsson et al, 2003). We would have also missed the inhibitory effect in response to PKN3 knockdown using 2D conditions, which is only evident on matrigel or collagen.
In contrast to PKN3, inhibition of the closely related kinases PKN1 and PKN2 did not interfere with matrigel growth (Supplementary Figure 4). This finding is unexpected, since the three proteins share a similar structural organization and extensive sequence homology (Mukai, 2003). A number of additional differences between PKN3 and PKN1/2 have been observed: (i) PKN1 and PKN2 appear to be ubiquitously expressed, whereas expression of PKN3 is low in normal adult tissue, but increased in human cancer cells (Oishi et al, 1999; Figure 2B) and in early mouse embryonic stages (not shown). (ii) The kinase domain fragments of PKN1/2 represent constitutively active forms (Mukai, 2003), whereas the corresponding PKN3 fragment exhibited significantly less catalytic activity than the full-length molecule (Figure 4A). (iii) The finding that the N-terminal domain of PKN3 is required for full enzymatic activity (Figure 4A) implies that the N-terminal region has a function different from the corresponding regions in PKN1/2. These regions determine the association between PKN1/2 and the small GTPases Rac and Rho, which regulate cytoskeletal remodeling (Vincent and Settleman, 1997). Whether PKN3 is also regulated by small GTPases remains to be elucidated.
Phosphorylation at the T-loop site was necessary, but not sufficient for PKN3 activity, since inhibition of the activity could occur uncoupled of T718 dephosphorylation (Supplementary Figure 6, Figure 4D). Also, insulin-stimulated T-loop phosphorylation was observed before kinase activity could be detected (Figure 4C). T-loop phosphorylation was reduced in ATP-binding site mutants (Figures 4B and 5C, Supplementary Figure 6), indicating that PKN3 can autophosphorylate to some extent or that a feedback loop between PKN3 and PDK1, the likely T-loop kinase (Newton, 2003), may exist. T-loop phosphorylation cannot be mimicked by acidic residues as in other kinase molecules; similarly, the presumed phosphorylation site within the turn motif of the kinase domain (Newton, 2003), T860, appears to be invariant (not shown).
PKN3 appears to be controlled by PI3K at several levels: Its mRNA was expressed in a PI3K-dependent manner only in cells grown on matrigel (Supplementary Figure 2A). However, PI3K-dependent expression at the protein level was detectable in cells grown on plastic, indicating a second level of control. For example, activated PI3K was sufficient for upregulating endogenous PKN3 in stable cell systems as well as recombinant PKN3, when transiently coexpressed (Figures 2A and 5C). In the latter case, this must be caused post-transcriptionally, since recombinant PKN3 expression was directed from a heterologous promoter. It is possible that the PI3K-directed phosphorylation at the T loop and other sites leads to increased stabilization of PKN3, as shown for other PKC family kinases (Newton, 2003).
The catalytic activity of PKN3 appears to be PI3K-dependently regulated as well, and this effect was independent of Akt in the systems investigated here (Figures 4C,D and 5A,B). Inhibition of the mTOR-p70S6K pathway by Rapamycin reversed M-p110*-induced PKN3 expression (Figure 2A) and reduced the level of basal PKN3 activity (Figure 4D), but did not affect PKN3 mRNA expression in PC-3 cells (Supplementary Figure 2B). This suggests that this effector arm downstream of PI3K can contribute to the regulation to PKN3 levels and activity to some extent. Also, PI3K-independent signals that mediate malignant growth can regulate PKN3 expression in certain cell types as well (Figure 6A).
We have identified, validated and characterized PKN3 as a novel effector of chronically active PI3K that appears to contribute to invasive prostate cancer. Our data suggest that the development of inhibitors against PKN3 holds great promise for therapeutic intervention in metastasis regulated by activated PI3K.
Materials and methods
Reagents
The reagents used were LY294002, rapamycin and U0126 (Calbiochem), doxycycline hydrochloride, 4-OHT and blasticidin (Sigma) and Geneticin (PAA laboratories).
Antibodies
The following antibodies were used: Akt, phospho-Akt (S473), MAP kinase, phospho-MAP kinase (T202/Y204), phospho-PKN1/2 (T778/T816) and phospho-p70S6 kinase (T389) antibody (Cell Signalling Technology); phospho-FOXO3a (T32) antibody (Upstate); p110β (H-239) and H-Ras antibodies (C-20) (Santa Cruz Biotechnology); PKN1 and 2 antibodies (Becton Dickinson); Penta-His antibody (Qiagen); TetR antibody (MoBiTec); β-tubulin (AB-1) antibody (Calbiochem). Rabbit PKN3 antibodies were raised against the 281 C-terminal amino acids overexpressed and purified from bacterial extracts (see Supplementary data). p110α and p85 antibodies have been described (Klippel et al, 1994). Immunoprecipitations were carried out using HA antibody (F-7) (Santa Cruz Biotechnology); mERα (#06–935) antibody (Upstate); ascites fluid-containing Myc-tag antibody (Klippel et al, 1996).
Orthotopic mouse prostate tumor/metastasis model
Male Shoe:NMRI-nu/nu mice (DIMED, Germany), 8-week old, were inoculated with 2 × 106 pools of stably transfected PC-3 cells into the left dorsolateral lobe of the prostate gland under total body anesthesia (Stephenson et al, 1992; Czauderna et al, 2003b). Doxycycline hydrochloride was administered as a 0.1% solution via drinking water with 3% sucrose from the day of surgery. Animals were killed 56 days post operation and tumors (prostate gland), regional lymph node metastases (caudal, lumbar, renal) and distant metastases were measured in two dimensions by means of a pair of calipers; the volume was calculated according to V (mm3)=ab2/2 with b<a. All animals were completely dissected and photographically documented. All experiments (surgery, cell transplantation, measurement of tumor volume) were performed double-blinded and independently by two researchers, and in compliance with the guidelines of the Landesamt für Arbeits-, Gesundheitsschutz and technische Sicherheit Berlin, Germany (institutional approval no. G0264/99).
Preparation of cell extracts and immunoblotting
Cell lysis and immunoblot analysis were carried out essentially as described (Klippel et al, 1998).
For details on 2D and 3D culture conditions, plasmid constructions, sequences of GBs and other oligonucleotides, in vivo and in vitro protein kinase assays, in situ hybridization and immunohistochemistry, see Supplementary information (available at The EMBO Journal Online).
Supplementary Material
Supplementary Information
Supplementary Video 1
Supplementary Video 2
Acknowledgments
We thank Ralf Sägebarth and Steve Lack for bioinformatics, Oliver Keil and Jens Endruschat for transfection reagent support. We are grateful to Heike Heuer and Stephanie Christ from the Max-Planck Institute Hannover for help with the in situ hybridizations, Gerald Fisch and Birgit Durieux for animal experimental support. We thank Katharina Ahrens, Bert Pronk, Frank Gebhardt, and especially Sabine Werner and Manfred Gossen for many helpful comments on the manuscript. This study was supported by a grant from the Bundesministerium für Forschung und Technologie (No. 0311830/9).
References
- Bissell MJ, Rizki A, Mian IS (2003) Tissue architecture: the ultimate regulator of breast epithelial function. Curr Opin Cell Biol 15: 753–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukierman E, Pankov R, Stevens DR, Yamada KM (2001) Taking cell–matrix adhesions to the third dimension. Science 294: 1708–1712 [DOI] [PubMed] [Google Scholar]
- Czauderna F, Fechtner M, Aygun H, Arnold W, Klippel A, Giese K, Kaufmann J (2003a) Functional studies of the PI(3)-kinase signaling pathway employing synthetic and expressed siRNA. Nucleic Acids Res 31: 670–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czauderna F, Santel A, Hinz M, Fechtner M, Durieux B, Fisch G, Leenders F, Arnold W, Giese K, Klippel A, Kaufmann J (2003b) Inducible shRNA expression for application in a prostate cancer mouse model. Nucleic Acids Res 31: E127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi PP (1998) Pten is essential for embryonic development and tumor suppression. Nat Genet 19: 348–355 [DOI] [PubMed] [Google Scholar]
- Flynn P, Mellor H, Casamassima A, Parker PJ (2000) Rho GTPase control of protein kinase C-related protein kinase activation by 3-phosphoinositide-dependent protein kinase. J Biol Chem 275: 11064–11070 [DOI] [PubMed] [Google Scholar]
- Grunert S, Jechlinger M, Beug H (2003) Diverse cellular and molecular mechanisms contribute to epithelial plasticity and metastasis. Nat Rev Mol Cell Biol 4: 657–665 [DOI] [PubMed] [Google Scholar]
- Gudjonsson T, Ronnov-Jessen L, Villadsen R, Bissell MJ, Petersen OW (2003) To create the correct microenvironment: three-dimensional heterotypic collagen assays for human breast epithelial morphogenesis and neoplasia. Methods 30: 247–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez C, Jones DR, Rodriguez-Viciana P, Gonzalez-Garcia A, Leonardo E, Wennstrom S, von Kobbe C, Toran JL, Borlado LR, Calvo V, Copin SG, Albar JP, Gaspar ML, Diez E, Marcos MA, Downward J, Martinez AC, Merida I, Carrera AC (1998) Identification and characterization of a new oncogene derived from the regulatory subunit of phosphoinositide 3-kinase. EMBO J 17: 743–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katso R, Okkenhaug K, Ahmadi K, White S, Timms J, Waterfield MD (2001) Cellular function of phosphoinositide 3-kinases: implications for development, homeostasis, and cancer. Annu Rev Cell Dev Biol 17: 615–675 [DOI] [PubMed] [Google Scholar]
- Kaufmann J, Pronk G, Giese K, Klippel A (2004) Identification of novel effectors of invasive cell growth downstream of phosphoinositide 3-kinase. Biochem Soc Trans 32: 355–359 [DOI] [PubMed] [Google Scholar]
- Klippel A, Escobedo JA, Hirano M, Williams LT (1994) The interaction of small domains between the subunits of phosphatidylinositol 3-kinase determines enzyme activity. Mol Cell Biol 14: 2675–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klippel A, Escobedo M, Wachowicz MS, Apell G, Brown TW, Giedlin M, Kavanaugh WM, Williams LT (1998) Activation of phosphatidylinositol 3-kinase is sufficient for cell cycle entry and promotes cellular changes characteristic of oncogenic transformation. Mol Cell Biol 18: 5699–5711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klippel A, Reinhard C, Kavanaugh WM, Apell G, Escobedo M, Williams LT (1996) Membrane localization of phosphatidylinositol 3-kinase is sufficient to activate multiple signal-transducing kinase pathways. Mol Cell Biol 16: 4117–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Nagata S, Iwasaki T, Yanagihara K, Saitoh I, Karouji Y, Ihara S, Fukui Y (1999) Dedifferentiation of adenocarcinomas by activation of phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA 96: 4874–4879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik G, Klippel A, Weber MJ (1997) Antiapoptotic signaling by the insulin-like growth factor I receptor, phosphatidylinositol 3-kinase, and Akt. Mol Cell Biol 17: 1595–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Manning BD, Cantley LC (2003) Targeting the PI3K–Akt pathway in human cancer: rationale and promise. Cancer Cell 4: 257–262 [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002) The protein kinase complement of the human genome. Science 298: 1912–1934 [DOI] [PubMed] [Google Scholar]
- Metzger E, Muller JM, Ferrari S, Buettner R, Schule R (2003) A novel inducible transactivation domain in the androgen receptor: implications for PRK in prostate cancer. EMBO J 22: 270–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai H (2003) The structure and function of PKN, a protein kinase having a catalytic domain homologous to that of PKC. J Biochem 133: 17–27 [DOI] [PubMed] [Google Scholar]
- Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS (2001) ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol 3: 785–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton AC (2003) Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J 370: 361–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi K, Mukai H, Shibata H, Takahashi M, Ona Y (1999) Identification and characterization of PKNβ, a novel isoform of protein kinase PKN: expression and arachidonic acid dependency are different from those of PKNα. Biochem Biophys Res Commun 261: 808–814 [DOI] [PubMed] [Google Scholar]
- Parekh DB, Ziegler W, Parker PJ (2000) Multiple pathways control protein kinase C phosphorylation. EMBO J 19: 496–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ (1992) Interaction with basement membrane serves to rapidly distinguish growth and differentiation pattern of normal and malignant human breast epithelial cells. Proc Natl Acad Sci USA 89: 9064–9068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roymans D, Slegers H (2001) Phosphatidylinositol 3-kinases in tumor progression. Eur J Biochem 268: 487–498 [DOI] [PubMed] [Google Scholar]
- Sahai E, Marshall CJ (2003) Differing modes of tumor cell invasion have distinct requirements for Rho/ROCK signaling and extracellular proteolysis. Nat Cell Biol 5: 711–719 [DOI] [PubMed] [Google Scholar]
- Stephenson RA, Dinney CP, Gohji K, Ordonez NG, Killion JJ, Fidler IJ (1992) Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J Natl Cancer Inst 84: 951–957 [DOI] [PubMed] [Google Scholar]
- Sternberger M, Schmiedeknecht A, Kretschmer A, Gebhardt F, Leenders F, Czauderna F, Von Carlowitz I, Engle M, Giese K, Beigelman L, Klippel A (2002) GeneBlocs are powerful tools to study and delineate signal transduction processes that regulate cell growth and transformation. Antisense Nucleic Acid Drug Dev 12: 131–143 [DOI] [PubMed] [Google Scholar]
- Stolarov J, Chang K, Reiner A, Rodgers L, Hannon GJ, Wigler MH, Mittal V (2001) Design of a retroviral-mediated ecdysone-inducible system and its application to the expression profiling of the PTEN tumor suppressor. Proc Natl Acad Sci USA 98: 13043–13048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotman LC, Pandolfi PP (2003) PTEN and p53: who will get the upper hand? Cancer Cell 3: 97–99 [DOI] [PubMed] [Google Scholar]
- Vazquez F, Sellers WR (2000) The PTEN tumor suppressor protein: an antagonist of phosphoinositide 3-kinase signaling. Biochim Biophys Acta 1470: M21–M35 [DOI] [PubMed] [Google Scholar]
- Vincent S, Settleman J (1997) The PRK2 kinase is a potential effector target of both Rho and Rac GTPases and regulates actin cytoskeletal organization. Mol Cell Biol 17: 2247–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Weaver VM, Petersen OW, Larabell CA, Dedhar S, Briand P, Lupu R, Bissell MJ (1998) Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures. Proc Natl Acad Sci USA 95: 14821–14826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, Liu X, Wu H (2003) Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4: 209–221 [DOI] [PubMed] [Google Scholar]
- Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, Bissell MJ (1997) Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol 137: 231–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Brocker EB, Friedl P (2003) Compensation mechanism in tumor cell migration: mesenchymal–amoeboid transition after blocking of pericellular proteolysis. J Cell Biol 160: 267–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymann MP, Pirola L (1998) Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta 1436: 127–150 [DOI] [PubMed] [Google Scholar]
- Wymann MP, Zvelebil M, Laffargue M (2003) Phosphoinositide 3-kinase signaling-which way to target? Trends Pharmacol Sci 24: 366–376 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Supplementary Video 1
Supplementary Video 2