

Abstract
Atomic force microscopy is a powerful and widely used imaging technique that can visualize single molecules and follow processes at the single-molecule level both in air and in solution. For maximum usefulness in biological applications, atomic force microscopy needs to be able to identify specific types of molecules in an image, much as fluorescent tags do for optical microscopy. The results presented here demonstrate that the highly specific antibody–antigen interaction can be used to generate single-molecule maps of specific types of molecules in a compositionally complex sample while simultaneously carrying out high-resolution topographic imaging. Because it can identify specific components, the technique can be used to map composition over an image and to detect compositional changes occurring during a process.
Atomic force microscopy (AFM) is unique in its ability to image single biomolecules and follow biomolecular processes in fluid with nanometer resolution (1); however, images of complex samples can be remarkably hard to interpret, because AFM yields only the shape and volume of the molecule, with no discrimination for the precise types of molecules being imaged. For example, in a sample of chromatin (nucleosomes) plus other proteins, DNA can be identified by its thread-like appearance, but the various protein components look similar, with an image size that depends only marginally on molecular weight. Techniques such as chemical force microscopy (2), force-volume mapping (3), and force curves (4) give information about the specific nature of the molecules being imaged, but they lack the important visual component provided by simultaneous imaging. Here we describe a technique that allows recognition of a specific type of molecule (histone H3) in a complex sample (chromatin) while simultaneously yielding high-resolution topographic images of the same sample. Recognition is efficient, reproducible, and specific. This technique extends the capability of AFM in much the same way as fluorescent tags have extended optical microscopy.
The technique uses an antibody tethered to the AFM tip and depends on the highly specific antibody–antigen recognition reaction between the tip-tethered antibody and its antigen in the sample to identify a specific type of molecule. Antibodies tethered to an AFM tip have been shown previously to bind to specific target molecules during scanning (5), but that work offered no way to separate composition-sensitive signals from topography signals. This difficulty arises because it is difficult to extract a signature of binding while the imaging servo acts to keep the amplitude of oscillation of the probe constant during a scan. The method described here detects antibody–antigen binding through small changes in the absolute (dc) level of the cantilever-deflection signal.
Materials and Methods
Preparation of Chromatin Samples. Nucleosomal arrays containing the mouse mammary tumor virus (MMTV) promoter region were salt-reconstituted to various subsaturated (for clarity in image analysis) levels of nucleosome occupation with HeLa histones exactly as described (6). The arrays were deposited on glutaraldehyde aminopropyltriethoxysilane (GD-APTES)-treated mica, derivatized at 1 μM levels with GD (7), and allowed to adsorb for ≈40 min.
Human (h)Swi-Snf was prepared as described (8). The preparation contains BSA in a 4:1 molar ratio with hSwi-Snf (further reduction in BSA concentration diminishes remodeling activity). For remodeling studies, nucleosomal arrays were preincubated with hSwi-Snf at stoichiometries of 15 nucleosomal array molecules per hSwi-Snf molecule (8). After deposition, fields are scanned twice. The second scan assesses the effect of the AFM-scanning process on chromatin structure and thus provides the background (tip-induced) level of change. Thus, this important control is carried out on the same samples that will be analyzed for remodeling. After activation of hSwi-Snf by ATP addition, the same fields (and the same set of tethered molecules) are scanned again to determine the changes induced by hSwi-Snf remodeling (8).
Tethering of Antibodies to AFM Tips. Polyclonal anti-histone H3 antibodies (Upstate Company, Charlottesville, VA) were thiolated and attached to a polyethylene glycol (PEG) tether on the end of an AFM probe as described (4). Amination of the probe was carried out by exposing a UV-cleaned silicon-nitride probe (Microlever, Veeco, Santa Barbara, CA, coated for MacMode AFM by Molecular Imaging, Tempe, AZ) to aminopropyltriethoxysilane vapor for 1 h.
The H3 antibody was produced against the whole histone, but the N-terminal tails of H3, which project out from the compact nucleosome core, are likely to provide the major antigenic determinant in the chromatin sample. Tips were selected for a strong recognition signal when imaging chromatin, and these signals were invariably blocked by the peptide corresponding to the histone tethered to the tip. For more details, see Protocol for Modifying AFM Tips with Antibodies, which is published as supporting information on the PNAS web site.
AFM Imaging and Separation of the Recognition Signal. Magnetized cantilevers are driven by a small solenoid using a MacMode dynamic-force microscope from Molecular Imaging. Images were taken in 10 mM NaCl/5 mM phosphate buffer, pH 7.5, with 3-nm peak-to-peak amplitude oscillation at 8 kHz, imaging at 70% set point, and scanning at 1 Hz. Recognition contrast decreased with increasing salt, disappearing at ≈0.8 M for this antibody.
The recognition signal was obtained by passing the raw deflection signal from the AFM-scanning head to a PicoTREC signal-processing system (Molecular Imaging). The instrument extracts rapid changes in the value of the positive-going peak signal, filtering the output with a bandpass of ≈150 Hz, while compensating for slow changes in dc level in the input caused by instrumental drift. The signal is passed to an auxiliary input to the microscope controller for simultaneous display alongside the topographical image. The microscope servo operates from the deflection signal in the normal way, which introduces a negligible error (on the order of ΔA or 0.06 nm in the present case) into the height display when an antibody binds.
Filtering and Signal to Noise. In the limit of heavy damping, where the mechanical Q factor of the cantilever approaches unity, the rms fluctuation amplitude 〈x〉 of a cantilever of resonant frequency ν0 measured in a bandwidth B Hz is given by (9)
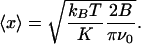 |
Thus, a signal-to-noise ratio of 3:1 [with a 0.06-nm signal (see Results and Discussion)] requires B ≈ 0.016 ν0, which is 130 Hz for the cantilevers used in this work.
Testing Recognition Specificity and Efficiency. To check recognition specificity, the solution in the sample cell was completely replaced with a solution of the imaging buffer plus 30 μg/ml of BSA (kept at the same temperature as the microscope to avoid drift), and the sample was reimaged. This buffer was then replaced with a solution of 30 μg/ml of an antigen (to the antibody on the tip), and the sample was imaged again. Recognition efficiency is defined conservatively as the ratio of total recognition events to total features in the topographic image that may be nucleosomes. False hits (see Fig. 4, red dot and circle) occur in ≈2% of all recognition features. The efficiency depends on the antibody used and imaging conditions such as salt concentration, and thus each new antibody must be calibrated in the imaging environment. The effective antibody concentration at the surface is high (5), and thus the technique is sensitive to even small amounts of crossreactivity.
Fig. 4.

The efficiency and repeatability of recognition imaging. Spots from a recognition image are superimposed (as green dots) on its corresponding topographic image. Features present in the topographic image but not in the recognition image are marked by blue dots. A recognition feature with no corresponding topographic feature (near middle of the image) is marked by a red spot. Changes in recognition events after rescanning are indicated by the circles color-coded with the same scheme (no circle equals no change after rescanning). For example, a feature recognized in the first scan but not in the second is shown by a green dot (recognition in the first scan) surrounded by a blue circle (not recognized on the second scan). The recognition efficiency generally remains >90%.
Results and Discussion
The Technique of Recognition Imaging. The method described in this report is based on the detection of small shifts in the absolute dc level of the cantilever-deflection signal that occur when an antibody tethered to the AFM tip (Fig. 1a) binds to its antigen in the sample being imaged (Fig. 1b), thus interfering with the normal oscillation of the AFM tip (Fig. 1c). All of the imaging in this work is carried out in solution, i.e., in situ. The antibody is attached near the apex of the tip by means of a 6-nm-long flexible PEG linker (10). This linker leaves the antibody free enough to find and bind properly to its target antigen, but it is short enough to permit accurate localization of the antigen site in the sample (5).
Fig. 1.

Recognition imaging. When an AFM tip-tethered antibody (blue blob in a) binds to its antigen in the sample being scanned (b), there is a transient reduction in the oscillation amplitude of the tip (green curve to red curve in c). The imaging servo restores the signal amplitude but with the peak signal shifted downward by an amount ΔA (blue curve in c). This peak shift provides the recognition signal for a specific antigen–antibody recognition event. A topographic image of MMTV arrays and the corresponding recognition image (a “map” of the change in peak signal, ΔA) are shown in d and e, respectively. These images were obtained simultaneously from one scan of the sample by using an H3-specific antibody tethered to the AFM tip. A plot of the peak signal, ΔA, for the portion of the recognition image between the green arrows (e) is shown in f. The dips in signal correspond well with the location of nucleosomes, and the changes in peak signal (traced in f) are in quantitative agreement with theory (see text). The x–y scale is shown in d. The highest features in d are ≈5 nm.
The origin of a recognition signal can be understood by considering the sequence of events that occurs when the oscillating AFM cantilever containing the tip-tethered antibody binds to its antigen in the sample. The green trace in Fig. 1c shows the displacement pattern of the cantilever as it taps on a surface submerged in solution (no antigen–antibody interaction). The cantilever motion is heavily damped and is sustained with a sinusoidal magnetic driving force (11). The bottom of the waveform is cut off where the tip hits the hard surface, but the top part of the waveform remains sinusoidal (12). When the tethered antibody binds to an antigen on the surface, the upward extent of the cantilever swing is restricted by the PEG tether, as shown in the red trace. This reduction of the oscillation amplitude on binding is compensated for by the microscope servo, which is engaged (and required) for normal topographic imaging. The servo pulls the probe away from the surface to restore the amplitude to its previous value (blue trace), but now the peak signal is displaced downward by the amount of the original amplitude reduction, ΔA (Fig. 1c). Thus, a map of the absolute value (i.e., dc-coupled) of this peak voltage, produced simultaneously with the topographic image, shows the location of the antibody-binding events as dark patches.
In principle, the downward-motion displacement of the sample by the microscope servo could cause further reduction of the upper peak amplitude, but the effect is very small because the PEG tether is so much less stiff than the surface.
The amplitude of tip oscillation is adjusted such that the antibody remains bound for most of the time that the tip is in the vicinity of the antigen. Binding and unbinding events occur on each scan line, giving rise to some fluctuation in image width from line to line.
The samples analyzed in this application of the technique are nucleosomal arrays made by reconstituting human histones on a specific DNA fragment containing the MMTV long terminal repeat promoter (6). Samples are deposited onto GD-functionalized mica (GD-APTES) in a flow cell linked to the AFM. The GD-APTES surface tethers the arrays to the surface, allowing repetitive scanning of the same sets of molecules as various reagents are flowed into the cell. The ability to reimage the same molecules allows the clearest possible comparisons of the effects of changing conditions on the sample molecules. Fig. 1d shows a topographic image (two nucleosomal arrays) taken with an AFM tip tethered to an antibody raised against histone H3, one of the proteins that constitute the core of each nucleosome. The corresponding recognition image is shown in Fig. 1e. The dark recognition patches and the pattern in which they occur clearly coincide with the positions of the nucleosomes in the topographic image (Fig. 1d). Fig. 1f is a plot of the recognition signal (ΔA/A) versus distance between the two green arrows in Fig. 1e. The dips coincide with recognition spots. They are a few percent of the oscillation amplitude, in agreement with calculations (see below).
The Basis of Recognition Imaging. The imaging mechanism can be understood quantitatively as follows. In the absence of structural relaxation, the reduction of the “free” oscillation amplitude, A0, to a new value, Az, after binding of the antibody is given by (13)
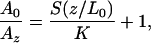 |
[1] |
where S(z/L0) is the stiffness of the PEG tether at a fractional extension, z/L0, of its maximum extended length, L0, and K is the spring constant of the AFM cantilever. An approximate result for the force generated by stretching a worm-like chain is (14)
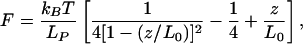 |
[2] |
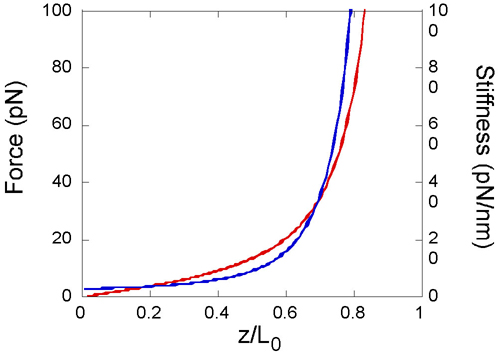
where Lp is the persistence length, and kBT can be conveniently taken as 4.1 pN·nm at room temperature. This formula may be differentiated with respect to z to obtain the stiffness, S(z/L0). For the PEG linker used here, Lp = 0.38 nm and L0 ≈ 6 nm (15). A plot of the force generated by stretching a molecule, and its consequent stiffness as a function of z/L0, is given in Fig. 6, which is published as supporting information on the PNAS web site. The force rises rapidly above 20 pN for z/L0 > 0.5, leading to potentially increased frequency of breaking of a typical antibody–antigen bond (16). Near this optimum value of z/L0, S(z/L0) is ≈2pN/nm. Thus, with a cantilever of stiffness K = 100 pN/nm, Eq. 1 predicts that the recognition events will cause a dip in peak amplitude of a few percent of the total amplitude, which is confirmed experimentally by the line trace across the recognition image shown in Fig. 1f. In this case, the oscillation amplitude is ≈3 nm (corresponding to z/L0 ≈ 0.5), and so the absolute value of the recognition signal is on the order of 0.06 nm. This value is smaller than broad-band thermal fluctuations of the cantilever, and thus the recognition signal requires filtering (see Materials and Methods).
The Specificity and Efficiency of Recognition Imaging. The specificity of the recognition process was tested by flowing exogenous proteins into the liquid flow cell in which the sample was being imaged. If the recognition signal is specific, only (added) proteins that are antigenic to the antibody on the AFM tip used for imaging will be able to bind to the tip, block the recognition events, and thus disrupt the signal. Such a test is shown in Fig. 2. A typical topographic image of MMTV chromatin and the accompanying recognition image (taken with an anti-histone H3 functionalized tip) are shown in Fig. 2 a and b. Again, recognition spots closely mirror the locations of nucleosomes in the topographic image. When a BSA solution (30 μg/ml) is flowed into the sample flow cell, the recognition image is unaffected (Fig. 2c). However, if a similar concentration of a peptide corresponding to amino acids 1–20 of the N-terminal tail of histone H3 (sequence shown in the Fig. 2 legend) is then flowed into this sample, the recognition signal is completely abolished (Fig. 2d). Thus, the ability of the tip-tethered antibody to produce a recognition signal (Fig. 2b) is blocked by an excess of its specific antigen in solution, demonstrating that the recognition signals come from specific interactions between the antibody on the tip and its antigen on the surface. The inability of BSA to alter the recognition signal (Fig. 2c) shows that nonspecific protein-binding effects do not affect specific recognition. Note that the images in Fig. 2 a–d are obtained by sequential rescanning of the same individual molecules in the same sample, which ensures that trivial, sample-dependent variations cannot affect the analysis. Fig. 2 e and f show another aspect of imaging specificity. When BSA is added (through the flow cell) to these samples, aggregates can accrue on the surface (Fig. 2e, yellow arrowheads on the topographic image). In Fig. 2f, the recognition image obtained from this field before BSA addition is shown as green dots, which are superimposed on the recognition signal obtained after BSA was adsorbed onto the surface. Clearly, the H3 antibody-tethered tip does not recognize the BSA aggregates, nor do the BSA aggregates interfere with recognition imaging. Thus, the level of nonspecific recognition appears to be minimal.
Fig. 2.

The specificity of recognition imaging. Topographic (a) and recognition (b) images of MMTV nucleosomal arrays similar to those shown in Fig. 1 are shown. Recognition images from the same region of the sample taken after addition of BSA into the sample in the flow cell (c) and then after the addition of ARTKQTARKSTGGKAPRKQLC (which corresponds to the N-terminal tail of histone H3) (d) are also shown. (e) Topographic image of a field with BSA aggregates pointed to by yellow arrowheads. (f) Recognition images of this same field after BSA was added. The recognition image obtained before BSA addition is superimposed on the recognition signal after BSA addition as green dots. The coincidence of the green dots and dark spots demonstrates that the added BSA did not interfere with the H3 recognition.
To observe the types of specific binding effects discussed above, it must be the case that nonspecific adhesion forces are smaller than the specific antibody–antigen binding force. To test this theory directly, we have measured the adhesion by using force-distance curves (Fig. 3). We find that an antibody-tethered tip in the presence of blocking antigen shows almost no adhesion to the molecules or surfaces used in these experiments. The average adhesion force is 57 ± 20 pN in the absence of the tail peptide but ≈6 pN in its presence. This lower average force reflects both a decrease in the value of adhesion and an increased number of curves in which no adhesion was measured at all. Adhesion forces without the antibody (PEG tether alone) are 116 ± 63 or 118 ± 134 pN for a clean (bare) tip. Thus, the blocked antibody is more of a “nonstick” surface than the tip or PEG alone. Perhaps this is a special property of blocked antibodies, because proteins are generally rather sticky.
Fig. 3.

Force curves for antibody-tethered tips. Histograms of the measured adhesion forces for an antibody-tethered tip probing a field of MMTV nucleosomal array molecules (a) and the same tip scanning in the presence of 30 μg/ml of a peptide from the N-terminal tail of histone H3, showing how the blocked antibody does not adhere to the sample surface (b). Two hundred force curves were analyzed for each histogram (bars at the origins are the numbers of force curves in which no adhesion force was measured). (Insets) Typical examples of force-versus-distance curves obtained. The curve in a shows the characteristic single-molecule binding curve characteristic of PEG stretching, whereas the curve in b shows negligible adhesion. The average adhesion forces are 57 ± 20 pN (absence of H3 tail peptide), 6 pN (presence of tail peptide), 116 ± 63 pN (PEG tether alone), and 118 ± 134 pN for the bare (clean) tip. Thus, the tethered antibody plays an important role in reducing nonspecific adhesion.
The efficiency and repeatability of recognition imaging are illustrated in Fig. 4. Here, features in the recognition image are superimposed (as green dots) on the topography image. Most features that look like nucleosomes in the topographic image are associated with a recognition signal. A few exceptions are described in the Fig. 4 legend (see also Materials and Methods). Some changes occur when the same sample is rescanned (highlighted by circles), but the recognition efficiency is generally high and remains so on subsequent rescans (data not shown). An average over several images yields 96 ± 2% recognition efficiency on the first scan and 92 ± 2% after scanning over the same area again.
We also used tips tethered with antibodies to histone H4 or histone H2A to image these nucleosomal array samples (data not shown). Whereas the H3 antibody was raised against the whole protein, the H4 antibody was raised against the N-terminal tail, and the H2A antibody was raised against part of the “acidic patch” on the 11-nm nucleosomal face (17). All antibodies gave similar results with respect to specificity, efficiency of recognition, and reproducibility, indicating a general applicability of the technique.
Following Compositional Changes During a Biologically Relevant Process. The major impact of this technique will derive from its ability to monitor specific components in heterogeneous samples while they are undergoing biological processes and at the same time as topographic images are being acquired. An example of this application is presented in Fig. 5 and is taken from AFM studies of the action of the human Swi-Snf (hSwi-Snf) ATP-dependent nucleosome remodeling complex (18) on MMTV promoter nucleosomal arrays (8). These studies can help us understand how nucleosome remodeling by these types of complexes might take place in vivo on this and other promoters.
Fig. 5.

Using recognition imaging to study a complex, biologically relevant process. Some examples of the changes seen after hSwi-Snf activation (by ATP) in deposited MMTV nucleosomal arrays are shown. a and b Upper shows topographic images taken before (Left) and after (Right) ATP addition. a and b Lower shows corresponding recognition images obtained with an anti-H3 tip. Protein loss on ATP addition (yellow arrows in the –ATP images) is accompanied by loss of the recognition signal showing that the lost features were likely not hSwi-Snf. Molecules labeled 2 and 3 in a appear unchanged in the topographic image but show loss of recognition signal after ATP addition. (The scale shown at the bottom applies to all the images.)
In these experiments, hSwi-Snf and chromatin are incubated together and then deposited onto GD-APTES in the flow cell linked to the AFM. As described previously, the GD-APTES surface tethers the array molecules, allowing in situ images of the same nucleosomal arrays to be taken before ATP is added (hSwi-Snf is inactive) and after ATP addition (hSwi-Snf is activated). The progress of remodeling, therefore, can be followed on individual molecules. Topographic imaging studies using this approach have shown that array molecules can undergo a variety of substantial ATP/hSwi-Snf-dependent conformational changes and have suggested that hSwi-Snf might also trigger compositional changes such as histone loss (8).
Comparison of recognition images before and after ATP addition shows a variety of remodeling changes and confirms the occurrence of compositional changes. A few examples are shown to illustrate the technique. In Fig. 5a, the three molecules in the field all show remodeling, as evidenced by release of DNA after ATP addition (8). Recognition images (using H3 antibody-tethered tips) indicate a substantial loss of histone (H3) for all three molecules, including a complete loss of histone recognition for molecule one, and new arrangements of the remaining recognition signal pattern in molecules two and three. Fig. 5b shows an example of complete loss of a protein (pointed to by the yellow arrow in the –ATP image) accompanied by a significant change in the H3 recognition images, suggesting that the protein was a nucleosome and not hSwi-Snf. hSwi-Snf (data not shown) and BSA (see above) are not recognized by the anti-histone antibodies. These various types of changes have been quantified and systematically analyzed (8), but these examples demonstrate the significant enhancement of information obtained when recognition is coupled to topographic imaging.
In conclusion, the addition of recognition imaging gives the atomic force microscope a capability for the identification of specific types of molecules at the single-molecule level without compromising its topographic imaging performance. The technique can be used to analyze the distribution of a specific component in heterogeneous samples or to follow compositional changes that occur during complex processes, thus revealing features that are not evident from topographic images alone. This capability will significantly enhance the usefulness of AFM as a tool for studying biologically relevant samples and processes.
Supplementary Material
Acknowledgments
We thank Gordon Hager for supplying hSwi-Snf and the plasmid containing the MMTV long terminal repeat and Jaya Yodh for supplying HeLa histones. This work was supported by the National Cancer Institute.
Abbreviations: AFM, atomic force microscopy; MMTV, mouse mammary tumor virus; GD-APTES, glutaraldehyde aminopropyltriethoxysilane; h, human; PEG, polyethylene glycol.
References
- 1.Drake, B., Prater, C. B., Weisenhorn, A. L., Gould, S. A. C., Albrecht, T. R., Quate, C. F., Cannell, D. S., Hansma, H. G. & Hansma, P. K. (1989) Science 243, 1586–1589. [DOI] [PubMed] [Google Scholar]
- 2.Noy, A., Vezenov, D. V. & Lieber, C. M. (1997) Annu. Rev. Mater. Sci. 27, 381–421. [Google Scholar]
- 3.A-Hassan, E., Heinz, W. F., Antonik, M. D., D'Costa, N. P., Nageswaran, S., Schoenenberger, C. & Hoh, J. H. (1998) Biophys. J. 74, 1564–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hinterdorfer, P., Baumgartner, W., Gruber, H. J., Schilcher, K. & Schindler, H. (1996) Proc. Natl. Acad. Sci. USA 93, 3477–3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raab, A., Han, W., Badt, D., Smith-Gill, S. J., Lindsay, S. M., Schindler, H. & Hinterdorfer, P. (1999) Nat. Biotechnol. 901–905. [DOI] [PubMed]
- 6.Bash, R., Wang, H., Yodh, J., Hager, G., Lindsay, S. M. & Lohr, D. (2003) Biochemistry 42, 4681–4690. [DOI] [PubMed] [Google Scholar]
- 7.Wang, H., Bash, R., Yodh, J. G., Hager, G. H., Lohr, D. & Lindsay, S. M. (2002) Biophys. J. 83, 3619–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang, H., Bash, R., Yodh, J. G., Hager, G., Lohr, D. & Lindsay, S. M. (2004) Biophys. J., in press. [DOI] [PMC free article] [PubMed]
- 9.Sarid, D. (1992) Atomic Force Microscopy (Oxford Univ. Press, New York).
- 10.Haselgrübler, T., Amerstorfer, A., Schindler, H. & Gruber, H. J. (1995) Bioconjugate Chem. 6, 242–248. [DOI] [PubMed] [Google Scholar]
- 11.Han, W., Lindsay, S. M. & Jing, T. (1996) Appl. Phys. Lett. 69, 4111–4114. [Google Scholar]
- 12.Lantz, M., Liu, Y. Z., Cui, X. D., Tokumoto, H. & Lindsay, S. M. (1999) Surf. Interface Anal. 27, 354–360. [Google Scholar]
- 13.Liu, Y. Z., Leuba, S. & Lindsay, S. M. (1999) Langmuir 14, 8547–8548. [Google Scholar]
- 14.Bustamante, C., Marko, J. F., Siggia, E. D. & Smith, S. (1994) Science 265, 1599–1600. [DOI] [PubMed] [Google Scholar]
- 15.Kienberger, F., Pastushenko, V., Kada, G., Gruber, H., Riener, C., Schindler, H. & Hinterdorfer, P. (2000) Single Mol. 1, 123–128. [Google Scholar]
- 16.Wilert-Badt, S., Hinterdorfer, P., Gruber, H., Lin, G.-T., Badt, D., Wimmer, B., Schindler, H. & Kinne, R. K.-H. (2002) Biophys. J. 82, 2767–2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F. & Richmond, T. J. (1997) Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- 18.Schnitzler, G. R., Cheung, C. L., Hafner, J. H., Saurin, A. J., Kingston, R. E. & Lieber, C. M. (2001) Mol. Cell. Biol. 21, 8504–8511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}