

Abstract
Unique, two-state modulating current signatures are observed when a cytosine-cytosine mismatch pair is confined at the 2.4 nm latch constriction of the α-hemolysin (αHL) nanopore. We have previously speculated that the modulation is due to base flipping at the mismatch site. Base flipping is a biologically significant mechanism in which a single base is rotated out of the DNA helical stack by 180°. It is the mechanism by which enzymes are able to access bases for repair operations without disturbing the global structure of the helix. Here, temperature dependent ion channel recordings of individual double-stranded DNA duplexes inside α-HL are used to derive thermodynamic (ΔH, ΔS) and kinetic (Ea) parameters for base flipping of a cytosine at an unstable cytosine-cytosine mismatch site. The measured activation energy for flipping a cytosine located at the latch of αHL out of the helix (18 ± 1 kcal mol−1) is comparable to that previously reported for base flipping at mismatch sites from NMR measurements and potential mean force calculations. We propose that the αHL nanopore is a useful tool for measuring conformational changes in dsDNA at the single molecule level.
Introduction
The DNA helix is a flexible molecule that continually undergoes a series of conformational changes over a range of timescales.1–4 The dynamics of DNA are extremely important in regulating overall function and recognition of individual base pairs.5, 6 On the nanosecond timescale, DNA undergoes a series of bending and twisting,7–9 breathing6, 10, 11 and fraying12, 13 motions that affect DNA protein interactions and consequently have an important role in gene expression.5
On the millisecond timescale, more significant conformational changes in DNA are observed. For example, individual bases are capable of rotating out of a DNA helical stack, in a process known as ‘base flipping’.14, 15 A growing body of evidence has shown that base flipping is a biologically important mechanism that plays a fundamental role in DNA maintenance and repair,16–19 primarily because the rotation of a single base out of a helical stack exposes the base to the solvent and permits access to proteins. In the context of a flipped-out base, these proteins are able to carry out repair (e.g., base recognition and excision)19, 20 or maintenance (e.g., methylation)16 without disrupting the global helical structure.
In the standard B-form conformation of double-stranded DNA (dsDNA), the DNA bases are held together in pairs through hydrogen bonding and π-π stacking interactions.21 Evidence from fluorescence,22 NMR23–25 and potential mean force calculations26–28 suggests that the energy required to disrupt the interactions that stabilize bases within the helix, and thus rotate an individual base out into the surrounding solvent, is large, between 10 and 20 kcal mol−1. The precise energy required depends on the nature of the base-pair23, 29 (mismatched base-pairs are less stable) and the sequence context,25 (which controls the strength of the stacking interactions.)
Among the most common method used to study spontaneous base flipping and base-pair opening are NMR measurements.23–25, 30–33 In these measurements, rotation of a DNA base out of the helical stack and exposure to solution permit the exchange of the imino protons on the bases guanine and thymine with protons in solution, resulting in measurable changes that indicate intrahelical lifetimes on the order of milliseconds31, 32 and extrahelical lifetimes on the order of microseconds.20 In a biological context, a number of proteins critical for DNA repair and function are believed to make direct use of base flipping to access individual bases. Because the lifetime of the extrahelical state in spontaneous base flipping is short, many proteins are believed to be actively involved in stabilizing the extrahelical state34, 35 and/or lowering the kinetic energy barrier for base flipping. However, experimentally measuring such protein-DNA interactions is extremely challenging.
In our recent work, we have discovered that ion channel recordings with the protein nanopore αHL can be used to quantitatively measure conformational changes in dsDNA36. These ion channel recordings can be used to identify changes to DNA structure such as damage,37–39 methylation40 and mismatch36 sites. In a typical experiment, a DNA helix, modified with a threading poly-T tail at the end of one strand, is driven into the αHL nanopore (embedded in a lipid bilayer) under an applied potential (Figure 1A). As the diameter of dsDNA (2 nm) 41 is greater than the diameter of the central constriction (1.4 nm),42 the DNA is held within the nanopore for tens of milliseconds up to seconds before ‘unzipping’ into its constituent single-stranded components. 43
Figure 1.

A duplex within the αHL nanopore gives unique modulating current signatures when a CC mismatch is located at the latch constriction. (A) A dsDNA duplex with a 5′-poly-T tail is driven into αHL up to the central constriction under an applied potential, with the mismatch site located in proximity to the 2.4 nm latch constriction. (B) (top) i-t trace for the capture and analysis of dsDNA containing a single CC mismatch (3 capture and unzipping events are shown in this trace). When a CC mismatch pair is present at the latch, the residual blocking current modulates between two distinct current states, I1 and I2. (bottom) i-t trace recorded in a solution containing a mixture of dsDNA molecules with either the CC mismatch or the CG Watson-Crick base pair. Current modulation is not observed for a fully-complementary duplex (only the I1 state is observed, e.g., the second capture event in the trace). Data were recorded in a 10 mM phosphate buffer (pH 7.5) with 0.25 M KCl at 25 °C. A potential of 120 mV was applied across the αHL channel.
The residual current during DNA residence within αHL is highly dependent on the composition of the bases situated at the latch constriction of αHL (Figure 1A).37, 38, 40 A DNA duplex containing a single mismatched base-pair positioned at the latch constriction results in a current signature that modulates between two distinct states (I1 and I2). In contrast, for a fully-complementary reference duplex, no modulation is observed (Figure 1B). We have speculated that the modulating signatures are a result of a cytosine base flipping in and out of the helical structure.36 Base flipping is expected to be significantly more prominent at mismatch sites because of their dynamic structure relative to a fully complementary duplex. 22, 23, 31
Herein, we present measurements of the temperature dependence of base flipping at a mismatch site in a DNA duplex held within the protein nanopore αHL. We extract kinetic (activation energy, EA) and thermodynamic (enthalpy, ΔH, and entropy, ΔS) parameters for the observed DNA conformational changes inside αHL. We demonstrate that these measured base pair lifetimes and activation energies are in good agreement with previously published base flipping dynamics data from NMR and fluorescence measurements. We discuss the evidence that supports our speculative theory of base flipping, and the potential for using αHL to measure conformational changes in DNA at the single molecule level.
Results
Measuring base flipping kinetics for a DNA mismatch site at the latch constriction of αHL
When a single DNA helix is captured inside the α-hemolysin (αHL) nanopore, there is significant attenuation of the ion flux and the measured electrical current decreases. Furthermore, when a DNA helix containing a cytosine-cytosine (CC) mismatch is positioned at the αHL latch constriction during capture, a distinct modulation of the electrical signal is observed that is described by two well-defined residual current states (I1 and I2), as shown in Figures 1 and 2. These states represent two distinct configurations of the dsDNA inside the αHL channel that result from base flipping. The focus of our discussion is on the energetics and dynamics of transitioning between these two chemical states via single molecule measurements.
Figure 2.

Effect of temperature on measuring base flipping kinetics inside the αHL nanopore. (A) Representative current-time traces showing the effect of temperature on measuring base flipping of DNA molecules captured with αHL. (B) Expanded view of one event each at (i) 15 °C (ii) 25 °C and (iii) 35 °C highlighting the significant change in lifetimes of states I1 and I2 as a function of temperature. Further examples are shown in Figures S1 – S5. Data were recorded in a 10 mM phosphate buffer (pH 7.5) with 0.25 M KCl. A potential of 120 mV was applied across the αHL channel.
The sequence of the two dsDNA molecules used in these studies, shown in Figure 1A, differ by a single-base change that results in a CC mismatch at position 9, as measured from the end of the duplex captured within the ion channel vestibule. A poly-T tail, 24 nucleotides long, is attached to the 5′ end of one ssDNA strand in order to thread the DNA by electrophoresis into the αHL channel under an applied voltage of 120 mV using Ag/AgCl electrodes placed on opposite sides of the channel. All measurements were performed in aqueous solutions containing 0.25 M KCl, with temperature control of ±1 °C.
The latch of αHL, which is just 0.4 nm larger in diameter than DNA, constitutes the tightest constriction relative to the duplex and is a major source of resistance in the protein channel.39, 42 Assuming that the measured residual current is primarily determined by the DNA conformation at the latch constriction, we assign I1 in Figure 1 to a DNA conformation in which all of the base-pairs are intrahelical and contained within the duplex. This assignment is based on the identical residual currents observed for state I1 for the duplex CC9 and the sole state observed for a fully complementary reference duplex, as shown in Figure 1B. The residual current for the duplexes CC9 and CG9 are expected to be very similar when the conformation/structure of the base-pairs at the latch constriction is similar.
We observe that the lifetimes of the I1 and I2 states are highly dependent on temperature (Figure 2), with a 100-fold decrease in the lifetimes of each state as the temperature is increased from 15 °C to 35°C. The overall dwell times of DNA inside the αHL nanopore prior to unzipping also increase at lower temperatures. At lower temperatures, the dsDNA within αHL is stabilized and less likely to unzip into its constituent single-stranded components.44 The temperature also affects the measured current of the two blocked current states (I1 and I2) and the open channel current (I0). In each case, the current rises approximately linearly with temperature (Figure S6), because of an increase in ion mobility.37
The lifetimes of the two residual current states I1 and I2 are well described by first-order rate kinetics at all temperatures. We extracted lifetime constants (τ) for each state from histograms of the state lifetimes, examples of which are shown in Figure 3. For a given experiment, the fraction (n/nT) of the measured states Ix (x = 1 or 2) with lifetime τ is given by:
| (Equation 1) |
where n(Ix) is the number of times the residual current is in state x, and n(Ix(T)) is the total number of measured states. The rate k(s−1) describes the kinetics of transitions between states, and is inversely related to τ.
Figure 3.

Effect of temperature on the lifetimes of states I1 and I2. Representative lifetime histograms (examples shown above for 18°C and 35°C) were used to extract first-order lifetimes (τ) and thus, rate constants (k) as a function of temperature. Lifetime histograms for 15, 20, 25 and 30 °C temperatures are shown in Figure S7.
| (Equation 2) |
While states I1 and I2 equate to possible conformations of DNA inside the protein pore, transitions between the states represent transformations from one conformation to another (Figure 4A). For state I1, two transitions are possible, either base flipping into state I2, which we have assigned to a DNA conformation in which one of the cytosine bases is extrahelical, or I0, which corresponds to unzipping of the dsDNA into the constituent single-stranded components, leading to the open channel without DNA inside (Figure 4B). The transition from I1 to I0 thus describes the well-known unzipping process for dsDNA inside αHL.36 The dwell time of the DNA inside αHL prior to unzipping is dependent on the duplex stability, and for 17 base-pair duplexes used in this study, unzipping times of 10 – 100 ms are expected. 45–47
Figure 4.

The effect of temperature on the kinetics of base flipping and unzipping. (A) Model for the base flipping and unzipping processes. Transitions between states I1 and I0 are unzipping of the DNA, while transitions between states 1 and 2 represent base flipping. Unzipping from state I1 is not observed. (B) Effect of temperature on extracted time constants for base flipping and unzipping.
As discussed previously, we speculate that the transition from I1 to I2 represents a base flipping out of the helix. In our model, the rotation of a single cytosine base out of the helical stack constitutes a conformational change in the DNA structure that gives rise to a measurable change in the residual current. The extrahelical base has a lifetime constant τ2, with the I2 to I1 transition representing the base flipping back into the helical structure. Curiously, we never observe duplex unzipping from state I1 (to I0), implying that the extrahelical base is interacting with the latch constriction in such a way as to raise the energy barrier for unzipping the helix. We discuss this topic in more detail later.
The intrahelical lifetime (the time before the base flips out of the helix) can be separated from the unzipping time (the dwell time in the pore prior to unzipping) using simple probability analysis. The time constant for state I1 (τ1) comprises both the unzipping time and the intrahelical lifetime:
| (Equation 3) |
The unzipping and intrahelical lifetime constants can be separated from one another by calculating the fraction of transitions along a particular pathway as a fraction of all the transitions. The intrahelical lifetime constant is given by:
| (Equation 4) |
where n1→2 represents transitions between states I1 and I2 (the base flipping out of the helix) and n1→0 represents transitions between states I1 and I0 (unzipping of the helix). Because the only transition from state I2 is to I1, the lifetime constant τ1 is precisely the extrahelical lifetime:
| (Equation 5) |
The intrahelical, extrahelical and unzipping lifetime constants are shown for the CC-mismatch containing duplex as a function of temperature in Figure 4B. In all three cases, the change in lifetime constants with temperature is described by an exponential function. The lifetime constants that describe the base flipping process, τextrahelical and τintrahelical, converge at a value of approximately 6 ms at 35 °C. Although αHL is stable over a wide temperature range,48 we were unable to resolve base flipping at higher temperatures because the dwell time of DNA inside the pore prior to unzipping becomes comparable to the intrahelical lifetime (τunzipping ≈ τintrahelical), and in many cases the DNA unzips before the base flips out. In addition, the increased noise associated with the current measurement at higher temperatures39 increases the difficulty of distinguishing between the two residual current states I1 and I2.
The measured unzipping times as a function of temperature are comparable to those published in other unzipping experiments with duplexes of a similar length and stability.49 The unzipping time decreases as a function of increasing temperature because of changes to the global stability of the helix, where higher temperatures provide more energy required to break the stacking and hydrogen-bond interactions that hold the helical structure together.21 Overcoming these interactions is a pre-requisite to unzipping the helical structure into its constituent components.
The lifetime constant I1, which we speculate represents the intrahelical lifetime, ranges from 50 ms at 15 °C to 6 ms at 35 °C. Values for the intrahelical lifetimes reported from NMR measurements, which are generally performed at low temperatures to improve resolution, range from 3 ms up to 300 ms at 15 °C, consistent with our measurements within the αHL nanopore. Such a wide range of intrahelical lifetimes are reported from NMR measurements because the stability of individual base-pairs is highly dependent on both the surrounding sequence context25 and the nature of the base-pair itself,23, 31 with, for example, a GC pair being more stable than AT because of the additional hydrogen bond.
It should be noted that recently published values from florescence measurements have suggested that base-pair lifetimes may be considerably longer, with values in the tens of seconds reported at 25 °C.22 The discrepancy is attributed to differences in each measurement, with shorter lifetimes reported from NMR measurements because imino-proton exchange, which occurs when a base is exposed to the surrounding solvent, can occur before a base is flipped fully out of the helix.50, 51 However, whether an individual base must rotate out by 180 ° to fulfil the biological function of ‘base flipping’, that is, to permit access to maintenance and repair proteins, is debatable.
The extrahelical lifetimes measured from the αHL nanopore measurements range from 250 ms at 15 °C to 6 ms at 35 °C. These times are significantly longer than those reported in bulk solution from NMR measurements,23 but comparable to those reported by Zhao and co-workers who measured extrahelical lifetimes between 6 and 30 ms at 25°C using florescence, comparable to the values we report here.
Extracting Kinetic and Thermodynamic Parameters for Base flipping
The derived time constants for the base flipping process were used to extract activation energies for flipping a base into or out of the DNA helix. The activation energy, EA, is related to the rate constant, k, at a given temperature, T, by the Arrhenius equation:
| (Equation 6) |
where k is the rate constant for flipping the base in or out of the helix, R is the gas constant and C is a the constant ‘pre-exponential’ factor.
The activation energy required to flip a cytosine base out of the helix at the latch constriction of αHL was found to be EA (flip-out) = 18 ± 1 kcal mol−1, which is within the range of base flipping activation energies experimentally measured by NMR and fluorescence techniques (6 to 21 kcal mol−1). 22, 23, 25 Activation energies between 18 and 20 kcal mol−1 have also been predicted from potential mean force calculations.26–28 The high activation energy barriers for the base flipping process are indicative of the significant molecular forces that must be disrupted to remove a base from the helical stack, including breaking of hydrogen bond(s) and overcoming the base-stacking interactions.19
Within the αHL nanopore, we determined the flip-in activation energy to be EA (flip-in) = 34 ± 1 kcal mol−1. The higher activation energy for flipping the base into the helix, relative to flipping the base out, indicates that the extrahelical state is the more stable conformation, which is not the case in the absence of a protein.
Base flipping in and out within the αHL nanopore occurs readily on the experimental timescale of our measurement, and as such, the derived rate constants can be used to compute an equilibrium constant, Kopen.
| (Equation 7) |
in eq 7, kin and kout are the rate constants for flipping the base in and out of the helix, respectively, and K is the equilibrium constant in the direction of flipping out the base.
From the temperature-dependent equilibrium constants, we determined the enthalpy and entropy of the base flipping process using the linearized version of the van’t Hoff equation (Figure 6). The van’t Hoff equation relates the equilibrium constant to the changes in the enthalpy, ΔH, and entropy, ΔS, between the flipped-in and flipped-out states:
| (Equation 8) |
Figure 6.
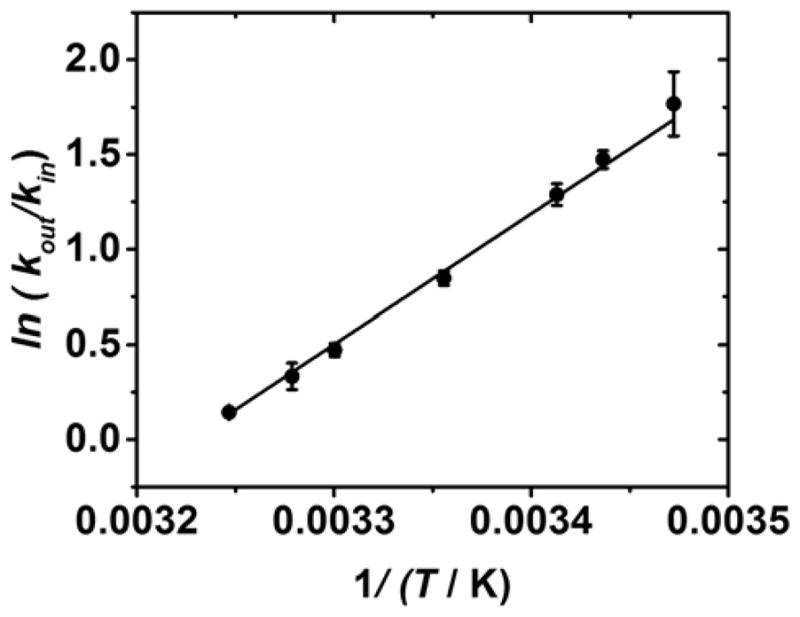
Van’t Hoff plot used to determine values for the enthalpy and entropy of base flipping, which were calculated as ΔH = −13.6 ± 0.3 kcal mol−1 and −44 ± 1 kcal mol−1 K−1.
Values for the enthalpy and entropy changes that occur when a base flips out of the helix at the αHL latch were determined as ΔH = −13.6 ± 0.3 kcal mol−1 and ΔS = −44 ± 1 kcal mol−1 K−1, respectively, from the slope and intercept of Figure 6. The positive slope of the Van’t Hoff plot (Figure 6) indicates that flipping out the base inside the αHL is exothermic, while the negative intercept indicates that the process is entropically disfavored. The overall change in Gibbs free energy that accompanies the base flipping out at 25 °C the helix is −0.50 ± 0.02 kcal mol−1, indicating that the process is marginally spontaneous. The difference in stability of the DNA when all of the bases are intrahelical, or one cytosine base is flipped out of the helical stack is small, equivalent to the Gibbs free energy that accompanies the formation of a single hydrogen bond between water molecules (ca. 0.5 kcal mol−1 at 25 °C),52 or the free energy of stacking between a CC mismatch and a neighbouring base pair (0.6 – 1.2 mol−1 at 25 °C).53
Discussion
Before attributing the modulating current states observed in our experiments to base flipping, we considered a number of other conformational changes that might give rise to a measurable change in ion flux through αHL while DNA is inside the pore. Based on our observations, several criteria must be met for any proposed mechanism:
The conformational change must be large because of the significant energy barrier (as measured from the activation energy) that separates the two states.
Each of the two conformations must be relatively stable to account for the millisecond lifetimes.
The conformational change must be specific to the identity of the base-pair of the latch, and localized to the latch constriction itself.
While we cannot completely discount that conformational changes other than base flipping give rise to the observed modulating current signatures, there are no other conformational changes in DNA that meet all three of the above criteria. The majority of DNA conformational changes, such as bending, breathing and twisting, are on very short timescales.1, 2 While interactions between the inner ion channel surface and the DNA backbone are expected,54 it seems unlikely that they would change the DNA conformation significantly enough to generate a measurable change in the ion flux. In any case, interactions with the phosphate backbone should be the same regardless of the base-pair identity. As another potential mechanism, we did also consider DNA sliding vertically within the pore, but found the kinetics to be independent of voltage;36 the driving force against diffusion that holds the DNA within the pore. Thus, our discussion herein assumes base flipping as the process observed from our measurements of DNA inside the αHL nanopore.
The lifetime of a base outside the helical stack for spontaneous base flipping (that is, in the absence of an interaction that stabilizes the extrahelical state) is shorter than the lifetime of the conformation in which all of the bases are held within the helical stack.34, 35 This is not surprising given that stacking and hydrogen-bonding interactions will stabilize intrahelical base pairs.21, 53 However, we observe long extrahelical lifetimes for the cytosine base in the confined environment of the latch constriction of αHL. Taken together with the large activation energy barrier required to flip the base back into the helix, our data strongly suggest a stabilizing interaction between the internal surface of the αHL latch and an extrahelical cytosine base.
Analysis of the crystal structure of αHL indicates that the latch constriction consists of a ring of lysine residues that will be positively charged at pH 7.5.42 Interactions between the nucleobase cytosine and lysine amino acid residue have been previously reported.55–57 We speculate here that the latch constriction is capable of stabilizing the extrahelical cytosine base, possibly through hydrogen bonding interactions. The precedent for such an interaction is found in the proteins that are known to utilize base flipping to repair or alter DNA base-pairs as a part of cell functions.34, 35 Examples of such proteins include UDG (uracil-DNA glycosylase) and DNMT (DNA methyltransferase). NMR measurements have suggested that extrahelical lifetimes in bulk solution last less than a microsecond at 25 °C, and so, for proteins such as UDG to be able to carry out their function, they must be capable of interacting and stabilizing the extrahelical base.34, 35
Base flipping in the presence of repair proteins has been found in some cases to be active, and in others, passive. In active cases, the kinetic barrier for base flipping is lowered, while in the passive case, the protein stabilizes the extrahelical state without altering the activation barrier. UDG falls into the latter category, with a body of evidence demonstrating that the enzyme can alter the equilibrium constant for opening base-pairs by stabilizing the extrahelical state by up to 23-fold.20, 34, 35 The activation energies we measure for outward base flipping inside αHL are comparable to those in bulk solution. We therefore speculate that the lysine residues at the latch constriction are not actively flipping the base out of the helix, but interactions between cytosine and lysine stabilize the extrahelical base once it is already flipped out. In this scenario, the resulting interactions between the flipped-out base and the αHL latch replace the base-stacking and hydrogen bonding that stabilizes the intrahelical state, result in a shift in the equilibrium constant that stabilizes the extrahelical base relative to the intrahelical conformation.
Conclusions
We have measured the temperature-dependent kinetics of base flipping at a DNA cytosine-cytosine mismatch site located at the latch constriction of α-hemolysin. Values for the intrahelical lifetime of the base-pairs, as well as the activation energy required to flip a base out of the helix, are in agreement with values previously reported from NMR measurements of DNA in bulk solution. We have speculated that an extrahelical cytosine is stabilized by interactions with the positively charged lysine residues that comprise the latch constriction in αHL. Thus, the latch constriction of α-hemolysin constitutes a useful tool for the measurement of biologically relevant conformational changes in double-stranded DNA at the single molecule level.
Experimental
DNA Preparation & Purification
DNA was prepared from commercially available phosphoramidites (Glen Research, Sterling, VA) by the DNA Core Facility at the University of Utah. Purification was via an anion-exchange HPLC column running a linear gradient of B from 25% to 100% over 30 min while monitoring UV absorbance at 260 nm (A = 20 mM NaPi, 1 M NaCl, pH 7 in 10% CH3CN/90% ddH2O, B = 10% CH3CN/90% ddH2O, flow rate = 3 mL/min).
Chemicals & Materials for Nanopore Measurements
Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich and used without further purification. The buffer solutions were prepared as 10 mM phosphate, 0.25 M KCl at pH 7.5. Wild-type αHL was purchased from List Biological Laboratories in the monomer form of lyophilized powder and dissolved in water at 1 mg/mL. 1,2-Diphytanoyl-sn-glycero-3-phospho-choline (DPhPC) was purchased from Avanti and dissolved in decane at 10 mg/mL and used to form the bilayer. The bilayer was supported by a glass nanopore membrane of diameter 600 nm, the fabrication of which has been described previously.58, 59 Glass nanopore membranes were modified with a 2% (v/v) (3-cyanopropyl) dimethylchlorosilane in acetonitrile to create a moderately hydrophobic surface. The DNA duplexes were annealed by mixing the 41-mer and 17-mer at a 1:1 mole ratio, followed by heating in a 90 °C water bath for 5 min and then cooling to room temperature over 3 hours.
Current-Time Recordings
Current-time (i-t) recordings were performed using the low noise Nanopatch system (Electronic BioSciences, Inc., San Diego, CA). A voltage was applied across the GNM between two Ag/AgCl electrodes placed inside and outside of the capillary. A lipid bilayer was deposited across the GNM orifice as previously described. A pressure of 60 to 80 mmHg was applied to the inside of the GNM capillary using a syringe, allowing the lipid bilayer to be functional for the protein channel reconstitution.59 Next, 0.2 μL of αHL monomer solution at 1 mg/mL was added to the cis (external) side of the GNM (a volume of 350 μL). Protein reconstitution into the lipid bilayer was indicated by a single jump in the current of approximately 0.3 pA/mV at 25 °C in 0.25 M KCl (pH 7.5) electrolyte. The duplex DNA (15 μM) was added to the cis (external) side of the GNM prior to measurement. A voltage of 120 mV was applied trans vs. cis, (Ag/AgCl electrode inside the capillary vs. Ag/AgCl electrode placed in the external solution (cis negative)).
Temperature control was achieved by a Peltier heater/cooler (custom PID control of a small-scale thermoelectric cooler (CUI Inc., CP20151)) situated directly below the cis (external reservoir). The temperature is measured by a thermocouple (0.1 °C precision) residing in the same solution as the pore (volume 350 μL). The distance between the pore orifice and the thermocouple is <2 mm. Thermal equilibrium at each desired temperature was reached in approximately 2 min, and each measurement reported was taken after leaving a minimum of 5 min to equilibrate, after which the solution (internal and external) and thermocouple is expected to be at the same temperature.
Data Collection and Analysis
I-t blockades that lasted longer than 1 ms in total were identified as DNA unzipping events, and those less than 1 ms were identified as single-stranded translocations. Events were extracted using QuB version 2.0.023 (freeware, www.qub.buffalo.edu). QUB was used to assign one of three states (0, 1 or 2) to continuous current-time data using the software’s inbuilt ‘idealization’ SKM (segmental K-means) method, which uses mathematical models to predict changes in state based on the current amplitude. State 0 corresponds to the open channel current. States 1 and 2 are the two modulating states observed when the duplex is inside αHL. The lifetimes and average current of each state were used to plot the histograms shown in this article. The number of transits between each state (e.g. 1 to 2 or 1 to 0) was extracted using QUB’s ‘transit’ function. Origin Pro 2015 (OriginLab) was used to plot histograms of the event time and determine lifetime constants.
Supplementary Material
Figure 5.

Arrhenius plots were used to derive the activation energy required flip a base out of the helix (red line), and the activation energy required to overcome interactions between the latch constriction of αHL and the extrahelical cytosine base in order flip the base back into the helical stack (black line). Values of the activation energy were EA (flip-out) = 18 ± 1 kcal mol−1 and EA (flip-in) = 34 ± 1 kcal mol−1.
Acknowledgments
RPJ acknowledges funding from a Marie Curie International Outgoing Fellowship under the EU FP7 program (Project No. 625984). This work was funded in part by a grant from the NIH (R01 GM093099). The authors thank Electronic BioSciences Inc. (San Diego, CA) for donating the ion-channel recording instrument and software, and Dr. M. A. Edwards (University of Utah) for assistance with the statistical and probability analysis.
Footnotes
Contributor Information
Cynthia J. Burrows, Email: burrows@chem.utah.edu.
Henry S. White, Email: white@chem.utah.edu.
References
- 1.Galindo-Murillo R, Roe DR, Cheatham TE., III Nat Commun. 2014;5 doi: 10.1038/ncomms6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Galindo-Murillo R, Roe DR, Cheatham TE., III Biochim Biophys Acta. 2015;1850:1041–1058. doi: 10.1016/j.bbagen.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frank-Kamenetskii MD, Prakash S. Phys Life Rev. 2014;11:153–170. doi: 10.1016/j.plrev.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 4.Vologodskii A. Phys Life Rev. 2006;3:119–132. [Google Scholar]
- 5.Lavelle C. Curr Opin Genet Dev. 2014;25:74–84. doi: 10.1016/j.gde.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 6.von Hippel PH, Johnson NP, Marcus AH. Biopolymers. 2013;99:923–954. doi: 10.1002/bip.22347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okonogi TM, Reese AW, Alley SC, Hopkins PB, Robinson BH. Biophys J. 1999;77:3256–3276. doi: 10.1016/S0006-3495(99)77157-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rüdisser S, Hallbrucker A, Mayer E. J Am Chem Soc. 1997;119:12251–12256. [Google Scholar]
- 9.Szyc Ł, Yang M, Nibbering ETJ, Elsaesser T. Angew Chemie Int Ed. 2010;49:3598–3610. doi: 10.1002/anie.200905693. [DOI] [PubMed] [Google Scholar]
- 10.Krueger A, Protozanova E, Frank-Kamenetskii MD. Biophys J. 2006;90:3091–3099. doi: 10.1529/biophysj.105.078774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fei J, Ha T. Proc Natl Acad Sci USA. 2013;110:17173–17174. doi: 10.1073/pnas.1316493110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andreatta D, Sen S, Pérez Lustres JL, Kovalenko SA, Ernsting NP, Murphy CJ, Coleman RS, Berg MA. J Am Chem Soc. 2006;128:6885–6892. doi: 10.1021/ja0582105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nonin S, Leroy JL, Gueron M. Biochem. 1995;34:10652–10659. doi: 10.1021/bi00033a041. [DOI] [PubMed] [Google Scholar]
- 14.Roberts RJ, Cheng X. Ann Rev Biochem. 1998;67:181–198. doi: 10.1146/annurev.biochem.67.1.181. [DOI] [PubMed] [Google Scholar]
- 15.Stivers JT, Jiang YL. Chem Rev. 2003;103:2729–2760. doi: 10.1021/cr010219b. [DOI] [PubMed] [Google Scholar]
- 16.Huang N, Banavali NK, ADM Proc Natl Acad Sci USA. 2003;100:68–73. doi: 10.1073/pnas.0135427100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang N, Banavali NK, MacKerell AD. Proc Natl Acad Sci USA. 2003;100:68–73. doi: 10.1073/pnas.0135427100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Larivière L, Moréra S. J Biol Chem. 2004;279:34715–34720. doi: 10.1074/jbc.M404394200. [DOI] [PubMed] [Google Scholar]
- 19.Stivers JT. Prog Nucleic Acid Res. 2004;77:37–65. doi: 10.1016/S0079-6603(04)77002-6. [DOI] [PubMed] [Google Scholar]
- 20.Stivers JT. Chem Eur J. 2008;14:786–793. doi: 10.1002/chem.200701501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.SantaLucia J. Proc Natl Acad Sci USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yin Y, Yang L, Zheng G, Gu C, Yi C, He C, Gao YQ, Zhao XS. Proc Natl Acad Sci USA. 2014;111:8043–8048. doi: 10.1073/pnas.1400667111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moe JG, Russu IM. Biochem. 1992;31:8421–8428. doi: 10.1021/bi00151a005. [DOI] [PubMed] [Google Scholar]
- 24.Coman D, Russu IM. Biophys J. 2005;89:3285–3292. doi: 10.1529/biophysj.105.065763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Folta-Stogniew E, Russu IM. Biochem. 1994;33:11016–11024. doi: 10.1021/bi00202a022. [DOI] [PubMed] [Google Scholar]
- 26.Bouvier B, Grubmüller H. Biophys J. 2007;93:770–786. doi: 10.1529/biophysj.106.091751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuxreiter M, Luo N, Jedlovszky P, Simon I, Osman R. J Mol Biol. 2002;323:823–834. doi: 10.1016/s0022-2836(02)00999-3. [DOI] [PubMed] [Google Scholar]
- 28.Banavali NK, MacKerell AD., Jr J Mol Biol. 2002;319:141–160. doi: 10.1016/S0022-2836(02)00194-8. [DOI] [PubMed] [Google Scholar]
- 29.Krosky DJ, Schwarz FP, Stivers JT. Biochem. 2004;43:4188–4195. doi: 10.1021/bi036303y. [DOI] [PubMed] [Google Scholar]
- 30.Every AE, Russu IM. J Mol Recogn. 2013;26:175–180. doi: 10.1002/jmr.2262. [DOI] [PubMed] [Google Scholar]
- 31.Bhattacharya PK, Cha J, Barton JK. Nuc Acids Res. 2002;30:4740–4750. doi: 10.1093/nar/gkf601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gueron M, Kochoyan M, Leroy JL. Nature. 1987;328:89–92. doi: 10.1038/328089a0. [DOI] [PubMed] [Google Scholar]
- 33.Guéron M, Leroy JL. Methods in Enzymology. 1995;261:383–413. doi: 10.1016/s0076-6879(95)61018-9. [DOI] [PubMed] [Google Scholar]
- 34.Cao C, Jiang YL, Stivers JT, Song F. Nat Struct Mol Biol. 2004;11:1230–1236. doi: 10.1038/nsmb864. [DOI] [PubMed] [Google Scholar]
- 35.Cao C, Jiang YL, Krosky DJ, Stivers JT. J Am Chem Soc. 2006;128:13034–13035. doi: 10.1021/ja062978n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson RP, Fleming AM, Beuth LR, Burrows CJ, White HS. J Am Chem Soc. 2016;138:594–603. doi: 10.1021/jacs.5b10710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Johnson RP, Fleming AM, Jin Q, Burrows CJ, White HS. Biophys J. 2014;107:924–931. doi: 10.1016/j.bpj.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin Q, Fleming AM, Johnson RP, Ding Y, Burrows CJ, White HS. J Am Chem Soc. 2013;135:19347–19353. doi: 10.1021/ja410615d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson RP, Fleming AM, Burrows CJ, White HS. J Phys Chem Lett. 2014;5:3781–3786. doi: 10.1021/jz502030e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ding Y, Fleming AM, White HS, Burrows CJ. ACS Nano. 2015;9:11325–11332. doi: 10.1021/acsnano.5b05055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drew HR, Wing RM, Takano T, Broka C, Tanaka S, Itakura K, Dickerson RE. Proc Natl Acad Sci USA. 1981;78:2179–2183. doi: 10.1073/pnas.78.4.2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song L, Hobaugh MR, Shustak C, Cheley S, Bayley H, Gouaux JE. Science. 1996;274:1859–1866. doi: 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- 43.Sauer-Budge AF, Nyamwanda JA, Lubensky DK, Branton D. Phys Rev Lett. 2003;90:238101. doi: 10.1103/PhysRevLett.90.238101. [DOI] [PubMed] [Google Scholar]
- 44.Angevine CE, Seashols-Williams SJ, Reiner JE. Anal Chem. 2016;88:2645–2651. doi: 10.1021/acs.analchem.5b03631. [DOI] [PubMed] [Google Scholar]
- 45.Jin Q, Fleming AM, Ding Y, Burrows CJ, White HS. Biochem. 2013;52:7870–7877. doi: 10.1021/bi4009825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sutherland TC, Dinsmore MJ, Kraatz HB, Lee JS. Biochem Cell Biol. 2004;82:407–412. doi: 10.1139/o04-005. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, Tian K, Hunter LL, Ritzo B, Gu LQ. Nanoscale. 2014;6:11372–11379. doi: 10.1039/c4nr03195d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kang XF, Gu LQ, Cheley S, Bayley H. Angew Chemie Int Ed. 2005;44:1495–1499. doi: 10.1002/anie.200461885. [DOI] [PubMed] [Google Scholar]
- 49.Jin Q, Fleming AM, Burrows CJ, White HS. J Am Chem Soc. 2012;134:11006–11011. doi: 10.1021/ja304169n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Giudice E, Várnai P, Lavery R. Nuc Acids Res. 2003;31:1434–1443. doi: 10.1093/nar/gkg239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Priyakumar UD, MacKerell AD. Chem Rev. 2006;106:489–505. doi: 10.1021/cr040475z. [DOI] [PubMed] [Google Scholar]
- 52.Silverstein KAT, Haymet ADJ, Dill KA. J Am Chem Soc. 2000;122:8037–8041. [Google Scholar]
- 53.Peyret N, Seneviratne PA, Allawi HT, SantaLucia J. Biochem. 1999;38:3468–3477. doi: 10.1021/bi9825091. [DOI] [PubMed] [Google Scholar]
- 54.Vercoutere W, Winters-Hilt S, Olsen H, Deamer D, Haussler D, Akeson M. Nat Biotech. 2001;19:248–252. doi: 10.1038/85696. [DOI] [PubMed] [Google Scholar]
- 55.Luscombe NM, Laskowski RA, Thornton JM. Nuc Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bruskov V. Stud Biophys (Berlin) 1978;67S:43–44. [Google Scholar]
- 57.Akeson M, Branton D, Kasianowicz JJ, Brandin E, Deamer DW. Biophys J. 1999;77:3227–3233. doi: 10.1016/S0006-3495(99)77153-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang B, Galusha J, Shiozawa PG, Wang G, Bergren AJ, Jones RM, White RJ, Ervin EN, Cauley CC, White HS. Anal Chem. 2007;79:4778–4787. doi: 10.1021/ac070609j. [DOI] [PubMed] [Google Scholar]
- 59.White RJ, Ervin EN, Yang T, Chen X, Daniel S, Cremer PS, White HS. J Am Chem Soc. 2007;129:11766–11775. doi: 10.1021/ja073174q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.