

Abstract
Background
The switch from photosynthetic or predatory to parasitic life strategies by apicomplexans is accompanied with a reductive evolution of genomes and losses of metabolic capabilities. Cryptosporidium is an extreme example of reductive evolution among apicomplexans, with losses of both the mitosome genome and many metabolic pathways. Previous observations on reductive evolution were largely based on comparative studies of various groups of apicomplexans. In this study, we sequenced two divergent Cryptosporidium species and conducted a comparative genomic analysis to infer the reductive evolution of metabolic pathways and differential evolution of invasion-related proteins within the Cryptosporidium lineage.
Results
In energy metabolism, Cryptosporidium species differ from each other mostly in mitosome metabolic pathways. Compared with C. parvum and C. hominis, C. andersoni possesses more aerobic metabolism and a conventional electron transport chain, whereas C. ubiquitum has further reductions in ubiquinone and polyisprenoid biosynthesis and has lost both the conventional and alternative electron transport systems. For invasion-associated proteins, similar to C. hominis, a reduction in the number of genes encoding secreted MEDLE and insulinase-like proteins in the subtelomeric regions of chromosomes 5 and 6 was also observed in C. ubiquitum and C. andersoni, whereas mucin-type glycoproteins are highly divergent between the gastric C. andersoni and intestinal Cryptosporidium species.
Conclusions
Results of the study suggest that rapidly evolving mitosome metabolism and secreted invasion-related proteins could be involved in tissue tropism and host specificity in Cryptosporidium spp. The finding of progressive reduction in mitosome metabolism among Cryptosporidium species improves our knowledge of organelle evolution within apicomplexans.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3343-5) contains supplementary material, which is available to authorized users.
Keywords: Reductive evolution, Genomics, Mitosome metabolism, Apicomplexa, Cryptosporidium
Background
The evolution of life generally proceeds towards bigger genomes and increased complexity, as the organisms adapt to new niches and environment. Recent evolutionary reconstructions, however, have shown a common occurrence of genome reduction, especially in parasitic and symbiotic organisms [1]. Among alveolates, a group of unicelluar eukaryotes consisted of photosynthetic protozoa, free-living predators, and obligate intracellular parasitic protozoa, reductive evolution is often observed in parasitic apicomplexans. For example, compared with the closely related chromerids, the photosynthetic algae, a significant reduction in genome sizes has occurred in apicomplexans [2]. Among apicomplexans, Cryptosporidium spp. and gregarines have lost the apicoplast, a plastid without photosynthetic functions, and depend on host cells for basic nutrients [3–6]. It is generally accepted that Cryptosporidium spp. as the based branch of Apicomplexa have also lost many other metabolic capabilities during the reductive evolution, especially the mitochondria-like organelle-derived energy metabolism, such as the tricarboxylic acid (TCA) cycle and cytochrome-based electron transport chain [4, 5, 7]. Cryptosporidium muris, however, has been shown recently to have all enzymes associated with the TCA cycle and a conventional respiratory chain system [8].
Cryptosporidium spp. are major causes of diarrhea in human and other animals, is [9]. To date, about 30 Cryptosporidium species have been recognized in humans, livestock, companion animals, and wild vertebrates [10]. They differ from each other in host specificity and predilection sites [10]. Among them, C. parvum and C. hominis are intestinal species and common causes of human cryptosporidiosis [11]. Although C. hominis is largely a pathogen of humans and nonhuman primates, C. parvum is also a major pathogen in ruminants. Recently, another intestinal Cryptosporidium species, C. ubiquitum, has been detected in humans in industrialized nations [12, 13]. Like C. parvum, this species has a broad host range and can infect other primates, domestic and wild ruminants, and rodents [12, 13]. In contrast, C. andersoni is a gastric species in cattle and has only been detected occasionally in other animal species [10, 14]. It is genetically related to another gastric species, C. muris, which infects a broad range of mammals and occasionally birds [15]. Like C. hominis, most other recognized Cryptosporidium species have some host specificity [10].
The genomes of C. parvum [5] and C. hominis [4] were sequenced using the Sanger technology and published in 2004. C. muris was also sequenced subsequently and its genome has been available in GenBank and CryptoDB (release 3.5) since 2007. All Cryptosporidium genomes presumably have 8 chromosomes, are around 9 Mb in size, and are more compact and efficient than genomes of most other apicomplexans [4, 5]. The predicted proteomes are highly similar between the two intestinal species C. parvum and C. hominis. However, a preliminary analysis of the C. muris genomic data has shown significant divergence in mitosome carbon and energy metabolism [8]. Because of the overall nucleotide sequence divergence between the C. parvum and C. hominis genomes is just ~3%, it has been suggested that differences in phenotypic characteristics between the two species, such as host range [11] and host cell invasion [16], may be caused by subtle sequence variations in coding regions or differences in expression levels of key genes rather than genome rearrangements and structural alterations [17]. Recently, several major insertions and deletions in gene content have been identified between the two closely related intestinal species, and it was suggested that subtelomeric gene duplications and deletions in two secreted protein families (MEDLE and insulinase-like proteins) in chromosomes 5 and 6 could be responsible for some of the observed biologic differences between C. parvum and C. hominis [18].
Although the first two genomes of Cryptosporidium spp. were sequenced over a decade ago, studies on genome evolution within the Cryptosporidium lineage is practically non-existent. As a result, we still have very limited knowledge of the evolution and invasion of Cryptosporidium spp. This is largely the result of only a limited number of Cryptosporidium species sequenced at the whole genome level. Here, we sequenced the genomes of six C. ubiquitum and C. andersoni isolates and conducted a comparative genomic analysis of Cryptosporidium spp. and other well-studied apicomplexans. We focused on reductive evolution in energy metabolism and differential evolution of invasion-related proteins among Cryptosporidium species, as they may be involved in tissue tropism and host specificity.
Results
Genome reduction in Cryptosporidium
The genomes of three isolates each of C. ubiquitum and C. andersoni were sequenced. The best assembly was 8.97 Mb in 27 contigs for C. ubiquitum and 9.10 Mb in 96 contigs for C. andersoni (Additional file 1: Table S1).
Genomes of all Cryptosporidium spp. have similar structural features, including genome size, GC content, number of tRNA genes, codon usages, and over-represented DNA sequence motifs in upstream regions of protein-coding genes (Table 1 and Additional file 2: Figure S1). Gene organization of chromosomes is in almost complete synteny and sequences identity is higher between the genomes of C. ubiquitum and C. parvum. In contrast, the genome sequence of C. andersoni is very different from that of C. parvum (Additional file 3: Figure S2A). Altogether, 3767 and 3905 protein-coding genes were identified in the C. ubiquitum and C. andersoni genomes, respectively (Table 1).
Table 1.
Genomic features of Cryptosporidium ubiquitum and C. andersoni in comparison with other apicomplexan parasitesa
| P. falciparum | T. gondii | C. parvum | C. hominis | C. ubiquitum | C. andersoni | |
|---|---|---|---|---|---|---|
| Total length (Mb) | 22.85 | 65.67 | 9.10 | 8.74 | 8.97 | 9.09 |
| No. of super contigs | 16 | 2,263 | 8 | 1,422 | 27 | 135 |
| GC content (%) | 20.0 | 48.5 | 30.3 | 30.9 | 30.8 | 28.5 |
| No. of genes | 5,542 | 8,322 | 3,805 | 3,886 | 3,767 | 3,905 |
| Total length of CDS (Mb)b | 12.58 | 20.03 | 6.83 | 5.28 | 6.94 | 6.86 |
| GC content in CDS (%) | 25.0 | 56.0 | 31.9 | 32.7 | 33.0 | 30.1 |
| GC content at 3rd position in codons (%) | 18.0 | 59.0 | 18.0 | 19.0 | 20.0 | 14.0 |
| Mean length of genes (bp) | 2,271 | 2,407 | 1,720 | 1,360 | 1,841 | 1,757 |
| Gene density (gene/Mb) | 242.5 | 126.7 | 418.1 | 444.6 | 420.0 | 429.6 |
| Percent coding (%) | 55.1 | 30.5 | 75.0 | 60.4 | 77.4 | 75.5 |
| No. of genes with intron | 3,055 | 6,729 | 163 | 8 | 758 | 832 |
| Genes with intron (%) | 55.1 | 80.9 | 4.2 | 0.02c | 20.1 | 21.3 |
| No. of tRNA | 72 | 174 | 45 | 46 | 45 | 44 |
| No. of tRNAmet | 2 | 8 | 2 | 2 | 2 | 2 |
| Proteins with signal peptide | 638 | 759 | 397 | 421 | 399 | 309 |
| Proteins with transmembrane domain | 1,754 | 1,103 | 832 | 769 | 772 | 839 |
| Proteins with GPI anchor | 62 | 255 | 63 | 51 | 50 | 47 |
aSource of data: Plasmodium falciparum: PlasmoDB release-11.1; Toxoplasma gondii: ToxoDB release-11.0; C. parvum and C. hominis: CryptoDB release-6.0
bCoding regions excluding intron sequences
cEstimated to be 5–20% by Xu et al., [4]
The Cryptosporidium species under analysis share a large group of orthologs, with only a small number of species-specific genes. As expected, the number of species-specific genes in C. andersoni is apparently bigger than that among intestinal species (Additional file 3: Figure S2B). The divergent nature of C. andersoni is supported by phylogenetic analysis of 100 orthologs from Cryptosporidium spp. and other common apicomplexan parasites (Fig. 1a). All Cryptosporidium species have similar numbers and components of major protein families, except for C. andersoni, which has fewer genes encoding insulinase-like peptidases (Fig. 1b).
Fig. 1.
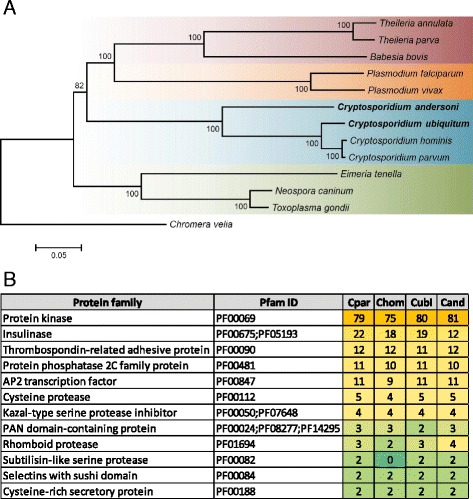
Genomic features and phylogenetic relationship of Cryptosporidium spp. a Phylogenetic relationship between Cryptosporidium spp. and other common apicomplexan parasites based on neighbor-joining analysis of sequences of 100 shared proteins. A concatenated sequences from the free-living photosynthetic chromerid, Chromera velia, was used to root the tree. b Comparison of major invasion-related protein families among Cryptosporidium species. The number of each protein family was identified based on Pfam domain search only, which may differs from the result of ortholog comparisons. Abbreviation of Cryptosporidium spp.: Cpar: Cryptosporidium parvum; Chom: C. hominis; Cubi: C. ubiquitum; Cand: C. andersoni
The metabolic capabilities of the four Cryptosporidium spp. are dramatically reduced compared with P. falciparum and T. gondii. This is especially reflected in carbohydrate, amino acid, and energy metabolism, with a limited de novo biosynthesis and overall reliance on the hosts for basic nutrients by Cryptosporidium spp. (Table 2). Other reductive evolutionary features in Cryptosporidium spp. include reduced gene numbers and length and increased gene density (Table 1), which were previously observed in C. parvum and C. hominis genomes [4, 5]. As expected, no apicoplast and mitochondrial genomes were detected in any Cryptosporidium species.
Table 2.
Comparison of essential metabolic pathways among Cryptosporidium spp. and other common apicomplexan parasitesa
| Category | Metabolic pathway | Cpar | Chom | Cubi | Cand | Pfal | Tgon |
|---|---|---|---|---|---|---|---|
| Carbohydrate and energy metabolism | Glycolysis | + | + | + | + | + | + |
| Methylcitrate cycle | - | - | - | - | - | + | |
| TCA cycle | - | - | - | + | + | + | |
| Pentose phosphate pathway | - | - | - | - | + | + | |
| Shikimate biosynthesis | - | - | - | - | + | + | |
| Folate biosynthesis | - | - | - | - | + | + | |
| Synthesis of tetrahydrobiopterin/dihydrobiopterin/molybdopterin | - | - | - | - | - | + | |
| Galactose metabolism | - | - | - | - | - | + | |
| Synthesis of starch | + | + | + | + | - | + | |
| Synthesis of trehalose | + | + | + | + | - | + | |
| Synthesis of 1,3-beta-glucan | - | - | - | - | - | + | |
| UDP-Glc < - > UDP-Gal | + | + | + | + | - | + | |
| GDP-Man < - > GDP-Fuc | - | - | - | - | + | + | |
| UDP-Glc - > UDP-GlcA - > UDP-Xyl | + | + | + | + | - | - | |
| Synthesis of mannitol from fructose | + | + | + | + | - | - | |
| Fatty acid biosynthesis in cytosol (FAS I) | + | + | + | + | - | + | |
| Fatty acid biosynthesis in apicoplast (FAS II) | - | - | - | - | + | + | |
| Fatty acid degradation | - | - | - | - | - | + | |
| Oxidative phosphorylation (NADH dehydrogenase) | + | + | + | + | + | + | |
| Oxidative phosphorylation (Complex II) | - | - | - | + | + | + | |
| Oxidative phosphorylation (Complex III) | - | - | - | 1 subunit | + | + | |
| Oxidative phosphorylation (Complex IV) | - | - | - | - | + | + | |
| F-ATPase | 2 subunit | 2 subunit | 2 subunit | + | + | + | |
| Alternative oxidase (AOX) | + | + | + | + | - | - | |
| Glyoxalase metabolism producing D-lactate | - | - | - | - | + | + | |
| Synthesis of isoprene (MEP/DOXP) | - | - | - | - | + | + | |
| Synthesis of farnesyl/polyprenyl diphosphate | + | + | - | + | + | + | |
| Nucleotide metabolism | Synthesis of purine rings de novo | - | - | - | - | - | - |
| IMP - > XMP - > GMP | + | + | + | - | + | + | |
| Synthesis of pyrimidine de novo | - | - | - | - | + | + | |
| Amino acid metabolism | Synthesis of alanine from pyruvate | - | - | - | - | - | + |
| Synthesis of glutamate from nitrite/nitrate | - | - | - | - | + | + | |
| Conversion from glutamate to glutamine | + | + | + | + | + | + | |
| Synthesis of aspartate from oxaloacetate and glutamate | - | - | - | - | + | + | |
| Conversion from aspartate to asparagine | + | + | + | - | + | + | |
| Conversion from glutamate to proline | + | + | + | + | - | + | |
| Synthesis of serine from glycerate/glycerol phosphate | - | - | - | - | - | + | |
| Conversion from serine to cysteine | - | - | - | - | - | + | |
| Conversion from serine to glycine | + | + | + | + | + | + | |
| Recycle homocysteine into methionine | - | - | - | - | + | + | |
| Synthesis of lysine from aspartate | - | - | - | - | - | + | |
| Synthesis of threonine from aspartate | - | - | - | - | - | + | |
| Synthesis of ornithine from arginine | - | - | - | - | + | - | |
| Synthesis of ornithine from proline | - | - | - | - | + | + | |
| Synthesis of polyamine from ornithine | - | - | - | - | + | - | |
| Polyamine pathway backward | + | + | + | + | - | + | |
| Degradation of branch-chain amino acids | - | - | - | - | - | + | |
| Synthesis of tryptophan | + | + | + | - | - | - | |
| Aromatic amino acid hydroxylases (AAAH) | - | - | - | - | - | + | |
| Vitamin and others | Synthesis of ubiquinone (Coenzyme Q) | + | + | - | + | + | + |
| Synthesis of Fe-S cluster | + | + | + | + | + | + | |
| Synthesis of heme | - | - | - | - | + | + | |
| Synthesis of thiamine (Vitamin B1) | - | - | - | - | + | - | |
| Conversion from thiamine to thiamine pyrophosphate (TPP) | - | - | - | - | + | + | |
| Synthesis of FMN/FAD from riboflavin | - | - | - | - | + | + | |
| Synthesis of pyridoxal phosphate (Vitamin B6) de novo | - | - | - | - | + | + | |
| Synthesis of NAD(P) + de novo from nicotinate/nicotinamide | - | - | - | - | + | + | |
| Synthesis of pantothenate from valine | - | - | - | - | - | + | |
| Synthesis of CoA from pantothenate | + | + | + | + | + | + | |
| Synthesis of lipoic acid de novo in apicoplast | - | - | - | - | + | + | |
| Salvage of lipoic acid in mitochondria | - | - | - | + | + | + | |
| Synthesis of porphyrin/cytochrome proteins | - | - | - | - | + | + |
aPlus symbol denotes that the essential enzymes for the pathway were identified, whereas minus symbol denotes that the essential enzymes for pathways were absent
Cpar Cryptosporidium parvum, Chom C. hominis, Cubi C. ubiquitum, Cand C. andersoni, Pfal Plasmodium falciparum Pfal, Tgon Toxoplasma gondii
Divergent mitosome metabolism among Cryptosporidium spp.
Compared with the canonical aerobic mitochondrion in most eukaryotic organisms, the mitosome of Cryptosporidium spp. has undergone remarkable reduction in size and function probably because of the anaerobic environment in the digestive tract [19]. The comparative analysis of metabolism of C. ubiquitum, C. andersoni and other Cryptosporidium spp. shows that mitosome metabolism, including the electron transport chain, has gone through progressive reductions within the Cryptosporidium genus (Fig. 2).
Fig. 2.
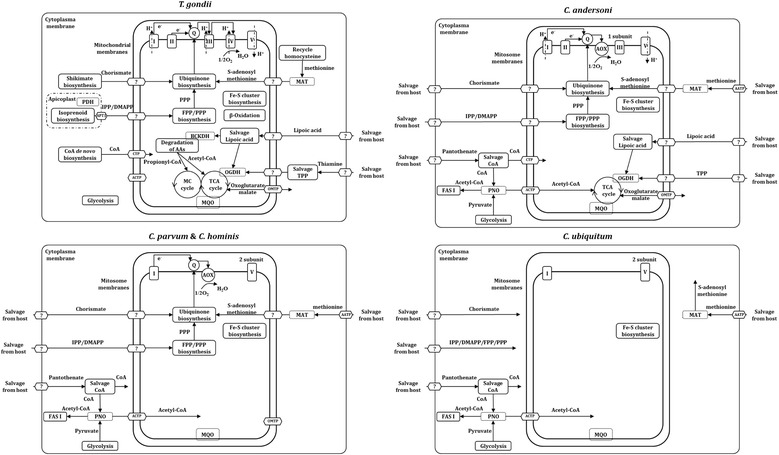
Reductive evolution in mitosome metabolism among Cryptosporidium spp. in comparison with Toxoplasma gondii. Abbreviation of enzymes: AOX: alternative oxidase; PDH: pyruvate dehydrogenase complex; PNO: pyruvate:NADP(+) oxidoreductase; MAT: methionine adenosyl transferase; OGDH: oxoglutarate dehydrogenase complex; MQO: malate:quinone oxidoreductase; BCKDH: branched-chain ketoacid dehydrogenase complex. Abbreviation of metabolites: Q: ubiquinone (coenzyme Q); CoA: coenzyme A; IPP: isopentenyl diphosphate; DMAPP: dimethylallyl diphosphate; FPP: farnesyl diphosphate; PPP: polyprenyl diphosphate; MC: 2-methylcitrate; TCA: tricarboxylic acid; TPP: thiamine pyrophosphate. Abbreviation of transporter proteins: APT1: apicoplast phosphate translocator; CTP: CoA transporter protein; ACTP: acetyl-CoA transporter protein; OMTP: oxoglutarate/malate transporter protein
TCA cycle and related metabolism in C. andersoni
Unlike intestinal Cryptosporidium spp., C. andersoni possesses a complete set of enzymes involved in the TCA cycle and related metabolism, such as the substrate transport system and salvage of the cofactor lipoic acid (Table 2, Fig. 2). In Plasmodium falciparum and Toxoplasma gondii, pyruvate derived from glycolysis is oxidated into acetyl-CoA by a multi-protein enzyme in the apicoplast, pyruvate dehydrogenase complex (PDH) [6]. Cryptosporidium spp. do not have this complex, but possess an alternative pyruvate:NADP+ oxidoreductase (PNO) localized in the cytosol [20]. The product, acetyl-CoA presumably is delivered into mitosomes through the transporter protein embedded on the mitosome membrane in C. andersoni. In other Cryptosporidium spp., acetyl-CoA is primarily involved in metabolic pathways in the cytosol, such as type I fatty acid biosynthesis, although this pathway was also detected in C. andersoni. The oxoglutarate dehydrogenase complex (OGDH) from the TCA cycle can become active when it couples with two cofactors, thiamine pyrophosphate (TPP) and lipoic acid. Among apicomplexans, only P. falciparum has the ability to synthesize thiamine de novo [21]. C. andersoni has to salvage TPP directly from the host, although its transporter has not been identified. In addition to synthesizing lipoic acid de novo in the apicoplast, P. falciparum and T. gondii can salvage lipoic acid from the host into the mitochondrion and conjugate it onto OGDH through the lipoate-protein ligase [22]. The gene encoding this ligase is present in the C. andersoni genome, indicating that C. andersoni salvages host lipoic acid and utilizes it presumably in mitosomes. Unlike T. gondii, which possesses de novo biosynthesis of CoA, all Cryptosporidium spp. salvage pantothenate from host cells and convert it into CoA.
Absence of polyisoprenoid biosynthesis in C. ubiquitum
In T. gondii, two 5-carbon isoprene units, isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), are synthesized de novo in the apicoplast from glycolysis-derived phosphonel pyruvate and dihydroxyacetone phosphate through the chloroplast-type MEP/DOXP pathway [6]. Both IPP and DMAPP are transported into the mitochondrion and condensed into farnesyl diphosphate (FPP) and polyprenyl diphosphate (PPP) [21, 23]. All Cryptosporidium spp. lack not only the apicoplast but also the genes coding enzymes for de novo isoprenoid biosynthesis (Table 2, Fig. 2). Within the subsequent mitosome polyisoprenoid anabolic pathway, two essential enzymes, FPP synthase and polyprenyl synthase, are absent in the predicted proteome of C. ubiquitum but present in other Cryptosporidium spp. The genes (cgd4_2550, cgd7_3730) encoding these two enzymes were shown transcribed in C. parvum in vitro, especially during early (2–6 h) infection [24]. Artz et al. suggested that the enzyme encoded by cgd4_2550 is a non-specific polyprenyl pyrophosphate synthase rather than FPP synthase only [25]. These two enzymes may cooperatively synthesize pyrophosphoric polyisoprenoids in C. parvum. As PPP is an essential substrate for the biosynthesis of ubiquinone, the absence of polyisprenoid biosynthesis in C. ubiquitum is consistent with its loss of ubiquinone biosynthesis (see below). In addition to participating in the biosynthesis of ubiquinone, isoprenoids such as FPP and PPP are involved in signaling pathways, post-translational modifications of proteins, and isoprenylation of tRNAs [6]. C. ubiquitum possesses enzymes involved in the isoprenylation of both proteins and tRNAs, suggesting that it may salvage FPP and PPP in addition to IPP and DMAPP from the host.
The absence in C. ubiquitum of three mitosome carrier proteins possessed by C. parvum further demonstrates that mitosome metabolic capabilities in C. ubiquitum are more limited than those of other Cryptosporidium spp. (Table 3). Because of the existence of more metabolic pathways within mitosomes, C. andersoni has several more mitosome transporter proteins than other Cryptosporidium spp. In addition, C. andersoni possesses two more amino acid transporters and one more potassium transporter than other Cryptosporidium spp. (Table 3).
Table 3.
Putative transporters in Cryptosporidium spp. and other common apicomplexan parasitesa
| Substrates | Cellular location | Cpar | Chom | Cubi | Cand | Pfal | Tgon |
|---|---|---|---|---|---|---|---|
| Hexose | 2 | 2 | 2 | 2 | 2 | 5 | |
| Triose phosphate | Plasma/Apicoplast membrane | 8 | 5 | 8 | 8 | 4 | 4 |
| Amino acids | Plasma membrane | 10 | 10 | 10 | 12 | 1 | 6 |
| Nucleobase/nucleoside | Plasma membrane | 1 | 1 | 1 | 1 | 4 | 4 |
| Nucleotide-sugar | Plasma membrane | 3 | 3 | 3 | 2 | 1 | 4 |
| Folate/pterine | Plasma membrane | 1 | 1 | 1 | 1 | 2 | 7 |
| Formate/nitrite | 0 | 0 | 0 | 0 | 1 | 3 | |
| GABA (aminobutanoate) | Plasma/Mitochondrial membrane | 0 | 0 | 0 | 0 | 2 | 5 |
| Acetyl-CoA | 1 | 0 | 1 | 1 | 1 | 1 | |
| Chloride | 0 | 0 | 0 | 0 | 0 | 2 | |
| Inorganic phosphate | 0 | 0 | 0 | 0 | 1 | 1 | |
| Sulfate | 1 | 1 | 1 | 1 | 1 | 4 | |
| Sodium/potassium/calcium | 2 | 2 | 2 | 3 | 0 | 9 | |
| Zinc | 2 | 2 | 2 | 2 | 2 | 4 | |
| Copper | 1 | 1 | 1 | 1 | 2 | 3 | |
| Choline | Plasma membrane | 0 | 0 | 0 | 0 | 1 | 2 |
| Cadmium/zinc/cobalt (efflux) | Plasma membrane | 1 | 1 | 1 | 1 | 1 | 1 |
| Glycerol/water | Plasma membrane | 0 | 0 | 0 | 0 | 2 | 2 |
| ABC transporterb | Plasma membrane | 21 | 18 | 21 | 21 | 16 | 24 |
| Mitochondrial carrierb | Mitochondrial membrane | 9 | 7 | 6 | 13 | 14 | 21 |
aThe detection of these transporter proteins was based on the Pfam search results
bABC transporters and mitochondrial carriers have a broad range of substrates
Cpar Cryptosporidium parvum, Chom C. hominis, Cubi C. ubiquitum, Cand C. andersoni, Pfal Plasmodium falciparum Pfal, Tgon Toxoplasma gondii
Progressive reduction in electron transport chain in Cryptosporidium spp.
Within the oxidative phosphorylation pathway on the inner mitochondrial membrane, the classical NADH dehydrogenase multi-protein complex, named complex I, is substituted by an alternative single NADH dehydrogenase in most apicomplexan parasites [21]. Similar to P. falciparum and T. gondii, which use three other multi-protein complexes (II-IV) to transfer electrons to oxygen, C. andersoni has the complete complex II, succinate dehydrogenase, which is also involved in the TCA cycle, and one subunit of complex III, ubiquinol-cytochrome c oxidoreductase. In contrast, C. parvum, C. hominis, and C. ubiquitum have lost these enzyme complexes entirely (Table 2, Fig. 2). With electrons transported through the chain, a proton gradient is generated across the inner mitochondrial membrane and the energy generated can be used to produce ATP by the ATP synthase (also known as complex V). Similar to P. falciparum and T. gondii, C. andersoni possesses a complete F-type ATP synthase, but only two subunits (subunit α and β) of this enzyme are present in C. parvum, C. hominis and C. ubiquitum.
Ubiquinone, also known as coenzyme Q, is responsible for transferring electrons from complex I or complex II to complex III. It is synthesized from chorismate, which has to be salvaged from the host because of the absence of the shikimate pathway in Cryptosporidium spp., together with two other substrates polyisoprenoid and adenosyl methionine [21]. C. parvum, C. hominis, and C. andersoni possess all enzymes and proteins involved in the ubiquinone biosynthesis [21]. Most of these enzymes have experimental expression evidence in C. parvum and show a higher expression level at the end (36–72 h) of the in vitro infection [24]. The absence of five additional enzymes suggests that C. ubiquitum lacks the capability of ubiquinone biosynthesis (Table 2, Fig. 2).
In addition to the conventional mitochondrial electron transport system, there is also a cytochrome-independent system executed by a cyanide-insensitive alternative oxidase (AOX), passing electrons from ubiquinone directly to oxygen to form water in higher plants, fungi and several protozoa [26–28]. All Cryptosporidium spp. possess orthologs of AOX [4, 5], with the exception of C. ubiquitum. This alternative pathway does not couple the proton transport across the inner mitosome membrane, indicating that energy production is reduced in intestinal Cryptosporidium spp. AOX in C. parvum can be inhibited by salicylhydroxamic acid and 8-hydroxyquinoline, affecting the growth of the parasite [27]. The mammalian bloodstream form of Trypanosoma brucei depends entirely on the AOX pathway for electron transport, making AOX an attractive target for drug development [29, 30]. The absence of AOX in C. ubiquitum suggests that the electron transport chain is totally lost in this apicomplexan parasite (Table 2, Fig. 2).
Unexpectedly, the gene encoding malate:quinone oxidoreductase (MQO), the enzyme involved in both the TCA cycle and electron transport chain, is present in most apicomplexans including C. parvum, C. hominis and C. ubiquitum (Fig. 2). MQO was presumably gained by the ancestors of apicomplexans, chromerids, and dinoflagellates from bacteria though lateral gene transfer [8].
Reductive evolution in biosynthesis of N-glycan and GPI-anchor precursors in Cryptosporidium spp.
Asparagine (N)-linked glycosylation is a common post-translational modification of proteins and the biosynthesis of N-glycans has been shown to be different among apicomplexan parasites [31]. Due to the secondary loss of Alg genes, apicomplexans differ from each other in the length of N-glycan precursors; the length of oligosaccharide chain of N-glycan precursors in Cryptosporidium spp. is shorter than that in T. gondii but longer than in P. falciparum (Fig. 3a). Among Cryptosporidium spp., intestinal species C. parvum, C. hominis and C. ubiquitum possess nine sugars in N-glycan precursors, whereas the gastric species C. andersoni has only five sugars. The addition of two mannose molecules and two glucose molecules onto the core structure of N-glycan precursor is lost in C. andersoni.
Fig. 3.
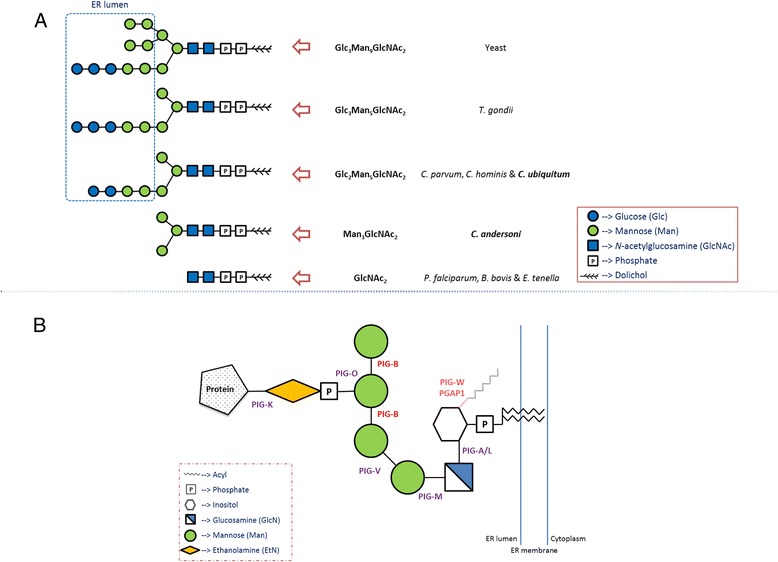
Diversity in post-translational modifications of proteins among Cryptosporidium spp. A) Divergent N-glycan precursors in different Cryptosporidium spp. and other apicomplexan parasites. B) Core structure of GPI-anchor and the critical enzymes involved in its biosynthesis. PIG-B is absent in Cryptosporidium ubiquitum but present in other Cryptosporidium spp., whereas PIG-W and PGAP1 are absent in all Cryptosporidium spp.
A large number of surface proteins attach to the cell membrane via the glycosylphosphatidylinositol (GPI) anchor [32]. The construction of GPI-anchors occurs in three stages: i) biosynthesis of a GPI precursor in the endoplasmic reticulum (ER) membrane, ii) attachment of the GPI to the C-terminus of a newly synthesized protein in the ER lumen, and iii) lipid remodeling and/or carbohydrate side-chain modifications in ER and Golgi lumens. The critical mannosyltransferase (PIG-B) catalyzing the addition of the third and fourth mannose onto the GPI-anchor precursor located in the ER lumen is present in C. parvum, C. hominis and C. andersoni but absent in C. ubiquitum (Fig. 3b). The transcription of the PIG-B encoding gene (cgd3_3590) has been demonstrated in C. parvum, with a high level of expression at the end of in vitro infection [24]. In addition, unlike in other apicomplexan parasites, the acylation (mediated by PIG-W) and de-acylation (mediated by PGAP1) of inositol during the construction of GPI-anchor precursors in the ER lumen are absent in all Cryptosporidium spp.
Reductive evolutions in other metabolic pathways
The gene encoding tryptophan synthase (cgd5_4560) catalyzing biosynthesis of tryptophan from serine and indole was identified in intestinal Cryptosporidium spp. but not in the gastric species C. andersoni. It has been suggested that intestinal bacteria can provide indole to C. parvum and C. hominis whereas this bacterial community does not exist in the stomach of the host, leading to the absence of tryptophan biosynthesis in gastric Cryptosporidium spp. [21]. Similarly, the gene encoding asparagine synthase A (AsnA) (cgd5_4540), which converts aspartate into asparagine, is only absent in C. andersoni.
All apicomplexan parasites are unable to synthesize purine rings de novo and have to salvage them from the host. Only one purine nucleotide transporter was detected in Cryptosporidium spp. and it probably transports adenosine only [21]. C. parvum, C. hominis and C. ubiquitum convert AMP into GMP through IMP dehydrogenase (cgd6_20) and GMP synthase (cgd5_4520), which have not been identified in C. andersoni.
Differential evolution in invasion-related proteins
Apicomplexan parasites possess several unique secretory organelles such as the rhoptry, microneme, and dense granules, which secret various catalytic proteins for host cell adhesion and invasion [33]. Among secreted proteins of Cryptosporidium spp., two protein families, mucin-type glycoproteins and thrombospondin-related adhesive proteins (TRAPs), are considered essential to host cell attachment [34, 35]. The genes encoding mucin-type glycoproteins associated with adhesion, such as gp900, gp60/40/15, P23, P30, CP2, and Clec, were compared among the four Cryptosporidium species and results of the comparison have shown a high divergence of C. andersoni from other species in compositions of these genes (Fig. 4 and Additional file 4: Table S2). In C. parvum, the gp60/40/15 complex, which is absent in C. andersoni, is probably translated from one single mRNA, glycosylated, and proteolytically processed into two smaller glycoproteins. The latter are localized at the surface or apical region of the parasite and thought to be involved in attachment and invasion [34]. P23, a surface protein that induces antibody responses in animal hosts, is absent in C. andersoni but present in other Cryptosporidium spp. [34]. CP2, the membrane associated protein that is also absent in C. andersoni, has been shown to be localized in the parasitophorous vacuole membrane (PVM) and is probably involved in host cell invasion or PVM integrity [36]. In addition, seven small mucin genes (muc1-7) located in tandem in chromosome 2 of C. parvum are expressed during intracellular development and products of at least two of them, Muc4 and Muc5, are associated with host cell attachment and invasion [37]. Among them, Muc6 is absent in C. ubiquitum and none of them are present in C. andersoni. A major characteristic of these mucin proteins is their low similarity in amino acid sequences. Among the species-specific proteins, 5 and 30 are O-linked mucin-type glycoproteins in C. ubiquitum and C. andersoni, respectively (Additional file 5: Table S3). Some of them are probably responsible for host specificity in Cryptosporidium spp. In addition to host cell attachment, mucin-type glycoproteins may contribute to the tethering of sporozoites to the inner surface of the oocyst walls [38].
Fig. 4.
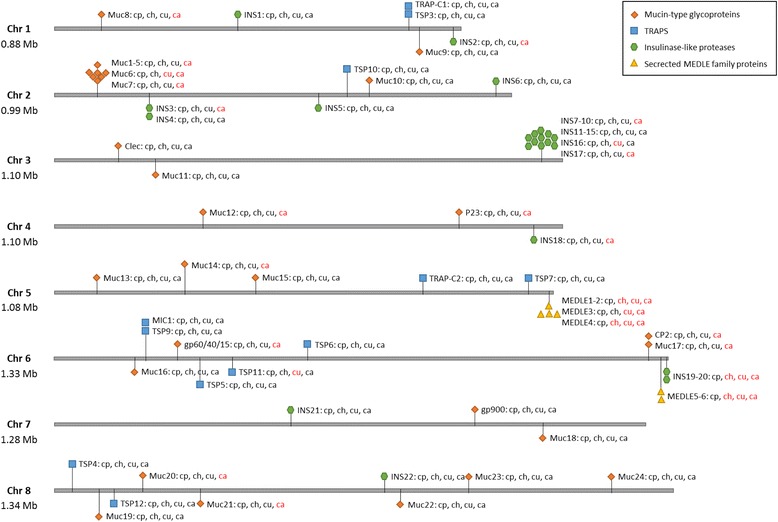
Diversity in invasion-related secreted protein and peptidase families among Cryptosporidium species. Abbreviation of Cryptosporidium spp.: cp: Cryptosporidium parvum; ch: C. hominis; cu: C. ubiquitum; ca: C. andersoni. The species name in red represents the absence of this family member
TRAPs, the best-characterized microneme proteins for gliding motility and host cell invasion in apicomplexans, usually contain two types of adhesive domains: the von Willebrand factor type A (VWA) domain and thrombospondin 1 (TSP1) domain, such as the TRAP of P. falciparum and MIC2 of T. gondii [39]. A screening of the TSP1 domain revealed that C. parvum possesses 12 paralogous genes (CpTSP1-12), which all have putative signal peptides and one or more TSP1 domains [40]. There are no VWA domains but other modules such as Kringle and epidermal growth factor (EGF) were detected within these TRAP genes. CpTSP8, previously known as CpMIC1 containing three TSP1 repeats and one EGF domain, is translocated to the surface of the parasite together with other microneme proteins [41]. All of these TRAP genes are present in the C. ubiquitum and C. andersoni genomes with the exception of TSP11, which is located in chromosome 6 and highly expressed in both early and late stages of C. parvum infection [40], is absent in C. ubiquitum (Fig. 4 and Additional file 4: Table S2).
A variety of secreted proteases and protein kinases in secretory organelles of apicomplexans are involved in processing invasion-related proteins or modifying host cell activities, enabling the evasion of host immune system during invasion [42]. Two members of the insulinase-like proteases containing the peptidase family M16 domain located in the subtelomeric region of chromosome 6 of C. parvum have been shown to be lost in the C. hominis genome. This is one of the few differences in gene content between these two closely related species with different host specificity [18]. In addition to the absences of these two subtelomeric genes on chromosome 6, one member (the ortholog of cgd3_4270, which has 90% nucleotide sequence similarity to cgd3_4260) of another insulinase gene cluster in the subtelomeric region of chromosome 3 of C. parvum is absent in C. ubiquitum. Additional paucity of this gene family was seen in C. andersoni, especially in genes located in subtelomeric regions. No new insulinase-like genes were identified in C. ubiquitum, although C. andersoni appears to have two or three new ones (Fig. 4 and Additional file 4: Table S2).
Another major genetic difference between genomes of C. parvum and C. hominis is in the number of genes encoding the MEDLE family surface proteins in the 3′ subtelomeric regions of chromosomes 5 and 6. Five of the six MEDLE family protein genes in C. parvum have no orthologs in C. hominis [18]. The two subtelomeric regions are both absent in genomes of C. ubiquitum and C. andersoni (Fig. 4 and Additional file 4: Table S2). Although the specific functions of these proteins have not been determined, most of them have a signal peptide.
Rhomboids, a family of intra-membrane serine proteases, are ubiquitously present in apicomplexan parasites and responsible for the cleavage of secreted adhesive proteins [43]. One additional rhomboid gene was detected in the genome of C. andersoni compared with other Cryptosporidium spp. (Additional file 4: Table S2).
Discussion
Whole genome sequencing and comparative genomic analysis have revealed a progressive reduction of mitosome metabolism in the genus Cryptosporidium, including major energy production mechanism, the electron transport chain, and the associated ancillary pathways. As shown in this study, like the genetically related C. muris [8], C. andersoni uses both the TCA cycle and glycolysis for energy metabolism and has a near conventional oxidative phosphorylation system. In contrast, C. parvum and C. hominis do not have the TCA cycle and possess an alternative oxidative phosphorylation chain, whereas C. ubiquitum has further lost the entire electron transport chain and the associated biosynthesis of ubiquinone and polyisoprenoids. It has been shown that Cryptosporidium spp. and gregarines differ from other apicomplexans in energy metabolism because of the absence of the intracellular organelle apicoplast and the presence of an alternative electron transport system. Data from this study now show a progressive reductive evolution in energy metabolism within the Cryptosporidium/gregarine lineage of apicomplexans. Thus far, energy metabolism and electron transport chain appear remarkably conserved in other lineages of apicomplexans (Fig. 5).
Fig. 5.
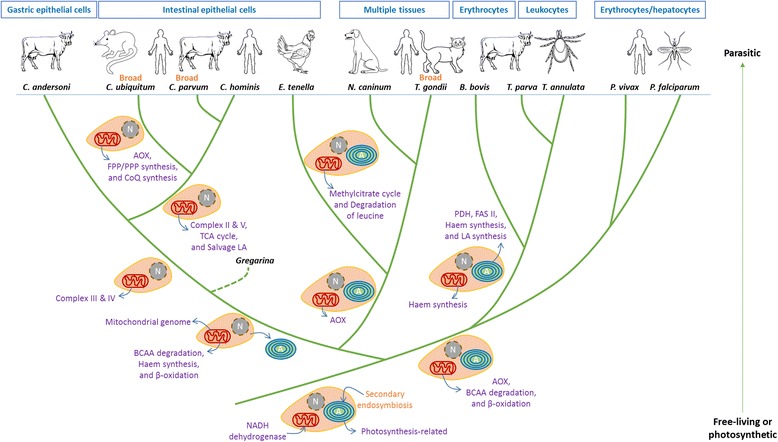
Reductive evolution of organelle-derived metabolism among major apicomplexan lineages. Abbreviation: AOX: alternative oxidase; FPP: farnesyl diphosphate; PPP: polyprenyl diphosphate; CoQ: coenzyme Q; TCA: tricarboxylic acid; LA: lipoic acid; BCAA: branched-chain amino acid; PDH: pyruvate dehydrogenase complex; FAS: fatty acid synthase
It is generally accepted that the ancestor of apicomplexan parasites obtained the mitochondrion and apicoplast through primary and secondary endosymbiosis, respectively [22]. During the reductive evolution process, most of the organelle metabolism-related genes have been progressively lost or horizontally transferred to nuclear genomes as different lineages adjust to new life strategies that differ from the photosynthetic or free-living lifestyle [2, 7]. Cryptosporidium spp. represent the extreme of apicomplexans, with the total loss of the apicoplast and having only a mitochondrial replica without the associated genome. The progressive losses of energy generation pathways and electron transport chains in Cryptosporidium spp. are probably results of adaptation to different niches of parasitism. As gregarines have retained several common features, such as the presence of a TCA cycle and a conventional electron transport chain, they probably represent ancestry members of the Cryptosporidium/gregarine lineage. Within the Cryptosporidium genus, C. andersoni appears to be an earlier member of the genus Cryptosporidium, whereas C. ubiquitum is probably a recent one (Fig. 5). Studies with molecular clocks are needed to confirm this theory.
The loss of enzymes involved in the biosynthesis of ubiquinone and polyisoprenoids in C. ubiquitum supports the conclusion on progressive evolution of energy metabolism and electron transport systems in the Cryptosporidium/gregarine lineage. Accompanying the loss of the conventional and alternative electron transport systems, C. ubiquitum has lost the ability to synthesize polyisoprenoids, which are substrates in ubiquinone biosynthesis, and ubiquinone itself, which is part of the conventional electron transport chain. By inspection of Mauve genome alignments, most of the genes that are absent in C. ubiquitum but present in C. parvum and C. hominis have syntenic nucleotide sequences in the C. ubiquitum genome, suggesting the occurrence of secondary losses of these functional genes in C. ubiquitum during species evolution. In agreement with this, C. andersoni, which has a TCA cycle, possesses several transporters for TCA substrates (such as acetyl CoA, oxoglutarate malate, and OGDH), more mitosome carrier proteins, and a salvage system for the cofactor lipoic acid. C. ubiquitum probably needs to salvage isoprenoids from the host for tRNA isoprenylation via enzyme miaA, which together with its downstream enzyme, miaB, is present in all Cryptosporidium genomes. Because isoprenylation of tRNA in apicomplexans occur mostly in the apicoplast [6], the ancestor of Cryptosporidium spp. probably had an apicoplast.
Reductive evolution apparently occurs in other metabolism processes in Cryptosporidium spp. Highly divergent N-glycan precursors have been seen among protists due to the secondary loss of Alg genes during reductive evolution [31]. Within the Cryptosporidium genus, the N-glycan precursor of the gastric C. andersoni has four fewer oligosaccharides than intestinal Cryptosporidium spp. This is the first time divergent N-glycan precursors are seen within a single genus of Apicomplexa, suggesting that the Alg genes are progressively lost after the emergence of Cryptosporidium spp. Similarly, the enzyme involved in the addition of the third mannose to the GPI anchor core structure, mannosyltransferase, is absent from the C. ubiquitum genome. Previously, mannosyltransferase was shown to be missing in Giardia and Entamoeba, while all enzymes in this pathway are absent in Trichomonas [31]. Recently, an absence of mannosyltransferases was detected in Eimeria falciformis [44].
Cryptosporidium spp. differ from each other in host specificity and tissue tropism. Presumably, the attachment and invasion of host cells are determinants for host specificity and tissue tropism, although differences in mitosome metabolism are probably also involved in the latter. Due to the relatively simple oral-fecal life cycle and the epicellular position of parasitism in restricted host cell type, the repertoire of invasion-related proteins is significantly reduced in Cryptosporidium spp. compared with other apicomplexans [45]. Thus far, the most obvious genetic differences between C. parvum and C. hominis are in the number of paralogous genes encoding secreted MEDLE family proteins and insulinase-like proteases in 3’subtelomeric regions of chromosomes 5 and 6 [18]. The role of these proteins in host specificity of Cryptosporidium spp. is supported by observations in this study, as C. ubiquitum and C. andersoni have lost altogether regions encoding these proteins. C. andersoni has further lost many genes of the insulinase-like proteases elsewhere in its genome. Mucin-type glycoproteins probably also play an important role in host specificity and tissue tropism, as they have shown high diversity in gene composition and sequences among Cryptosporidium spp., especially between the intestinal and gastric Cryptosporidium species. Other surface proteins such as TRAPs are known to be involved in invasion, but their role in host specificity determination is probably limited, as all Cryptosporidium spp. largely have the same class and number of TRAPs.
Results of this comparative genomic analysis have revealed drastic genetic differences between gastric and intestinal Cryptosporidium species. For example, C. andersoni differs from C. parvum, C. hominis, and C. ubiquitum significantly in not only genome organizations but also mitosome metabolism and invasion-associated secreted proteins and peptidases. Previously, gastric and intestinal Cryptosporidium species formed separate clades in phylogenetic analysis of the small subunit rRNA gene [46]. In addition to taxonomic implications, these findings suggest that the two groups of pathogens may require different strategies in the development of therapeutic agents. For example, Cryptosporidium spp. were known to use mainly glycolysis for energy metabolism and an alternative AOX pathway for electron transport. Based on findings from this study, drugs targeting these pathways probably are effective against only intestinal Cryptosporidium, as gastric species such as C. andersoni and C. muris also use TCA cycle for energy metabolism and have a near conventional oxidative phosphorylation pathway for electron transport. Although C. ubiquitum has genomic features and gene content very similar to C. parvum and C. hominis, it may respond to drugs differently. For example, the absence of the AOX electron transport system in C. ubiquitum suggests that ascofuranone, which is a well-known AOX inhibitor in T. brucei, a protozoon relying on the AOX pathway for electron transport [29], may not be effective against this intestinal Cryptosporidium species. Similarly, although the synthesis and metabolism of isoprenoids have been targets for drug development in apicomplexan parasites [22, 47], the absence of the apicoplast in Cryptosporidium spp. and polyisoprenoid biosynthesis in C. ubiquitum indicates that effectiveness of this approach is probably limited against Cryptosporidium spp., especially C. ubiquitum. Currently, there is no specific anti-parasitic agent against Cryptosporidium spp. The diversity in mitochondrial metabolism and invasion-related proteins within the Cryptosporidium genus suggests that different approaches may be needed in the development of therapeutic agents against gastric and intestinal Cryptosporidium spp.
Conclusions
In conclusion, we sequenced the genomes of six C. ubiquitum and C. andersoni isolates and performed a comparative genomics analysis of Cryptosporidium spp. and other apicomplexans. Results of this analysis suggest that C. ubiquitum and C. andersoni share many genomic features with C. parvum and C. hominis, but have divergent mitosome metabolism, electron transport chains, and invasion-related surface or secreted proteins and peptidases. This indicates the occurrence of a progressive reductive evolution in mitosome metabolism in the Cryptosporidium lineage within Apicomplexa. Associated with this is the differential evolution of invasion-related proteins, especially between the intestinal and gastric groups. With improved cultivation and animal models and development of genetic manipulation tools in recent years [48, 49], the biologic importance of these genetic differences among Cryptosporidium species could be verified. This in turn may lead to the development of intervention strategies against these important waterborne and zoonotic pathogens in both developing countries and industrialized nations.
Methods
Sample processing
C. ubiquitum isolates 39668, 39725, and 39726 were collected from sporadic cases of human cryptosporidiosis in Wisconsin, USA in summer, 2013, whereas C. andersoni isolates 30847, 31729, and 37034 were collected from beef cattle in Alberta, Canada in 2009, dairy cattle in Henan, China in 2010, and water buffalo in Kafr El Sheikh, Egypt in 2011, respectively. They were diagnosed to Cryptosporidium species by PCR-RFLP analysis and DNA sequencing of the small subunit rRNA gene [46]. C. ubiquitum was further subtyped by DNA sequence analysis of the gp60 gene [12]. Among the three C. ubiquitum isolates, 39668 and 39726 belonged to the XIIb subtype family whereas 39725 belonged to the XIIc subtype family. Cryptosporidium oocysts were purified from the specimens using a combination of sucrose and cesium chloride gradient centrifugation and immunomagnetic separation [50]. Total genomic DNA was extracted from purified oocysts using the QIAamp®DNA Mini Kit (Qiagen Sciences, Germantown, Maryland), after the oocysts were subjected to five freeze-thaw cycles and overnight digestion with proteinase K. Extracted DNA was amplified using REPLI-g Midi Kit (Qiagen).
Genome sequencing and assembly
Genomes of C. ubiquitum and C. andersoni were sequenced on an Illumina Genome Analyzer IIX using the Illumina TruSeq (v3) library protocol, with 100 × 100 bp paired-end reads generated. Sequence reads with quality score below 30 were trimmed using CLC Genomics Workbench 7.03 (http://www.clcbio.com/products/clc-genomics-workbench). Using the same software, they were assembled de novo into contigs with word size of 40, bubble size of 50, and minimum contig length of 500 bp. Assemblies of C. ubiquitum isolate 39726 and C. andersoni isolate 30847 were selected for gene prediction, genome annotation and comparative genomics analysis.
Analysis of genome structure
After the de novo assembly of sequencing reads, Mauve 2.3.1 [51] and MUMmer 3.2.3 [52] were used to map C. ubiquitum contigs to the published C. parvum IOWA genome [5] and C. andersoni contigs to the published C. muris RN66 genome (CryptoDB) using default parameters. Unmapped contigs represented C. ubiquitum- or C. andersoni-unique ones or those from contaminating microbes. BLASTN [53] analysis of GenBank nucleotides database was used to identify contigs from contaminants using e-value threshold as 1e-10. The syntenic relationship between the C. ubiquitum or C. andersoni genome and C. parvum genome was visualized by using Circos 0.67 [54], with sequence identities of >75% shown.
Software packages tRNAscan-SE 1.3.1 [55] and ARAGORN 1.2.36 [56] were used to identify tRNAs. Both of them were executed at the default settings using the general tRNA model or standard genetic codon. Ribosomal RNA genes were predicted using RNAmmer 1.2 [57] and BLASTN. Other genomic features such as mean length, N50 and N90 of contigs and GC content were calculated.
Gene prediction
Protein-coding genes were predicted using a pipeline of three software packages, including AUGUSTUS 2.7 [58], SNAP [59], and GeneMark-ES [60]. All three software packages were run suing the default settings. Prior to their use in gene prediction, AUGUSTUS and SNAP were trained with the gene model of the published C. parvum IOWA genome (CryptoDB release-6.0) for the prediction of C. ubiquitum genes, and gene model of the published C. muris RN66 genome (CryptoDB release-6.0) for the prediction of C. andersoni genes. The length and identities of gene sequences predicted by the three approaches were compared with the reference genomes. As the outcome from AUGUSTUS agreed mostly with the reference genomes, the gene set predicted by AUGUSTUS was supplemented with new genes predicted by GeneMark-ES and SNAP. Only genes predicted by both software package were added to the proteome predicted by AUGUSTUS.
The frequency of protein translation codon usage for each genome was calculated using INCA 2.1 [61]. The entire coding region and 500 bp upstream were extracted to search the most conserved or overrepresented motifs in each genome by using MEME 4.9 [62] with the width threshold as 6-8 bp. SignalP 4.1 [63] and TMHMM 2.0 [64] were used to identify signal peptides and the transmembrane domains in predicted proteins, respectively. Proteins with GPI anchor sites were identified by using the GPI-SOM webserver [65]. The mucin-type O-glycoproteins were predicted by using NetOGlyc 3.1 [66]. These software packages were run using the default parameters.
Functional annotation and comparative genomics analysis proteins
The predicted proteomes of C. ubiquitum and C. andersoni were compared with those of other Cryptosporidium spp. using OrthoMCL [67], BLASTP [53] and in-house scripts to identify shared orthologs and potential species-unique proteins in each genome. OrthoMCL and BLASTP were run with e-value thresholds of 1e-1 and 1e-3, respectively. The in-house scripts were used to extract synthenic genes located in the genome but not identified by OrthoMCL or BLASTP. The genes located in the non-syntenic regions were considered as potential species-unique protein-encoding genes. Protein domains in them were identified using Pfam (http://pfam.xfam.org/) with the default setting [68].
Comparative metabolism analyses of C. ubiquitum and C. andersoni were performed using the web server KAAS [69] with the BBH (Bi-directional Best Hit) method and eukaryote gene model. BLASTP search of the GenBank NR database was used as a supplemental analysis. Data on metabolic pathways, catalytic enzymes, and functional proteins of other apicomplexans were retrieved primarily from online databases LAMP (Library of Apicomplexan Metabolic Pathways, release-2) [21], KEGG (http://www.genome.jp/kegg/), and Pfam [68], with the EuPathDB (http://eupathdb.org/eupathdb/) as the supplement. Comparisons of transporter proteins and invasion-related proteins among Cryptosporidium species were mainly based on Pfam search results.
Phylogenetic analysis
A venn diagram of shared orthologs and species-specific genes of C. parvum, C. hominis, C. ubiquitum and C. andersoni was drawn using the web-tool Venny (http://bioinfogp.cnb.csic.es/tools/venny/). The amino acids sequences of 100 orthologs shared by C. ubiquitum, C. andersoni and other common apicomplexan parasites, including C. parvum, C. hominis, Eimeria tenella, Neospora caninum, T. gondii, P. falciparum, P. vivax, Babesia bovis, Theileria annulata, and T. parva, were extracted to construct a neighbor-joining tree with the maximum composite likelihood mode for distance calculation and 1000 replications for bootstrapping. The concatenated amino acid sequences were aligned with ClustalX [70], trimmed off un-aligned region using Gblocks [71], and used in MEGA 6 [72] for the construction of the neighbor-joining tree. A concatenated sequence from the free-living photosynthetic chromerid, Chromera velia, was used to root the tree.
Acknowledgements
The C. muris annotation in CryptoDB was used in guiding the annotation of the C. andersoni genome. We thank investigators of the C. muris genome sequencing project for this valuable dataset:
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the US Centers for Disease Control and Prevention.
Funding
This work was supported by the National Natural Science Foundation of China (31425025, 31229005, 31110103901 and 31302078); Open Funding Project of the State Key Laboratory of Veterinary Etiological Biology, Lanzhou, China (SKLVEB2014KFKT008); and the US Centers for Disease Control and Prevention.
Availability of data and materials
The datasets supporting the conclusion of this article, including all Sequence Read Archive (SRA) data, assembled contigs, and genome annotations, were submitted to NCBI BioProject under accession No. PRJNA246478 and PRJNA308889.
Authors’ contributions
YF and LX conceived and designed the experiments; SL, DMR, YG, NL, MAF, and LZ performed the experiments; SL, KT, YF, and LX analyzed the data; SL, YF and LX wrote the paper. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The genome sequencing was done on delinked residual diagnostic specimens from Human Subjects Protocol No. 990115 “Use of residual human specimens for the determination of frequency of genotypes or sub-types of pathogenic parasites,” which was reviewed and approved by the Institutional Review Board of the Centers for Disease Control and Prevention (CDC). No personal identifiers were associated with the specimens at the time of submission for diagnostic service at CDC.
Abbreviations
- AOX
Alternative oxidase
- DMAPP
Dimethylallyl diphosphate
- EGF
Epidermal growth factor
- FPP
Farnesyl diphosphate
- GPI
Glycosylphosphatidylinositol
- IPP
Isopentenyl diphosphate
- MQO
Malate:quinone oxidoreductase
- OGDH
Oxoglutarate dehydrogenase complex
- PDH
Dehydrogenase complex
- PNO
Pyruvate:NADP+ oxidoreductase
- PPP
Polyprenyl diphosphate
- PVM
Parasitophorous vacuole membrane
- TCA
Tricarboxylic acid
- TPP
Thiamine pyrophosphate
- TRAP
Thrombospondin-related adhesive proteins
- TSP
Thrombospondin
- VWA
Willebrand factor type A
Additional files
Summary of Cryptosporidium ubiquitum and C. andersoni genomes sequenced in this study. (DOCX 19 kb)
A) Similarity in codon usage frequency among Cryptosporidium parvum, C. ubiquitum and C. andersoni. As expected, the third position of the most commonly used codons mostly has A or T, except for the UGG codon for tryptophan and UTG codon for methionine. B) The most over-represented sequence motifs in upstream regions of protein-encoding genes of C. parvum, C. ubiquitum and C. andersoni. The E2F-like motif, 5′-TGGCGCCA-3′, is the dominant one in all Cryptosporidium species. (DOCX 429 kb)
A) Syntenic relationship between the genomes of Cryptosporidium parvum and C. ubiquitum or C. andersoni. Syntenic sequences (identity >75%) are connected with lines. The colors of lines represent different chromosomes of C. parvum. B) Venn diagram of shared orthologs and species-specific genes among four Cryptosporidium species. Because of the use of different gene prediction approaches in genome annotation, species-specific genes are generally over-estimated. (DOCX 2376 kb)
Orthologs of potential invasion-related mucin-type glycoproteins, thrombospondin-related adhesive proteins (TRAPs), insulinase-like proteases, secreted MEDLE family proteins, and rhomboid-like proteases of Cryptosporidium spp. (DOCX 21 kb)
Sequences of putative Cryptosporidium ubiquitum- and C. andersoni-specific mucin-type glycoproteins. O-linked glycosylation sites (indicated by single-letter amino acid codes) are shown in the second sequence of each mucin-type glycoprotein. (DOCX 27 kb)
Contributor Information
Shiyou Liu, Email: lsy7@outlook.com.
Dawn M. Roellig, Email: iyd4@cdc.gov
Yaqiong Guo, Email: guoyaqiong1987@sina.com.
Na Li, Email: nli@ecust.edu.cn.
Michael A. Frace, Email: mff3@cdc.gov
Kevin Tang, Email: kht7@cdc.gov.
Longxian Zhang, Email: zhanglongxian8999@foxmail.com.
Yaoyu Feng, Email: yyfeng@ecust.edu.cn.
Lihua Xiao, Email: lxiao@cdc.gov.
References
- 1.Wolf YI, Koonin EV. Genome reduction as the dominant mode of evolution. Bioessays. 2013;35(9):829–37. doi: 10.1002/bies.201300037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Woo YH, Ansari H, Otto TD, Klinger CM, Kolisko M, Michalek J, Saxena A, Shanmugam D, Tayyrov A, Veluchamy A, et al. Chromerid genomes reveal the evolutionary path from photosynthetic algae to obligate intracellular parasites. Elife. 2015;4:e06974. doi: 10.7554/eLife.06974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Templeton TJ, Enomoto S, Chen WJ, Huang CG, Lancto CA, Abrahamsen MS, Zhu G. A genome-sequence survey for Ascogregarina taiwanensis supports evolutionary affiliation but metabolic diversity between a Gregarine and Cryptosporidium. Mol Biol Evol. 2010;27(2):235–48. doi: 10.1093/molbev/msp226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu P, Widmer G, Wang Y, Ozaki LS, Alves JM, Serrano MG, Puiu D, Manque P, Akiyoshi D, Mackey AJ, et al. The genome of Cryptosporidium hominis. Nature. 2004;431(7012):1107–12. doi: 10.1038/nature02977. [DOI] [PubMed] [Google Scholar]
- 5.Abrahamsen MS, Templeton TJ, Enomoto S, Abrahante JE, Zhu G, Lancto CA, Deng M, Liu C, Widmer G, Tzipori S, et al. Complete genome sequence of the apicomplexan, Cryptosporidium parvum. Science. 2004;304(5669):441–5. doi: 10.1126/science.1094786. [DOI] [PubMed] [Google Scholar]
- 6.Seeber F, Soldati-Favre D. Metabolic pathways in the apicoplast of apicomplexa. Int Rev Cell Mol Biol. 2010;281:161–228. doi: 10.1016/S1937-6448(10)81005-6. [DOI] [PubMed] [Google Scholar]
- 7.Seeber F, Limenitakis J, Soldati-Favre D. Apicomplexan mitochondrial metabolism: a story of gains, losses and retentions. Trends Parasitol. 2008;24(10):468–78. doi: 10.1016/j.pt.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 8.Mogi T, Kita K. Diversity in mitochondrial metabolic pathways in parasitic protists Plasmodium and Cryptosporidium. Parasitol Int. 2010;59(3):305–12. doi: 10.1016/j.parint.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 9.Striepen B. Parasitic infections: Time to tackle cryptosporidiosis. Nature. 2013;503(7475):189–91. doi: 10.1038/503189a. [DOI] [PubMed] [Google Scholar]
- 10.Ryan U, Fayer R, Xiao L. Cryptosporidium species in humans and animals: current understanding and research needs. Parasitology. 2014;141(13):1667–85. doi: 10.1017/S0031182014001085. [DOI] [PubMed] [Google Scholar]
- 11.Xiao L. Molecular epidemiology of cryptosporidiosis: an update. Exp Parasitol. 2010;124(1):80–9. doi: 10.1016/j.exppara.2009.03.018. [DOI] [PubMed] [Google Scholar]
- 12.Li N, Xiao L, Alderisio K, Elwin K, Cebelinski E, Chalmers R, Santin M, Fayer R, Kvac M, Ryan U, et al. Subtyping Cryptosporidium ubiquitum, a zoonotic pathogen emerging in humans. Emerg Infect Dis. 2014;20(2):217–24. doi: 10.3201/eid2002.121797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fayer R, Santin M, Macarisin D. Cryptosporidium ubiquitum n. sp. in animals and humans. Vet Parasitol. 2010;172(1-2):23–32. doi: 10.1016/j.vetpar.2010.04.028. [DOI] [PubMed] [Google Scholar]
- 14.Liu A, Zhang J, Zhao J, Zhao W, Wang R, Zhang L. The first report of Cryptosporidium andersoni in horses with diarrhea and multilocus subtype analysis. Parasit Vectors. 2015;8:483. doi: 10.1186/s13071-015-1102-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng Y, Yang W, Ryan U, Zhang L, Kvac M, Koudela B, Modry D, Li N, Fayer R, Xiao L. Development of a multilocus sequence tool for typing Cryptosporidium muris and Cryptosporidium andersoni. J Clin Microbiol. 2011;49(1):34–41. doi: 10.1128/JCM.01329-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hashim A, Mulcahy G, Bourke B, Clyne M. Interaction of Cryptosporidium hominis and Cryptosporidium parvum with primary human and bovine intestinal cells. Infect Immun. 2006;74(1):99–107. doi: 10.1128/IAI.74.1.99-107.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazurie AJ, Alves JM, Ozaki LS, Zhou S, Schwartz DC, Buck GA. Comparative genomics of Cryptosporidium. Int J Genomics. 2013;2013:832756. doi: 10.1155/2013/832756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo Y, Tang K, Rowe LA, Li N, Roellig DM, Knipe K, Frace M, Yang C, Feng Y, Xiao L. Comparative genomic analysis reveals occurrence of genetic recombination in virulent Cryptosporidium hominis subtypes and telomeric gene duplications in Cryptosporidium parvum. BMC Genomics. 2015;16:320. doi: 10.1186/s12864-015-1517-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makiuchi T, Nozaki T. Highly divergent mitochondrion-related organelles in anaerobic parasitic protozoa. Biochimie. 2014;100:3–17. doi: 10.1016/j.biochi.2013.11.018. [DOI] [PubMed] [Google Scholar]
- 20.Ctrnacta V, Ault JG, Stejskal F, Keithly JS. Localization of pyruvate:NADP+ oxidoreductase in sporozoites of Cryptosporidium parvum. J Eukaryot Microbiol. 2006;53(4):225–31. doi: 10.1111/j.1550-7408.2006.00099.x. [DOI] [PubMed] [Google Scholar]
- 21.Shanmugasundram A, Gonzalez-Galarza FF, Wastling JM, Vasieva O, Jones AR. Library of apicomplexan metabolic pathways: a manually curated database for metabolic pathways of apicomplexan parasites. Nucleic Acids Res. 2013;41(Database issue):D706–13. doi: 10.1093/nar/gks1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seeber F, Feagin JE, Parsons M. Chapter 9 - The apicoplast and mitochondrion of Toxoplasma gondii. In: Weiss LM, Kim K, editors. Toxoplasma Gondii (Second Edition) Boston: Academic; 2014. pp. 297–350. [Google Scholar]
- 23.Ling Y, Li ZH, Miranda K, Oldfield E, Moreno SN. The farnesyl-diphosphate/geranylgeranyl-diphosphate synthase of Toxoplasma gondii is a bifunctional enzyme and a molecular target of bisphosphonates. J Biol Chem. 2007;282(42):30804–16. doi: 10.1074/jbc.M703178200. [DOI] [PubMed] [Google Scholar]
- 24.Mauzy MJ, Enomoto S, Lancto CA, Abrahamsen MS, Rutherford MS. The Cryptosporidium parvum transcriptome during in vitro development. PLoS ONE. 2012;7(3):e31715. doi: 10.1371/journal.pone.0031715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Artz JD, Dunford JE, Arrowood MJ, Dong A, Chruszcz M, Kavanagh KL, Minor W, Russell RG, Ebetino FH, Oppermann U, et al. Targeting a uniquely nonspecific prenyl synthase with bisphosphonates to combat cryptosporidiosis. Chem Biol. 2008;15(12):1296–306. doi: 10.1016/j.chembiol.2008.10.017. [DOI] [PubMed] [Google Scholar]
- 26.McIntosh L. Molecular biology of the alternative oxidase. Plant Physiol. 1994;105(3):781–6. doi: 10.1104/pp.105.3.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts CW, Roberts F, Henriquez FL, Akiyoshi D, Samuel BU, Richards TA, Milhous W, Kyle D, McIntosh L, Hill GC, et al. Evidence for mitochondrial-derived alternative oxidase in the apicomplexan parasite Cryptosporidium parvum: a potential anti-microbial agent target. Int J Parasitol. 2004;34(3):297–308. doi: 10.1016/j.ijpara.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 28.Suzuki T, Hashimoto T, Yabu Y, Kido Y, Sakamoto K, Nihei C, Hato M, Suzuki S, Amano Y, Nagai K, et al. Direct evidence for cyanide-insensitive quinol oxidase (alternative oxidase) in apicomplexan parasite Cryptosporidium parvum: phylogenetic and therapeutic implications. Biochem Biophys Res Commun. 2004;313(4):1044–52. doi: 10.1016/j.bbrc.2003.12.038. [DOI] [PubMed] [Google Scholar]
- 29.Chaudhuri M, Ott RD, Hill GC. Trypanosome alternative oxidase: from molecule to function. Trends Parasitol. 2006;22(10):484–91. doi: 10.1016/j.pt.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 30.Shiba T, Kido Y, Sakamoto K, Inaoka DK, Tsuge C, Tatsumi R, Takahashi G, Balogun EO, Nara T, Aoki T, et al. Structure of the trypanosome cyanide-insensitive alternative oxidase. Proc Natl Acad Sci U S A. 2013;110(12):4580–5. doi: 10.1073/pnas.1218386110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Samuelson J, Robbins PW. Effects of N-glycan precursor length diversity on quality control of protein folding and on protein glycosylation. Semin Cell Dev Biol. 2015;41:121–8. doi: 10.1016/j.semcdb.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ferguson MAJ, Kinoshita T, Hart GW. Glycosylphosphatidylinositol anchors. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Essentials of Glycobiology. 2. NY: Cold Spring Harbor; 2009. [Google Scholar]
- 33.Butcher BA, Reese ML, Boothroyd JC, Denkers EY. Interactions between Toxoplasma effectors and host immune responses. In: Toxoplasma Gondii: The Model Apicomplexan - Perspectives and Methods: Amsterdam: 2nd ed. Elsevier Ltd.; 2013. p. 505–19.
- 34.Bouzid M, Hunter PR, Chalmers RM, Tyler KM. Cryptosporidium pathogenicity and virulence. Clin Microbiol Rev. 2013;26(1):115–34. doi: 10.1128/CMR.00076-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wanyiri J, Ward H. Molecular basis of Cryptosporidium-host cell interactions: recent advances and future prospects. Future Microbiol. 2006;1(2):201–8. doi: 10.2217/17460913.1.2.201. [DOI] [PubMed] [Google Scholar]
- 36.O’Hara SP, Yu JR, Lin JJ. A novel Cryptosporidium parvum antigen, CP2, preferentially associates with membranous structures. Parasitol Res. 2004;92(4):317–27. doi: 10.1007/s00436-003-1057-5. [DOI] [PubMed] [Google Scholar]
- 37.O’Connor RM, Burns PB, Ha-Ngoc T, Scarpato K, Khan W, Kang G, Ward H. Polymorphic mucin antigens CpMuc4 and CpMuc5 are integral to Cryptosporidium parvum infection in vitro. Eukaryot Cell. 2009;8(4):461–9. doi: 10.1128/EC.00305-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chatterjee A, Banerjee S, Steffen M, O’Connor RM, Ward HD, Robbins PW, Samuelson J. Evidence for mucin-like glycoproteins that tether sporozoites of Cryptosporidium parvum to the inner surface of the oocyst wall. Eukaryot Cell. 2010;9(1):84–96. doi: 10.1128/EC.00288-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paing MM, Tolia NH. Multimeric assembly of host-pathogen adhesion complexes involved in apicomplexan invasion. PLoS Pathog. 2014;10(6):e1004120. doi: 10.1371/journal.ppat.1004120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Deng M, Templeton TJ, London NR, Bauer C, Schroeder AA, Abrahamsen MS. Cryptosporidium parvum genes containing thrombospondin type 1 domains. Infect Immun. 2002;70(12):6987–95. doi: 10.1128/IAI.70.12.6987-6995.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Putignani L, Possenti A, Cherchi S, Pozio E, Crisanti A, Spano F. The thrombospondin-related protein CpMIC1 (CpTSP8) belongs to the repertoire of micronemal proteins of Cryptosporidium parvum. Mol Biochem Parasitol. 2008;157(1):98–101. doi: 10.1016/j.molbiopara.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 42.Hunter CA, Sibley LD. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat Rev Microbiol. 2012;10(11):766–78. doi: 10.1038/nrmicro2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dowse TJ, Soldati D. Rhomboid-like proteins in Apicomplexa: phylogeny and nomenclature. Trends Parasitol. 2005;21(6):254–8. doi: 10.1016/j.pt.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 44.Heitlinger E, Spork S, Lucius R, Dieterich C. The genome of Eimeria falciformis-reduction and specialization in a single host apicomplexan parasite. BMC Genomics. 2014;15:696. doi: 10.1186/1471-2164-15-696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anantharaman V, Iyer LM, Balaji S, Aravind L. Adhesion molecules and other secreted host-interaction determinants in Apicomplexa: insights from comparative genomics. Int Rev Cytol. 2007;262:1–74. doi: 10.1016/S0074-7696(07)62001-4. [DOI] [PubMed] [Google Scholar]
- 46.Xiao L, Escalante L, Yang C, Sulaiman I, Escalante AA, Montali RJ, Fayer R, Lal AA. Phylogenetic analysis of Cryptosporidium parasites based on the small-subunit rRNA gene locus. Appl Environ Microbiol. 1999;65(4):1578–83. doi: 10.1128/aem.65.4.1578-1583.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Imlay L, Odom AR. Isoprenoid metabolism in apicomplexan parasites. Curr Clin Microbiol Rep. 2014;1(3-4):37–50. doi: 10.1007/s40588-014-0006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Morada M, Lee S, Gunther-Cummins L, Weiss LM, Widmer G, Tzipori S, Yarlett N. Continuous culture of Cryptosporidium parvum using hollow fiber technology. Int J Parasitol. 2016;46(1):21–9. doi: 10.1016/j.ijpara.2015.07.006. [DOI] [PubMed] [Google Scholar]
- 49.Vinayak S, Pawlowic MC, Sateriale A, Brooks CF, Studstill CJ, Bar-Peled Y, Cipriano MJ, Striepen B. Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature. 2015;523(7561):477–80. doi: 10.1038/nature14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo Y, Li N, Lysen C, Frace M, Tang K, Sammons S, Roellig DM, Feng Y, Xiao L. Isolation and enrichment of Cryptosporidium DNA and verification of DNA purity for whole-genome sequencing. J Clin Microbiol. 2015;53(2):641–7. doi: 10.1128/JCM.02962-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010;5(6):e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 54.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–64. doi: 10.1093/nar/25.5.0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laslett D, Canback B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004;32(1):11–6. doi: 10.1093/nar/gkh152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35(9):3100–8. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stanke M, Steinkamp R, Waack S, Morgenstern B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 2004;32(Web Server issue):W309–12. doi: 10.1093/nar/gkh379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Korf I. Gene finding in novel genomes. BMC Bioinformatics. 2004;5:59. doi: 10.1186/1471-2105-5-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005;33(20):6494–506. doi: 10.1093/nar/gki937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Supek F, Vlahovicek K. INCA: synonymous codon usage analysis and clustering by means of self-organizing map. Bioinformatics. 2004;20(14):2329–30. doi: 10.1093/bioinformatics/bth238. [DOI] [PubMed] [Google Scholar]
- 62.Bailey TL, Williams N, Misleh C, Li WW. MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Res. 2006;34(Web Server issue):W369–73. doi: 10.1093/nar/gkl198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 64.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–80. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 65.Fankhauser N, Maser P. Identification of GPI anchor attachment signals by a Kohonen self-organizing map. Bioinformatics. 2005;21(9):1846–52. doi: 10.1093/bioinformatics/bti299. [DOI] [PubMed] [Google Scholar]
- 66.Steentoft C, Vakhrushev SY, Joshi HJ, Kong Y, Vester-Christensen MB, Schjoldager KT, Lavrsen K, Dabelsteen S, Pedersen NB, Marcos-Silva L, et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013;32(10):1478–88. doi: 10.1038/emboj.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–89. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. Pfam: the protein families database. Nucleic Acids Res. 2014;42(Database issue):D222–30. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35(Web Server issue):W182–5. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–8. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 71.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17(4):540–52. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 72.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–9. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets supporting the conclusion of this article, including all Sequence Read Archive (SRA) data, assembled contigs, and genome annotations, were submitted to NCBI BioProject under accession No. PRJNA246478 and PRJNA308889.