

Abstract
Drug-induced cholestasis is a leading cause of drug withdrawal. However, the use of primary human hepatocytes (PHHs), the gold standard for predicting cholestasis in vitro, is limited by their high cost and batch-to-batch variability. Mature hepatocyte characteristics have been observed in human induced hepatocytes (hiHeps) derived from human fibroblast transdifferentiation. Here, we evaluated whether hiHeps could biosynthesize and excrete bile acids (BAs) and their potential as PHH alternatives for cholestasis investigations. Quantitative real-time PCR (qRT-PCR) and western blotting indicated that hiHeps highly expressed BA synthases and functional transporters. Liquid chromatography tandem mass spectrometry (LC-MS/MS) showed that hiHeps produced normal intercellular unconjugated BAs but fewer conjugated BAs than human hepatocytes. When incubated with representative cholestatic agents, hiHeps exhibited sensitive drug-induced bile salt export pump (BSEP) dysfunction, and their response to cholestatic agent-mediated cytotoxicity correlated well with that of PHHs (r2 = 0.8032). Deoxycholic acid (DCA)-induced hepatotoxicity in hiHeps was verified by elevated aspartate aminotransferase (AST) and γ-glutamyl-transferase (γ-GT) levels. Mitochondrial damage and cell death suggested DCA-induced toxicity in hiHeps, which were attenuated by hepatoprotective drugs, as in PHHs. For the first time, hiHeps were reported to biosynthesize and excrete BAs, which could facilitate predicting cholestatic hepatotoxicity and screening potential therapeutic drugs against cholestasis.
In the process of drug discovery and development, drug-induced cholestasis has gradually emerged as a major cause of drug-induced liver injury (DILI) and has led to drug withdrawal from the market or the termination of candidate compounds1. Hepatic transporter dysfunction, especially drug-induced inhibition of the bile salt export pump (BSEP), can lead to the accumulation of bile acids (BAs) in hepatocytes1,2,3, potentially resulting in mitochondrial dysfunction, oxidative stress, and other types of intrahepatic cholestatic hepatotoxicity4,5.
Because of species differences6, primary human hepatocytes (PHHs) are still considered the gold standard in vitro model for investigating the risk of cholestasis in pharmaceutical research7. These cells contain the full set of BA synthesis and transport systems8,9, but their limited availability, short lifespan, high cost and batch-to-batch variability limit their use, especially during the drug discovery phase10. Other currently available in vitro models have been applied to investigate cholestatic liver damage, but multiple limitations also restrict their use. For example, HepG2 is the most extensively characterized hepatic carcinoma cell line, and it has been used to study cholestatic hepatotoxicity11; however, HepG2 cells lack BA transport function10,12. In addition, HepaRG cells have been used to evaluate cholestatic features, but their low BSEP activity and time-consuming differentiation procedure limit their use13,14. Both human embryonic stem cell- and induced pluripotent stem cell-derived hepatocytes are promising cell models for studying drug hepatotoxicity15; however, their high cost and associated complications restrict their application16,17. To the best of our knowledge, no study has reported the use of stem cell-derived hepatocytes to study cholestatic toxicity to date.
Direct lineage reprogramming is defined as the direct induction of one specialized cell type into another lineage, with avoidance of the intermediate pluripotent state18,19,20. A previous study has reported the generation of highly proliferative human induced hepatocytes (hiHeps) by linear conversion that have characteristic functions of mature hepatocytes, including glycogen accumulation, albumin excretion, cytochrome P450 (CYP) enzymatic activity and biliary drug excretion20. The hiHeps were derived from human fibroblasts induced by the three transcription factors: FOXA3, HNF1A and HNF4A. HNF1A and HNF4A have been reported to promote the mRNA expression of BA synthases21 and transporters22, implying that this novel cell line may have the capacity for BA synthesis/excretion. However, the ability of these cells to biosynthesize and excrete BA and their potential use for the evaluation of cholestatic liver toxicity have not been yet explored. Thus, in this study, we investigated whether hiHeps could be applied to predict the risk of cholestatic liver toxicity as a hepatocyte-like alternative model to PHHs. We first compared the capacity of hiHeps to biosynthesize and excrete BA with that of PHHs. Then, we examined the potential inhibitory effects of 6 representative cholestatic agents on activity and expression of the BA efflux transporter BSEP in hiHeps. BA-induced direct toxicity was also characterized in both hiHeps and PHHs. Finally, the therapeutic activities of representative hepatoprotective drugs against cholestasis were verified in hiHeps.
Results
Expression of the enzymes responsible for BA biosynthesis
Under phase-contrast microscopy, cultured hiHeps displayed an epithelial morphology similar to that of PHHs (Fig. 1A). hiHeps expressed the major enzymes responsible for BA synthesis (i.e., cholesterol 7α-hydroxylase (CYP7A1), sterol 12-alpha-hydroxylase (CYP8B1) and sterol 27-hydroxylase (CYP27A1)), as determined by measurement of mRNA levels (238.41%, 52.88% and 199.16% of the levels in PHHs, respectively) (Fig. 1B). The mRNA levels of the nuclear factors farnesoid X receptor (FXR), constitutive androstane receptor (CAR) and pregnane X receptor (PXR) in hiHeps were lower than those in PHHs. In addition, the protein expression of CYP7A1 in hiHeps was 24.44% of that in PHHs and 41.26% of that in sandwich-cultured human hepatocytes (SCHHs) (Fig. 1C). The FXR protein level was close to 50% of that in PHHs (Fig. 1C).
Figure 1. The expression levels of BA synthases in hiHeps and PHHs.

(A) The typical epithelial morphologies of hiHeps and PHHs, as determined by light phase contrast microscopy. (B) The mRNA expression levels of BA synthases (i.e., CYP7A1, CYP8B1, and CYP27 A1) and the upstream nuclear factors (i.e., FXR, CAR, and PXR) in hiHeps. as determined by qRT-PCR. The data are expressed as the mean ± SD (n = 3). *p < 0.05 relative to PHHs. (C) Comparison of the protein expression levels of CYP7A1 and FXR among hiHeps, PHHs and SCHHs by western blotting (left) and gray intensity analysis (right). β-ACTIN was used as a reference control.
Expression of the efflux and influx transporters responsible for BA excretion
hiHeps expressed the efflux transporter BSEP (27.75% of the expression in PHHs), multidrug resistance-associated proteins (e.g., MRP2, MRP3 and MRP4, expressed at 37.5%, 84.54% and 271.88% of the levels in PHHs, respectively) and multidrug resistance proteins (e.g., MDR1 and MDR3, expressed at 54.92% and 22.02% of the levels in PHHs, respectively), as determined by measurement of the mRNA levels (Fig. 2A). The mRNA expression related to influx transporters, including the Na(+)-taurocholate cotransport protein (NTCP) and organic anion-transporting polypeptides (e.g., OATP1B1 and OATP1B3), was lower in hiHeps than in PHHs (Fig. 2A). Further, the protein expression of two important efflux transporters BSEP and MRP2 in hiHeps was comparable to that in PHHs and SCHHs (Fig. 2B). The protein level of the influx transporter NTCP was higher in hiHeps than in PHHs and SCHHs (Fig. 2B). Moreover, the biliary excretion index (BEI) values for BSEP and MRP2 in hiHeps were comparable to those in SCHHs (BSEP: 41.36% ± 15.63% vs 31% ± 2.06%; MRP2: 20.17% ± 10.22% vs 26.82% ± 13.72%) (Fig. 3A). Polarization of the efflux transporters was apparent in cultured hiHeps and SCHHs (Fig. 3B). The fluorescent signals in both hiHeps and SCHHs were abolished by an MRP2 inhibitor 5-(3-(2-(7-chloroquinolin-2-yl) ethenyl) phenyl)-8-dimethylcarbamyl-4,6-dithiaoctanoic acid sodium salt hydrate (MK571) (Fig. 3B). The accumulation of deuterium-labelled sodium taurocholate (d8-TCA) in hiHeps was approximately half of that in PHHs. Further, the accumulation of d8-TCA in both cell types was significantly reduced by an NTCP inhibitor, troglitazone (Fig. 3C).
Figure 2. The expression levels of BA transporters in hiHeps and human hepatocytes.

(A) The mRNA levels of BA efflux (i.e., BSEP, MRPs and MDRs) and influx (i.e., NTCP, OATP1B1 and OATP1B3) transporters in hiHeps determined by qRT-PCR. The data are expressed as the mean ± SD (n = 3). *p < 0.05 relative to PHHs. (B) Comparison of the protein expression levels of MRP2, BSEP and NTCP among hiHeps, PHHs and SCHHs by western blotting (left) and gray intensity analysis (right). β-ACTIN was used as a reference control.
Figure 3. Comparison of the activities of BA transporters between hiHeps and human hepatocytes.
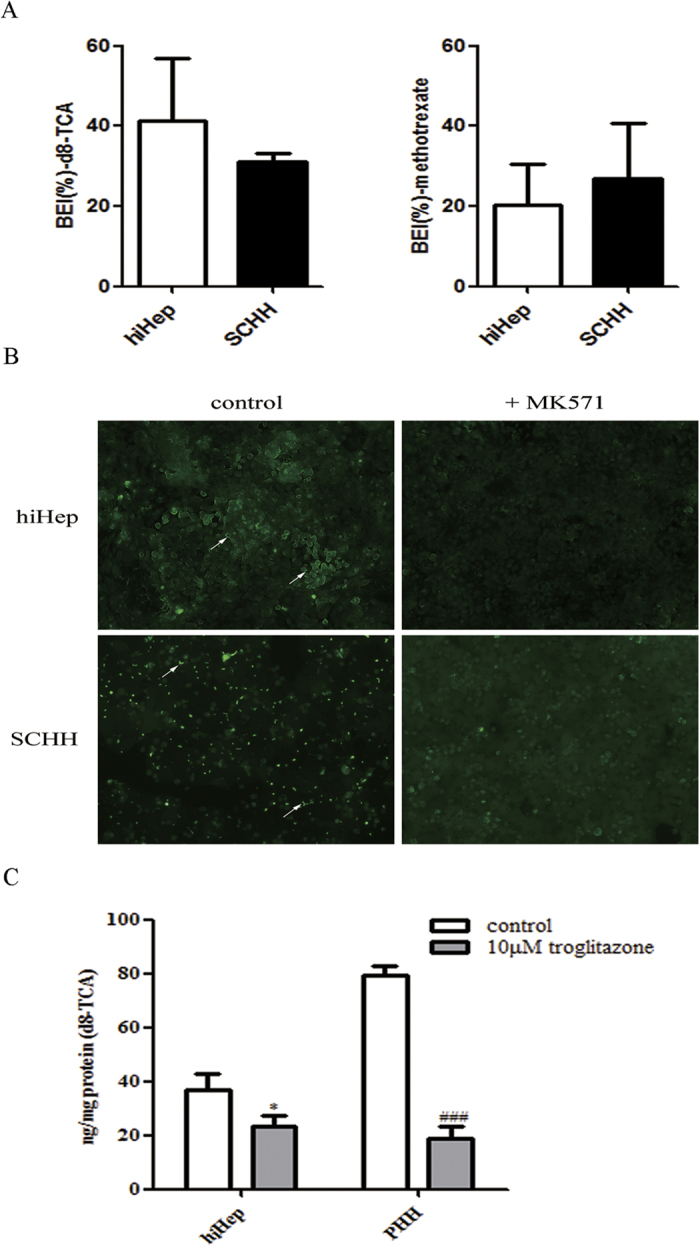
(A) The efflux transporter activities of BSEP and MRP2 were determined by calculating the BEI values of d8-TCA (the substrate of BSEP) and methotrexate (the substrate of MRP2) for hiHeps (white columns) and SCHHs (black columns). The data are expressed as the mean ± SD (n = 3). (B) The polarized locations of efflux transporters in the bile canaliculi of both hiHeps and SCHHs, as determined by fluorescence microscopy. In the absence or presence of MK571 (20 μmol/L), an MRP2 inhibitor, bile canaliculi were labelled with an MRP2 fluorescent substrate, CDF, which was formed from non-fluorescent CDFDA via intracellular esterases. (C) The influx transporter activity of NTCP was determined by accumulation assay of an NTCP substrate, d8-TCA. Troglitazone (10 μmol/L) was used as a positive control. The data are expressed as the mean ± SD (n = 3). *p < 0.05 vs control in hiHeps, #p < 0.05 vs control in PHHs.
BA biosynthesis and excretion
The concentrations of individual BAs in cell lysates and supernatants were quantitated by liquid chromatography tandem mass spectrometry (LC-MS/MS). The amounts of unconjugated BAs (i.e., cholic acid [CA], chenodeoxycholic acid [CDCA], deoxycholic acid [DCA], and lithocholic acid [LCA]) were comparable between hiHeps and SCHHs (Fig. 4A). Ursodeoxycholate (UDCA) was not detected in either cell type. The concentrations of conjugated BAs (i.e., glycine-conjugated species [GCA, GCDCA, GDCA, and GLCA] and taurine-conjugated species [TCDCA, TCA and TLCA]) were lower in the cell lysate of hiHeps than in that of SCHHs (Fig. 4A). However, the amounts of conjugated BAs (except for GCA, TCA, and GCDCA) were much higher in the supernatant of hiHeps than in that of SCHHs (Fig. 4A). GUDCA, TUDCA and TDCA were not detected in either the cell lysates or the supernatants. The total BA concentration was similar between hiHep and SCHH supernatants but was lower in the hiHep lysate than in the SCHH lysate (Fig. 4B).
Figure 4. Comparisons of the BA concentrations in cell lysates and supernatants of hiHeps and SCHHs.

. (A) The concentration of each BA (i.e., CA, GCA, TCA, CDCA, GCDCA, TCDCA, DCA, GDCA, LCA, GLCA and TLCA) in both the cell lysates and supernatants of hiHeps and SCHHs was determined by LC-MS/MS. (B) The total BA concentrations in the cell lysates and supernatants were measured using a total BA reagent kit.
Inhibition of the efflux BA transporter BSEP by representative cholestatic agents
The six representative cholestatic agents assessed (troglitazone, ketoconazole, rifampicin, bosentan, glibenclamide and omeprazole) all significantly inhibited BSEP activity following incubation for 15 min, reducing the BEI value from 40.13% to <10% in hiHeps; in contrast, only troglitazone significantly inhibited BSEP activity in SCHHs, reducing the BEI value from 29.91% to <10% at the same concentration (Fig. 5A). In addition to the functional inhibition of BSEP, its mRNA expression was significantly suppressed in hiHeps after incubation with these 6 cholestatic agents. The cholestatic agents also inhibited BSEP mRNA expression in SCHHs, but only troglitazone exhibited significant inhibitory potency (Fig. 5B). Moreover, the responses to the cytotoxicity of these cholestatic agents in hiHeps were positively correlated with those in human hepatocytes (r2 = 0.8032) (Fig. 5C).
Figure 5. Cholestatic agent-induced BSEP inhibition and cytotoxicity in hiHeps and human hepatocytes.

(A) BSEP function was inhibited by cholestatic drugs in hiHeps compared with SCHHs, resulting in a decrease in the BEI value of d8-TCA. The cholestatic drugs included 10 μmol/L troglitazone, 30 μmol/L ketoconazole, 25 μmol/L rifampicin, 25 μmol/L bosentan, 10 μmol/L glibenclamide and 100 μmol/L omeprazole. The data are expressed as the mean ± SD (n = 3). (B) BSEP expression was inhibited by cholestatic drugs in hiHeps (white columns) compared with SCHHs (grey columns). The data are expressed as the mean ± SD (n = 3). *p < 0.05 vs control. (C) Correlation analysis of hiHeps and human hepatocytes based on the log(IC50) determined by MTT assay. The calculated r2 value of the linear correlation between hiHeps and human hepatocytes was 0.8032.
BA-induced hepatotoxicity in hiHeps
The results of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay revealed that the individual BAs decreased the viabilities of hiHeps and PHHs at certain concentrations after 24 hours of incubation (Fig. 6). In contrast to conjugated BAs, the cytotoxicity of the unconjugated BAs (i.e., CA, CDCA, DCA and LCA) was substantially more similar between hiHeps and PHHs (Fig. 6, Table 1). Moreover, the abnormal aspartate aminotransferase (AST) and γ-glutamyl transferase (γ-GT) levels suggested concentration-dependent hepatotoxicity in hiHeps and PHHs after 24 hours of incubation with DCA. The alkaline phosphatase (ALP) level did not obviously change in hiHeps, whereas in PHHs, only treatment with 1000 μmol/L DCA resulted in a slight increase in ALP activity. The alanine aminotransferase (ALT) level did not change in hiHeps, even in the presence of 1000 μmol/L DCA, whereas the ALT activity was significantly increased in PHHs in the presence of 500 or 1000 μmol/L DCA (Fig. 7A and B).
Figure 6. Comparison of the BA concentration-dependent cytotoxicities between hiHeps and PHHs.

The BAs included CA, TCA, and GCA (top row); CDCA, TCDCA, and GCDCA (middle row); and DCA and LCA (bottom row). The data are expressed as the mean ± SD (n = 5).
Table 1. Comparison of the cytotoxicities of BAs between hiHeps and PHHs based on MTT assay.
| BAs | HiHeps (IC50, μM) | PHHs (IC50, μM) |
|---|---|---|
| Unconjugated BAs | ||
| CA | 2207 | 1745 |
| CDCA | 341.1 | 249.2 |
| DCA | 314.9 | 237.3 |
| LCA | 168.4 | 679.2 |
| Conjugated BAs | ||
| GCA | 4328 | 775.8 |
| TCA | 998.6 | 1144 |
| GCDCA | 1073 | 374.2 |
| TCDCA | 1224 | 537 |
Figure 7. Blood biochemical analysis of DCA-mediated hepatotoxicity in hiHeps and PHHs.

(A) The enzymatic activities of ALT, AST, ALP and γ-GT in hiHeps. The data are expressed as the mean ± SD (n = 3), *p < 0.05 vs 0 group. (B) The enzymatic activities of ALT, AST, ALP and γ-GT in PHHs. The data are expressed as the mean ± SD (n = 2), *p < 0.05 vs 0 group.
Cytoprotective effects of representative hepatoprotective agents in hiHeps vs PHHs
Treatment of hiHeps and PHHs with DCA at certain concentrations exposure resulted in decreases in cell viability (Fig. 6), the ATP concentration (Fig. 8A), and the mitochondrial membrane potential (MMP) (Fig. 8B) and increases in the production of reactive oxygen species (ROS) (Fig. 8C), caspase 3/7 activity (Fig. 8D) and lactate dehydrogenase (LDH) release (Fig. 8E). Co-incubation with representative hepatoprotective drugs (i.e., quercetin, silymarin, curcumin and metformin) for 24 h protected hiHeps and PHHs against DCA-induced decreases in cell viability and the ATP level (Fig. 9A and B). The MMP was enhanced by quercetin in hiHeps, whereas it was significantly increased by silymarin and curcumin in PHHs (Fig. 9C). All four of these compounds significantly reduced DCA-induced ROS production and apoptosis (Fig. 9D and E), but they did not prevent the DCA-induced LDH release in hiHeps or PHHs (Fig. 9F).
Figure 8. The mechanisms of DCA-mediated toxicity in hiHeps and PHHs.

The mechanism of DCA-mediated toxicity was examined by performing ATP (A), MMP (B), ROS (C), apoptosis (D) and LDH (E) assays. The data are expressed as the mean ± SD (n = 3–5). *p < 0.05 in hiHeps, #p < 0.05 in PHHs.
Figure 9. Hepatoprotective effects of therapeutic drugs in hiHeps and PHHs.

The hepatic roles of the hepatoprotective drugs quercetin (40 μmol/L), silymarin (25 μmol/L), curcumin (15 μmol/L) and metformin (200 μmol/L) in hiHeps and PHHs were elucidated via MTT (A), ATP (B), MMP (C), ROS (D), apoptosis (E) and LDH (F) assays. The data are expressed as the mean ± SD (n = 3–5). *p < 0.05 vs DCA group in hiHeps, #p < 0.05 vs DCA group in PHHs.
Discussion
BAs play significant roles in the digestion of lipids, nutrients and vitamins and the regulation of cholesterol homeostasis23,24,25,26. The biosynthesis of BAs from cholesterol in the liver is mainly mediated by CYP7A1 (the rate-limiting enzyme for BA synthesis), CYP8B1 and CYP27A127,28. The transcriptional activation of BA synthases is primarily mediated by nuclear factors, including FXR, PXR and CAR29. The excretion of BAs proceeds readily via glycine or taurine conjugation23, which is mediated by a broad range of efflux and influx transporters30. BA efflux transporters include BSEP, MRPs and MDRs13,31,32. Hepatocellular BA influx is mediated predominantly by NTCP and OATPs32. Our study is the first report of the comparable expression of BA synthases and transporters between hiHeps and PHHs (Figs 1 and 2). To compare protein levels and determine the influence of hepatic polarity on protein expression33, we used both PHHs and SCHHs as positive controls to assess the protein expression levels of BA synthases and transporters in hiHeps. The BA synthases and efflux and influx transporters normally govern hepatic BA concentration. Thus, we measured the total BA amount in hiHeps and found that the amount in the cell lysate attained to 30% of that in the SCHH lysate. The lower total intercellular BA amount in hiHeps might be attributable to the relatively low CYP7A1 protein level in these cells (Fig. 1C). Interestingly, the CYP7A1 mRNA level was significantly increased in hiHeps compared with PHHs, while the CYP7A1 protein level was significantly decreased. This apparent uncoupling of CYP7A1 mRNA and protein levels might be due to different mechanisms for the transcriptional and translational regulation of the gene and protein expression. The precise reason for this difference needs to be further explored. In addition, CYP7A1 expression is regulated by FXR, and it is decreased when the FXR level is increased34. Our results showed that reduced FXR mRNA expression was correlated with increased CYP7A1 mRNA expression in hiHeps. However, a reduction in the FXR protein level was not correlated with an increase in the CYP7A1 protein level in these cells. The possible reason for this finding was that the CYP7A1 protein level was still lower in hiHeps than in SCHHs following FXR-mediated negative regulation. However, the total BA amount in the hiHep supernatants was similar to that in the SCHH supernatants (Fig. 4B), consistent with the comparable BA efflux transport activities detected between these two cell types (Fig. 3A). Interestingly, the LC-MS/MS data revealed that the levels of unconjugated BAs in the hiHep lysate were comparable to those in the PHH lysate, whereas the levels of conjugated BAs were lower in the hiHep lysate (Fig. 4A). These lower levels of conjugated BAs might have been due to reduced expression of the enzymes (e.g., BA-CoA synthetase [BACS] and BA-CoA: amino acid N-acetyltransferase [BAT]) responsible for BA conjugation (data not shown). Because the serum concentration of unconjugated BAs is significantly elevated during cholestasis35, it was speculated that the unconjugated BA concentration in cells might influence hepatotoxicity to a greater extent than the conjugated BA concentration. In this study, we found that the response to unconjugated BA toxicity in hiHeps was more similar to that in PHHs than the response to conjugated BA toxicity (Fig. 6). This consistent toxic response to unconjugated BAs in hiHeps and PHHs implied that hiHeps could serve as an alternative to PHHs for the prediction of potential cholestatic toxicity.
Cholestasis induced by the over-accumulation of BAs is thought to be a chronic condition, ultimately resulting in liver fibrosis and cirrhosis31. One of the most important mechanisms of drug-induced cholestasis is BSEP inhibition36. In the clinic, some human cholestatic liver diseases are related to BSEP malfunction, including progressive familial intrahepatic cholestasis (PFIC) and benign recurrent intrahepatic cholestasis (BRIC)1,37. Importantly, in humans, no compensatory mechanism for the loss of BSEP function exists31,38. Therefore, the assessment of BSEP inhibition by candidate drugs is extremely important during drug discovery and development36. In in vitro studies, a BEI value of 10% is the recommended cut-off value for compounds with obvious bile clearance potentials39. Thus, we used a BEI value of 10% in our study as the cut-off value for evaluating the potential for drug-induced BSEP inhibition. Our results demonstrated that hiHeps significantly reflected the effects of cholestatic drugs on the inhibition of BSEP function (BEI < 10%) and expression (Fig. 5A and B), reflecting direct and indirect mechanisms of transporter dysfunction, respectively23. However, only troglitazone significantly reduced BSEP activity (BEI < 10%) and expression in SCHHs. These results indicated that hiHeps were more susceptible than SCHHs to cholestatic agent-mediated inhibition of both BSEP function and expression. With regard to BSEP function, we think that the increased BSEP sensitivity was potentially due to the reduced uptake of d8-TCA (BSEP substrate) by hiHeps, while in terms of BSEP expression, the increased sensitivity was possibly attributed to the lower BSEP expression in these cells. Thus, hiHeps could be utilized to sensitively predict potential drug-induced BSEP inhibition and the consequent cholestasis risk, especially during the early stages of drug discovery.
To further investigate the potential application of hiHeps in cholestatic research, the cytotoxic hydrophobic compound DCA, which causes hepatocyte cell death during intrahepatic cholestasis40, was chosen to induce hepatotoxicity in hiHeps. The results showed that the level of AST, but not that of ALT was increased by DCA in hiHeps, whereas the activities of both of these enzymes were increased in PHHs (Fig. 7A and B). The reason for this difference might be that DCA did not alter the ALT gene or protein level in hiHeps, and thus, ALT activity was not affected41. However, an increased AST level alone is not enough to confirm BA-induced hepatotoxicity; thus, we analysed other liver-specific markers, such as ALP and γ-GT. γ-GT is an enzyme that is involved in the first step of glutathione catabolism42, and it was significantly elevated in a concentration-dependent manner in hiHeps and PHHs. In addition, the ALP level was slightly elevated in hiHeps and was significantly increased in PHHs following exposure to 1000 μmol/L DCA in PHHs, suggesting the presence of a biliary tract disorder. Taken together, these data suggested that DCA induced hepatotoxicity. Additionally, DCA caused mitochondrial damage, and promoted ROS generation and cell death in both hiHeps and PHHs (Fig. 8), as reported previously40. However, the caspase 3/7 activity was decreased shown in Fig. 8D at the higher DCA concentration (500 μmol/L) in PHHs. It might be due to the loss of cellular viability that occurred when the DCA concentration reached 500 μmol/L. Similarly, in Fig. 8E, LDH release was reduced at the highest DCA concentration (5000 μmol/L) observed at in PHHs. The reason for this result was not clear but it might be because of DCA’s limited solubility at that concentration in the PHH medium. These above results indicated that hiHeps could be a good alternative to PHHs for the evaluation of cholestatic toxicity.
We further investigated the protective potentials of hepatoprotective drugs in hiHeps. Quercetin, silymarin and curcumin are antioxidants that have been reported to protect hepatocytes against cholestatic liver damage43,44,45. Our study showed that these compounds also protected hiHeps against DCA-mediated cholestatic toxicity (Fig. 9A–E). However, although quercetin has been reported to mitigate the ethanol-induced LDH increase in rat primary hepatocytes46, our results indicated that it had no effect on DCA-mediated LDH release in either cell type (Fig. 9F). One potential reason for these discrepant findings could be that the previous study employed ethanol, whereas DCA was used in our study. Metformin, an effective diabetes drug, has been reported to protect rat primary hepatocytes against GCDCA-induced apoptosis rather than necrosis47. Our study demonstrated that metformin effectively protected hiHeps and PHHs against DCA-mediated apoptosis (Fig. 9E) but not necrosis, as evaluated by LDH release (Fig. 9F). These results indicated that hiHeps could be used to screen potential anti-cholestatic candidates.
Our study has provided the first evidence that hiHeps not only have the capacity to biosynthesize and excrete BAs but are also sensitive enough to be used in the evaluation of BSEP inhibition for assessing cholestatic drug-induced toxicity. Additionally, hiHeps could be potentially used in comprehensive assessments of the risk of BA-mediated cytotoxicity and the underlying mechanism, and they could also be developed into a potential in vitro model for the screening of drug candidates with anti-cholestatic activity.
Methods
Experimental design
Initially, we examined the expression of the key enzymes and transporters responsible for BA biosynthesis and excretion at both the mRNA and protein levels. BA efflux transporter activity was assessed by the BEI assay and polarization location, whereas BA influx transporter activity was evaluated by accumulation assay. To comprehensively assess BA biosynthesis and excretion in hiHeps, we measured the BA concentrations in cell lysates and supernatants by LC-MS/MS or using a commercial total BA reagent kit. To further determine whether hiHeps could be used to study the mechanism of cholestasis, we examined the inhibitory potency of 6 representative cholestatic agents against BSEP, a BA efflux transporter, as well as their cytotoxicities, in both hiHeps and PHHs. Then, we assessed the direct toxicity of individual BAs and further explored the representative DCA-induced BA hepatotoxicity. Finally, we assessed the effects of 4 representative hepatoprotective agents on DCA-induced cytotoxicity in hiHeps. At least 3 replicates were performed per experiment for hiHeps, while 2–3 replicates were performed per experiment for PHHs.
Cell culture
hiHeps
hiHeps transdifferentiated from human fibroblasts induced by FOXA3, HNF1A and HNF4A were obtained from Lijian Hui’s lab and were cultured in rat tail collagen-pre-coated dishes according to a previously described protocol20. Cells were routinely passaged when they were almost confluent. The cells were cultured on collagen-coated plates at a seeding density of 100,000 cells per mL for approximately four days and were then used for further assays. In contrast, for BA qualification assay, 1 ml of hepatocyte maintenance medium (HMM) used to culture hiHeps in 6-well plates was collected on day 5 after fresh medium had been added to the cells for 24 h; the remaining medium was simultaneously aspirated and then the plates were washed with phosphate-buffered saline (PBS). Both the collected medium and 6-well plates were frozen for further BA measurement by LC-MS/MS.
Cryopreserved PHHs
Cryopreserved PHHs were purchased from the Research Institute for Liver Diseases (RILD) (Shanghai) Co. Ltd. and cultured according to an industrial culturing method. Briefly, PHHs were thawed and seeded at a density of 0.70 × 106 viable cells per mL on collagen-coated plates. The cells were allowed to attach overnight, and then the medium was replaced with fresh medium. On day 3, the PHHs were incubated with toxic drugs or used for accumulation assay. SCHHs have been recommended as the most appropriate in vitro model to mimic the hepatobiliary secretory process48,49; thus, we compared hiHeps with SCHHs rather than with PHHs in terms of the activity and polarization localization of the efflux transporters and the BA amounts in the cell lysates and supernatants. PHHs were overlaid with 0.25 mg/mL Matrigel (BD Biosciences, CA, USA) to form SCHHs. To determine the BA concentrations, the medium in which the SCHHs was cultured in 6-well plates was replaced with fresh medium for 24 hours. On day 5, at 24 h after the addition of fresh medium,1 ml of the SCHH medium was collected; the remaining medium was aspirated and then the plates were washed with phosphate-buffered saline (PBS). Both the collected medium and 6-well plates were frozen for further BA quantification by LC-MS/MS.
Quantitative real-time PCR (qRT-PCR)
hiHeps were harvested at confluence, whereas PHHs were collected on day 3 after plating. Total RNA was extracted from the hepatocytes using TRIzol reagent (Life Technology, CA, USA), and cDNA synthesis was performed via the reverse transcription of 1 μg RNA using a Primescript RT Reagent Kit (Takara, Shiga, Japan). Gene expression levels were quantified using a real-time PCR kit (Qiagen, Hilden, Germany). Primers for CYP7A1, CYP8B1, CYP27A1, FXR, CAR, PXR, BSEP, MRP2, MRP3, MRP4, MDR1, MDR3, NTCP, OATP1B1, OATP1B3, and β-ACTIN were synthesized according to the primer sequences listed in Table 2. β-ACTIN was used as a reference gene. The targeted genes were amplified using the Qiagen Roter Gene Q instrument (Qiagen, Germany).
Table 2. Primer sequences for qRT-PCR analysis.
| Gene | Forward (5′-3′) | Reverse(5′-3′) |
|---|---|---|
| β-ACTIN | ATCGCTGACAGGATGCAGAA | TAGAGCCACCAATCCACACAG |
| CYP7A1 | AGAAGCATTGACCCGATGGAT | AGCGGTCTTTGAGTTAGAGGA |
| CYP8B1 | TGGCTTTCCGGAAGAATATG | CTTGGTGCTGGCTGAGTGTA |
| CYP27A1 | AAGCGATACCTGGATGGTTG | TGTTGGATGTCGTGTCCACT |
| FXR | AACCATACTCGCAATACAGCAA | ACAGCTCATCCCCTTTGATCC |
| CAR | GTGCTCCTGTGCGGAGTAG | ATGGCAGATAGGCAGTTTCCC |
| PXR | AAGCCCAGTGTCAACGCAG | GGGTCTTCCGGGTGATCTC |
| BSEP | GTCGGACCTGCATTGTCATTG | ATGTGTGTCTGAGATTCTTGCATT |
| MRP2 | AGCAGCCATAGAGCTGGCCCTT | AGCAAAACCAGGAGCCATGTGCC |
| MRP3 | GGCGTCTATGCTGCTTTAGG | CCTTGGAGAAGCAGTTCAGG |
| MRP4 | ACTGCACCGTGCTAACCATT | CTTCTGCCTTGCCAAGTTGT |
| MDR1 | ATGAAGTTGAATTAGAAAATGCAG | GGAAACTGGAGGTATACTTTCATC |
| MDR3 | TTGATGGGCAGGATATTAGGA | GTTGGCCTCTTTGACAGCTT |
| NTCP | GTGGCAATCAAGAGTGGTGTC | ACTGGTCCTGGTTCTCATTCC |
| OATP1B1 | TTGGAGGTGTTTTGACTGCTT | ACAAGTGGATAAGGTCGATGTTG |
| OATP1B3 | GTCCAGTCATTGGCTTTGCA | CAACCCAACGAGAGTCCTTAGG |
Western blot analysis
Total proteins were extracted from hiHeps, PHHs and SCHHs with radio-immunoprecipitation assay (RIPA) lysis buffer (Beyotime, Haimen, China) containing 1 mmol/L phenylmethylsulfonyl fluoride (PMSF) (Beyotime, Haimen, China). Then, the proteins were separated on 8% polyacrylamide gels and electrophoretically transferred to polyvinylidene difluoride (PVDF) membranes (GE-Amersham, USA). Subsequently, the membranes were blocked with Tris-buffered saline containing 5% defatted milk for two hours and incubated with primary antibodies (CYP7A1, 58 kDa, Absci, 1:1000; FXR, 69 kDa, Santa Cruz, 1:200; MRP2, 174 kDa, Proteintech, 1:600; BSEP, 146 kDa, Abcam, 1:1000; and NTCP, 38 kDa, Abcam, 1:1000) overnight at 4 °C. β-ACTIN (1:4000, Santa Cruz) was selected as an internal reference. Next, the membranes were rinsed, incubated with horseradish peroxidase (HRP)-labelled secondary antibodies for 1 h, rinsed again, and visualized by electrochemiluminescence (ECL) (Tanon, Shanghai, China) using a Tanon-5200 automatic chemiluminescence image analysis system (Tanon, Shanghai, China). Gray intensity analysis of the bands was performed by Image J software (National Institute of Health, MD, USA).
BEI assay
hiHeps or SCHHs were rinsed three times and pre-incubated with warm Hank’s balanced salt solution (HBSS) with (standard buffer) or without calcium (to disrupt cell tight junctions) at 37 °C for 15 min. Then, the HBSS was removed, and the hepatocytes were incubated for 15 min with 5 μmol/L d8-TCA (Martrex, Inc., Minnesota, USA) in the presence or absence of a BSEP inhibitor (i.e., 10 μmol/L troglitazone, 30 μmol/L ketoconazole, 25 μmol/L rifampicin, 25 μmol/L bosentan, 10 μmol/L glibenclamide and 100 μmol/L omeprazole) or 20 μmol/L methotrexate (Sigma-Aldrich, St. Louis, MO) alone in 300 μL of standard buffer. After incubation, the solution was aspirated, and the cells were washed three times with ice-cold PBS and frozen at −80 °C for LC-MS/MS analysis. The BEI, defined as the proportion of accumulated substrates excreted into the bile canaliculi, was calculated using B-CLEAR® technology as follows: BEI = [Aplus_Ca++ − Aminus_Ca++]/Aplus_Ca++ × 100%, where A is the amount of substrate accumulation in the cells, Aplus_Ca++ is the amount of substrate accumulation in standard buffer-treated cells (cells + bile) and Aminus_Ca++ is the substrate accumulation in calcium-free buffer-treated cells (hepatocytes)39,50.
Functional polarization analysis
hiHeps or SCHHs were washed with standard buffer and pre-incubated for 20 min in the presence or absence of 20 μmol/L MK571 (Sigma-Aldrich, St. Louis, MO), an MRP2 inhibitor, before the addition of 2 μmol/L 5(6)-carboxy-2′, 7′-dichlorofluorescein diacetate (CDFDA) (BD Biosciences, Palo Alto, CA) for 30 min51. Fluorescent CDF formed from non-fluorescent CDFDA was visualized in the bile canaliculi for both hiHeps and SCHHs using a fluorescence microscope (Olympus, Japan).
Accumulation assay
hiHeps or PHHs were rinsed three times and pre-incubated with standard buffer for 15 min at 37 °C. After aspiration of the standard buffer, d8-TCA uptake was initiated by the addition of standard buffer containing 5 μmol/L d8-TCA and incubation for another 15 min. Troglitazone (Sigma-Aldrich, St. Louis, MO; 10 μmol/L) was selected as a positive control for taurocholate accumulation because it inhibits d8-TCA uptake. After incubation, d8-TCA uptake was terminated by washing the cells three times with ice-cold PBS. Subsequently, the samples were frozen at −80 °C for LC-MS/MS analysis.
LC-MS/MS analysis
The d8-TCA concentration was analysed by LC-MS/MS (LCMS-8030; Shimadzu, Kyoto, Japan) according to a previously described method52. The methotrexate concentration was determined in electrospray ionization (ESI) mode with an Inertsil ODS-4 column (100 mm × 2.10 mm, 3 μm, GL Sciences Inc., Tokyo, Japan). The selected reaction monitoring transitions were 454.6.1 m/z > 308.4 m/z for methotrexate and 180.0 m/z > 110.10 m/z for the internal standard (phenacetin). The column temperature was maintained at 40 °C. The mobile phase consisted of acetonitrile (organic phase) and ultrapure water with 0.1% formic acid (aqueous phase). The substrate amounts were normalized to the protein amounts using a bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL).
To simultaneously quantify 15 BA constituents, a Shimadzu LC-20AD HPLC system coupled with an AB Sciex API 4000 triple quadrupole mass spectrometer was used as previously described53. The total BA concentration was determined using a total BA reagent kit (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Blood biochemistry
hiHeps were treated with different concentrations (100, 500 and 1000 μmol/L) of DCA at 4 days after plating, whereas PHHs were treated at 3 days after plating according to the manufacturer’s protocol. After 24 h of treatment, the supernatants of hiHeps and PHHs were harvested for biochemical analyses, and the enzymatic activities of ALT, AST, ALP and γ-GT were determined using an Automatic Clinical Analyser (AU5800, Beckman Coulter, Inc., USA).
Toxicity evaluation
MTT assay
First, 96-well plates were seeded with hiHeps (10,000 cells per well) or PHHs (70,000 cells per well). At the indicated time point, the medium was replaced with 100 μl of fresh medium containing five different concentrations of BSEP inhibitors or BA constituents, and the cells were incubated for 24 hours. Cell viability was assessed using 5 mg/mL MTT (Sigma-Aldrich, St. Louis, MO) reagent. In addition, absorbance was measured with a microplate reader (Biotek, Winooski, VT, USA) at 570 nm.
ATP assay
The cellular ATP level was determined using a CellTiter-Glo® Luminescent Cell Viability Assay Kit (Promega, Madison, WI, USA). Luminescence was measured with a microplate reader (Biotek, Winooski, VT, USA).
MMP measurement
The MMP was evaluated with 1 μmol/L tetramethylrhodamine ethylester (TMRE) (Sigma-Aldrich, St. Louis, MO)54. Briefly, after incubation with TMRE for 30 min, the cells were washed once with PBS containing 0.2% bovine serum albumin (BSA), and fluorescence was measured with a microplate reader (Biotek, Winooski, VT, USA) at excitation and emission wavelengths of 549 and 575 nm, respectively.
ROS assay
Cellular ROS production was assessed by monitoring the cellular conversion of 10 μmol/L 2′,7′-dichlorofluorescein diacetate (DCFDA) (Sigma-Aldrich, St. Louis, MO) to dichlorofluorescein (DCF)55. Fluorescence intensity was measured at excitation and emission wavelengths of 485 and 530 nm, respectively.
Apoptosis assay
Apoptosis was determined by measuring caspase 3/7 activity using a Caspase-Glo® 3/7 Assay Kit (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Luminescence was measured with a microplate reader (Biotek, Winooski, VT, USA).
LDH release
LDH was measured using a CytoTox-ONETM Homogeneous Membrane Integrity Assay Kit (Promega, Madison, WI, USA). Fluorescence intensity was measured at excitation and emission wavelengths of 560 and 590 nm, respectively. LDH release indicates potential cellular necrosis56.
To establish the protective potentials of hepatoprotective drugs in hiHeps, hiHeps and PHHs were co-incubated for 24 hours with 4 hepatoprotective compounds (quercetin (40 μmol/L), silymarin (25 μmol/L), curcumin (15 μmol/L) and metformin (200 μmol/L)) and DCA in different assays at toxic concentrations (MTT assay: 1000 μmol/L; ATP assay: 500 μmol/L; MMP measurement: 1000 μmol/L; ROS assay: 1000 μmol/L; apoptosis assay: 200 μmol/L; and LDH release assay: 500 μmol/L).
Data analysis
Statistical analysis was performed using GraphPad Prism 5.03 software (GraphPad Software Inc., La Jolla, CA). The data are expressed as the mean ± SD. Differences between two groups were analysed using the t-test. One-way analysis of variance (ANOVA) was performed to determine the statistical significance among groups. In addition, correlation analysis was conducted using a linear correlation method. In all analyses, differences were considered significant at a p value of <0.05.
Additional Information
How to cite this article: Ni, X. et al. Functional human induced hepatocytes (hiHeps) with bile acid synthesis and transport capacities: A novel in vitro cholestatic model. Sci. Rep. 6, 38694; doi: 10.1038/srep38694 (2016).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Acknowledgments
We thank Lijian Hui’s lab for providing hiHeps and the cell culturing protocol. We are also grateful to Fandi Kong from Xiaoyan Chen’s lab for performing bile acid quantification. This work was funded by the National Science Foundation of China (Grant No. 81302836 and 81573499) and the National High Technology Research and Development Program of China (Grant No. 2013AA032202).
Footnotes
Author Contributions Xuan Ni designed the study, carried out the experiments and wrote the manuscript. Guoyu Pan designed the experiments and reviewed the manuscript. Yimeng Gao, performed data analysis. Zhitao Wu was involved in designing the study. Lijian Hui reviewed the manuscript. Leilei Ma, Chen Chen, Le Wang and Yunfei Lin provided technical assistance.
References
- Dawson S., Stahl S., Paul N., Barber J. & Kenna J. G. In vitro inhibition of the bile salt export pump correlates with risk of cholestatic drug-induced liver injury in humans. Drug Metab. Dispos. 40, 130–138, doi: 10.1124/dmd.111.040758 (2012). [DOI] [PubMed] [Google Scholar]
- Susukida T., Sekine S., Nozaki M., Tokizono M. & Ito K. Prediction of the clinical risk of drug-induced cholestatic liver injury using an in vitro sandwich cultured hepatocyte assay. Drug Metab. Dispos. 43, 1760–1768, doi: 10.1124/dmd.115.065425 (2015). [DOI] [PubMed] [Google Scholar]
- Woodhead J. L. et al. Exploring BSEP inhibition-mediated toxicity with a mechanistic model of drug-induced liver injury. Front. Pharmacol. 5, 240, doi: 10.3389/fphar.2014.00240 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez M. J. & Briz O. Bile-acid-induced cell injury and protection. World J. Gastroenterol. 15, 1677–1689 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S. et al. Progressive stages of mitochondrial destruction caused by cell toxic bile salts. Biochim. Biophys. Acta 1828, 2121–2133, doi: 10.1016/j.bbamem.2013.05.007 (2013). [DOI] [PubMed] [Google Scholar]
- Chiang J. Y. Bile acids: regulation of synthesis. J. Lipid Res. 50, 1955–1966, doi: 10.1194/jlr.R900010-JLR200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolbright B. L., McGill M. R., Yan H. & Jaeschke H. Bile acid-induced toxicity in HepaRG cells recapitulates the response in primary human hepatocytes. Basic Clin. Pharmacol. Toxicol. 118, 160–167, doi: 10.1111/bcpt.12449 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telbisz A. & Homolya L. Recent advances in the exploration of the bile salt export pump (BSEP/ABCB11) function. Expert Opin. Ther. Targets 20, 501–514, doi: 10.1517/14728222.2016.1102889 (2016). [DOI] [PubMed] [Google Scholar]
- Sharanek A. et al. Cellular accumulation and toxic effects of bile acids in cyclosporine a-treated HepaRG hepatocytes. Toxicol. Sci. 147, 573–587, doi: 10.1093/toxsci/kfv155 (2015). [DOI] [PubMed] [Google Scholar]
- Rodrigues R. M. et al. Toxicogenomics-based prediction of acetaminophen-induced liver injury using human hepatic cell systems. Toxicol. Lett. 240, 50–59, doi: 10.1016/j.toxlet.2015.10.014 (2016). [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rubio S. et al. AP-1 Inhibition by SR 11302 Protects Human Hepatoma HepG2 Cells from Bile Acid-Induced Cytotoxicity by Restoring the NOS-3 Expression. PloS one 11, e0160525, doi: 10.1371/journal.pone.0160525 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper A. D., Craig W. Y., Taniguchi T. & Everson G. T. Characteristics and Regulation of Bile-Salt Synthesis and Secretion by Human Hepatoma Hepg2 Cells. Hepatology 20, 1522–1531, doi: DOI 10.1002/hep.1840200623 (1994). [DOI] [PubMed] [Google Scholar]
- Le Vee M., Noel G., Jouan E., Stieger B. & Fardel O. Polarized expression of drug transporters in differentiated human hepatoma HepaRG cells. Toxicol. In Vitro 27, 1979–1986, doi: 10.1016/j.tiv.2013.07.003 (2013). [DOI] [PubMed] [Google Scholar]
- Braeuning A. et al. Comparative analysis and functional characterization of HC-AFW1 hepatocarcinoma cells: cytochrome P450 expression and induction by nuclear receptor agonists. Drug Metab. Dispos. 43, 1781–1787, doi: 10.1124/dmd.115.064667 (2015). [DOI] [PubMed] [Google Scholar]
- Yokohira M. et al. Long-term chronic toxicity and mesothelial cell reactions induced by potassium octatitanate fibers (TISMO) in the left thoracic cavity in A/J female mice. Int. J. Toxicol. 34, 325–335, doi: 10.1177/1091581815587744 (2015). [DOI] [PubMed] [Google Scholar]
- Avior Y. et al. Microbial-derived lithocholic acid and vitamin K2 drive the metabolic maturation of pluripotent stem cells-derived and fetal hepatocytes. Hepatology 62, 265–278, doi: 10.1002/hep.27803 (2015). [DOI] [PubMed] [Google Scholar]
- Kelaini S., Cochrane A. & Margariti A. Direct reprogramming of adult cells: avoiding the pluripotent state. Stem cells and cloning: advances and applications 7, 19–29, doi: 10.2147/SCCAA.S38006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf T. & Enver T. Forcing cells to change lineages. Nature 462, 587–594, doi: 10.1038/nature08533 (2009). [DOI] [PubMed] [Google Scholar]
- Xu J., Du Y. & Deng H. Direct lineage reprogramming: strategies, mechanisms, and applications. Cell stem cell 16, 119–134, doi: 10.1016/j.stem.2015.01.013 (2015). [DOI] [PubMed] [Google Scholar]
- Huang P. et al. Direct reprogramming of human fibroblasts to functional and expandable hepatocytes. Cell stem cell 14, 370–384, doi: 10.1016/j.stem.2014.01.003 (2014). [DOI] [PubMed] [Google Scholar]
- Crestani M., Galli G. & Chiang J. Y. Genomic cloning, sequencing, and analysis of the hamster cholesterol 7 alpha-hydroxylase gene (CYP7). Arch. Biochem. Biophys. 306, 451–460, doi: 10.1006/abbi.1993.1537 (1993). [DOI] [PubMed] [Google Scholar]
- Martovetsky G., Bush K. T. & Nigam S. K. Kidney versus liver specification of SLC and ABC drug transporters, tight junction molecules, and biomarkers. Drug Metab. Dispos. 44, 1050–1060, doi: 10.1124/dmd.115.068254 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K., Kock K., Sedykh A., Tropsha A. & Brouwer K. L. An updated review on drug-induced cholestasis: mechanisms and investigation of physicochemical properties and pharmacokinetic parameters. J. Pharm. Sci. 102, 3037–3057, doi: 10.1002/jps.23584 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt H. S. et al. Bile acids in drug induced liver injury: key players and surrogate markers. Clin. Res. Hepatol. Gastroenterol. 40, 257–266, doi: 10.1016/j.clinre.2015.12.017 (2016). [DOI] [PubMed] [Google Scholar]
- Hofmann A. F. Bile acids: the good, the bad, and the ugly. News Physiol. Sci. 14, 24–29 (1999). [DOI] [PubMed] [Google Scholar]
- Hofmann A. F. & Hagey L. R. Bile acids: chemistry, pathochemistry, biology, pathobiology, and therapeutics. Cell. Mol. Life Sci. 65, 2461–2483, doi: 10.1007/s00018-008-7568-6 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. et al. Comparative regulation of major enzymes in the bile acid biosynthesis pathway by cholesterol, cholate and taurine in mice and rats. Life Sci. 77, 746–757, doi: 10.1016/j.lfs.2004.11.036 (2005). [DOI] [PubMed] [Google Scholar]
- Chiang J. Y. Bile acid metabolism and signaling. Compr. Physiol. 3, 1191–1212, doi: 10.1002/cphy.c120023 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo G. L. et al. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J. Biol. Chem. 278, 45062–45071, doi: 10.1074/jbc.M307145200 (2003). [DOI] [PubMed] [Google Scholar]
- Kosters A. & Karpen S. J. Bile acid transporters in health and disease. Xenobiotica; the fate of foreign compounds in biological systems 38, 1043–1071, doi: 10.1080/00498250802040584 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor F. A shift in paradigm towards human biology-based systems for cholestatic-liver diseases. J. Physiol. 593, 5043–5055, doi: 10.1113/JP271124 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier P. J. & Stieger B. Bile salt transporters. Annu. Rev. Physiol. 64, 635–661, doi: 10.1146/annurev.physiol.64.082201.100300 (2002). [DOI] [PubMed] [Google Scholar]
- Deharde D. et al. Bile canaliculi formation and biliary transport in 3D sandwich-cultured hepatocytes in dependence of the extracellular matrix composition. Arch. Toxicol., doi: 10.1007/s00204-016-1758-z (2016). [DOI] [PubMed] [Google Scholar]
- Goodwin B. et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell 6, 517–526 (2000). [DOI] [PubMed] [Google Scholar]
- Benedetti A. et al. Cytotoxicity of bile salts against biliary epithelium: a study in isolated bile ductule fragments and isolated perfused rat liver. Hepatology 26, 9–21, doi: 10.1002/hep.510260102 (1997). [DOI] [PubMed] [Google Scholar]
- Morgan R. E. et al. Interference with bile salt export pump function is a susceptibility factor for human liver injury in drug development. Toxicol. Sci. 118, 485–500, doi: 10.1093/toxsci/kfq269 (2010). [DOI] [PubMed] [Google Scholar]
- Chatterjee S., Richert L., Augustijns P. & Annaert P. Hepatocyte-based in vitro model for assessment of drug-induced cholestasis. Toxicol. Appl. Pharmacol. 274, 124–136, doi: 10.1016/j.taap.2013.10.032 (2014). [DOI] [PubMed] [Google Scholar]
- Kis E. et al. Effect of membrane cholesterol on BSEP/Bsep activity: species specificity studies for substrates and inhibitors. Drug Metab. Dispos. 37, 1878–1886, doi: 10.1124/dmd.108.024778 (2009). [DOI] [PubMed] [Google Scholar]
- Pan G., Boiselle C. & Wang J. Assessment of biliary clearance in early drug discovery using sandwich-cultured hepatocyte model. J. Pharm. Sci. 101, 1898–1908, doi: 10.1002/jps.23070 (2012). [DOI] [PubMed] [Google Scholar]
- Sousa T. et al. Deoxycholic acid modulates cell death signaling through changes in mitochondrial membrane properties. J. Lipid Res. 56, 2158–2171, doi: 10.1194/jlr.M062653 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thulin P., Bamberg K., Buler M., Dahl B. & Glinghammar B. The peroxisome proliferator-activated receptor alpha agonist, AZD4619, induces alanine aminotransferase-1 gene and protein expression in human, but not in rat hepatocytes: correlation with serum ALT levels. Int. J. Mol. Med. 38, 961–968, doi: 10.3892/ijmm.2016.2681 (2016). [DOI] [PubMed] [Google Scholar]
- Song S. H. & Lim C. J. Nitrogen depletion causes up-regulation of glutathione content and gamma-glutamyltranspeptidase in Schizosaccharomyces pombe. J. Microbiol. 46, 70–74, doi: 10.1007/s12275-007-0244-y (2008). [DOI] [PubMed] [Google Scholar]
- Rawat N. et al. Curcumin abrogates bile-induced NF-kappaB activity and DNA damage in vitro and suppresses NF-kappaB activity whilst promoting apoptosis in vivo, suggesting chemopreventative potential in Barrett’s oesophagus. Clin. Transl. Oncol. 14, 302–311, doi: 10.1007/s12094-012-0799-x (2012). [DOI] [PubMed] [Google Scholar]
- Peres W. et al. The flavonoid quercetin ameliorates liver damage in rats with biliary obstruction. J. Hepatol. 33, 742–750 (2000). [DOI] [PubMed] [Google Scholar]
- Muriel P. & Moreno M. G. Effects of silymarin and vitamins E and C on liver damage induced by prolonged biliary obstruction in the rat. Basic Clin. Pharmacol. Toxicol. 94, 99–104 (2004). [DOI] [PubMed] [Google Scholar]
- Li Y. et al. Quercetin protects rat hepatocytes from oxidative damage induced by ethanol and iron by maintaining intercellular liable iron pool. Hum. Exp. Toxicol. 33, 534–541, doi: 10.1177/0960327113499168 (2014). [DOI] [PubMed] [Google Scholar]
- Woudenberg-Vrenken T. E., Conde de la Rosa L., Buist-Homan M., Faber K. N. & Moshage H. Metformin protects rat hepatocytes against bile acid-induced apoptosis. PloS one 8, e71773, doi: 10.1371/journal.pone.0071773 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bruyn T. et al. Sandwich-cultured hepatocytes: utility for in vitro exploration of hepatobiliary drug disposition and drug-induced hepatotoxicity. Expert Opin. Drug Metab. Toxicol. 9, 589–616, doi: 10.1517/17425255.2013.773973 (2013). [DOI] [PubMed] [Google Scholar]
- LeCluyse E. L., Audus K. L. & Hochman J. H. Formation of extensive canalicular networks by rat hepatocytes cultured in collagen-sandwich configuration. Am. J. Physiol. 266, C1764–1774 (1994). [DOI] [PubMed] [Google Scholar]
- Liu X., Chism J. P., LeCluyse E. L., Brouwer K. R. & Brouwer K. L. Correlation of biliary excretion in sandwich-cultured rat hepatocytes and in vivo in rats. Drug Metab. Dispos. 27, 637–644 (1999). [PubMed] [Google Scholar]
- Bachour-El Azzi P. et al. Comparative localization and functional activity of the main hepatobiliary transporters in HepaRG cells and primary human hepatocytes. Toxicol. Sci. 145, 157–168, doi: 10.1093/toxsci/kfv041 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C. et al. Alpha-naphthylisothiocyanate modulates hepatobiliary transporters in sandwich-cultured rat hepatocytes. Toxicol. Lett. 224, 93–100, doi: 10.1016/j.toxlet.2013.09.019 (2014). [DOI] [PubMed] [Google Scholar]
- Li X., Zhong K., Guo Z., Zhong D. & Chen X. Fasiglifam (TAK-875) inhibits hepatobiliary transporters: a possible factor contributing to fasiglifam-induced liver injury. Drug Metab. Dispos 43, 1751–1759 (2015). [DOI] [PubMed] [Google Scholar]
- Wu Z. T. et al. Timosaponin A3 induces hepatotoxicity in rats through inducing oxidative stress and down-regulating bile acid transporters. Acta Pharmacol. Sin. 35, 1188–1198, doi: 10.1038/aps.2014.65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B. & Whiteman M. Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br. J. Pharmacol. 142, 231–255, doi: 10.1038/sj.bjp.0705776 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchini-Pascal N., de Sousa G., Pizzol J. & Rahmani R. Pregnane X receptor activation protects rat hepatocytes against deoxycholic acid-induced apoptosis. Liver Int. 30, 284–297, doi: 10.1111/j.1478-3231.2009.02108.x (2010). [DOI] [PubMed] [Google Scholar]