

Abstract
Perfringolysin O (PFO) is a prototype of the large family of pore-forming cholesterol-dependent cytolysins (CDCs). A central enigma of the cytolytic mechanism of the CDCs is that their membrane-spanning β-hairpins (the transmembrane amphipathic β-hairpins (TMHs)) appear to be ∼40 Å too far above the membrane surface to cross the bilayer and form the pore. We now present evidence, using atomic force microscopy (AFM), of a significant difference in the height by which the prepore and pore protrude from the membrane surface: 113±5 Å for the prepore but only 73±5 Å for the pore. Time-lapse AFM micrographs show this change in height in real time. Moreover, the monomers in both complexes exhibit nearly identical surface features and these results in combination with those of spectrofluorimetric analyses indicate that the monomers remain in a perpendicular orientation to the bilayer plane during this transition. Therefore, the PFO undergoes a vertical collapse that brings its TMHs to the membrane surface so that they can extend across the bilayer to form the β-barrel pore.
Keywords: AFM, perfringolysin, pore, toxin
Introduction
Pathogenic bacteria often secrete water-soluble protein toxins that can insert into cellular membranes and form aqueous pores (Bhakdi et al, 1996; van der Goot, 2001). Unlike most proteins that fold into conformations that are stable only in aqueous solution or only in lipid bilayers, these toxins have the remarkable ability to convert from a conformation that is water-soluble to one that spans a membrane in an oligomeric pore-forming complex. Many of these toxins are critical pathological factors (van der Goot, 2001) and some are used for biotechnological applications (Schnepf et al, 1998; Bayley and Cremer, 2001). Understanding the mechanisms by which these remarkable proteins convert between their two seemingly incompatible conformations has become the focus of a great deal of attention in the last decade (Heuck et al, 2001; Zakharov and Cramer, 2002).
Of the pore-forming mechanisms, one of the more common is a three-step process involving a prepore intermediate (Walker et al, 1992) in which the water-soluble monomers first bind to the membrane and then assemble into oligomeric prepore complexes on the surface. These prepore oligomers then convert into pore-forming complexes as a region of each monomer inserts one or more amphipathic β-hairpins that combine to form a transmembrane β-barrel pore (Walker et al, 1992; Sellman et al, 1997; Miller et al, 1999; Shepard et al, 2000; Nguyen et al, 2002; Kawate and Gouaux, 2003; Melton et al, 2004). Structural data indicate that some of these toxins undergo only a limited change in their structure when they convert from their water-soluble to transmembrane conformation (Song et al, 1996; Petosa et al, 1997; Olson et al, 1999; Pedelacq et al, 2000). In fact, to date, the only region of these toxins that has been found to undergo significant structural change and/or movement during pore formation is the region of the protein that forms the transmembrane amphipathic β-hairpin.
However, it is not yet clear whether members of the large family of cholesterol-dependent cytolysins (CDCs) (Tweten et al, 2001) share this property with other prepore-forming toxins. The CDCs are a family of pore-forming toxins that form an unusually large membrane pore (250–300 Å in diameter), consisting of up to 50 monomers (Olofsson et al, 1993). The CDCs also form a prepore complex that then converts to a pore-forming complex by the insertion of two transmembrane β-hairpins per monomer (Shepard et al, 1998, 2000; Shatursky et al, 1999; Hotze et al, 2001). The crystal structure of the water-soluble monomer of one member of this family, perfringolysin O (PFO), has been solved (Rossjohn et al, 1997). PFO is an 115 Å long molecule comprised of four domains, two of which have regions that directly interact with the bilayer lipids (Figure 1). First, in domain 4, there are three hydrophobic loops and a conserved undecapeptide at the tip of this domain that anchor the oligomer to the membrane (Nakamura et al, 1998; Heuck et al, 2000, 2003; Ramachandran et al, 2002). These residues are not deeply inserted within the bilayer and do not form the amphipathic wall of the pore in the membrane (Ramachandran et al, 2002; Heuck et al, 2003). Instead, a number of spectrofluorimetric studies (Shepard et al, 1998; Shatursky et al, 1999) have shown that the wall of the β-barrel is formed by a pair of transmembrane amphipathic β-hairpins (TMHs) that are derived from six α-helices in domain 3 of the monomer (Figure 1).
Figure 1.
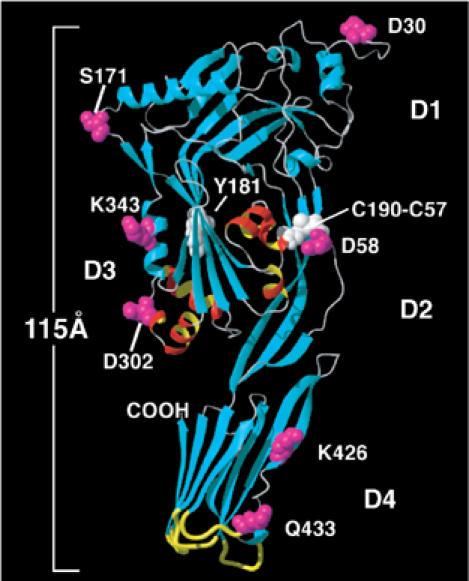
Structure of the water-soluble PFO monomer. A ribbon representation of the crystal structure of PFO is shown (Rossjohn et al, 1997). The α-helical bundles (TMH1 and TMH2) located in domain 3 (D3) that insert into the membrane in the pore complex are colored red and yellow. The six domain 3 α-helices unfurl into two amphipathic β-hairpins that insert into the membrane to form the transmembrane β-barrel (Shepard et al, 1998; Shatursky et al, 1999). Domain 4 (D4) has been shown to anchor PFO to the membrane in the prepore and pore complexes via the undecapeptide and three other short hydrophobic loops at its tip (Heuck et al, 2000; Ramachandran et al, 2002) (shown in yellow). This domain exists in a perpendicular orientation to the membrane and, except for its tip, is surrounded by the aqueous milieu (Ramachandran et al, 2002). Also shown in a space-filled mode are the positions of various residues studied herein (magenta) and the location of residues Y181 and the C190-C57 disulfide (white). The representations were generated using MOLMOL (Koradi et al, 1996).
However, a cryo-electron microscopy (cryo-EM) study of the structure of the putative pore-forming complex of another CDC, pneumolysin (PLY), concluded that its TMHs are approximately 40 Å too far from the membrane surface to span the bilayer and that this complex does not actually form a pore in the membrane (Gilbert et al, 1999). The cross-sectional electron density profile of PLY suggested that its structure in the ‘pore' remains similar to that of the water-soluble monomer (of PFO). Yet, this model is irreconcilable with the aforementioned spectroscopic results which clearly show that the PFO TMHs span the lipid bilayer and line the pore (Shepard et al, 1998; Shatursky et al, 1999). Therefore, how the TMHs cross the bilayer when they are ostensibly too far from the membrane surface is a central enigma in the pore-forming mechanism of the CDCs.
Herein we use atomic force microscopy (AFM) (Hansma and Hoh, 1994; Czajkowsky et al, 2000; Fisher et al, 2000; Viani et al, 2000; Zlatanova et al, 2000) and fluorescence spectroscopy to investigate the structures of the PFO prepore and pore complexes in model lipid membranes. Our results clearly show that the structures of the PFO prepore and pore complexes are strikingly different in one key feature: the height of the pore complex from the membrane surface is approximately 40 Å less than that of the prepore complex. Thus, unlike any other prepore-forming toxin described to date, the prepore-to-pore conversion of PFO is associated with a dramatic, vertical collapse of its structure by approximately 40 Å. These results not only provide a resolution to the aforementioned structural paradox, but also illustrate a novel mechanistic feature in the membrane insertion process of a pore-forming toxin.
Results
The surface topology of the prepore complex
Since the prepore intermediate is only a short-lived state of the wild-type toxin (Shepard et al, 2000; Hotze et al, 2001), we investigated the membrane-associated complexes formed by mutants of PFO that were previously found to be trapped at the prepore stage. In one, PFOS190C-G57C, the introduction of a disulfide bridge between residues 57 in domain 2 and 190 in domain 3 crosslinks these domains together and prevents the insertion of the TMHs into the membrane (Hotze et al, 2001). Subsequent reduction of the disulfide bond results in the penetration of the TMHs into the bilayer and the conversion of the prepore complex into the pore. Two other mutations, in which tyrosine at residue 181 has been substituted with either cysteine (PFOY181C) or alanine (PFOY181A), produces a toxin that assembles into oligomeric complexes, but does not insert its TMHs (Hotze et al, 2002). By studying the structures formed by each of these prepore mutants, we ensured that the observed structural features are properties of the prepore state and are not particular to a specific protein mutant.
As the large scan size image in Figure 2A shows, the addition of PFOY181C to the supported lipid bilayer results in a high density of protein complexes associated with the membrane. The majority of these complexes are circular rings, each with roughly the same diameter, while the remaining complexes consist of incomplete rings, or arcs, with a similar radius of curvature as the ring complexes. The outer diameter of the circular structures, determined from the center-to-center distance between neighboring complexes within hexagonally packed regions, is 38±2 nm (n=190), while their height from the membrane surface, as measured from regions where there were only clusters of complexes within otherwise protein-free bilayer (Figure 2B), is 113±5 Å (n=240).
Figure 2.
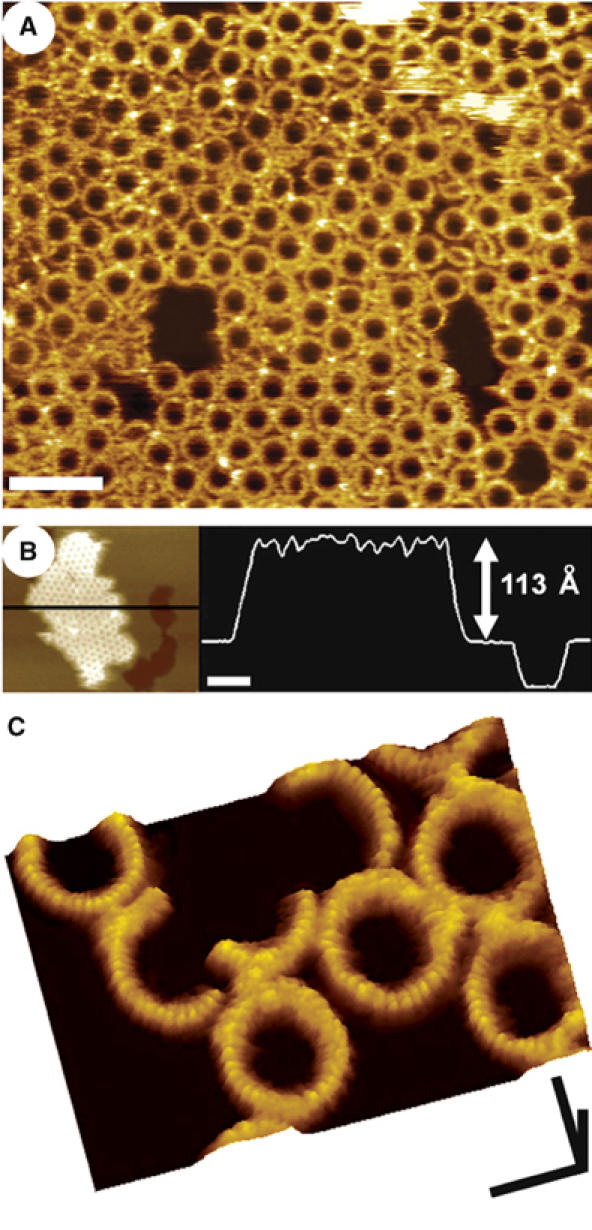
AFM images of the PFO prepore complex associated with supported lipid bilayers containing cholesterol. (A) The prepore-trapped PFOY181C complexes form a largely uniform population of ring structures, 38 nm in outer diameter. Scale bar: 100 nm. (B) These complexes measure 113±5 Å high from the top of the membrane surface, similar to the height of the water-soluble monomer (Figure 1). The darkest region in this figure is a large defect in the membrane, where the tip is directly in touch with the mica substrate. Scale bar: 100 nm. (C) At smaller scan sizes, higher resolution features are clearly discerned, including the 25 Å periodic arrangement of subunits in the complex and a slight 7 Å decrease in height of each subunit from its outermost to its innermost edge. Scale bars: x, y, 25 nm; z, 10 nm.
Higher resolution images obtained at smaller scan sizes show that each subunit within the complex has an ellipsoidal surface contour, with its longer axis directed along the radius of the complex (Figure 2C). In this direction, the contour has a length of 65±10 Å (n=90), which is probably slightly larger than the actual length because of tip broadening, and decreases slightly in height from the outermost to the innermost edge by 7±3 Å (n=82). In the perpendicular direction, along the circumference, the periodicity of the complex is 25±3 Å (n=106), which, assuming that there is very little interdigitation between the monomers within the complex, is also a reasonable estimate of the width of the monomer at the top of this complex.
The surface topology of the pore complex
The addition of pore-forming PFO to cholesterol-containing membranes also produces a high density of complexes associated with the bilayer (Figure 3A), although more of these complexes consist of arcs, of various sizes, rather than complete rings. The center-to-center distance between the neighboring ring complexes is 37±3 nm (n=49), similar to the size of the rings of the prepore complexes and consistent with earlier electron microscopy observations of the PFO pore complexes (Olofsson et al, 1993; Shepard et al, 2000; Hotze et al, 2001).
Figure 3.
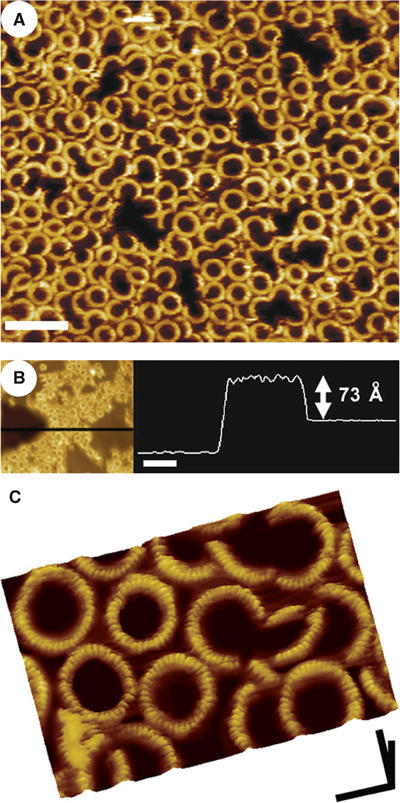
AFM images of the PFO pore complexes in supported lipid bilayers that contain cholesterol. (A) Pore-forming PFO self-assembles into both ring and arc-shaped complexes similar in size to the prepore complexes. Scale bar: 100 nm. (B) The pore complexes protrude 73±5 Å from the bilayer, approximately 40 Å less than the prepore complexes. The darkest region on the left in this figure is a large defect in the membrane, where the tip is directly in touch with the mica substrate. Scale bar: 100 nm. (C) Smaller scan size images of these complexes reveal a number of finer features of the complexes, including a similar 25 Å periodicity of subunits in the complex and 7 Å decrease in the height of each subunit from its outer to inner edge. Comparison with the prepore complexes (Figure 2) shows that the surface contours of the protein in the prepore and pore complexes are the same. Scale bar: x, y, 25 nm; z, 10 nm.
However, as is evident from the cross-sectional profile in Figure 3B, these pore complexes protrude only 73±5 Å (n=178) from the membrane surface, a distance that is markedly smaller (40 Å) than that measured of the prepore complexes (Figure 2B).
It should be noted that we have previously determined (Shepard et al, 1998; Shatursky et al, 1999) that it is unlikely that more than one residue near the predicted turn in the transmembrane β-hairpins of PFO extends beyond the membrane since these residues are flanked by membrane-embedded residues. Therefore, it is unlikely that the insertion of the pore structure is significantly affected by the presence of the mica support on the far side of the bilayer since there is more than 10 Å of water between the mica and lipid bilayer (Bayerl and Bloom, 1990; Johnson et al, 1991).
At smaller scan sizes, each subunit in the pore complex is found to exhibit an ellipsoidal surface contour, with its longest axis directed along the radius of the complex (Figure 3C), similar to the subunit in the prepore complex (Figure 2C). The length of the monomers along the radius of the complex (63±7 Å, n=73) and the width of each subunit along the circumference (24±3 Å, n=234) are also essentially the same as that observed in the prepore complexes. Moreover, the height of the subunit surface, measured along the radius of the complex, also decreases slightly from the outer to the inner edge by 7±2 Å (n=50), as observed with the prepore complexes.
These AFM images show that the structures of the prepore and pore complexes have the same surface morphology, within the resolution limits of AFM, but differ in the height by which they project from the membrane surface by 40 Å. The prepore-to-pore transition in PFO therefore appears to be associated with an essentially, if not exclusively, vertical collapse of the structure by 40 Å.
Time-lapse images of the prepore-to-pore conversion
We previously showed that if the disulfide bridge of PFOS190C-G57C is reduced after the formation of the prepore complexes, the majority of complexes insert into the membrane and form a pore (Hotze et al, 2001). Thus, the addition of DTT to prepore complexes of PFOS190C-G57C should change their topographic profiles only in the height by which they project from the bilayer. Figure 4 shows a time-lapse series of images of PFOS190C-G57C obtained after the addition of DTT. In each image, the population of complexes consists of structures with one of two heights: the taller complexes protrude ∼113 Å from the membrane surface while the shorter complexes project only ∼73 Å, as shown for the prepore and pore complexes, respectively (Figures 1 and 2). Moreover, it is clear that almost all of the complexes that were taller at an early time period became shorter at a later time period. These results show that changing the conditions in a manner that has been previously shown to convert these prepore complexes into pores also causes complexes with a height of 113 Å to convert into those with a height of 73 Å. Also, the closely packed prepore complexes converted into pore complexes without any detectable change in the packing of the complexes, indicating that the outer diameter of the pore complex cannot be significantly greater than that of the prepore complex, which is consistent with the measurements presented above.
Figure 4.

Direct observation of the change in height upon the conversion of prepore complexes to pores. Sequential images (from left to right) of the same oligomeric complexes of PFOS190C-G57C obtained after the addition of DTT are shown. The first image (0 min) was obtained 30 min after the application of the reducing agent. The complexes are initially at a height of ∼113 Å and decrease to a height of ∼73 Å over time. Scale bar: x, y, 100 nm; z, 10 nm.
The orientation of PFO on the membrane surface
We have previously shown that collisional quenchers that are localized to either the lipid acyl chains or the aqueous solution can be used to clearly identify whether a fluorescent dye-labeled residue of PFO faces the hydrophobic region of the bilayer or the aqueous milieu (Shepard et al, 1998; Shatursky et al, 1999). The AFM observations described above indicate that the surface structure of the oligomeric complex does not change as the prepore converts into the pore. This observation suggests that the PFO monomers within the oligomeric complex do not change their orientation with respect to the membrane surface, that is, they do not tilt over and lie down on the bilayer to insert their TMHs. To test this prediction, collisional quenching was used to determine whether residues from different domains of PFO are in contact with the membrane surface either in the prepore or pore complex. We choose several residues that are present on the surface of PFO based on crystal structure of the soluble monomer. Residues D58, K426 and Q433 are located along the predicted outside face of the oligomer in domains 1, 2 and 4, whereas residues S171, K343 and D302 are located along the face of the monomer predicted to face the pore in the oligomer (Figure 1). Hence, if the monomer tilts towards the inside of the oligomer in the prepore complex, we would predict that those residues facing the inside would be quenched, but those residues facing the outside would not, whereas the opposite would be true if the monomer tilts towards the outside of the complex. In the pore complex though, only the quenching of those residues facing the outside of the complex could be used to assess molecular orientation with this assay, since the lipid is absent from the interior of the pore.
Collisional quenching requires physical contact of the fluorophore and a collisional quenching agent. The quenching agent (a nitroxide) was thus placed near the membrane surface by replacing a fraction of the phosphatidylcholine with 1,2-diacyl-sn-glycero-3-phosphoTEMPOcholine (TEMPO-PC) in PC-cholesterol liposomes. The extent to which the TEMPO-PC reduced the fluorescence from fluorescent probes located on residues mutated to cysteine in the disulfide-trapped mutant PFOS190C-G57C was determined in both the prepore (oxidized disulfide) and pore (reduced disulfide) states (Figure 5).
Figure 5.
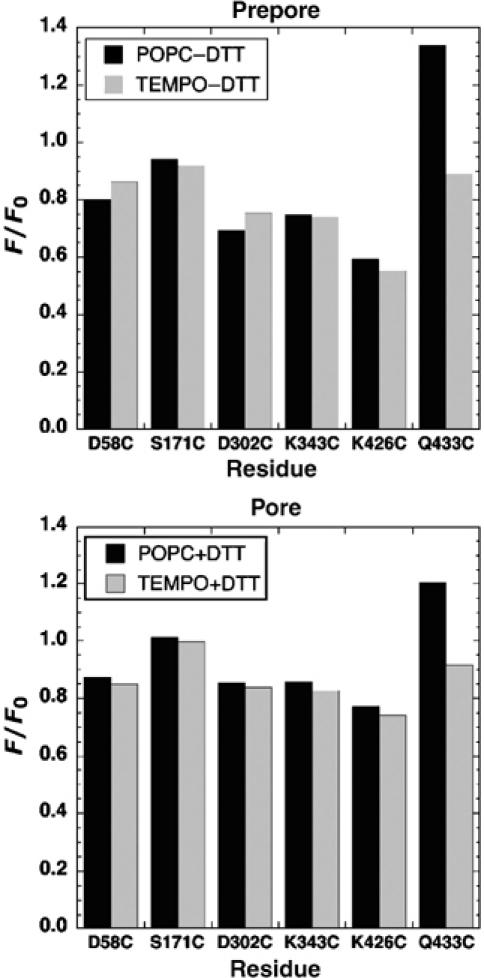
Determination of residues in close proximity to the membrane surface in both prepore and pores by collisional quenching. The extent by which the fluorescence from selected labeled residues is reduced in bilayers that contain lipids with a quenching moiety in the headgroup region (TEMPO-PC), compared with the fluorescence from similarly modified proteins in bilayers without the TEMPO-PC (POPC), is shown. The residues denoted on the X-axis were mutated to cysteine and labeled with the fluorescent probe in the disulfide-locked mutant, PFOS190C-G57C. The extent of quenching was determined in both prepore and pore complexes of each dye-labeled mutant. Fluorophores located near the surface of the membrane surface will be quenched by the TEMPO-PC present in the liposomes. Legend: POPC−DTT, PFOS190C-G57C incubated with POPC-cholesterol liposomes as a prepore complex (disulfide remains oxidized); POPC+DTT, PFOS190C-G57C incubated with POPC-cholesterol liposomes as a pore complex (disulfide is reduced to allow prepore-to-pore conversion); TEMPO−DTT and TEMPO+DTT (same as POPC−DTT and POPC+DTT, except that 10% of the total lipid is replaced with TEMPO-labeled lipid). F/F0, ratio of fluorescence of membrane-bound PFO (F) to that for the soluble monomer (F0).
The residues in domain 3 (K343C and D302C), where the TMHs are located, and in domain 1 (S171C) are not quenched to any appreciable degree in the prepore or pore conformation. However, as mentioned above, since these residues are predicted to face the pore lumen in the pore, we did not expect them to be quenched when in the pore conformation since the lipid in the channel is lost. The fact that these are not quenched to any significant degree in the prepore indicates that they are not in contact with the membrane surface in the prepore complex. Therefore, the toxin monomers within the prepore complex do not appear to be tilted forward, lowering domain 3 closer to the membrane in the prepore prior to their insertion into the membrane. In addition, we found that the fluorescence from a residue near the middle of domain 2 (D58C) is not quenched in either the prepore or pore complex, consistent with a location distal from the membrane surface at both prepore and pore stages.
There was a marked reduction (25–35%) of probe fluorescence when it was attached to the domain 4 residue Q433C in both the prepore and pore conformations, but no significant change in the fluorescence from another domain 4 residue, K426C, which is ∼15 Å above residue Q433 in the crystal structure. These findings suggest that domain 4 interacts with the bilayer only through a limited contact via the residues near its very tip, which is consistent with the previous observations of Ramachandran et al (2002), who showed that this domain in both the prepore and pore complexes is in a perpendicular orientation with the membrane surface. Residues (i.e., D58 and K426) along the outside surface of domains 2 and 4 of the oligomer are predicted to face away from the pore and do not come into contact with the surface upon conversion of the prepore to the pore complex.
Taken together, these results suggest that only domain 4 near its tip is juxtaposed to the membrane surface in the prepore complex and that the PFO monomer appears to maintain an upright position in the prepore and does not ‘tip over' such that domains 1, 2, or 3 are in contact with membrane surface in the prepore complex.
Localization of N-terminal residues in the prepore and pore complexes
The significant changes in the structure of the two complexes that are suggested by the dramatic difference in height must occur in a region of the protein that, even in the pore conformation, is far from the membrane surface. The surface profiles of the prepore and pore complexes in the AFM images are similar, but this could be coincidental and there could be significantly different structures in the protein at its topmost surface that nonetheless give similar contours. We therefore investigated whether there were molecules that are large enough to be detected by AFM which would specifically bind to the surface of either the prepore or pore complex, and then determined if these molecules would bind to both complexes.
The AFM images and spectroscopic data of the prepore complex both suggest that the surface of the protein that is in direct contact with the AFM tip is the top of domain 1 that includes the amino terminus (Figure 1). If true, then the addition of molecules large enough to resolve by AFM that bind to residues near the amino terminus should change the surface topography of prepore and/or pore complexes. In the first set of experiments, we examined the topography of the PFOY181A prepore complexes after the addition of an antibody specific for an epitope tag at the amino terminus of the recombinant PFO molecules (see Materials and methods). In a separate experiment, the prepore-forming mutant, PFOY181A/D30C, which also contains a mutation of the amino-terminal residue Asp-30 to cysteine, was biotinylated after the prepore complex was formed. Streptavidin was then incubated with PFOY181A/D30C. Before the addition of the labels, the complexes of both PFOY181A and PFOY181A/D30C project ∼113 Å from the bilayer surface in the AFM images (data not shown).
Figure 6A shows the sample of PFOY181A after the addition of the amino-terminal epitope-specific antibodies. The middle region was scraped away prior to obtaining this image by applying a greater tip force and scanning speed. The addition of the antibodies clearly changes the surface topography of the sample by ∼10 nm, consistent with the size of antibodies (Han et al, 1995). In control experiments, the addition of antibodies that recognize a heterologous epitope did not cause any change to the AFM images of these prepore complexes (data not shown).
Figure 6.
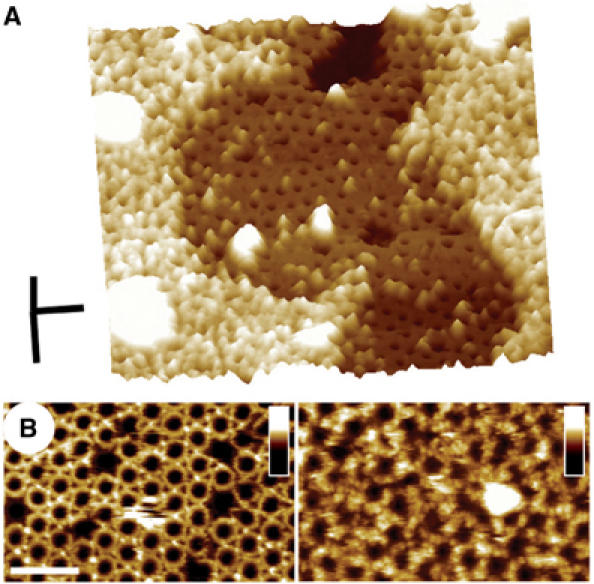
Location of residues near the N-terminus of PFO of the prepore complex. The amino-terminal residues of PFO, as well as the amino-terminal polyhistidine tag, are predicted to be near the top of the oligomeric complex. The locations of the histidine tag and residue D30C were each identified in the oligomeric complex by specifically tagging each with a large protein and imaging with AFM. (A) The addition of antibodies that recognize the amino-terminal polyhistidine epitope near the N-terminus results in the appearance of a number of large globular particles on the surface of the oligomers. The particles in the middle region were scraped away by the application of a greater force prior to obtaining this image. The difference in height between the middle and the unperturbed region is ∼10 nm, which is consistent with the size of antibodies (Han et al, 1995). Scale bar: x, y, 100 nm; z, 25 nm. (B) Addition of a thiol-reactive biotin label to PFOY181A/D30C prepore complex (left panel) followed by the addition of streptavidin (right panel) results in the appearance of a number of ∼8 nm globular particles (right image) that are distributed in a pattern which resembles the surfaces the prepore complexes. Vertical scale: 20 nm. Scale bar: 100 nm.
Figure 6B shows the sample of prepore complexes of PFOY181A/D30C before (left panel) and after (right panel) biotinylation and the addition of streptavidin. The surface topography of the sample treated with streptavidin clearly shows a number of globular particles that are distributed in a pattern that resembles the close-packed arrangement of the surfaces of the prepore complexes. These globular particles are ∼8 nm in diameter, consistent with the expected (slightly tip broadened) size of streptavidin (Hendrickson et al, 1989). Moreover, these particles could be scraped away by applying a greater tip force and scanning speed, leaving just the topmost surface of the prepore complexes and revealing the height of the molecules to be ∼4 nm, as expected for streptavidin (data not shown) (Hendrickson et al, 1989). Neither biotinylation nor the addition of streptavidin, alone, produced any changes in the AFM images of PFOY181A/D30C, nor did following a similar biotinylation/streptavidin procedure change the AFM images of the single point mutant PFOY181A that lacked a cysteine residue. Thus, taken together, these results clearly show that N-terminal residues are indeed localized to the top of the prepore complex.
We next added the N-terminal antibodies to the wild-type pore complexes and followed the same biotinylation/streptavidin procedure with the mutant, PFOD30C, which forms pores and projects ∼73 Å from the membrane surface in the AFM images (Figure 7). As the images in Figure 7 show, these topographic labels change the surface topography and height of the pore complexes to an extent similar to that of the prepore complexes. Similar control experiments as described above with the prepore complexes yielded no change to the AFM images of the pore complexes (data not shown). Hence, the N-terminal residues are near the top of both the prepore and pore complexes, and these residues are as accessible in the pore complex as they are in the prepore complex.
Figure 7.
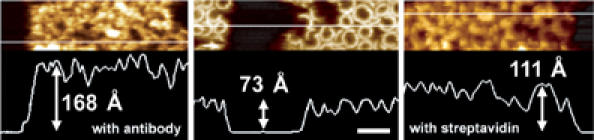
Location of residues near the N-terminus of PFO in the pore complex. Addition of the same antibodies described for Figure 6A produces a change in the sample topography in the PFO pore complexes (left image) as found with the prepore complexes. Likewise, following the same biotinylation/streptavidin labeling procedure as in Figure 6B with the pore-forming mutant PFOD30C produced a similar change in topography in the pore complexes (right image) as observed with prepore complexes. Thus, residues near the N-terminus are located at the top of both prepore and pore complexes and are similarly accessible to these structural labels in both complexes. Scale bar: 100 nm.
Discussion
Structural implications for the mechanism of pore formation by PFO
The AFM results presented here, together with the previously obtained crystal structure of the PFO monomer (Rossjohn et al, 1997), provides the first structural comparison of a prepore-forming toxin in each of its biochemically defined states: water-soluble, prepore and pore. The results of these studies show that prepore-to-pore conversion results in a dramatic 40 Å vertical collapse of the oligomeric structure. This observation provides a resolution to the long-standing enigma of how PFO brings its transmembrane domain within striking distance of the membrane surface.
The water-soluble PFO monomer is a highly asymmetric molecule, roughly 115 Å × 55 Å × 30 Å (Rossjohn et al, 1997). The N-terminus is located at the opposite end of the molecule from domain 4, and, with the molecular orientation depicted in Figure 1, the topmost surface of domain 1, on moving from the N-terminus to domain 3, is tilted down slightly. The AFM images of the prepore complex show that each subunit in this oligomer stands 113 Å from the membrane surface, and exhibits a surface contour that measures roughly 65 Å (but probably less, owing to tip broadening) in the radial direction and 25 Å along the circumference. There is also a slight decrease in the height of the surface contour along the radial direction on moving from the outermost to innermost edge of the ring, and N-terminal residues are located at the top of the complex (that is, on the face of the protein opposite to that which interacts the membrane surface).
These observations are consistent with a model of the prepore structure in which the PFO monomer is simply attached to the membrane in a largely perpendicular orientation by the end of domain 4, with domain 1 directed along the radius of the complex and domain 3 located inside the prepore complex (Figure 8). This model is consistent with previous studies that have clearly shown that domain 4, at the opposite end of the molecule from domain 1, is attached to the membrane via its tip and is oriented in a perpendicular fashion to the membrane in the prepore complex (Ramachandran et al, 2002). In addition, of the six residues examined in the various domains of PFO, only residue Q433 within domain 4 of the PFO structure is quenched by a collisional quencher when PFO assembles into the prepore complex. The lack of quenching of other residues, predicted to be located at the back and front of domain 1, is also consistent with a perpendicular orientation of the entire PFO monomer, attached to the membrane by the end of domain 4, in the prepore oligomer.
Figure 8.
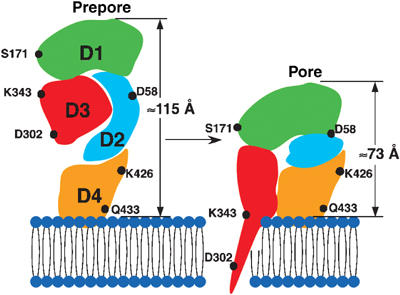
Schematic structural model of the prepore-to-pore transition of PFO. The data presented here suggest that the conformation of PFO in the prepore complex is largely the same as that of the water-soluble monomer, attached to the membrane in a perpendicular orientation via the tip of its domain 4. At the prepore stage of formation, the TMHs are ∼40 Å from the membrane surface. Upon converting to the pore, there is a vertical collapse of the structure by 40 Å, which changes neither the outer diameter of the complex nor the structure of its topmost surface significantly. We propose that the vertical collapse is a consequence of a disruption of the extended domain 2 structure. As a result of this collapse, the TMHs are brought close enough to the membrane surface to be able to span the bilayer and line the pore.
Therefore, the transition of PFO from a water-soluble monomer to a membrane-associated prepore subunit is probably not associated with significant changes to its structure. Inspection of the monomeric structure in Figure 1 shows that, with the attachment of this monomer to the membrane surface by the tip of domain 4 in a vertical orientation, the TMHs of such a prepore subunit would be 40 Å from the membrane surface. Yet, if this region were in a similar position in the pore conformation, the amphipathic β-hairpins of the TMHs would be too far from the bilayer to span the membrane, which would conflict with the previous spectroscopic data clearly indicating that these β-hairpins line the pore in the bilayer (Shepard et al, 1998; Shatursky et al, 1999). Hence, according to this model of the prepore structure and the earlier spectroscopic results, the conversion from prepore to pore must be associated with structural changes that result in the movement of the TMHs, and thus domain 3, nearer to the membrane surface by approximately 40 Å. Moreover, since domain 3 is connected to the rest of the protein only through the domain at the top of the complex (domain 1), these structural changes must also change the topographic profile of the complex.
The AFM images presented here confirm this prediction, and show that this different topographic profile is characterized by an essentially vertical collapse of the prepore complex by 40 Å, with no significant change in the outer diameter of the complex nor in the surface contour or location of the N-terminus. In addition, the collisional quenching results show that the conversion to the pore does not bring the various residues examined in domains 2 and 4 within the range of the surface-anchored collisional quencher and so the monomer in this complex does not tilt over such that these residues rest on the membrane surface. As expected, this quenching assay did not show any change in quenching in those residues that are predicted to face the channel (S171, D302, K343) after conversion to the pore, since the lipid is lost from the interior of the channel upon forming the pore. Previous data have in fact shown that residue D302 is near the core of the membrane, facing the pore channel, with its flanking residues, T301 and I303, embedded in the core of the bilayer in the pore complex (Shatursky et al, 1999).
What structural changes in PFO could bring the TMHs within striking distance of the membrane? Domain 4 does not tilt over or embed more deeply into the membrane upon prepore-to-pore conversion (Figure 5; Ramachandran et al, 2002) and so changes in the structure of domain 4 or its orientation with the membrane are not involved in the observed collapse of the prepore structure. Alternatively, the surface of domain 1 that faces the pore could rotate downward while at the same time the bent β-strands between domains 1 and 3 could straighten out. However, this scenario would not result in a significant difference in the vertical height of the prepore and pore complexes. Moreover, the similar surface features of the prepore and pore oligomers in these AFM images suggest that there is no major change in the orientation of domain 1. Thus, taken together, these observations suggest that the change in height described here most likely results from a change in the structure of domain 2 (Figure 8).
Domain 2 in the water-soluble (and probably prepore) conformation consists of two long β-strands, which make considerable contact with one of the domain 3 α-helical bundles that eventually convert to one of the two extended transmembrane β-hairpins (TMH1) that line the pore (Figure 1) (Shepard et al, 1998). Thus, it is likely that, without these contacts which must be broken for the TMHs to span the bilayer (Shepard et al, 1998; Shatursky et al, 1999; Hotze et al, 2001), this conformation is unstable and a more compact structure may be energetically favored (Figure 8). The two β-strands that comprise domain 2 may thus behave as a ‘stretched spring' that is stabilized by interactions with TMH1; without these, the domain 2 ‘spring' contracts to a more compact form, consequently bringing the TMHs close enough to the membrane so that they may span the bilayer.
Comparison with other prepore-forming toxins
The previous cryo-EM study of the membrane-associated oligomer of the related PLY described a complex that does not form a pore in the bilayer, and whose subunits exhibit a cross-sectional electron density profile that resembles the structure of the water-soluble monomer of PFO that is attached to the membrane surface in a vertical orientation by the tip of its domain 4 (Gilbert et al, 1999). It was suggested that the transmembrane β-hairpins were 40 Å above the membrane surface and therefore could not penetrate the membrane. However, as shown herein, the prepore complex vertically collapses this same distance upon pore formation. Although the PLY complex was suggested to correspond to the pore-forming state of this toxin, the results presented here suggest that the proposed structure of the PLY oligomer (Gilbert et al, 1999) was likely that of the prepore and not the pore complex.
The analyses of the structures of other pore-forming toxins suggest that they do not undergo a vertical collapse as seen here for PFO. The structure of the pore complex and monomer structure of α-hemolysin and the related leukocidin, respectively (Song et al, 1996; Olson et al, 1999; Pedelacq et al, 2000), showed that the β-hairpin that spans the membrane simply extends itself to penetrate the membrane, requiring no significant vertical movement of the protein to bring it nearer to the bilayer surface. Similarly, the anthrax protective antigen (PA) structure (Petosa et al, 1997) showed that predicted location of the transmembrane β-hairpin was close to the membrane surface in the prepore oligomer, suggesting that no significant change in the vertical height of the rest of the toxin was required for insertion of the β-barrel.
The studies herein show that an unprecedented change in the vertical height of the oligomeric complex of PFO occurs during the conversion of a prepore to pore. This structural change is consistent with a vertical collapse that would be required to bring the transmembrane β-hairpins of the prepore oligomer into close proximity to the membrane surface so that they may insert into and cross the bilayer to form the transmembrane β-barrel.
Materials and methods
Preparation of PFO and its derivatives
Derivatives of PFO in which one or more residues were converted to cysteine in the cysteine-less PFO derivative, PFOC459A, were generated by QuikChange PCR kit (Stratagene, La Jolla, CA) and oligonucleotide primers designed to introduce the TGT codon for cysteine at the required positions within the PFO structure. So as to avoid confusion, we shall omit the designation C459A in the text. The purified, recombinant forms of PFO and its derivatives were purified from Escherichia coli and modified with fluorescent probes as previously described (Hotze et al, 2001, 2002). All recombinant proteins contain an amino-terminal polyHistidine ‘Xpress' tag. The cysteine-substituted PFO mutants PFOD58C, PFOS171C, PFOD302C, PFOK343C, PFOK426C and PFOQ433C were generated from the disulfide-trapped derivative of PFOC459A in which residues G57 and S190 were substituted with cysteines that formed a disulfide bond that locks domain 3 to domain 2 (PFOS190C-G57C) (Hotze et al, 2001). Each mutant was labeled at the designated cysteine residues with the sulfhydryl-specific N-ethylmaleimide fluorescein (NEMF) (Molecular Probes, Eugene, OR). Each purified toxin (20 nmol) was labeled at the unique cysteine residue by incubating it with a 20 M excess of NEMF for 2 h at room temperature. The dye-labeled toxin was separated from unbound dye by passage over a 1.5 × 30 cm2 column packed with Sephadex G-50. The efficiency of labeling was determined spectroscopically (Shepard et al, 1998) and was determined to be 25–50% for each mutant. Disulfide formation in each mutant that was generated in the PFOC190-C57 mutant was assessed by the ratio of the hemolytic activity of unreduced (inactive, disulfide locked) to reduced toxin (active, reduced disulfide). Disulfide formation was determined to be >95% in all cases.
Liposome preparation
Liposomes were prepared as previously described by an extrusion system (Shepard et al, 1998) with the following modifications. Liposomes were prepared using 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and cholesterol at a ratio of 45:55 mol% or in which 10 mol% of the total lipid was substituted with the spin-labeled lipid 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho(TEMPO)choline (PC-TEMPO) (Avanti Polar Lipids, Alabaster, AL) so that the composition was 10:35:55 mol% PC-TEMPO:POPC:cholesterol. PC-TEMPO places a nitroxide spin probe near the surface of the membrane. After preparation, the liposomes were stored on ice under argon until used (within 3 days of preparation).
Fluorescence measurements
All fluorescence measurements were performed in an SLM-8100 photon-counting spectrofluorimeter (SLM Instruments) as described previously (Shepard et al, 1998). An excitation wavelength of 480 nm was used for fluorescein and the emission intensity was measured between 500 and 600 nm. The bandpass was 4 nm for all experiments. Emission scans of the fluorescein-labeled residues in the cysteine-substituted derivatives PFOD58C, PFOS171C, PFOD302C, PFOK343C, PFOK426C, and PFOQ433C of the disulfide-trapped mutant PFOG57C-S190C were recorded after incubation of each protein with POPC:cholesterol or the PC-TEMPO:POPC:cholesterol liposomes in both the presence and absence of 1 mM DTT in 2 ml of buffer A (100 mM NaCl, 50 mM HEPES, pH 7.5) at 37°C. In all experiments, the total toxin concentration was 44 nM. Emission spectra of controls in which the labeled toxin was replaced with unlabeled toxin were subtracted from each experimental emission spectrum. Also, except for the controls, experiments contained a mixture of 50 mol% dye-labeled toxin and 50 mol% unlabeled toxin (the controls contained 100% unlabeled toxin) to minimize the effects of self-quenching of the fluorescein dyes when the PFO molecules are juxtaposed in the oligomeric complexes.
In the absence of reduction, PFOS190C-G57C remains locked in the prepore state (i.e., the transmembrane β-barrel does not form) and when reduced the prepore complex is converted to the pore complex (the transmembrane β-hairpins insert into the membrane). Changes in the fluorescence intensity upon membrane binding of each labeled toxin were determined by the ratio (F/F0) of membrane-bound (F) and free monomer (F0). The extent of quenching by the TEMPO-PC can be determined by comparing the fluorescence intensity of the probe in the absence and presence of the TEMPO-PC, when the toxin is in the prepore (the G57C-S190C disulfide remains oxidized) or pore (the G57C-S190C disulfide is reduced) conformations.
Preparation of the supported bilayers and imaging with AFM
The supported membranes containing the PFO derivatives were formed by sequentially depositing two separately prepared lipid monolayers onto a mica substrate using small Teflon wells, followed by the injection of the protein into the wells, as described previously (Czajkowsky et al, 1998, 1999). The composition of the first monolayer (facing mica) was egg phosphatidylcholine (eggPC, Avanti Polar Lipids, Alabaster, AL), while that of the second monolayer was eggPC:cholesterol at 50:50 mol%. For all PFO derivatives, the final concentration of the protein in the well was ∼15 μg/ml, and, except for the samples with PFOS190C-G57C, the buffer in the well consisted of buffer B (10 mM sodium phosphate, pH 7) and 5 mM DTT; with PFOS190C-G57C, the buffer did not contain 5 mM DTT. After incubating for ∼45 min, the sample was extensively washed and then imaged in the AFM under the same buffer used during incubation.
Imaging was performed in the contact mode with a Nanoscope II AFM (Digital Instruments, Santa Barbara, CA) using oxide-sharpened ‘twin-tip' Si3N4 cantilevers with a spring constant of 0.06 N/m. The typical scan rate was 9 Hz, and the applied force was minimized to 0.1 nN. The piezoscanner (14 μm, D scanner, Digital Instruments, Santa Barbara, CA) was calibrated using a variety of samples including mica, the cholera toxin B subunit, and two-dimensional crystals of streptavidin on biotinylated supported bilayers. All images were reproducible with different tips and different fast-scan directions, and the lateral measurements were determined from the full-width at half-height in unprocessed images.
Modifications to the sample after membrane binding
For the time-series experiments with PFOS190C-G57C, after first preparing and evaluating the sample in buffer B, DTT was added to a final concentration of 5 mM. For the labeling experiments with antibodies, the antibodies were added to the sample to a final concentration of 7 μg/ml. After incubating for ∼2 h, the sample was washed extensively with buffer B and then imaged. The epitope for the antibodies (Invitrogen, CA) was either the ‘Xpress' tag found at the N-terminus of all of the PFO derivatives studied here or, in the control experiments, a C-terminal His tag that is not present in any of the proteins. For labeling with biotin and streptavidin, the sample was first extensively washed in buffer B, and then three cycles of biotinylation with 10 μg/ml (1-biotinamido)-4-(4′-[maleimidomethyl]-cyclohexanecarboxamido)-hexane (BMMCC, ProChem Inc., Rockford, IL), incubation for 1 h, and then extensive washing with buffer B were performed. Finally, streptavidin (Sigma) was added to the sample to a final concentration of 5 μg/ml, and, after incubating for 1 h, the sample was washed extensively with buffer B and then imaged.
Acknowledgments
These studies were supported by the NIH grants to RKT (AI37657) and ZS (EB002017). The technical assistance of Amy Marpoe was appreciated.
References
- Bayerl TM, Bloom M (1990) Physical properties of single phospholipid bilayers adsorbed to micro glass beads. A new vesicular model system studied by 2H-nuclear magnetic resonance. Biophys J 58: 357–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayley H, Cremer PS (2001) Stochastic sensors inspired by biology. Nature 413: 226–230 [DOI] [PubMed] [Google Scholar]
- Bhakdi S, Bayley H, Valeva A, Walev I, Walker B, Weller U, Kehoe M, Palmer M (1996) Staphylococcal alpha-toxin, streptolysin-O, and Escherichia coli hemolysin: prototypes of pore-forming bacterial cytolysins. Arch Microbiol 165: 73–79 [DOI] [PubMed] [Google Scholar]
- Czajkowsky DM, Iwamoto H, Cover TL, Shao Z (1999) The vacuolating toxin from Helicobacter pylori forms hexameric pores in lipid bilayers at low pH. Proc Natl Acad Sci USA 96: 2001–2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czajkowsky DM, Iwamoto H, Shao ZF (2000) Atomic force microscopy in structural biology: from the subcellular to the submolecular. J Electron Microsc 49: 395–406 [DOI] [PubMed] [Google Scholar]
- Czajkowsky DM, Sheng S, Shao Z (1998) Staphylococcal alpha-hemolysin can form hexamers in phospholipid bilayers. J Mol Biol 276: 325–330 [DOI] [PubMed] [Google Scholar]
- Fisher TE, Marszalek PE, Fernandez JM (2000) Stretching single molecules into novel conformations using the atomic force microscope. Nat Struct Biol 7: 719–724 [DOI] [PubMed] [Google Scholar]
- Gilbert RJC, Jiménez JL, Chen S, Tickle IJ, Rossjohn J, Parker M, Andrew PW, Saibil HR (1999) Two structural transitions in membrane pore formation by pneumolysin, the pore-forming toxin of Streptococcus pneumoniae. Cell 97: 647–655 [DOI] [PubMed] [Google Scholar]
- Han W, Mou J, Sheng J, Yang J, Shao Z (1995) Cryo atomic force microscopy: a new approach for biological imaging at high resolution. Biochemistry 34: 8215–8220 [DOI] [PubMed] [Google Scholar]
- Hansma HG, Hoh JH (1994) Biomolecular imaging with the atomic force microscope. Annu Rev Biophys Biomol Struct 23: 115–139 [DOI] [PubMed] [Google Scholar]
- Hendrickson W, Pähler AA, Smith JL, Satow Y, Merritt EA, Phizackerly RP (1989) Crystal structure of core streptavidin determined from multiwavelength anomalous diffraction of synchrotron radiation. Proc Natl Acad Sci USA 86: 2190–2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuck AP, Hotze E, Tweten RK, Johnson AE (2000) Mechanism of membrane insertion of a multimeric β-barrel protein: perfringolysin O creates a pore using ordered and coupled conformational changes. Mol Cell 6: 1233–1242 [DOI] [PubMed] [Google Scholar]
- Heuck AP, Tweten RK, Johnson AE (2001) Beta-barrel pore-forming toxins: intriguing dimorphic proteins. Biochemistry 40: 9065–9073 [DOI] [PubMed] [Google Scholar]
- Heuck AP, Tweten RK, Johnson AE (2003) Assembly and topography of the prepore complex in cholesterol-dependent cytolysins. J Biol Chem 278: 31218–31225 [DOI] [PubMed] [Google Scholar]
- Hotze EM, Heuck AP, Czajkowsky DM, Shao Z, Johnson AE, Tweten RK (2002) Monomer–monomer interactions drive the prepore to pore conversion of a beta-barrel-forming cholesterol-dependent cytolysin. J Biol Chem 277: 11597–11605 [DOI] [PubMed] [Google Scholar]
- Hotze EM, Wilson-Kubalek EM, Rossjohn J, Parker MW, Johnson AE, Tweten RK (2001) Arresting pore formation of a cholesterol-dependent cytolysin by disulfide trapping synchronizes the insertion of the transmembrane beta-sheet from a prepore intermediate. J Biol Chem 276: 8261–8268 [DOI] [PubMed] [Google Scholar]
- Johnson SJ, Bayerl TM, McDermott DC, Adam GW, Rennie AR, Thomas RK, Sackmann E (1991) Structure of an adsorbed dimyristoylphosphatidylcholine bilayer measured with specular reflection of neutrons. Biophys J 59: 289–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawate T, Gouaux E (2003) Arresting and releasing Staphylococcal alpha-hemolysin at intermediate stages of pore formation by engineered disulfide bonds. Protein Sci 12: 997–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koradi R, Billeter M, Wuthrich K (1996) MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph 14: 51–55, 29–32. [DOI] [PubMed] [Google Scholar]
- Melton JA, Parker MW, Rossjohn J, Buckley JT, Tweten RK (2004) The identification and structure of the membrane-spanning domain of the Clostridium septicum alpha Toxin. J Biol Chem 279: 14315–14322 [DOI] [PubMed] [Google Scholar]
- Miller CJ, Elliot JL, Collier RL (1999) Anthrax protective antigen: prepore-to-pore conversion. Biochemistry 38: 10432–10441 [DOI] [PubMed] [Google Scholar]
- Nakamura M, Sekino-Suzuki N, Mitsui K, Ohno-Iwashita Y (1998) Contribution of tryptophan residues to the structural changes in perfringolysin O during interaction with liposomal membranes. J Biochem (Tokyo) 123: 1145–1155 [DOI] [PubMed] [Google Scholar]
- Nguyen VT, Higuchi H, Kamio Y (2002) Controlling pore assembly of staphylococcal gamma-haemolysin by low temperature and by disulphide bond formation in double-cysteine LukF mutants. Mol Microbiol 45: 1485–1498 [DOI] [PubMed] [Google Scholar]
- Olofsson A, Hebert H, Thelestam M (1993) The projection structure of perfringolysin-O (Clostridium perfringens theta-toxin). FEBS Lett 319: 125–127 [DOI] [PubMed] [Google Scholar]
- Olson R, Nariya H, Yokota K, Kamio Y, Gouaux E (1999) Crystal structure of Staphylococcal LukF delineates conformational changes accompanying formation of a transmembrane channel. Nat Struct Biol 6: 134–140 [DOI] [PubMed] [Google Scholar]
- Pedelacq JD, Prevost G, Monteil H, Mourey L, Samama JP (2000) Crystal structure of the F component of the Panton–Valentine leucocidin. Int J Med Microbiol 290: 395–401 [DOI] [PubMed] [Google Scholar]
- Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC (1997) Crystal structure of the anthrax toxin protective antigen. Nature 385: 833–838 [DOI] [PubMed] [Google Scholar]
- Ramachandran R, Heuck AP, Tweten RK, Johnson AE (2002) Structural insights into the membrane-anchoring mechanism of a cholesterol-dependent cytolysin. Nat Struct Biol 9: 823–827 [DOI] [PubMed] [Google Scholar]
- Rossjohn J, Feil SC, McKinstry WJ, Tweten RK, Parker MW (1997) Structure of a cholesterol-binding thiol-activated cytolysin and a model of its membrane form. Cell 89: 685–692 [DOI] [PubMed] [Google Scholar]
- Schnepf E, Crickmore N, Van Rie J, Lereclus D, Baum J, Feitelson J, Zeigler DR, Dean DH (1998) Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol Mol Biol Rev 62: 775–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellman BR, Kagan BL, Tweten RK (1997) Generation of a membrane-bound, oligomerized pre-pore complex is necessary for pore formation by Clostridium septicum alpha toxin. Mol Microbiol 23: 551–558 [DOI] [PubMed] [Google Scholar]
- Shatursky O, Heuck AP, Shepard LA, Rossjohn J, Parker MW, Johnson AE, Tweten RK (1999) The mechanism of membrane insertion for a cholesterol dependent cytolysin: a novel paradigm for pore-forming toxins. Cell 99: 293–299 [DOI] [PubMed] [Google Scholar]
- Shepard LA, Heuck AP, Hamman BD, Rossjohn J, Parker MW, Ryan KR, Johnson AE, Tweten RK (1998) Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O: an α-helical to β-sheet transition identified by fluorescence spectroscopy. Biochemistry 37: 14563–14574 [DOI] [PubMed] [Google Scholar]
- Shepard LA, Shatursky O, Johnson AE, Tweten RK (2000) The mechanism of assembly and insertion of the membrane complex of the cholesterol-dependent cytolysin perfringolysin O: formation of a large prepore complex. Biochemistry 39: 10284–10293 [DOI] [PubMed] [Google Scholar]
- Song LZ, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE (1996) Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 274: 1859–1866 [DOI] [PubMed] [Google Scholar]
- Tweten RK, Parker MW, Johnson AE (2001) The cholesterol-dependent cytolysins. In Pore-Forming Toxins, van der Goot G (ed), Vol. 257, pp 15–33. Heidelberg: Springer-Verlag [DOI] [PubMed] [Google Scholar]
- van der Goot FG (ed) (2001) Pore-forming Toxins. Heidelberg: Springer-Verlag [Google Scholar]
- Viani MB, Pietrasanta LI, Thompson JB, Chand A, Gebeshuber IC, Kindt JH, Richter M, Hansma HG, Hansma PK (2000) Probing protein–protein interactions in real time. Nat Struct Biol 7: 644–647 [DOI] [PubMed] [Google Scholar]
- Walker B, Krishnasastry M, Zorn L, Bayley H (1992) Assembly of the oligomeric membrane pore formed by Staphylococcal alpha-hemolysin examined by truncation mutagenesis. J Biol Chem 267: 21782–21786 [PubMed] [Google Scholar]
- Zakharov SD, Cramer WA (2002) Colicin crystal structures: pathways and mechanisms for colicin insertion into membranes. Biochim Biophys Acta 1565: 333–346 [DOI] [PubMed] [Google Scholar]
- Zlatanova J, Lindsay SM, Leuba SH (2000) Single molecule force spectroscopy in biology using the atomic force microscope. Prog Biophys Mol Biol 74: 37–61 [DOI] [PubMed] [Google Scholar]