

Abstract
Constitutive activation of signal transducer and activator of transcription 3 (Stat3) has been found in a wide spectrum of human malignancies. Here, we have assessed the effect of Stat3 deficiency on skin tumor development using the 2-stage chemical carcinogenesis model. The epidermis of Stat3-deficient mice showed a significantly reduced proliferative response following treatment with the tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA) because of a defect in G1-to-S-phase cell cycle progression. Treatment with the tumor initiator 7,12-dimethylbenz[a]anthracene (DMBA) resulted in a significant increase in the number of keratinocyte stem cells undergoing apoptosis in the bulge region of hair follicles of Stat3-deficient mice compared with nontransgenic littermates. Notably, Stat3-deficient mice were completely resistant to skin tumor development when DMBA was used as the initiator and TPA as the promoter. Abrogation of Stat3 function using a decoy oligonucleotide inhibited the growth of initiated keratinocytes possessing an activated Ha-ras gene, both in vitro and in vivo. In addition, injection of Stat3 decoy into skin tumors inhibited their growth. To our knowledge, these data provide the first evidence that Stat3 is required for de novo epithelial carcinogenesis, through maintaining the survival of DNA-damaged stem cells and through mediating and maintaining the proliferation necessary for clonal expansion of initiated cells during tumor promotion. Collectively, these data suggest that, in addition to its emerging role as a target for cancer therapy, Stat3 may also be a target for cancer prevention strategies.
Introduction
Human cancers develop via a multistep process that involves the accumulation of at least 4–6 genetic mutations (1). Based on work in experimental animal model systems, the carcinogenesis process can be divided both operationally and mechanistically into the initiation, promotion, and progression stages (2, 3). The initiation stage is an irreversible event in which carcinogens damage DNA and induce mutations in critical genes in target stem cells. During the promotion stage, initiated cells undergo selective clonal expansion due to the acquisition of a proliferative advantage, and/or of the ability to evade growth inhibitory or apoptotic signals (3). This clonal expansion of initiated cells enhances the probability of additional genetic mutations that might lead to the development of malignant lesions (2).
Many classes of signaling molecules and pathways are altered during cancer development (4). One such signaling molecule, signal transducer and activator of transcription 3 (Stat3), is latent in the cytoplasm, until activated by a wide variety of cell surface receptors via tyrosine phosphorylation, leading to its dimerization and translocation into the nucleus (5). It is well documented that Stat3 is constitutively activated in a number of human epithelial malignancies, including prostate, breast, lung, head and neck, brain, and pancreas (6), as well as hematopoietic malignancies, including lymphomas, leukemias, mycosis fungoides, and multiple myelomas (6). However, naturally occurring mutations of Stat3 that lead to its constitutive activation have not been identified (6). It is therefore proposed that aberrant growth factor signaling, which is a frequent event in human cancers, may play an important role in the constitutive activation of Stat3 (6). In addition, Stat3 was proposed to be an oncogene, based on evidence that a constitutively dimerized/active form of Stat3 (Stat3C) transformed immortalized mouse and rat fibroblasts and bestowed upon these cells the ability to grow into tumors in nude mice (7). Abrogation of Stat3 function via various strategies leads to apoptosis or inhibition of cell proliferation in several epithelial cancer cell lines (8, 9). Thus, evidence is accumulating to suggest that Stat3 may play a critical role in proliferation and survival of cancer cells. Despite our current understanding, the exact requirement for Stat3 in the de novo development of tumors has not been directly addressed, and the biological functions of constitutively active Stat3 in cancers remain to be further elucidated.
Recently, we found that diverse classes of skin tumor promoters activated Stat3 in mouse epidermis following topical treatment (10), which suggested a possible role for this transcription factor in the tumor promotion stage of epithelial carcinogenesis. To further explore the possible roles of Stat3 in different stages of epithelial carcinogenesis, we have used mice that are deficient in Stat3 in epidermis generated via the Cre-LoxP system (11). Here, we report that Stat3-deficient mice did not develop any tumors after treatment with the carcinogen 7,12-dimethylbenz[a]anthracene (DMBA) followed by repeated applications of the tumor promoter 12-O-tetradecanoylphorbol-13-acetate (TPA). This result may be attributed, in part, to the observation that keratinocyte stem cells deficient in Stat3 are more sensitive to DMBA-induced apoptosis. In addition, Stat3-deficient mice exhibited a significant reduction in the epidermal proliferative response induced by TPA. Finally, abrogation of Stat3 function using a decoy oligonucleotide inhibited the growth of initiated keratinocytes both in vitro and in vivo and inhibited growth of papilloma cells in vivo. Collectively, these data demonstrate a requirement for Stat3 during the development of skin tumors in vivo. Furthermore, our results identify an important dual role for Stat3 both as an antiapoptotic molecule during tumor initiation and as a critical regulatory switch governing cell cycle progression from G1 to S phase during tumor promotion.
Results
Stat3 deficiency reduces TPA-mediated epidermal hyperproliferation.
During tumor promotion in mouse skin, initiated cells undergo selective clonal expansion to form precancerous lesions (i.e., papillomas) (3). Tumor promoters such as the phorbol ester TPA induce a dramatic proliferative response in mouse epidermis, which is necessary for the clonal expansion of initiated cells (3). To examine the role of Stat3 in tumor promoter–induced epidermal hyperproliferation in vivo, Stat3-deficient mice were treated topically with TPA. Following 4 topical treatments of 6.8 nmol TPA, the epidermis of control mice showed a dramatic hyperplasia (acanthosis) (Figure 1, B and E), while the epidermis of Stat3-deficient mice exhibited a significantly milder acanthosis (Figure 1, D and E). Stat3-deficient mice also showed a significantly reduced epidermal labeling index (LI) as revealed by a reduction (by ∼80%) in the number of BrdU-positive cells following treatment with TPA at a dose of 6.8 nmol (Figure 1F). These data indicate that functional Stat3 protein is necessary for TPA-induced epidermal hyperproliferation.
Figure 1.

Response of Stat3-deficient and control mice to TPA-induced epidermal hyperproliferation. (A–D) Groups of mice (n = 3) were treated topically with 4 applications of TPA and sacrificed 24 hours after the last treatment. BrdU was injected 30 minutes prior to sacrifice. H&E staining of epidermis from (A) untreated control mice, (B) control mice treated with TPA, (C) untreated Stat3-deficient mice, and (D) Stat3-deficient mice treated with TPA. (E) Quantitation of epidermal thickness from control (white bars) and Stat3-deficient (black bars) mice treated with different doses of TPA. (F) Percentage of BrdU-positive epidermal basal cells in control (white bars) and Stat3-deficient (black bars) mice treated with different doses of TPA. Scale bar: 50 μm. **P < 0.01 by Mann-Whitney U test.
Altered cell cycle progression in epidermis of Stat3 deficient mice following TPA treatment.
We next examined the time course for epidermal DNA synthesis in epidermis of Stat3-deficient mice following TPA treatment. In nontransgenic mice, the percentage of epidermal basal cells in S phase reached a peak (LI ∼70%) at about 17 hours following a single topical application of 6.8 nmol TPA (Figure 2A, solid line). In contrast, there was a delayed epidermal proliferative response in Stat3-deficient mice with a significantly reduced peak (LI ∼30%) at about 24 hours after a single TPA treatment (Figure 2A, broken line). To analyze the underlying mechanisms contributing to the altered response of Stat3-deficient mice to TPA-induced DNA synthesis, we examined the levels of several critical regulatory proteins governing G1-to-S-phase cell cycle progression. In recent experiments, we found that topical treatment with diverse skin tumor promoters, including TPA, led to rapid activation of Stat3 (10). Immunohistochemical analyses showed an intense nuclear staining for tyrosine-phosphorylated Stat3 (PYStat3) 4 hours after TPA treatment in both basal and suprabasal keratinocytes (Figure 2B, lower left panel) in nontransgenic mice. In contrast, PYStat3 was not detectable in the epidermal keratinocytes of Stat3-deficient mice and was only detected in the dermal compartment (e.g., in fibroblasts and macrophages) after TPA treatment (Figure 2B, arrows). Western blot analyses confirmed this activation of Stat3 in epidermis of nontransgenic mice treated with TPA (Figure 2C). The Stat3 and PYStat3 protein detected in the epidermal preparations from Stat3-deficient mice by Western blotting (Figure 2C) was likely due to (a) the presence of other cells that reside in the epidermis, including Langerhans cells, and melanocytes; and/or (b) contamination from the dermis.
Figure 2.

Analysis of cell cycle regulatory proteins in Stat3-deficient mice following treatment with TPA. (A–D) Groups of mice (n = 3) were treated with a single application of 6.8 nmol TPA and sacrificed at various times thereafter, as indicated. BrdU was injected 30 minutes prior to sacrifice. (A) Percentage of BrdU-positive epidermal basal cells in control (solid line) and Stat3-deficient (broken line) mice. (B) Immunohistochemical analysis of p-Stat3 localization in skin sections from Stat3-deficient mice (–/–) and control littermates (+/+) with (lower panels) or without (upper panels) TPA treatment. Positive staining for tyrosine-phosphorylated Stat3 in the nuclei of epidermal keratinocytes is shown by brown staining. Arrows, positive signals found in dermal cells such as fibroblasts and macrophages. Scale bar: 50 μm. (C) Western blot analyses of Stat3 and tyrosine-phosphorylated Stat3 protein levels in relation to levels of cyclin D1, cyclin E, and c-Myc at different time points in control and Stat3-deficient mouse epidermis following TPA treatment. (D) Semiquantitative analysis of the mRNA levels of cyclin D1 and c-Myc without treatment and 4 hours after TPA treatment. (E) Western blot analysis of Erk1/2 and phosphorylated Erk1/2 levels at different time points following TPA treatment in epidermis of control and Stat3-deficient mice.
Following TPA treatment, there was an initial drop in the levels of c-Myc, cyclin E, and cyclin D1 proteins (at 4 hours), followed by a recovery of these proteins to control or to slightly above control levels by 10–17 hours (Figure 2C). These data in control mice are similar to those reported by Rodriguez-Puebla et al. (12) and are consistent with the finding that cyclin D1–cdk4 and cyclin E–cdk2 complexes phosphorylate retinoblastoma protein (Rb), thus relieving its repression of E2F transcription factors and leading to activation of critical genes that drive cells from G1 into S phase (13). In epidermis of Stat3-deficient mice, we first noted reduced levels of cyclin D1, cyclin E, and c-Myc relative to those in epidermis from control mice (Figure 2C). Furthermore, following TPA treatment, the recovery of cyclin D1 and cyclin E protein levels was significantly delayed. Notably, c-Myc levels remained significantly reduced throughout the time course examined (Figure 2C). Semiquantitative RT-PCR analysis showed reduced c-Myc and cyclin D1 mRNA levels in epidermis of Stat3-deficient mice and a reduced induction of these mRNAs at 4 hours following TPA treatment (Figure 2D). These data are consistent with other data suggesting that c-Myc and cyclin D1 may be transcriptional targets of Stat3 (7). The delayed recovery of cyclin E in Stat3-deficient mice is most likely a direct effect of the delay in cyclin D1 expression. This conclusion is supported by the observation that knock-in of cyclin E into cyclin D1–deficient mice can rescue its phenotype (14). Collectively, the delayed recovery of cyclin D1 and cyclin E and the persistent downregulation of c-Myc are likely responsible for the delayed entry of Stat3-deficient epidermal cells into S phase following topical TPA treatment.
Next, we examined the impact of Stat3 deficiency on the MAPK pathway with and without tumor promoter treatment. The Erk-MAPK pathway has been implicated in regulating cyclin D1 expression and driving G1-to-S-phase cell cycle progression in murine keratinocytes (15). In Stat3-deficient mice, we found that the activity of Erk-MAPK remained intact in the epidermis, as shown by Western blot analysis of the phosphorylated form of Erk1/2 (Figure 2E). Following TPA treatment, the levels of phospho-Erk1/2 were increased in both control and Stat3-deficient mice at all the time points examined. However, the activation of Erk1/2 was enhanced in the Stat3-deficient mice treated with TPA. These data indicate that the delay in the recovery of cyclin D1 levels seen in Stat3-deficient mice treated with TPA is not due to a defect in the Erk-MAPK signaling pathway. Rather, enhanced Erk1/2 signaling could not compensate for the lack of Stat3 to drive entry from G1 to S phase following TPA treatment. Collectively, these data (Figure 2) suggest that Stat3 likely plays a critical role in the commitment to cell cycle progression in murine keratinocytes following treatment with TPA.
Stat3-deficient keratinocytes are more sensitive to DMBA-induced apoptosis.
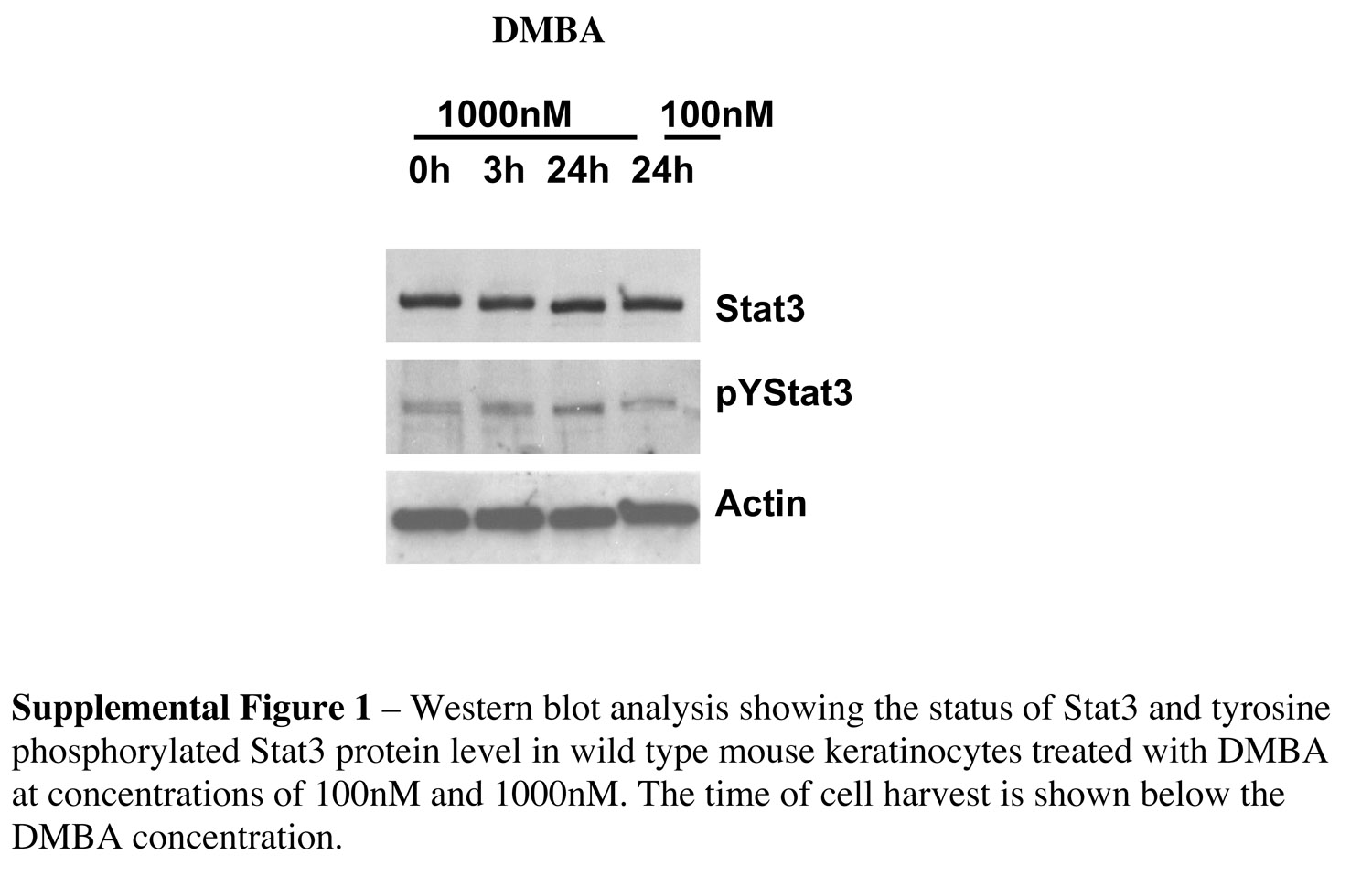
In mouse epidermis, the initiation of skin tumors occurs following treatment with a carcinogen such as DMBA. DMBA is metabolized to reactive diol-epoxides that covalently bind to DNA bases and induce mutations in certain target cells (16). Initiated cells are likely to have escaped the normal mechanisms (DNA repair, apoptosis) that provide protection against carcinogen-induced DNA damage and thus are available for selective clonal expansion during tumor promotion (17). To determine the effect of Stat3 deficiency on tumor initiation, we examined the survival of keratinocytes in response to DMBA treatment. In initial experiments using cultured keratinocytes in the absence of treatment, no significant differences were observed in the viability of Stat3-deficient keratinocytes compared with keratinocytes from nontransgenic littermates. In contrast, DMBA (30 nM) induced apoptosis in a significantly higher population of Stat3-deficient keratinocytes (Figure 3B) as compared with control keratinocytes (Figure 3A). Quantitation of DMBA-induced apoptosis in cultured keratinocytes is shown in Figure 3C. Western blot analyses of wild-type (control) keratinocytes did not reveal any significant changes in Stat3 protein levels or phosphotyrosine levels at any of the DMBA doses used (Supplemental Figure 1: supplemental material available at http://www.jci.org/cgi/content/full/114/5/720/DC1). In further experiments, topical application of DMBA to Stat3-deficient mice resulted in a significantly increased number of epidermal cells undergoing apoptosis compared with that in control mice, analyzed by both caspase-3–positive staining (Figure 3, D and E) and the presence of sunburn cells in H&E-stained sections (data not shown). Examination of the caspase-3–positive cells revealed that the majority of DMBA-induced apoptotic cells were located in a specific region of the hair follicles, with a smaller number appearing in the interfollicular epidermis (Figure 3D, arrows and arrowhead, respectively). The distribution of caspase-3–positive cells in the epidermis of nontransgenic littermates following DMBA treatment was similar to that observed in Stat3-deficient mice, although the total number was lower (Figure 3E).
Figure 3.

Response of Stat3-deficient keratinocytes to DMBA-induced apoptosis both in vitro and in vivo. (A and B) Cultures of primary keratinocytes from (A) control mice and (B) Stat3-deficient mice treated with DMBA. Scale bar: 200 μm. (C) Percentage of apoptotic cells in control (white bars) and Stat3-deficient (black bars) keratinocytes treated with DMBA. *P < 0.05 by Mann-Whitney U test. (D) Caspase-3 staining of epidermis from Stat3-deficient mice. Note that most of the caspase-3–positive cells are located in a restricted region of the hair follicles (arrows), while a smaller number are located in the interfollicular epidermis (arrowhead). IFE, interfollicular epidermis; HF, hair follicle; D, dermis. Scale bar: 100 μm. (E) Number of caspase-3–postive cells per centimeter of epidermis in control (white bars) and Stat3-deficient mice (black bars). *P < 0.05 and **P < 0.01 by Mann-Whitney U test. (F) Double staining of label-retaining cells (BrdU; red) and caspase-3–positive cells (green) in skin of Stat3-deficient mice treated with DMBA (25 nmol) and sacrificed 24 hours later. Scale bar: 20 μm. Dotted lines mark the margins of hair follicles, and “b” indicates the bulge region of hair follicle.
Evidence has accumulated suggesting that keratinocyte stem cells reside primarily in the bulge region of the hair follicle, where they are physically protected by the epidermis (18, 19). To determine whether DMBA-induced apoptotic cells in Stat3-deficient mice were primarily localized in the bulge region, keratinocyte stem cells were pulse-chase labeled in vivo using BrdU as previously described (18) and then the location of these cells and the DMBA-induced apoptotic cells was examined. Staining for BrdU (red) to trace the “label-retaining keratinocytes” showed that they were localized primarily in the bulge region of the hair follicle, as expected (Figure 3F). Furthermore, the DMBA-induced apoptotic cells (caspase-3–positive cells, shown in green) were also located in this region, usually adjacent to label-retaining cells (Figure 3F, “b”). These findings provide evidence that Stat3 may be critical for maintaining the survival of keratinocyte stem cells following DNA damage induced by DMBA at the time of tumor initiation.
Stat3-deficient mice are completely resistant to skin tumor development.
To further assess the role of Stat3 in multistage skin carcinogenesis, Stat3-deficient mice and nontransgenic littermates were treated with DMBA, followed by repetitive application of TPA. After 25 weeks of TPA treatment, Stat3-deficient mice did not develop any skin papillomas (Figure 4, A–C). At this point, over 90% of the nontransgenic littermates developed papillomas, with an average of 10 papillomas per mouse. These results were confirmed in a repeat experiment using a similar number of nontransgenic and Stat3-deficient mice (data not shown). Collectively, these data indicate a critical role for functional Stat3 protein in the development of skin tumors in mouse skin, using the DMBA-TPA, initiation-promotion protocol.
Figure 4.

Responsiveness of Stat3-deficient mice to 2-stage carcinogenesis. (A–C) Groups of mice (n = 15) were treated with 25 nmol DMBA and, starting 2 weeks later, treated with twice-weekly applications of TPA (6.8 nmol) for the duration of the experiment. (A) Percentage of mice with papillomas. (B) Average number of papillomas per mouse. Circles, control mice; triangles, Stat3-deficient mice. (C) Representative photograph of Stat3-deficient mice and tumor-bearing control mice at the end of the experiment shown in A and B.
Stat3 is required for clonal expansion of initiated keratinocytes and maintenance of tumor cell growth.
Further experiments were conducted to evaluate whether abrogation of Stat3 function inhibited growth of initiated keratinocytes and tumor cells harboring an activated Ha-ras gene. Keratinocytes transduced with v-Ha-ras were used as an in vitro model for initiated keratinocytes (20, 21). Treatment of v-Ha-ras keratinocytes with a Stat3 decoy oligonucleotide for 2 days in culture led to a marked reduction (70–90%) in cell number compared with those treated with a nonfunctional mutant control oligonucleotide (Figure 5A). Initial Western blot analysis demonstrated the presence of a significant level of PYStat3 protein in v-Ha-ras keratinocytes (data not shown). Further Western blot analyses demonstrated that Stat3 decoy significantly reduced the protein level of two Stat3 downstream targets in v-Ha-ras–transduced keratinocytes, cyclin D1 (6.7-fold reduction based on densitometry) and Bcl-xL (3.8-fold reduction) (Figure 5B). The mutant Stat3 oligonucleotide had no significant effects on the levels of cyclin D1 or Bcl-xL (Figure 5B). Stat3 protein was also downregulated (2.8-fold) by Stat3 decoy treatment (Figure 5B). A previous report has shown that Stat3 can regulate its own transcription (22). The reduced levels of Stat3, cyclin D1, and Bcl-xL proteins in Stat3 decoy–treated v-Ha-ras keratinocytes is consistent with inhibition of Stat3 function. Furthermore, these results suggested that Stat3 is required for growth of initiated keratinocytes, through its effects on both cell proliferation and survival.
Figure 5.

Effects of a Stat3 decoy oligonucleotide on growth of initiated keratinocytes in vitro and in vivo. (A) Effect of vehicle control (TE), mutant oligonucleotide control, and Stat3 decoy oligonucleotide on growth of v-Ha-ras–transduced keratinocytes 48 hours after treatment. Scale bar: 100 μm. (B) Western blot analysis of Stat3, Bcl-xL, and cyclin D1 levels in v-Ha-ras keratinocytes treated with vehicle control (con), mutant decoy (mut), or Stat3 decoy (decoy) for 48 hours. (C) Effect of mutant oligonucleotide control (left 2 mice) and Stat3 decoy oligonucleotide (right 3 mice) on TPA-induced papilloma formation in TG.AC mice at 6 weeks and 13 weeks after first TPA treatment. Arrows indicate papillomas developing in TG.AC mice that received the mutant oligonucleotide together with TPA. (D) Immunohistochemical stain of PYStat3 in a representative section from a skin papilloma. Scale bar: 25 μm. (E) Immunohistochemical stain of Ki67 in a representative section of a skin papilloma. Scale bar: 25 μm. (F) Effect of mutant oligonucleotide and Stat3 decoy oligonucleotide on growth of primary skin papillomas after 2 weeks of treatment. Arrows, tumors injected with mutant oligonucleotide; arrowhead, tumor injected with Stat3 decoy.
To further substantiate a role for Stat3 in clonal expansion of initiated keratinocytes in vivo, we used TG.AC mice. TG.AC mice are transgenic mice that were generated using a ζ-globin promoter linked to a v-Ha-ras gene (23). These mice are an in vivo model for initiated keratinocytes, since topical TPA treatment promotes the development of papillomas (23). As shown in Figure 5C, treatment (as described in Methods) with Stat3 decoy inhibited the early development of papillomas promoted with TPA (6 weeks, upper panels), while the mutant decoy did not. At 6 weeks, no tumors were detected in skin of decoy-treated TG.AC mice, while papillomas were already present in skin of mice treated with mutant decoy oligonucleotide (Figure 5C, arrows). At 13 weeks of observation (Figure 5C, lower panel), there was a marked inhibition of papilloma development in the decoy-treated mice compared with the mutant decoy–treated mice.
We have recently reported that Stat3 is activated (as assessed by Western blot analysis for PYStat3) in skin papillomas and squamous cell carcinomas that develop during 2-stage carcinogenesis (10). Immunohistochemical analyses of papillomas from control littermates showed an intense PYStat3 staining in the nucleus of both basal and suprabasal cells (Figure 5D), which represent the proliferative compartment for these tumors as demonstrated by intense Ki67 staining (Figure 5E). Thus, Stat3 is activated in the proliferative compartment of these tumors. The functional importance of Stat3 in maintaining growth of papilloma cells was examined by injection of Stat3 decoy into primary tumors. Papilloma cells initiated in mouse skin using DMBA have an Ha-ras mutation in codon 61 (3). As shown in Figure 5F, direct injection of Stat3 decoy into primary skin papillomas led to a significant reduction in the volume compared with that of papillomas injected with the mutant decoy. Table 1 provides a summary of data from 13 papillomas injected with Stat3 decoy and 10 papillomas injected with mutant decoy. While not all papillomas injected with Stat3 decoy responded to this treatment protocol, approximately 50% of the papillomas injected with Stat3 decoy underwent significant reduction in size (>60%) compared with those treated with the mutant decoy. Collectively, the data presented in this section demonstrate that inhibition of Stat3 function inhibits the growth of initiated keratinocytes in mouse skin.
Table 1.
Effects of Stat3 decoy oligonucleotide and mutant oligonucleotide control on skin tumor volume

Discussion
A growing number of studies in epithelial cancer cell lines have revealed that constitutive activation of Stat3 by oncogenic tyrosine kinases (e.g., v-Src, EGFR) is important for maintaining cancer cell proliferation (24–26). Furthermore, Bromberg et al. demonstrated that, in fibroblasts expressing Stat3C, mRNA levels of c-Myc and cyclin D1 were elevated at the transcriptional level, possibly contributing to the transforming potential of Stat3C (7). These earlier in vitro studies suggested a potential oncogenic role for Stat3. Recently, we found that Stat3 is activated in mouse epidermis by diverse types of skin tumor promoters (including TPA) and have provided evidence that this occurs via activation of EGFR (10). In addition, Stat3 was found to be constitutively activated in primary skin papillomas and squamous cell carcinomas generated by the 2-stage, DMBA-TPA carcinogenesis regimen (10). These data suggested a role for Stat3 at both early and later stages in the carcinogenesis process. Here, for the first time to our knowledge, we provide evidence that Stat3 is required for the de novo development of skin tumors and is essential for maintaining the proliferation and survival of skin tumor cells. Stat3 deficiency sensitized keratinocyte stem cells to carcinogen-induced apoptosis and significantly diminished the proliferative capacity of keratinocytes in response to tumor promoter treatment. Abrogation of Stat3 function also led to significant inhibition of growth of initiated keratinocytes and papilloma cells, all of which harbor an activated Ha-ras gene. These data indicate an important role for Stat3 early in the carcinogenesis process during both the initiation and the promotion stages. In addition, the data indicate a role for Stat3 in maintaining proliferation and survival in premalignant lesions (i.e., papillomas) that are induced in this model of epithelial carcinogenesis.
In the current study, we found that Stat3-deficient keratinocytes responded poorly to proliferation induced by the tumor promoter TPA. These data suggest that Stat3 activation may be critical for the transduction of extracellular signals to the nucleus that may induce cell cycle progression in response to external stimuli. In mammalian cells, G1-to-S-phase transition involves the integration of external growth-stimulatory or -inhibitory signals during early G1 phase (27). Once cells pass through the restriction point, they are committed to enter S phase for 1 round of the cell cycle (27). Cell cycle progression is tightly regulated by the coordinated control of cell growth and cell division (28). Accumulation of cell mass via protein synthesis, and the spatial and temporal expression of G1 cyclins, which interact with and are required for the catalytic activities of cyclin-dependent kinases (cdk’s), are essential components governing G1-to-S-phase transition. The cyclin D1–cdk4 catalytic complex is known to be required for phosphorylation of Rb and relief of its repression of E2F transcription factors (27), which subsequently activate critical genes that initiate DNA synthesis and the entry of cells into S phase. The reduction of cyclin D1 mRNA level and the delayed recovery of cyclin D1 protein level in epidermal cells of Stat3-deficient mice suggest a role of cyclin D1 in the reduced response of Stat3-deficient mice to TPA-induced epidermal hyperproliferation. Earlier studies have identified cyclin D1 protein expression during G1 as the direct result of a sustained activation of Erk-MAPK (29, 30). Our results demonstrate an intact Erk-MAPK signaling pathway in the Stat3-deficient mice. In addition, the intact and sustained elevation of Erk-MAPK activity in Stat3-deficient mice treated with TPA suggests that signaling through this pathway could not compensate for the loss of Stat3 in mediating TPA-induced epidermal hyperproliferation. In other studies, Erk-MAPK and Stat3 have been shown to have a reciprocal relationship in mediating proliferation (31). These data lead us to propose that Stat3 signaling may be an early requirement for elevation of cyclin D1 levels during G1 in response to external stimuli.
Cyclin D1–deficient mice have been generated (32) and respond to TPA treatment with a transient delay in epidermal DNA synthesis (M. Rodriguez-Puebla and C. Conti, personal communication), while Stat3-deficient mice exhibited a more persistent downregulation of BrdU incorporation following TPA treatment. These data indicate a more global defect in Stat3-deficient mice. The persistent downregulation of c-Myc protein level in the epidermis of Stat3-deficient mice following TPA treatment may contribute to the more prolonged delay observed in epidermal DNA synthesis. Recently, c-Myc deficiency in fibroblasts has been shown to prolong G1 phase four- to fivefold, and the progression from the restriction point into S phase has also been shown to be drastically delayed (33). These results are consistent with our observations in Stat3-deficient mice treated with TPA. c-Myc has been implicated in regulation of cyclin E–cdk2 catalytic activity through various mechanisms during the G1-to-S-phase transition (34–38), and it has been shown to regulate protein synthesis via transcriptional regulation of translation initiation factors (eIF4E and eIF2α) (39). Collectively, our data demonstrate an essential role of Stat3 activation in tumor promotion, possibly by modulation of G1-to-S-phase transition in response to tumor-promoting stimuli. Further experiments using a Stat3 decoy oligonucleotide indicated that abrogation of Stat3 function inhibited the growth of initiated cells and the growth of papilloma cells, both of which possess Ha-ras mutations. These data strengthen the conclusion that Stat3 plays an important role in clonal expansion of initiated cells during tumor promotion.
A defect in the proliferative response of the Stat3-deficient mice to TPA-induced epidermal hyperproliferation and inhibition of clonal expansion of initiated cells during the tumor promotion stage likely contributed to the resistance of these mice to skin carcinogenesis. However, the complete lack of tumors in Stat3-deficient mice led us to also explore the possible involvement of Stat3 during tumor initiation. In skin, emerging evidence suggests that carcinogens target keratinocyte stem cells during tumor initiation (40), whereby these DNA-damaged cells are converted to latent neoplastic cells (i.e., initiated cells) via mutation in specific genes (e.g., Ha-ras) that are critical in modulating cellular functions (41). Keratinocyte stem cells are proposed to reside in the interfollicular epidermis (42, 43), the germinal matrix of growing hair follicles (42, 43), and the major pool in the bulge region of hair follicles (18, 19, 43) underneath the protective layer of epidermis. These stem cells have been found to possess a slow cell-cycling property and thus to retain BrdU when pulse-chase labeled (43). Interestingly, these “label-retaining” cells were also proposed to be “carcinogen-retaining” cells and are located primarily in the bulge region of the hair follicles (44, 45). Our data demonstrate that keratinocytes deficient in Stat3 are more sensitive to DMBA-induced apoptosis in vitro, and in particular that bulge region keratinocytes deficient in Stat3 are more sensitive to DMBA-induced apoptosis in vivo. These data suggest a critical antiapoptotic role for Stat3 in maintaining the survival of keratinocyte stem cells following carcinogen-induced DNA damage during the initiation stage of mouse skin carcinogenesis. Although we did not detect any significant changes in Stat3 protein or phosphotyrosine levels following exposure of normal keratinocytes to DMBA, it is likely that the normal levels of Stat3-regulated antiapoptotic molecules are compromised in Stat3-deficient keratinocytes. It is known that several antiapoptotic molecules such as Bcl-xL (46), Bcl-2 (47), and Mcl-1 (48) are downstream targets for Stat3. In support of this hypothesis, mice overexpressing Bcl-xL in epidermis were resistant to DMBA-induced apoptosis and developed a greater number of papillomas following initiation with DMBA and promotion with TPA (49). Thus, 1 or more of these Stat3-regulated molecules may be important in keratinocyte stem cell survival following carcinogen-induced DNA damage.
In conclusion, based on our current data, we propose the following model for the role of Stat3 in epithelial carcinogenesis (Figure 6). This model depicts Stat3 as having a role in both the initiation and the promotion stages of carcinogenesis. During tumor initiation, Stat3 functions as an antiapoptotic molecule critical for maintaining the survival of DNA-damaged stem cells. During tumor promotion, Stat3 functions as a critical regulator transducing external signals to coordinate both cell proliferation and survival. Thus, Stat3 is required for epidermal proliferation in response to exogenous skin tumor promoters, and for the clonal expansion of initiated cells that harbor Ha-ras mutations during tumor promotion. Finally, once skin tumors develop and are able to grow autonomously, Stat3 is required for maintenance of this autonomous growth. Our findings have a number of significant clinical implications. First, targeting of Stat3 as an anticancer therapy is clearly an important goal (50); in addition, targeting of Stat3 together with DNA-damaging chemotherapies may specifically sensitize cancer cells to apoptosis. Secondly, our data also suggest that targeting of Stat3 at both the initiation and the promotion stages of the carcinogenic process may be a novel and effective strategy for the prevention of epithelial cancers. Finally, because Stat3 appears to play such a critical role in the response of keratinocytes to carcinogens and tumor promoters, variation in this gene could have significant impact on cancer susceptibility in this and other epithelial tissues.
Figure 6.

Model for the role of Stat3 in multistage skin carcinogenesis. Stat3 is proposed to play a dual role in both the initiation and the promotion stages of carcinogenesis. At the initiation stage, Stat3 is required for keratinocyte stem cell survival following carcinogen-induced DNA damage. At the promotion stage, Stat3 is proposed to mediate cell cycle progression in response to external tumor-promoting signals that leads to clonal expansion of initiated cells. This may be mediated by 1 or more cell cycle regulatory proteins (e.g., cyclin D1, c-Myc) as well as cell survival factors regulated by Stat3 (e.g., Bcl-xL). Finally, Stat3 plays a role in maintaining proliferation in skin papillomas.
Methods
Generation of K5Cre.Stat3fl/fl mice and 2-stage carcinogenesis experiments.
K5Cre transgenic mice and Stat3fl/fl mice were originally on 129/sv × C57BL/6 genetic background and were backcrossed to FVB/N congenic background for at least 5 generations. FVB/N is a sensitive genetic background for 2-stage carcinogenesis experiments (51, 52). Keratinocyte-specific Stat3-deficient mice were generated as previously described (11). For all in vivo experiments, the dorsal skin of each mouse was shaved 2 days prior to treatment; only those mice in the resting phase of the hair cycle were used. For the 2-stage carcinogenesis experiment, 6-week-old mice (n = 15) were treated with a single application of DMBA (25 nmol in 200 μl acetone; Sigma-Aldrich) followed by twice-weekly applications of TPA (6.8 nmol in 200 μl acetone; LC Laboratories). All treatment protocols followed NIH guidelines.
Analysis of epidermal thickness and cell proliferation following treatment with TPA.
Groups of mice (n = 3) were treated with 4 applications of TPA (1.7 nmol, 3.4 nmol, or 6.8 nmol) or the acetone vehicle and sacrificed 24 hours after treatment. For analysis of cell cycle kinetics, groups of mice (n = 3) were treated with 1 application of TPA (6.8 nmol) or the acetone vehicle and sacrificed at various times after treatment. Mice were injected intraperitoneally with BrdU (100 μg/g body weight; Sigma-Aldrich) in PBS 30 minutes prior to sacrifice. Dorsal skin was then fixed in formalin, embedded in paraffin, sectioned at 4 μm, stained with H&E and anti-BrdU antibody (BD Biosciences — Pharmingen), and then treated with biotinylated anti-mouse IgG and HRP-conjugated ABC reagent (Vector Laboratories Inc.). Epidermal cell proliferation (presented as the LI) was determined as follows: a minimum of 500 basal cells from 3 mice were counted, and the number of BrdU-positive cells per 500 basal cells was calculated and multiplied by 100.
Analysis of epidermal apoptosis following treatment with DMBA.
Groups of mice (n = 3) were treated on the dorsal skin with a single application of DMBA (25, 200, or 1,000 nmol) or acetone and sacrificed 24 hours after treatment. Skin sections were stained with an antibody to the active form of caspase-3 (R&D Systems Inc.) and then treated with biotinylated anti-rabbit IgG and HRP-conjugated ABC reagent (BD Biosciences — Pharmingen). Apoptotic keratinocytes were counted microscopically in at least 3 nonoverlapping fields in sections from 3 individual mice.
Analysis of follicular label-retaining cells.
Mice were given 4 rounds of BrdU (0.25 mg in 0.2 ml PBS per mouse) by intraperitoneal injection every 12 hours (total dose of 1.0 mg/mouse) starting on postnatal day 10. Dorsal skin was fixed in formalin and embedded in paraffin. Four-micrometer sections were stained with anti-BrdU antibody in a 1:300 dilution and anti–caspase-3 antibody in a 1:600 dilution and then treated with Cy3-conjugated anti-mouse IgG and FITC-conjugated anti-rabbit IgG (BD Biosciences — Pharmingen), respectively.
Preparation of protein lysates and Western blot analysis.
Mouse skin was placed on an ice-cold glass plate, and the epidermis was removed with a razor blade and placed into RIPA lysis buffer containing 50 mM Tris-HCl (pH 8.6), 1% NP-40, 0.25% Na-deoxycholate, 150 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1 μg/ml leupeptin, 1 μg/ml aprotinin, 1 mM Na3VO4, 1 mM NaF, and 10 μl/ml protease inhibitor cocktail (Sigma-Aldrich). The lysates were incubated on ice for 10 minutes, snap-frozen in liquid nitrogen, rethawed, and then centrifuged at 14,000 g for 15 minutes at 4°C. The supernatant was separated by electrophoresis on 8–12% SDS/polyacrylamide gels. Separated proteins were electrophoretically transferred onto PVDF membranes and blocked with 5% nonfat dry milk in PBS with 0.1% Tween-20 (TPBS) for 1 hour at room temperature. Blots were then incubated for 2 hours at room temperature with specific primary antibodies for Stat3 (C-20; Santa Cruz Biotechnology Inc.) and phospho-Stat3 (Cell Signaling Technology Inc.), cyclin D1 (RB-010-PO; NeoMarkers Inc.), cyclin E (M-20; Santa Cruz Biotechnology Inc.), and actin (I-19; Santa Cruz Biotechnology Inc.). Blots were washed with TPBS and subjected to corresponding HRP-conjugated secondary antibodies: rabbit (NA934V; Amersham Biosciences UK Ltd.), mouse (sc-2005; Santa Cruz Biotechnology Inc.), and goat (sc-2020; Santa Cruz Biotechnology Inc.). Blots were washed with TPBS and detected with the LumiGLO chemiluminescent kit (Cell Signaling Technology Inc.). Where indicated, relative changes in protein levels were determined by densitometry with normalization to β-actin.
Immunohistochemical analysis.
Formalin-fixed, paraffin-embedded tissues were deparaffinized and hydrated using standard procedures. Endogenous peroxidase activity was blocked with 0.03% hydrogen peroxide for 10 minutes. Sections were microwaved (10 minutes) in the presence of 10 mM citrate buffer (pH 6.0) containing 0.01% Tween-20 and then allowed to cool down for 20 minutes. Sections were then stained with anti–phospho-Stat3 at a 1:100 dilution or anti-Ki67 (DakoCytomation) according to procedures suggested by the manufacturer.
Keratinocyte cultures and in vitro treatment with DMBA.
Full-thickness skin taken from newborn mice was treated with 250 U/ml of dispase (Godo Shusei Co.) overnight at 4°C, and the epidermis was peeled off from the dermis and trypsinized to create a single-cell suspension. Cells were plated in MCDB153 medium (Kyokuto Pharmaceutical Co.) supplemented with 0.1 mM monoethanolamine, 0.1 mM phosphoryl ethanolamine, 0.5 μM hydrocortisone at 37°C under an atmosphere of 5% CO2 in dishes precoated with collagen type I (Iwaki Glass Co.) for 5 hours. Unattached cells were removed by washing with PBS, and attached cells were further cultured in the media in the absence of growth factors. Twenty-four hours later, DMBA was added to a final concentration of 30 nM. Twelve hours later, viable or apoptotic cells were counted under a phase-contrast microscope as previously described (53). Briefly, apoptotic keratinocytes detach from the dishes and undergo morphological changes such as cell ballooning, nuclear condensation, and bleb formation, while viable cells remain attached to dish. Cells were counted on photographs from 4 nonoverlapping fields, and the percentage of apoptosis was calculated as apoptotic cells/(apoptotic cells + viable cells) × 100.
In vitro and in vivo Stat3 decoy oligonucleotide treatment.
For in vitro studies, primary murine keratinocytes were infected with retroviral v-Ha-ras as previously described (20, 21). The Stat3 decoy and mutant decoy oligonucleotides were designed as described previously (9). Cells were treated with Stat3 decoy oligonucleotide (25 nM), mutant control oligonucleotide (25 nM), or vehicle control (Tris-HCl/EDTA buffer [TE]), and the corresponding effects were evaluated at 24 and 48 hours after treatment. For in vivo studies, TG.AC mice (23) were treated at 1 week of age with either TPA plus Stat3 decoy (3 mice) or TPA plus mutant decoy (2 mice). For the first 2 weeks of treatment, mice received TPA (17 nmol twice weekly) followed immediately by oligonucleotides in TE buffer (20 nmol/0.02 ml). To facilitate absorption, mice received tape stripping (10 times with Scotch Magic Tape 810, 3M) 5 minutes prior to TPA treatment. In addition, an occlusive dressing was applied to the decoy- or mutant decoy–treated area for 24 hours to further facilitate absorption. Beginning in the third week, mice received twice-weekly treatments with TPA followed immediately by subcutaneous injection (60 nmol/0.02 ml) of oligonucleotides in TE buffer in 3 separate areas of the TPA-treated skin for the next 5 weeks. Stat3 decoy or mutant decoy treatments together with TPA treatment were then discontinued, and the mice were observed for tumor development over an additional 6 weeks (13 weeks total). Finally, primary tumors that were generated from control littermates in the 2-stage carcinogenesis experiment were directly injected with 20 μl of 1 mM decoy oligonucleotide or mutant oligonucleotide control once per week. The effects were evaluated after 2 weeks of oligonucleotide injection. Tumor dimensions were measured with a caliper, and tumor volume was calculated as [(x + y)/2]3, where x and y represent the width and the length of the tumor, respectively.
RNA extraction and RT-PCR.
Total RNA from epidermis was extracted using TRI Reagent (Molecular Research Center Inc.) and reverse-transcribed by GeneAmp RNA PCR Core Kit (Applied Biosystems). Mouse Ha-ras was amplified with primers 5′-GGCGTGGGAAAGAGTG-3′ (sense) and 5′-TGTTGTTGATGGCAAATACA-3′ (antisense); mouse cyclin D1 with primers 5′-TGGAGCCCCTGAAGAAGAGC-3′ (sense) and 5′-TGGAAAGAAAGTGCGTTTGTG-3′(antisense); and mouse c-Myc with primers 5′-TCTTTCCCTACCCGCTCAAC-3′ (sense) and 5′-ACCCTGCCACTGTCCAACTT-3′ (antisense). The optimal annealing temperature was 60°C for all the primers with 35 reaction cycles.
Supplementary Material
Acknowledgments
We thank Joi Holcomb for preparing the graphics and figures, and Vanessa Edwards for her editorial assistance in preparing the manuscript. We also thank the staff of Histology Core and Animal Resources Facility for their excellent help in support of this work. This research was supported by NIH grants RO1 CA76520 (to J. DiGiovanni) and U01 ES11047, University of Texas M.D. Anderson Cancer Center Support Grant CA16672, and National Institute of Environmental Health Sciences Center Grant ES07784.
Footnotes
See the related Commentary beginning on page 619.
Shigetoshi Sano’s permanent address is: Department of Dermatology, Osaka University Graduate School of Medicine, Suita, Osaka, Japan.
Keith Syson Chan and Shigetoshi Sano contributed equally to this work.
Nonstandard abbreviations used: cdk, cyclin-dependent kinase; DMBA, 7,12-dimethylbenz[a]anthracene; LI, labeling index; PYStat3, tyrosine-phosphorylated signal transducer and activator of transcription 3; Rb, retinoblastoma protein; Stat3, signal transducer and activator of transcription 3; TPA, 12-O-tetradecanoylphorbol-13-acetate; TPBS, PBS with 0.1% Tween-20.
Conflict of interest: The authors have declared that no conflict of interest exists.
References
- 1.Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat. Rev. Cancer. 2002;2:331–341. doi: 10.1038/nrc795. [DOI] [PubMed] [Google Scholar]
- 2.Weinberg RA. Oncogenes, antioncogenes, and the molecular bases of multistep carcinogenesis. Cancer Res. 1989;49:3713–3721. [PubMed] [Google Scholar]
- 3.DiGiovanni J. Multistage carcinogenesis in mouse skin. Pharmacol. Ther. 1992;54:63–128. doi: 10.1016/0163-7258(92)90051-z. [DOI] [PubMed] [Google Scholar]
- 4.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 5.Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- 6.Bromberg J. Stat proteins and oncogenesis. J. Clin. Invest. 2002;109:1139–1142. doi:10.1172/JCI200215617. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bromberg JF, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- 8.Mora LB, et al. Constitutive activation of Stat3 in human prostate tumors and cell lines: direct inhibition of Stat3 signaling induces apoptosis of prostate cancer cells. Cancer Res. 2002;62:6659–6666. [PubMed] [Google Scholar]
- 9.Leong PL, et al. Targeted inhibition of Stat3 with a decoy oligonucleotide abrogates head and neck cancer cell growth. Proc. Natl. Acad. Sci. U. S. A. 2003;100:4138–4143. doi: 10.1073/pnas.0534764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan KS, et al. Epidermal growth factor receptor-mediated activation of Stat3 during multistage skin carcinogenesis. Cancer Res. 2004;64:2382–2389. doi: 10.1158/0008-5472.can-03-3197. [DOI] [PubMed] [Google Scholar]
- 11.Sano S, et al. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J. 1999;18:4657–4668. doi: 10.1093/emboj/18.17.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rodriguez-Puebla ML, Robles AI, Johnson DG, LaCava M, Conti CJ. Synchronized proliferation induced by 12-O-tetradecanoylphorbol-13-acetate treatment of mouse skin: an in vivo model for cell cycle regulation. Cell Growth Differ. 1998;9:31–39. [PubMed] [Google Scholar]
- 13.Hatakeyama M, Weinberg RA. The role of RB in cell cycle control. Prog. Cell Cycle Res. 1995;1:9–19. doi: 10.1007/978-1-4615-1809-9_2. [DOI] [PubMed] [Google Scholar]
- 14.Geng Y, et al. Rescue of cyclin D1 deficiency by knockin cyclin E. Cell. 1999;97:767–777. doi: 10.1016/s0092-8674(00)80788-6. [DOI] [PubMed] [Google Scholar]
- 15.Robles AI, et al. Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev. 1998;12:2469–2474. doi: 10.1101/gad.12.16.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Slaga TJ, Gleason GL, DiGiovanni J, Sukumaran KB, Harvey RG. Potent tumor-initiating activity of the 3,4-dihydrodiol of 7,12-dimethylbenz(a)anthracene in mouse skin. Cancer Res. 1979;39:1934–1936. [PubMed] [Google Scholar]
- 17.Perez-Losada J, Balmain A. Stem-cell hierarchy in skin cancer. Nat. Rev. Cancer. 2003;3:434–443. doi: 10.1038/nrc1095. [DOI] [PubMed] [Google Scholar]
- 18.Cotsarelis G, Sun TT, Lavker RM. Label-retaining cells reside in the bulge area of pilosebaceous unit: implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell. 1990;61:1329–1337. doi: 10.1016/0092-8674(90)90696-c. [DOI] [PubMed] [Google Scholar]
- 19.Taylor G, Lehrer MS, Jensen PJ, Sun TT, Lavker RM. Involvement of follicular stem cells in forming not only the follicle but also the epidermis. Cell. 2000;102:451–461. doi: 10.1016/s0092-8674(00)00050-7. [DOI] [PubMed] [Google Scholar]
- 20.Dlugosz AA, et al. Autocrine transforming growth factor alpha is dispensible for v-rasHa-induced epidermal neoplasia: potential involvement of alternate epidermal growth factor receptor ligands. Cancer Res. 1995;55:1883–1893. [PubMed] [Google Scholar]
- 21.Dlugosz AA, et al. Targeted disruption of the epidermal growth factor receptor impairs growth of squamous papillomas expressing the v-ras(Ha) oncogene but does not block in vitro keratinocyte responses to oncogenic ras. Cancer Res. 1997;57:3180–3188. [PubMed] [Google Scholar]
- 22.Ichiba M, Nakajima K, Yamanaka Y, Kiuchi N, Hirano T. Autoregulation of the Stat3 gene through cooperation with a cAMP-responsive element-binding protein. J. Biol. Chem. 1998;273:6132–6138. doi: 10.1074/jbc.273.11.6132. [DOI] [PubMed] [Google Scholar]
- 23.Spalding JW, Momma J, Elwell MR, Tennant RW. Chemically induced skin carcinogenesis in a transgenic mouse line (TG.AC) carrying a v-Ha-ras gene. Carcinogenesis. 1993;14:1335–1341. doi: 10.1093/carcin/14.7.1335. [DOI] [PubMed] [Google Scholar]
- 24.Grandis JR, et al. Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor–mediated cell growth in vitro. J. Clin. Invest. 1998;102:1385–1392. doi: 10.1172/JCI3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xi S, et al. Src kinases mediate STAT growth pathways in squamous cell carcinoma of the head and neck. J. Biol. Chem. 2003;278:31574–31583. doi: 10.1074/jbc.M303499200. [DOI] [PubMed] [Google Scholar]
- 26.Garcia R, et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene. 2001;20:2499–2513. doi: 10.1038/sj.onc.1204349. [DOI] [PubMed] [Google Scholar]
- 27.Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat. Rev. Cancer. 2001;1:222–231. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- 28.Neufeld TP, Edgar BA. Connections between growth and the cell cycle. Curr. Opin. Cell Biol. 1998;10:784–790. doi: 10.1016/s0955-0674(98)80122-1. [DOI] [PubMed] [Google Scholar]
- 29.Albanese C, et al. Transforming p21ras mutants and c-Ets-2 activate the cyclin D1 promoter through distinguishable regions. J. Biol. Chem. 1995;270:23589–23597. doi: 10.1074/jbc.270.40.23589. [DOI] [PubMed] [Google Scholar]
- 30.Lavoie JN, L’Allemain G, Brunet A, Muller R, Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 1996;271:20608–20616. doi: 10.1074/jbc.271.34.20608. [DOI] [PubMed] [Google Scholar]
- 31.Ernst M, et al. Defective gp130-mediated signal transducer and activator of transcription (STAT) signaling results in degenerative joint disease, gastrointestinal ulceration, and failure of uterine implantation. J. Exp. Med. 2001;194:189–203. doi: 10.1084/jem.194.2.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fantl V, Stamp G, Andrews A, Rosewell I, Dickson C. Mice lacking cyclin D1 are small and show defects in eye and mammary gland development. Genes Dev. 1995;9:2364–2372. doi: 10.1101/gad.9.19.2364. [DOI] [PubMed] [Google Scholar]
- 33.Schorl C, Sedivy JM. Loss of protooncogene c-Myc function impedes G1 phase progression both before and after the restriction point. Mol. Biol. Cell. 2003;14:823–835. doi: 10.1091/mbc.E02-10-0649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steiner P, et al. Identification of a Myc-dependent step during the formation of active G1 cyclin-cdk complexes. EMBO J. 1995;14:4814–4826. doi: 10.1002/j.1460-2075.1995.tb00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berns K, Hijmans EM, Bernards R. Repression of c-Myc responsive genes in cycling cells causes G1 arrest through reduction of cyclin E/CDK2 kinase activity. Oncogene. 1997;15:1347–1356. doi: 10.1038/sj.onc.1201280. [DOI] [PubMed] [Google Scholar]
- 36.Bouchard C, et al. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 1999;18:5321–5333. doi: 10.1093/emboj/18.19.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perez-Roger I, Kim SH, Griffiths B, Sewing A, Land H. Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1) EMBO J. 1999;18:5310–5320. doi: 10.1093/emboj/18.19.5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hermeking H, et al. Identification of CDK4 as a target of c-MYC. Proc. Natl. Acad. Sci. U. S. A. 2000;97:2229–2234. doi: 10.1073/pnas.050586197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rosenwald IB, Rhoads DB, Callanan LD, Isselbacher KJ, Schmidt EV. Increased expression of eukaryotic translation initiation factors eIF-4E and eIF-2 alpha in response to growth induction by c-myc. Proc. Natl. Acad. Sci. U. S. A. 1993;90:6175–6178. doi: 10.1073/pnas.90.13.6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morris RJ. Keratinocyte stem cells: targets for cutaneous carcinogens. J. Clin. Invest. 2000;106:3–8. doi: 10.1172/JCI10508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quintanilla M, Brown K, Ramsden M, Balmain A. Carcinogen-specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature. 1986;322:78–80. doi: 10.1038/322078a0. [DOI] [PubMed] [Google Scholar]
- 42.Ghazizadeh S, Taichman LB. Multiple classes of stem cells in cutaneous epithelium: a lineage analysis of adult mouse skin. EMBO J. 2001;20:1215–1222. doi: 10.1093/emboj/20.6.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Potten CS, Booth C. Keratinocyte stem cells: a commentary. J. Invest. Dermatol. 2002;119:888–899. doi: 10.1046/j.1523-1747.2002.00020.x. [DOI] [PubMed] [Google Scholar]
- 44.Morris RJ, Fischer SM, Slaga TJ. Evidence that a slowly cycling subpopulation of adult murine epidermal cells retains carcinogen. Cancer Res. 1986;46:3061–3066. [PubMed] [Google Scholar]
- 45.Morris RJ, Tryson KA, Wu KQ. Evidence that the epidermal targets of carcinogen action are found in the interfollicular epidermis of infundibulum as well as in the hair follicles. Cancer Res. 2000;60:226–229. [PubMed] [Google Scholar]
- 46.Grad JM, Zeng XR, Boise LH. Regulation of Bcl-xL: a little bit of this and a little bit of STAT. Curr. Opin. Oncol. 2000;12:543–549. doi: 10.1097/00001622-200011000-00006. [DOI] [PubMed] [Google Scholar]
- 47.Nielsen M, et al. Inhibition of constitutively activated Stat3 correlates with altered Bcl-2/Bax expression and induction of apoptosis in mycosis fungoides tumor cells. Leukemia. 1999;13:735–738. doi: 10.1038/sj.leu.2401415. [DOI] [PubMed] [Google Scholar]
- 48.Epling-Burnette PK, et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J. Clin. Invest. 2001;107:351–362. doi: 10.1172/JCI9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pena JC, Rudin CM, Thompson CB. A Bcl-xL transgene promotes malignant conversion of chemically initiated skin papillomas. Cancer Res. 1998;58:2111–2116. [PubMed] [Google Scholar]
- 50.Darnell JE., Jr Transcription factors as targets for cancer therapy. Nat. Rev. Cancer. 2002;2:740–749. doi: 10.1038/nrc906. [DOI] [PubMed] [Google Scholar]
- 51.Hennings H, et al. FVB/N mice: an inbred strain sensitive to the chemical induction of squamous cell carcinomas in the skin. Carcinogenesis. 1993;14:2353–2358. doi: 10.1093/carcin/14.11.2353. [DOI] [PubMed] [Google Scholar]
- 52.Matsumoto T, et al. Targeted expression of c-Src in epidermal basal cells leads to enhanced skin tumor promotion, malignant progression, and metastasis. Cancer Res. 2003;63:4819–4828. [PubMed] [Google Scholar]
- 53.Umeda J, et al. In vivo cooperation between Bcl-xL and the phosphoinositide 3-kinase-Akt signaling pathway for the protection of epidermal keratinocytes from apoptosis. FASEB J. 2003;17:610–620. doi: 10.1096/fj.02-0597com. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}