

The receptor for the growth factor progranulin has remained unclear. Neill et al. show that the Ephrin receptor tyrosine kinase EphA2 is a functional signaling receptor for progranulin and mediates its effects in capillary morphogenesis and autoregulation.
Abstract
Although the growth factor progranulin was discovered more than two decades ago, the functional receptor remains elusive. Here, we discovered that EphA2, a member of the large family of Ephrin receptor tyrosine kinases, is a functional signaling receptor for progranulin. Recombinant progranulin bound with high affinity to EphA2 in both solid phase and solution. Interaction of progranulin with EphA2 caused prolonged activation of the receptor, downstream stimulation of mitogen-activated protein kinase and Akt, and promotion of capillary morphogenesis. Furthermore, we found an autoregulatory mechanism of progranulin whereby a feed-forward loop occurred in an EphA2-dependent manner that was independent of the endocytic receptor sortilin. The discovery of a functional signaling receptor for progranulin offers a new avenue for understanding the underlying mode of action of progranulin in cancer progression, tumor angiogenesis, and perhaps neurodegenerative diseases.
Introduction
Progranulin (PGRN) is an evolutionarily conserved Cys-rich secreted glycoprotein easily measurable in blood and cerebral spinal fluid (Toh et al., 2011). Structurally, progranulin encompasses seven and a half repeats of the granulin module (arranged in the sequence, P-G-F-B-A-C-D-E) and is characterized by a unique protein architecture comprising a stack of β hairpins. Each granulin subdomain contains four β hairpins “stapled” together by six parallel disulfide bridges, with 12 Cys residues per granulin module, culminating in a distinctive ladder-shaped topological superstructure (Tolkatchev et al., 2008; Toh et al., 2011).
The expression of progranulin is ubiquitous and encompasses diverse cell types such as rapidly cycling epithelial cells (Serrero and Mills, 1991), leukocytes (Toh et al., 2011), microglial cells (Toh et al., 2011), bone marrow cells (Bhandari et al., 1992), and chondrocytes (Xu et al., 2007), as well as functioning as a key mitogen found in the secretome of Hobit osteoblastic and osteocytic cells (Romanello et al., 2014). The pleiotropic biological manifestations of progranulin may, in part, stem from its modular architecture, as each granulin can be liberated by secreted neutrophil proteases (e.g., elastase, matrix metalloproteinase [MMP]-12, MMP-14, proteinase 3), with each possessing distinct biological effector functions, presumably downstream of their cognate binding partner and/or receptor. Progranulin has roles beyond development and is central for maintaining organismal homeostasis (Bhandari et al., 1996; Cenik et al., 2012).
There is mounting evidence that progranulin overexpression is linked to cancer progression (Monami et al., 2006, 2009; Buraschi et al., 2016; Tanimoto et al., 2016), wound healing (He et al., 2003), aging (Ahmed et al., 2010), and inflammation (Toh et al., 2011), as well as obesity and insulin resistance (Matsubara et al., 2012). In contrast, low circulating levels of progranulin, resulting from mutations in the progranulin gene (GRN), cause frontotemporal dementia (Gijselinck et al., 2008). Moreover, the lysosomal storage disorder, neuronal ceroid lipofuscinosis, results from GRN homozygous mutations and is clinically recognized by cerebellar ataxia, progressive vision loss, seizures, and retinal dystrophy (Kohlschütter and Schulz, 2009; Smith et al., 2012). In both conditions, there is profound loss of circulating progranulin. Further, decreased levels of progranulin have been found in children diagnosed with autism (Al-Ayadhi and Mostafa, 2011). Progranulin may also play a larger role in other neurodegenerative disorders such as amyotrophic lateral sclerosis (Sleegers et al., 2008), Alzheimer’s disease (Minami et al., 2014), and Parkinson’s disease (Van Kampen et al., 2014).
Despite the fundamental understanding of progranulin action and the elucidation of shared core signal transduction pathways (MAPK and phosphoinositide 3-kinase [PI3K]/Akt/FAK; Zanocco-Marani et al., 1999), the signaling receptor is still elusive. Two candidate receptors, sortilin (gene symbol SORT1; Hu et al., 2010) and the TNF receptor (TNFR1; Tang et al., 2011), have recently emerged as the potential “missing links” for progranulin biology. Sortilin is a conventional single-pass transmembrane protein and member of the Vps family (Vps10) of cell surface, nonsignaling, endocytic receptors (Nykjaer and Willnow, 2012). Sortilin is used for extracellular progranulin internalization (Hu et al., 2010) that strictly relies on proper SORT1 mRNA splicing for the generation of a functional progranulin receptor (Prudencio et al., 2012). Sortilin loss might contribute to prostate cancer progression by enhancing progranulin action in castration-resistant prostate cancer cells (Tanimoto et al., 2015). Recently, we have shown that drebrin, an F-actin–binding protein, binds progranulin and is critical for progranulin-dependent activation of motility, invasion, and anchorage-independent growth of urothelial carcinoma cells (Xu et al., 2015). On the other hand, the discovery of TNFR1 as a receptor for progranulin has provided tantalizing insights and therapeutic promise regarding the mechanism governing the anti-inflammatory properties of progranulin (Tang et al., 2011). However, both sortilin and TNFR are currently the subject of contention, as progranulin can mediate axonal outgrowth independently of sortilin (Gass et al., 2012) and TNFR may or may not be a direct target (Chen et al., 2013; Wang et al., 2015).
Using the yeast two-hybrid system, we previously discovered that progranulin binds specifically to the C terminus of perlecan, termed endorepellin (Gonzalez et al., 2003), and mapped the binding to a region encompassing granulins B/A (Iozzo, 2005). We found that expression of progranulin and perlecan overlapped in a series of ovarian carcinomas (Gonzalez et al., 2003), especially within the tumor microvessels. Because perlecan is expressed in both vascular and avascular compartments (Iozzo, 2005; Zoeller et al., 2008; Farach-Carson et al., 2014; Lord et al., 2014a; Wilusz et al., 2014; Iozzo and Schaefer, 2015), as well as by various inflammatory cells (Lord et al., 2014b), it is likely that proteolytic processing of perlecan (Whitelock et al., 2008; Grindel et al., 2014) would release progranulin into the microenvironment. Moreover, progranulin promotes cell growth, migration, and invasion of prostate and bladder tumor cells, breast carcinomas, and multiple myelomas (He and Bateman, 2003; Monami et al., 2006, 2009; Bateman and Bennett, 2009; Lovat et al., 2009), as well as promoting angiogenesis (Toh et al., 2013).
Here, we have identified EphA2, a member of a large family of receptor tyrosine kinases (RTKs), as a functional signaling receptor for progranulin. Moreover, we provide evidence that progranulin regulates its own expression in an EphA2-dependent, but sortilin-independent, manner. The discovery of a functional progranulin receptor such as EphA2 provides avenues of investigation that might lead to a better molecular understanding of neurodegenerative diseases and new clues about cancer development.
Results
Progranulin activates EphA2
Based on the biological properties of progranulin and downstream activation of MAPK and PI3K/AKT pathways, it has been postulated that progranulin interacts with a classic RTK (Zanocco-Marani et al., 1999; He and Bateman, 2003). In addition, chemical cross-linking experiments with either progranulin or granulins have identified putative receptors ranging between 120 and 140 kD in a variety of cells (Culouscou et al., 1993; Xia and Serrero, 1998), consistent with the size of RTKs. To identify potential progranulin receptors, we used antibody arrays that simultaneously examine differential Tyr phosphorylation levels of 49 different human RTKs. We have successfully used this strategy to identify Met as a novel decorin receptor (Goldoni et al., 2009). Throughout our study, we used highly purified (99.7%) human recombinant progranulin free of copurifying contaminants (Fig. S1 A), as determined by colloidal Coomassie blue staining, which has a detection threshold of as little as 5 ng of protein. Full details regarding the purification of progranulin can be found in Materials and Methods. Initially, we tested T24 urothelial carcinoma cells, as they respond to progranulin (Lovat et al., 2009). After 10-min exposure of quiescent (serum-starved) T24 cells to progranulin, rapid phosphorylation of three members of the Eph family of RTKs, EphA2, EphA4, and EphB2, as well as epidermal growth factor receptor (EGFR; Fig. 1 A) was found. Using short exposures, the first receptor to be consistently phosphorylated was EphA2. Immunoprecipitation and immunoblotting experiments confirmed rapid and sustained activation of EphA2 by progranulin (Fig. 1 B). Similar activation was observed in human umbilical vein endothelial cells (HUVECs; Fig. 1 C), which are to express high levels of EphA2, and PC3 cells (Fig. S1 B), which also express high levels of EphA2 (Miao et al., 2000). We performed an experiment similar to that in Fig. 1 C, wherein we used BSA and EphrinA1-Fc as negative and positive controls, respectively. We found EphA2 activation with progranulin and EphrinA1-Fc, but no phosphorylation with BSA (Fig. S1 C). Because EphrinA1-Fc can stimulate EphA2 (Yang et al., 2011), we recapitulated EphA2 phosphorylation in comparison with progranulin (Fig. S1 D). Further, we evaluated doxazosin, a previously characterized EphA2 agonist (Petty et al., 2012), and found EphA2 activation in PC3 cells, but to a lesser degree than with progranulin (Fig. S1 D).
Figure 1.

Progranulin activates EphA2. (A) Phospho-RTK arrays of serum-starved T24 cells and T24 cells treated with progranulin for 10 min (100 nM). Note that after progranulin incubation EGFR, EphA2, EphA4, and EphB2 are phosphorylated. (B) Immunoprecipitation (IP) of EphA2 from quiescent T24 cells exposed to progranulin (100 nM) for the times indicated. The immunoprecipitated extracts were probed with PY20 and detected by chemiluminescence, stripped, and reprobed with anti-EphA2 or anti-Erk1/2. The Coomassie blue–stained markers, as detected by infrared, appear in lane 1. (C) Immunoprecipitation of EphA2 from HUVECs followed by EphA2 and phospho-Tyr immunoblots (IB; left and right, respectively) in the absence or presence of progranulin, as denoted by − or + above the lane. Cells were treated with progranulin for 30 min.
To ascertain the role of EphA2 in the concurrent activation of EGFR, EphA4, and EphB2, we verified depletion of EphA2 in T24 cells and evaluated the phosphorylation of each receptor in the presence of progranulin (Fig. S1 E). To this end, EGFR, EphA4, and EphB2 were immunoprecipitated and verified (Fig. S1 E), followed by immunoblotting with an anti-phosphotyrosine monoclonal antibody (clone 4G10) to detect tyrosine phosphorylation. We found that loss of EphA2 prevented progranulin-mediated activation of all three receptors, positing a central role for EphA2 in the receptor cross talk as a response to progranulin (Fig. S1 E).
Progranulin physically interacts with EphA2
To determine physical interaction between progranulin and EphA2, we performed solid-phase binding assays in which progranulin served as the immobilized substrate and EphA2-Fc, a chimerical protein containing the entire ectodomain of EphA2 fused to the Fc fragment of IgG, as the soluble ligand, or vice versa. EphA2-Fc bound progranulin in a concentration-dependent and saturable manner (Kd ∼18 nM; Fig. 2 A); progranulin bound EphA2-Fc with similar kinetics (Kd ∼35 nM; Fig. 2 B). EphrinA1, the natural ligand of EphA2, bound immobilized EphA2-Fc with an affinity comparable to that of progranulin (Kd ∼17 nM; Fig. 2 C). Next, we used lithocholic acid (LCA), a bile salt that acts at the level of Eph–Ephrin interactions by disrupting binding at the ligand/ectodomain interface (Giorgio et al., 2011). Increasing concentrations of LCA efficiently displaced progranulin bound to EphA2 (half-maximal inhibitory concentration [IC50] = 42 µM; Fig. 2 D). Surprisingly, LCA modestly displaced bound EphrinA1-Fc (IC50 = 850 µM; Fig. 2 E). We controlled for any potentially compounding effects conveyed by the Fc region by using a nonrelevant Fc binding protein (VEGFR1-Fc; Fig. 2 F, red triangles) and found no significant binding to immobilized EphA2-Fc (red triangles). We also used a similarly sized (85-kD) His6-tagged protein known as endorepellin that is produced in our laboratory with the same methodologies as progranulin and found no significant binding to EphA2-Fc (Fig. 2 F, black circles). Collectively, the lack of VEGFR1-Fc and endorepellin binding to EphA2-Fc substantially verifies the efficacy and specificity of our binding studies. Complementing the solid-phase binding assays, we used microscale thermophoresis (Wienken et al., 2010) to confirm binding of fluorescently labeled progranulin or EphrinA1 to EphA2-Fc. Progranulin bound EphA2 with high affinity (Kd = 1.2 ± 0.3 nM; Fig. 2 G), whereas the interaction of EphrinA1 with EphA2 was significantly weaker (Kd = 75.2 ± 15.8 nM; Fig. 2 H). As negative controls, no binding was detected between EphA2-Fc and BSA (Fig. 2 I) or progranulin and BSA (Fig. 2 J).
Figure 2.

Progranulin interacts with EphA2. (A and B) Solid-phase binding assays using progranulin (100 ng/well) as immobilized substrate and EphA2-Fc as soluble ligand (A) or EphA2-Fc (100 ng/well) as immobilized substrate with soluble progranulin (B). (C) Solid-phase binding assay using immobilized EphA2-Fc and EphrinA1-Fc as a soluble ligand. (D and E) Displacement of bound progranulin (100 ng/well; D) or bound EphrinA1-Fc (100 ng/well; E) from EphA2-Fc with LCA. (F) Solid-phase binding assay using immobilized EphA2-Fc (100 ng/well) and a nonrelevant Fc-fusion protein, VEGFR1-Fc (red triangles) or endorepellin (black circles). (G) Interaction between fluorescently labeled, purified progranulin (20 nM) with recombinant EphA2-Fc. (H) Interaction of fluorescently labeled EphrinA1-Fc (20 nM) with recombinant EphA2-Fc. Changes in thermophoresis at concentrations (of EphA2-Fc) from 1 µM to 0.4 nM were used. (I and J) BSA served as a negative control for binding EphA2-Fc (I) and progranulin (J). Labeled progranulin (20 nM) with increasing concentrations of nonlabeled BSA (up to 10 µM) were used, and changes in thermophoresis were measured. The reported Kd was calculated from four independent thermophoresis measurements. FNorm (%) indicates normalized fluorescence per million. Values represent the mean ± SEM of three independent experiments performed in triplicate.
We further extended the physical interaction of progranulin with EphA2-Fc in solution using pull-down experiments. EphA2-Fc was bound to protein A–Sepharose beads (PA-EphA2-Fc) to mimic a more physiologically oriented receptor surface. Under physiological salt and pH conditions, progranulin bound specifically to immobilized PA-EphA2-Fc (Fig. S1 F) and not in the absence of PA-EphA2-Fc. Comparable results were obtained in pull-down experiments in which equimolar amounts of progranulin and EphA2-Fc were first incubated individually and then precipitated with protein A–Sepharose beads (Fig. S1 G).
To further investigate progranulin–EphA2 interaction, we used affinity chromatography with His6-tagged progranulin bound to Ni-NTA beads and PC3 lysates (these cells express high levels of EphA2; see Progranulin binds EphA2 at the cell surface). After binding in a buffer containing low amounts of imidazole, the bound material was eluted using high imidazole. Under such conditions, we detected significant binding of endogenous EphA2 to the progranulin affinity column (Fig. S1 H).
To completely eliminate any confounding effects conferred by the fused Fc domain (Fig. 2 F), we used a cell-free coupled transcription/translation system to express full-length human EphA2 (Fig. S1 I). After incubation with recombinant progranulin, we could efficiently coimmunoprecipitate the two proteins (Fig. S1 J). Importantly, EphA2 was not detected in the immunoprecipitate of a negative IgG control or when EphA2 was omitted (Fig. S1 J).
Collectively, these reciprocal binding experiments and cell-free approaches substantiate a high-affinity, physical interaction between EphA2 and progranulin.
Progranulin colocalizes with EphA2
To corroborate the binding studies, we treated HUVECs with recombinant progranulin for various times and used confocal laser microscopy. Notably, under basal conditions, endogenous progranulin colocalized with EphA2 (Fig. 3, A–E). We imaged the field at low magnification for each time point (Fig. 3, A, F, and K) and captured high-magnification images from this representative view (Fig. 3, B–D, G–I, and L–N). Individual channels showed a codistribution of EphA2 (Fig. 3, B and C, green) and progranulin (red) puncta within cytosolic and membranous compartments. Semiquantitative line scanning confirmed significant overlap among the differentially labeled pixels (Fig. 3 E). The area measured for line scanning is designated by the dotted line between the white arrowheads (Fig. 3, D, I, and N). Exogenous progranulin (10 min) promoted intense colocalization with EphA2 (Fig. 3, F–I). Importantly, this relationship was clearly depicted within the individual channels (Fig. 3, G and H) and reinforced by line scanning (Fig. 3 J). Longer progranulin exposure (20 min) had a less colinear appearance and a more punctate morphology (Fig. 3, K–N). Colocalization was further confirmed via line scanning (Fig. 3 O). Importantly, we detected no signal when omitting the EphA2 primary antibody (Fig. 3 P), indicating specificity.
Figure 3.

Progranulin colocalizes with EphA2. (A–E) Representative confocal images of vehicle-treated cells evaluated at low magnification (A) for EphA2 (B), progranulin (C), and both (D) at high magnification from the same field as in A. (E) Line scanning profiles for colocalized pixels shown in D and measured between the white arrows, indicated by the dotted line. Exogenous progranulin for 10 min (F–J) and associated line scan analysis (J) and for 20 min (K–O) with line scanning (O). (P) EphA2 primary antibody was omitted as a negative control. All images were taken with the same exposure, gain, and intensity. Bar, ∼10 µm. (Q–S) Pearson’s coefficient of colocalization was calculated for EphA2 (Q), progranulin (R), and the degree of overlap (S) for each time point. Values represent the mean of three independent experiments in HUVECs and are represented as box plots. Statistics were generated by one-way ANOVA (***, P < 0.001).
The interaction of progranulin with EphA2 was quantified by calculating the Pearson coefficient of colocalization per individual channel and for the overlapped pixels (Misaki et al., 2010; Dunn et al., 2011; Zinchuk et al., 2011). As per the basal association of progranulin with EphA2, there were already several contributing pixels for EphA2 (Fig. 3 Q) and progranulin (Fig. 3 R), resulting in a moderate degree of overlap (Fig. 3 S). However, as exogenous progranulin was added, the proportion of individual and overlapped pixels involved in the colocalization for EphA2 and progranulin increased significantly (Fig. 3, Q–S). Further, a longer treatment time (30 min) had a similar proportion of colocalized pixels (Fig. 3, Q–S; unpublished data). Colocalization of progranulin and EphA2 was done by focusing on entire cells. The representative images and quantifications were not done solely for a particular cellular compartment, but rather for all colocalized pixels present. The provided images are representative of this method.
Because we did not observe robust staining of progranulin and EphA2 at the plasma membrane, we hypothesized that lack of colocalization at the cell surface may be caused by rapid uptake and internalization of progranulin. Therefore, we evaluated very short time points (1, 4, and 8 min) and found that at 1 min, we could see colocalization at the plasma membrane (Fig. S2 A, white arrows). However, at subsequent time points (4 and 8 min), progranulin was rapidly internalized together with EphA2 (Fig. S2 A).
Parallel results were obtained in PC3 cells stimulated with progranulin (Fig. S2 B). Similarly, progranulin elicited rapid, robust, and transient activation and clustering of EphA2 as measured by receptor phosphorylation in as little as 5 min (Fig. S2 C, white arrows). A hallmark of EphrinA1 activity is EphA2 internalization (2 h; Wykosky et al., 2008); therefore, we evaluated the same effect of progranulin on EphA2 in HUVECs. We found that progranulin internalizes EphA2 over time (Fig. S2 D) in a manner similar to EphrinA1-Fc (Fig. S2 C). Because EphA2 has a propensity to form receptor-rich clusters, we tested whether EphA2 can interact with the aforementioned progranulin receptors, TNFR1 or sortilin. We found that there is no interaction of EphA2 with either TNFR1 or sortilin in the absence or presence of progranulin in HUVECs (Fig. S2 E) or PC3 cells (Fig. S2 F). Collectively, these data further authenticate the results obtained with a visual and biochemical depiction of the tight and exclusive interaction involving only progranulin and EphA2.
Progranulin binds EphA2 at the cell surface
Next, we investigated whether progranulin would bind to the surface of PC3 cells. Using soluble progranulin conjugated with the near-infrared dye IR800 (Fig. 4 A), we found saturable binding to immobilized EphA2-Fc (Kd ∼50 nM, Fig. 4 B). Specificity was determined via in-cell binding assays in which IR800-progranulin was incubated with confluent PC3 cells for 15 min, followed by incubation with increasing concentrations of unlabeled EphrinA1-Fc. IR800-progranulin could be displaced by EphrinA1-Fc (Fig. 4 C) with an IC50 of ∼400 nM (Fig. 4 D). Using the same approach for LCA, IR800-progranulin was significantly displaced (Fig. 4, E and F), suggesting impaired binding of progranulin to EphA2 at the cell surface in the presence of LCA. It is important to note that LCA, at the doses used (50 or 100 µM), does not perturb the activity of other RTKs (Giorgio et al., 2011).
Figure 4.

Progranulin binding requires EphA2 at the cell surface and stimulates MAPK and Akt. (A) Gel of increasing concentrations of IR800-labeled progranulin (B) solid-phase binding assay of labeled progranulin to immobilized EphA2-Fc. (C) In-cell binding using equimolar equivalencies of either IR800-progranulin (C, top) or IR800-progranulin in combination with EphrinA1-Fc (C, bottom). (D) Resulting displacement curve and corresponding IC50 value after exposure to increasing concentrations of EphrinA1-Fc. (E) In-cell binding using IR800-progranulin and DRAQ5 (genomic DNA) in the absence or presence of LCA (100 µM). (F) Signal intensity of bound IR800-progranulin normalized to DRAQ5. (G) Immunoblotting depicting RNAi-mediated silencing of EphA2. (H). In-cell binding assay using IR800-progranulin after transfection of siRNA against EphA2. (I) Quantification of IR800-progranulin signal intensity after DRAQ5 normalization, as in F. (J–M) Representative immunoblots of phosphorylated MAPK in quiescent PC3 (J) and HUVECs (K) or phosphorylated Akt in PC3 (L) and HUVECs (M) after progranulin stimulation at the indicated time points. (N) Immunoblotting and quantification of phosphorylated Akt and MAPK in PC3 after combination treatment of progranulin with LCA, as designated. Bar, ∼1 cm (C, E, and H). Data are representative of at least three independent experiments and are reported as the fold change ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
The role of EphA2 as a putative high-affinity receptor for progranulin was further substantiated by depleting the receptor and evaluating progranulin binding at the cell surface. After authenticating EphA2 silencing (Fig. 4 G), we added IR800-progranulin and determined cell surface binding of the labeled ligand. In a manner that parallels pharmacological inhibition, overt loss of EphA2 substantially prevented cell surface binding of progranulin (Fig. 4, H and I). Thus, we provide three distinct and complementary methodologies that collectively establish EphA2 as an authentic cell surface receptor for progranulin.
Progranulin activation of MAPK and Akt is EphA2 dependent
Because progranulin activates canonical MAPK and Akt signaling, presumably downstream of an RTK (Zanocco-Marani et al., 1999), we tested the phosphorylation of MAPK and Akt in PC3 cells and HUVECs in response to progranulin over time. A transient increase in MAPK phosphorylation (Thr202/Tyr204) was seen at 10 min in PC3 cells and HUVECs (Fig. 4, J and K; and Fig. S3, A and B). Robust and sustained activation of Akt (Ser473) in PC3 cells, beginning at 10 min, was seen (Figs. 4 L and S3 C). However, no such induction of Akt signaling was found in HUVECs (Figs. 4 M and S3 D). As demonstrated in the Progranulin physically interacts with EphA2 section, LCA abrogates progranulin–EphA2 interactions at the cell surface. Therefore, we tested whether LCA could functionally block the robust activation of Akt and MAPK in PC3 cells. We found that LCA significantly prevented Akt and MAPK phosphorylation when combined with progranulin (Fig. 4 N). Moreover, RNAi-mediated silencing of EphA2 recapitulated the effects of LCA on Akt and MAPK phosphorylation, insofar as receptor loss prevented activation of these downstream signaling effectors (Fig. S3 E). Collectively, these data posit a functional signaling role mediated by progranulin–EphA2 interactions.
EphA2 is required for capillary morphogenesis
Progranulin induces endothelial cell migration during wound repair (He et al., 2003), stimulates exaggerated vessel growth in vivo (Toh et al., 2013), and evokes VEGF expression (Tangkeangsirisin and Serrero, 2004). Notably, EphA2 plays a key role in postnatal vascular function. Although Epha2−/− mice are viable with no overt developmental abnormalities, endothelial cells isolated from these mice do not form capillary-like structures in vitro in response to EphrinA1 (Brantley-Sieders et al., 2004). Moreover, Epha2−/− mice display an impaired tumor microenvironment that is not conducive for proper tumor development and metastatic spreading (Brantley-Sieders et al., 2005). Additional studies have shown that EphA2 is a key angiogenesis receptor (Kullander and Klein, 2002) and that soluble EphA2 blocks tumor angiogenesis (Brantley et al., 2002).
We sought to address the potential role of progranulin in in vitro angiogenesis using an RNAi approach. After efficient knockdown of EphA2 (>85%; Fig. 5 A), we performed in vitro capillary morphogenesis assays. Within 4 h, vehicle-treated HUVECs formed an anastomosing network of tube-like structures (Fig. 5 B), a morphogenetic process that was markedly enhanced by progranulin treatment (Fig. 5 C). The formation of capillary-like structures in HUVECs depleted of EphA2 was reduced, but still active (Fig. 5 D). However, progranulin had no further effects on the EphA2-deficient cells (Fig. 5 E). Quantification of the normalized tube area from five individual experiments showed a significant enhancement of capillary area by progranulin in the vehicle-treated cells (P < 0.001), but no significant effect on the EphA2-deficient endothelial cells (P = 0.69; Fig. 5 F). We complemented this approach with a collagen type I sandwich capillary morphogenesis assay after EphA2 depletion in the presence of progranulin (Fig. S4, A–D). We obtained comparable results, insofar as silencing EphA2 prevented progranulin-evoked capillary morphogenesis (Fig. S4 E). As LCA displaces progranulin from EphA2 and effectively blocks signaling (Figs. 2 D and 4 N), we hypothesized that LCA would phenocopy the loss of EphA2. We found that LCA significantly abrogated progranulin-mediated capillary morphogenesis, matching results obtained with EphA2 knockdown with cells on Matrigel or type I collagen (Fig. S4, F–J). Collectively, these results provide additional evidence for a functional relationship between progranulin and EphA2 in the process of capillary morphogenesis.
Figure 5.
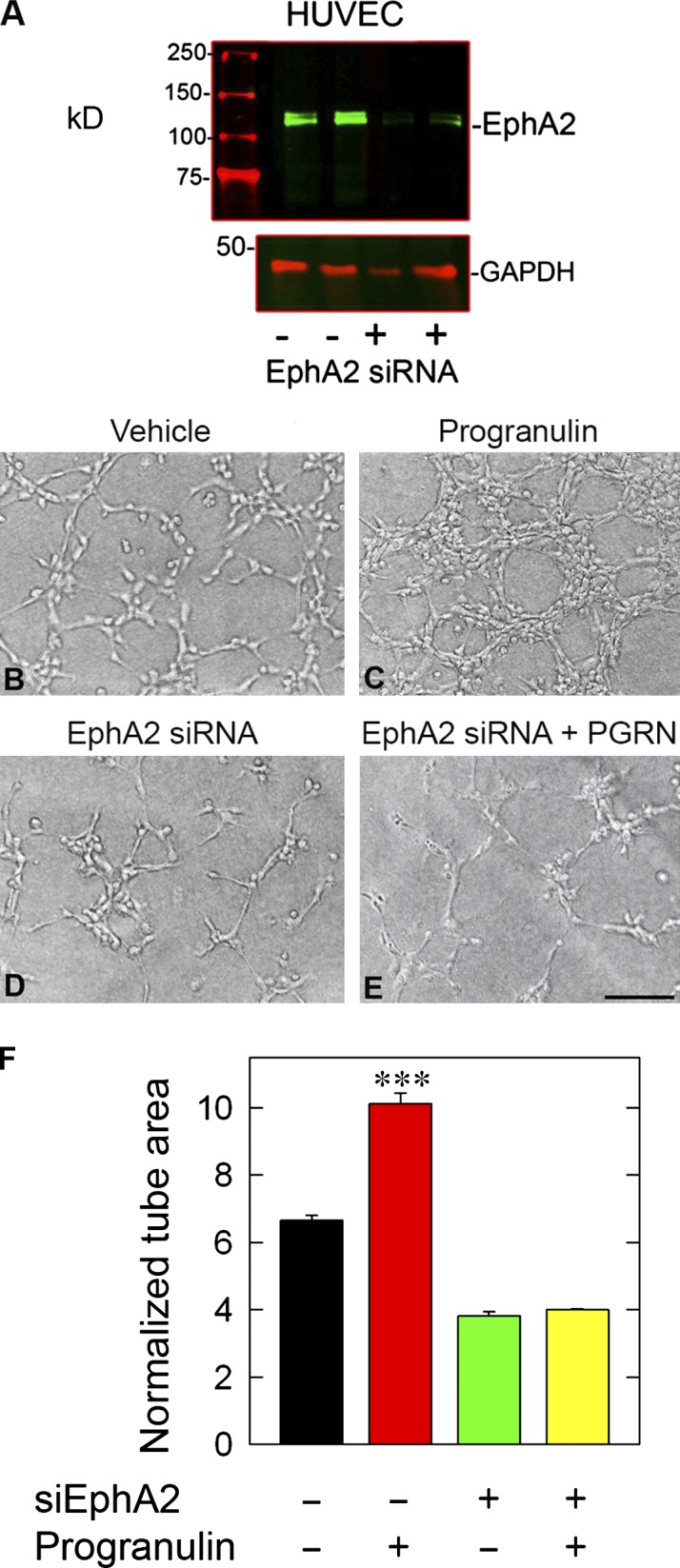
EphA2 is required for progranulin-evoked stimulation of capillary morphogenesis. (A) Immunoblotting verification of EphA2 silencing. (B–E) Representative capillary morphogenesis bright-field images of HUVECs embedded on Matrigel, transfected with siScr or siEphA2, and challenged with progranulin (100 nM). (F) Morphometric parameter quantification for surface area of the Matrigel capillary morphogenesis assay. The accompanying quantification represents mean tube surface area ± SEM from five experiments.
Soluble progranulin is an autocrine factor for GRN autoregulation
It is well established that several RTK ligands have a proclivity for autoregulation (Lemmon and Schlessinger, 2010). Thus, we tested the ability of progranulin self-regulation of GRN expression in HUVECs and PC3 cells. Progranulin evoked endogenous GRN expression in a dose-dependent manner in HUVECs (Fig. 6 A) and PC3 cells (Fig. 6 C). Significant GRN induction was achieved in both cells with as little as 12.5 nM of progranulin (∼1 µg/ml; Fig. 6, A and C). Addition of exogenous progranulin resulted in a time-dependent increase of GRN in HUVECs (Fig. 6 B) and PC3 cells (Fig. 6 D). The kinetics of GRN induction differed between the two cell types. In HUVECs, there was a rapid (∼1 h) and significant increase of GRN followed by sustained GRN abundance that was stably maintained for up to 6 h (Fig. 6 B). In contrast, GRN expression in PC3 cells peaked at 2 h, followed by a gradual decline to baseline levels (Fig. 6 D). Based on the kinetic profiles, we performed all subsequent experiments with HUVECs and PC3 cells for 6 and 2 h, respectively. To complement the autoregulatory functions of progranulin over the GRN locus, we evaluated additional genes known to be regulated (VEGFA) and not regulated (Myc; Brantley-Sieders and Chen, 2004) by EphA2. We found that VEGFA expression was suppressed by progranulin (Fig. 6 E), whereas MYC expression was unchanged (Fig. 6 F). We also analyzed the effect of EphrinA1-Fc on GRN expression and found a decrease of GRN mRNA (Fig. 6 G). Collectively, these data demonstrate a dynamic modulation of endogenous GRN levels upon stimulation with progranulin in two genetically distinct cellular models.
Figure 6.

Progranulin stimulates GRN expression in HUVECs and PC3 cells. (A and C) Dose–response of GRN in HUVECs (A) or PC3 cells (C) to progranulin (6 h) at the indicated concentrations. (B and D) Time course of GRN expression in HUVECs (B) or PC3 cells (D) after progranulin (50 nM) at the indicated time points. (E and F) Analysis of VEGFA (E) and MYC (F) expression after progranulin treatment (50 nM, 2 h). (G) Analysis of GRN mRNA after EphrinA1-Fc (50 nM, 2 h). Data are the result of at least three independent experiments and are reported as fold change ± SEM. ACTB served as an internal housekeeping gene for all expression analyses. Statistical significance for A–D was determined via one-way ANOVA (*, P < 0.05; **, P < 0.01).
Progranulin requires EphA2 signaling for its autoregulation
As a functional coupling between progranulin and EphA2, we tested whether progranulin-mediated autoregulation would require the presence of this receptor. First, we significantly depleted EPHA2 using three specific siRNAs targeting various regions of the gene in HUVECs (Fig. 7 A) and PC3 cells (Fig. 7 B). Loss of EphA2 substantially abrogated the autoregulatory property of progranulin on GRN expression in both cell types (Fig. 7, C and D). This effect was in stark contrast to that in vehicle-treated cells (Fig. 7, C and D). Individual loss of EPHA2 had no adverse effect on the basal GRN, suggesting that progranulin must first engage EphA2 for GRN autoregulation. To further corroborate these results, we used the EphA2 inhibitor, LCA. When LCA was concurrently administered with progranulin, there was a near-complete block of progranulin activity on GRN mRNA levels in both cell types (Fig. 7, E and F).
Figure 7.

EphA2 is required for progranulin-mediated autoregulation of GRN. (A and C) Depletion of EPHA2 (A) and resulting effect on GRN (C) in HUVECs after progranulin stimulation (6 h). (B and D) EphA2 silencing in PC3 cells (B) and ensuing effect on GRN after progranulin (2 h; D). (E and F) Pharmacological manipulation of HUVECs (E) and PC3 cells (F) with LCA in combination with progranulin and queried for GRN expression. (G and H) Evaluation of GRN expression after pretreatment (1 h) of PC3 cells (G) or HUVECs (H) with the MAPK inhibitor (U0129, 10 µM) or Akt inhibitor (LY294004, 10 µM) and stimulated with progranulin. Data result from at least three independent experiments and are reported as fold change ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Next, we evaluated the role of MAPK and Akt activity for GRN autoregulation by using known inhibitors of the MAPK (U0129) or Akt (LY294004) signaling pathways. Inhibiting either MAPK or Akt signaling blunted GRN autoregulation in PC3 cells (Fig. 7 G). Inhibiting with LY294004 did not prevent GRN induction in HUVECs (Fig. 7 H); however, U0129 did prevent GRN expression (Fig. 7 H). Interestingly, these results match the biochemical analyses of HUVECs (Fig. 4, K and M) insofar as MAPK activation was robustly activated with a lack of Akt phosphorylation. Collectively, these results identify EphA2 as a pivotal signaling receptor and implicate MAPK and Akt as key components of the signaling apparatus needed for progranulin-mediated induction of GRN expression.
Sortilin is not directly involved in progranulin autoregulation
Sortilin has been proposed as an endocytic receptor for progranulin (Hu et al., 2010). Thus, we tested whether sortilin could also be involved in GRN autoregulation. SORT1 knockdown was verified in HUVECs (Fig. 8 A) and PC3 cells (Fig. 8 C). As was the case for EPHA2, stimulation with exogenous progranulin did not modulate basal SORT1 (Fig. 8, A and C). Notably, knockdown of SORT1 evoked a threefold increase in GRN expression, comparable to levels evoked by exogenous progranulin in the presence or absence of sortilin (Fig. 8, B and D).
Figure 8.

EphA2, but not sortilin, allows GRN induction after progranulin stimulation. (A and B) In HUVECs, verification of SORT1 depletion (A) and evaluation of GRN (B). (C and D) Verification of SORT1 depletion in PC3 cells (C) and GRN expression (D) after challenge with progranulin. (E–G) Dual silencing and confirmation of EPHA2 loss (E) and SORT1 loss (F) and subsequent evaluation of dual knockdown on GRN (G) in HUVECs. (H–J) Identical experiment performed in PC3 cells. For E–J, siScr was transfected and stimulated with progranulin (50 nM). For dual silencing, siRNA for EPHA2 and SORT1 were mixed and transfected in tandem, without exogenous progranulin. (K and L) Verification of sortilin depletion (K) and quantification of in-cell binding assay using IR800-labeled progranulin in PC3 cells (L). Data are the result of at least four independent experiments and are reported as fold change ± SEM. Statistical analyses for conditions with three or more groups, one-way ANOVA (**, P < 0.01; ***, P < 0.001).
Because silencing of sortilin leads to a significant accumulation of extracellular progranulin (Hu et al., 2010), we postulated that loss of sortilin would trigger accumulation of extracellular progranulin, which in turn would signal through EphA2 for GRN autoregulation. Next, a dual-RNAi approach was used wherein EPHA2 and SORT1 were depleted simultaneously in HUVECs (Fig. 8, E and F) and PC3 cells (Fig. 8, H and I). Progranulin alone had no regulatory effects on EPHA2 or SORT1 (Fig. 8, E, F, H, and I). In agreement with the results shown in the previous section, progranulin increased GRN expression in the presence of nontargeting siRNA (Fig. 8, G and J). Moreover, induction of GRN was equivalent in magnitude to loss of SORT1 alone (Fig. 8, G and J). Importantly, dual loss of EPHA2 and SORT1 ablated GRN expression compared with individual SORT1 silencing in HUVECs (Fig. 8 G) and PC3 (Fig. 8 J). Because sortilin is a high-affinity endocytic receptor for progranulin, we tested whether loss of sortilin would subsequently increase progranulin binding to EphA2, resulting in enhanced autoregulation. We depleted sortilin (Fig. 8 K) and found that 60% of the IR800-labeled progranulin still bound to the cell surface, thereby indicating no augmented binding to EphA2 after sortilin knockdown (Fig. 8 L). Thus, in support of our hypothesis that loss of sortilin resulted in accumulated progranulin that is subsequently signaling via EphA2 for GRN autoinduction, PC3 cells were depleted of sortilin (Fig. S4 K), and progranulin levels in the conditioned media were measured via ELISA. A standard curve was generated for progranulin (Fig. S4 L) and used for calculating progranulin levels. As predicted, loss of sortilin resulted in a significant accumulation of progranulin in the conditioned media (Fig. S4 M). This increase in progranulin may be sufficient for activating EphA2 for GRN induction after sortilin depletion.
Collectively, these data indicate that loss of EphA2 prevents GRN autoinduction stemming from the single loss of sortilin, and that this is seemingly independent of exogenous progranulin. As such, this effect is presumably occurring as a result of compromised progranulin reuptake as coordinated by the dedicated endocytic functions of sortilin.
Progranulin stimulates GRN promoter activity
To further investigate the autoregulatory loop of progranulin, we generated two stably transfected PC3 cells (mass cultures) containing either a promoterless GFP construct (PC3GFP) or reporter cells (PC3GRN-GFP) expressing GFP driven by a 1.8-kb genomic fragment of the human GRN promoter (Fig. 9 A). In addition, we generated stably transfected PC3 cells (mass cultures) harboring the same human GRN promoter driving the firefly luciferase gene (PC3GRN−Luc; Fig. 9 A). Progranulin had no effect on GFP levels in the PC3GFP cells vis-à-vis vehicle-treated controls (Fig. 9 B). In contrast, progranulin evoked about a 12-fold increase of GFP in the PC3GRN-GFP cells, which was efficiently blocked by LCA (Fig. 9 B).
Figure 9.
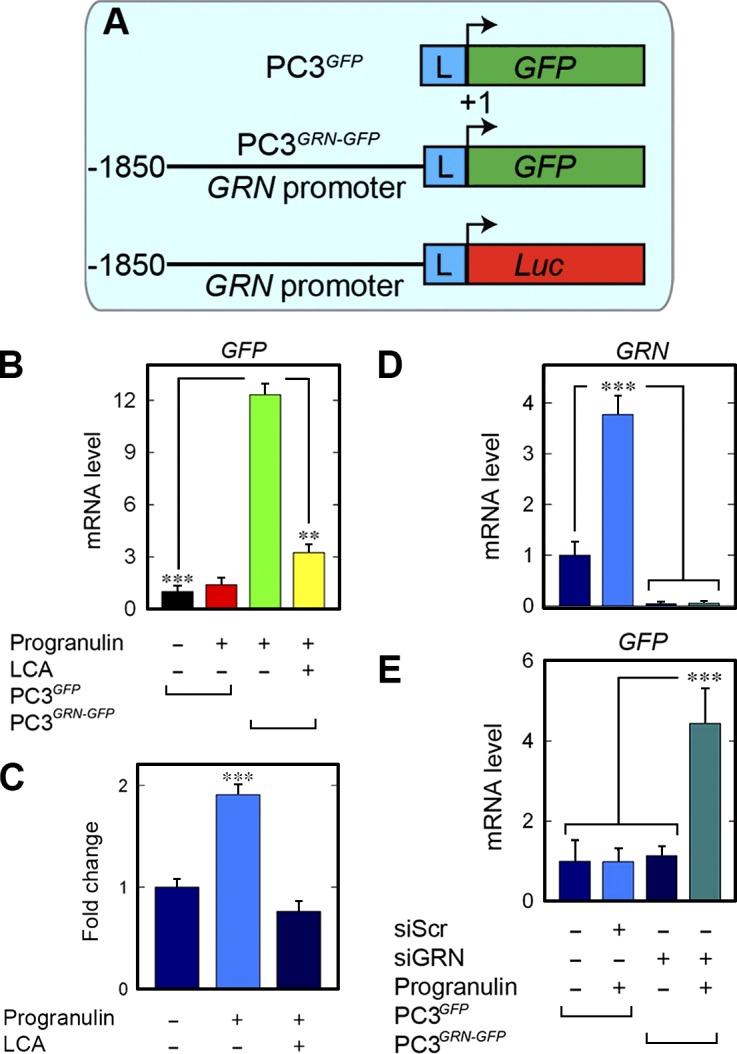
Progranulin stimulates a GRN-GFP reporter via EphA2. (A) Schematic of the GFP reporter constructs of empty GFP (top), GRN-GFP (middle), and GRN-Luc (bottom). L, linker region of the pGL3 backbone. (B) Quantification of GFP from PC3GFP or the active reporter PC3GRN-GFP cells after progranulin. (C) PC3 cells were transiently transfected with GRN-Luc and assayed for luciferase activity. Modulation of GRN promoter activity was evaluated after incubation with progranulin with or without LCA. Data are shown as mean ± SEM of three independent experiments; n = 6 for each condition. (D and E) Quantification of GRN and GFP levels after depletion of endogenous progranulin from the PC3 reporter cell lines. Data reported as mean ± SEM and represent at least three independent experiments. **, P < 0.01; ***, P < 0.001.
In support of these findings, progranulin evoked luciferase activity in PC3GRN-GFP (Fig. 9 C). Concurrent treatment with LCA and progranulin completely abrogated progranulin-mediated luciferase activity (Fig. 9 C). Because PC3 cells synthesize high levels of progranulin (He and Bateman, 2003; Cenik et al., 2012), we tested whether GRN autoregulation would be influenced by endogenous progranulin. As such, GRN was silenced from PC3GFP and PC3GRN-GFP cells (Fig. 9, D and E). As expected, progranulin increased endogenous GRN in the vehicle-treated cells (Fig. 9 D). Analysis of GFP after endogenous progranulin depletion revealed that despite loss of cellular progranulin, exogenous progranulin was sufficient for GRN-GFP activity (Fig. 9 E). Interestingly, loss of GRN alone had no considerable effect on basal GRN-GFP (Fig. 9 E). These findings suggest that progranulin may not maintain basal GRN activity, at least in the context of the 1.8-kb promoter fragment used for the reporter (Fig. 9 E).
Next, we validated the concept of progranulin-evoked self-induction via fluorescence imaging of the reporter GFP cells. Exogenous progranulin had no effect on GFP when applied to PC3GFP cells (Fig. S5, A and B); in contrast, progranulin induced GFP in PC3GRN-GFP (Fig. S5, C and D). Immunoblotting for GFP revealed a significant increase in GFP protein levels (Fig. S5, E and F), further confirming the reporter data. Loss of sortilin did not perturb activation of the GFP reporter (Fig. S5 J), as induction was comparable to progranulin alone (Fig. S5 H). Furthermore, silencing of sortilin alone did increase reporter activity (Fig. S5 I) compared with control (Fig. S5 G).
Finally, endogenous progranulin was depleted, and no significant induction after loss of progranulin alone was observed (Fig. S5, K and M). Stimulation of the GFP reporter with exogenous progranulin was similar in the presence (Fig. S5 L) or absence (Fig. S5 N) of endogenous progranulin. Collectively, these data reinforce the notion of GRN autoregulation and further underscore that GRN-driven GFP expression and luciferase activities are dependent on competent EphA2 signaling.
Discussion
In this study, we used an unbiased screening approach to identify a cell surface RTK whose activation, as determined by the degree of Tyr phosphorylation, is evoked by recombinant, soluble progranulin. Our main hypothesis is that the signaling cascade evoked by progranulin is likely mediated by a classic RTK, insofar as canonical transduction pathways triggered by RTKs include the MAPK/PI3K/Akt/FAK/paxillin signaling apparatus. Using this platform, we discovered three members of the Eph family of RTKs (EphA2, EphA4, and EphB2) and EGFR that were rapidly activated by progranulin. We focused our studies on EphA2 because this receptor gave the most robust Tyr phosphorylation and because it was also the first Eph receptor to be activated. Moreover, EphA2 seemingly plays a central role in receptor cross talk and mediated progranulin signal transduction with other RTKs.
Specific interactions between EphA2 and progranulin were validated by a multitude of complementary approaches such as solid-phase binding assays (Kd ∼18 nM), microscale thermophoresis (Kd ∼1.2 nM), pull-downs using EphA2-Fc, cell binding assays, and in vitro synthesized, label-free systems. Progranulin binding to EphA2 was further confirmed via affinity chromatography, in which progranulin was vectorially bound via a C-terminal His6-tag to Ni-NTA beads.
Indeed, the binding affinity between progranulin and EphA2 is similar to the reported interaction of progranulin and sortilin (∼15.4 nM; Hu et al., 2010). Intriguingly, using EphrinA1 as a positive control for EphA2 binding, we found a much tighter interaction (Kd ∼17 nM) for EphrinA1/EphA2 compared with previously published studies (e.g., Kd ∼330 nM for the monomer and Kd ∼144 nM for dimeric EphrinA1; Ferluga et al., 2013). The apparent differences may reside with the methodology used (sandwich ELISAs using Fc-fusion proteins versus surface plasmon resonance with data-fitting to a single site).
Cell surface binding proteins ranging between 120 and 140 kD have been previously cross-linked to 125I-labeled progranulin or granulins (Culouscou et al., 1993; Xia and Serrero, 1998). The 1:1 cross-linking and the overall molecular mass are in agreement with previous studies (EphA2 is ∼120 kD). It is widely accepted that Eph receptors require clustered, GPI-anchored ligands regulating intercellular and integrated cross talk among multiple signaling pathways. There is now evidence that secreted EphrinA1 can activate its cognate receptor EphA2 as a soluble monomer (Wykosky et al., 2008; Wykosky and Debinski, 2008). In further support of our previous studies, large multimolecular complexes were noted, suggesting the formation of multimeric aggregates of EphA2, and perhaps other Eph receptors excluding TNFR1 and sortilin, upon engagement with soluble progranulin. Moreover, in both cases, there were two binding sites with high and low affinity, ranging between 43–200 pM and 4–10 nM, respectively (Culouscou et al., 1993; Xia and Serrero, 1998). Notably, in both cell-free and live cell experiments, EphA2 bound to progranulin could be competed by EphrinA1-Fc and LCA. Moreover, loss of EphA2 resulted in a significant loss of progranulin binding at the cell surface.
We believe that the widely accepted notion that Eph receptors require cell surface Ephrin ligands to mediate either forward or reverse signaling has driven researchers away from focusing on this class of RTKs. In retrospect, the relationship between progranulin, a factor that regulates neurite outgrowth and enhances neuronal survival (Van Damme et al., 2008), and Eph receptors, which are highly expressed in the central nervous system and are often neurotrophic, should have been considered. Notably, in contrast to the large number of Eph receptors (n = 14) and Ephrin ligands (n = 8) in mammals, Drosophila melanogaster and Caenorhabditis elegans have only one Eph receptor and comparatively fewer Ephrins, suggesting that the remarkable expansion of Eph receptors during evolution could be linked to brain expansion in more complex mammalian systems (Yamaguchi and Pasquale, 2004). Moreover, the expression of most Eph RTKs is restricted to the central nervous system, where they regulate axonal guidance by mediating repulsive signals for improper synaptic connections (Yamaguchi and Pasquale, 2004). Deleting EphA2 disrupts proper alignment of equatorial epithelial cells and prevents constriction of the elongating epithelium within the lens fulcrum during lens morphogenesis (Cheng et al., 2013) and coincident with the development of cortical cataracts (Jun et al., 2009). Mutations common in congenital cataracts arise within the intracellular SAM domain of EphA2 and result in impaired receptor stability and aberrant localization (Park et al., 2012; Dave et al., 2016).
EphA2 has been implicated in promoting many types of cancer, including melanomas (Margaryan et al., 2009; Parri et al., 2009), castration-resistant prostate carcinomas (Taddei et al., 2009), and mammary and pancreatic adenocarcinomas (Duxbury et al., 2004; Brantley-Sieders et al., 2008; Wykosky and Debinski, 2008; Kessenbrock et al., 2015). EphA2 has also been implicated in promoting tumor neovascularization (Ogawa et al., 2000; Chan and Sukhatme, 2009) and vascular mimicry (Hess et al., 2001). Moreover, EphA2 coordinates CD8+ T cell tumor infiltration for increased tumorigenicity and altered tumor immunogenicity (Wesa et al., 2008). Conversely, Epha2−/− mice exhibit less tumorigenicity in the ApcMin/+ system (Bogan et al., 2009) and are characterized by a compromised tumor microenvironment (Brantley-Sieders et al., 2005) with impaired metastatic potential and angiogenesis (Brantley-Sieders et al., 2005). It is likely that the high-affinity interactions between progranulin and EphA2, with downstream activation of the MAPK/Akt cascade, JNK signaling (Song et al., 2014), and β-catenin (Huang et al., 2014), may underscore the mechanism by which progranulin mediates malignant transformation for a variety of solid tumors (He and Bateman, 2003; Monami et al., 2006, 2009; Lovat et al., 2009; Tanimoto et al., 2015). Moreover, the EphA2/Akt and EphA2/MAPK signaling pathways are sensitive to LCA, an inhibitor that competes for progranulin binding with the EphA2 ectodomain that is further mirrored by EphA2 depletion.
In a manner analogous to the tumor cell expression of progranulin, soluble EphrinA1 is coexpressed with EphA2 and is secreted, presumably as an autocrine and/or paracrine mediator, during tumorigenesis and angiogenesis (Ogawa et al., 2000; Wykosky and Debinski, 2008; Wykosky et al., 2008). Intriguingly, monomeric EphrinA1 exhibits anti-oncogenic properties via engagement of EphA2 via the G-H loop, a region of EphrinA1 critical for this potentially therapeutic property (Lema Tomé et al., 2012; Kessenbrock et al., 2015). The interplay among the regulatory properties of the EphrinA1/progranulin/EphA2 signaling system appear intricate and most likely cell type dependent.
In the present work, we have identified a previously unknown autocrine feedback mechanism wherein exogenously administered progranulin is sufficient for increasing its own expression. Stimulation of the GRN locus upon additional progranulin requires the presence of EphA2 and physical interaction between progranulin and EphA2. As of yet, the region of progranulin and/or the granulin responsible for autoinduction and EphA2 engagement remain unknown. It is known that granulin E, the most C-terminal granulin, is necessary for sortilin binding (Hu et al., 2010). As such, a similar requirement may be needed for progranulin/EphA2 and subsequent downstream activities.
Silencing or pharmacological inhibition of EphA2 significantly prevents progranulin-mediated autoregulation. The role of sortilin in progranulin-mediated GRN induction appears indirect, as SORT1 loss is sufficient for GRN expression. This concept aligns with previous findings that focused on the depletion of sortilin from the neuronal compartment (Hu et al., 2010), and from prostate cancer cells (Tanimoto et al., 2015). The genetic ablation of sortilin induces a twofold increase in extracellular progranulin levels (Hu et al., 2010); our findings reveal a similar induction of endogenous GRN expression upon sortilin loss that is wholly abrogated after the simultaneous silencing of SORT1 and EPHA2. A genome-wide association study has identified the single nucleotide polymorphism rs646776 as a key regulator of circulating progranulin (Carrasquillo et al., 2010). Intriguingly, this single nucleotide polymorphism maps in close proximity to the SORT1 locus (Carrasquillo et al., 2010).
In the present study, we validated our biochemical data with functional reporter assays (GRN-Luc, GRN-GFP) and found a similar transcriptional induction of GRN mRNA with a concurrent requirement for EphA2. Importantly, depletion of endogenous progranulin does not alter the ability of exogenous progranulin in promoting GRN-GFP promoter activity, underscoring the non–cell-autonomous function of progranulin-mediated GRN induction. Interestingly, endogenously synthesized progranulin is not sufficient to stimulate basal reporter activity above control conditions.
In conclusion, the discovery of a novel functional receptor for progranulin and its feedback autoregulatory loop may hold therapeutic promise and may provide the cornerstone for understanding progranulin pathobiology. For example, progranulin-mediated GRN induction could be blocked with specific antibodies directed to the receptor itself or with small molecules blocking EphA2 tyrosine kinase activity (Noberini et al., 2008). Given that progranulin is a crucial neurotrophic factor (Van Damme et al., 2008), and given the epidemiological and genetic evidence that reduced levels of progranulin are causatively associated with frontotemporal dementia (Baker et al., 2006; Cruts et al., 2006; Cruts and Van Broeckhoven, 2008; Ghidoni et al., 2008), abnormal EphA2 activity or loss of progranulin autoinduction could be involved in the pathogenesis of neurodegenerative diseases. Moreover, the role of EphA2 in tumorigenesis parallels that of progranulin and may represent a key mechanism for progranulin- and EphA2-mediated tumorigenic growth and progression. Thus, any pharmacological treatment that would differentially modulate progranulin levels and EphA2 tyrosine kinase activity could be beneficial for a broad spectrum of patients suffering from progranulin-related diseases.
Materials and methods
Cells and materials
HUVECs were obtained from Lifeline Cell Technology, grown in basal medium supplemented with the VascuLife EnGS LifeFactors kit (Lifeline Cell Technology), and used within the first five passages. T24 urothelial and PC3 prostate carcinoma cells were obtained from ATCC and maintained in Ham’s F12K-1X medium or DMEM (Corning) supplemented with 5% FBS. Dulbecco’s PBS (DPBS) was also purchased from Corning. Stably expressing PC3 cells were maintained in 200 µg/ml Hygromycin B (Thermo Fisher Scientific) supplemented in Ham’s F12K-1X medium. Primary antibodies for MAPK, Akt, phosphorylated MAPK and Akt, GFP, TNFR1, and GAPDH were purchased from Cell Signaling Technology. Anti-sortilin and mouse anti-phosphoEphA2/3/4 antibodies were purchased from Abcam. Primary antibodies for EphA2 are enumerated as follows: rabbit anti-EphA2 (C-20) and mouse anti-EGRF were purchased from Santa Cruz Biotechnology, Inc.; rabbit anti-EphA2 and mouse anti-EphA2 (8B6) from Cell Signaling Technology; and mouse anti-EphA2 (clone D7) from EMD Millipore and as published previously (Yang et al., 2011), as was the mouse anti-phosphotyrosine clone 4G10. Mouse anti-mouse anti-EphB2 was from Invitrogen. Mouse anti-phosphotyrosine PY20 was obtained from EMD Millipore. HRP-conjugated goat anti–rabbit and donkey anti–mouse secondary antibodies were obtained from EMD Millipore. SuperSignal West Pico Enhanced Chemiluminescence substrate and mouse anti-EphA4 were purchased from Thermo Fisher Scientific. Doxazosin, LCA, and rabbit anti-progranulin (C terminus) were purchased from Sigma-Aldrich.
Expression and purification of human recombinant progranulin
A pCEP-Pu vector bearing the sequence of the BM40 signal peptide and full-length human progranulin was electroporated into 0.5 × 106 HEK293-EBNA. Mass cultures were selected in media containing 250 µg/ml G418 and 2 µg/ml puromycin. Serum-free conditioned medium was concentrated in a dialysis bag with polyethylene glycol, dialyzed, and purified on Ni-NTA resin eluted with 500 mM imidazole. In all purification steps, we used PMSF (2 mM) and N-ethylmaleimide (2 mM) as protease inhibitors. Typically, ∼1 mg of progranulin was purified from 200 ml of conditioned media. Our progranulin preparations were verified by colloidal Coomassie blue (EZ Blue Gel Staining Reagent; Sigma-Aldrich), which can detect as little as 5 ng of protein, and thus 99.7% pure (Fig. S1 A). We did not detect any copurifying contaminants with this method. Throughout the study, progranulin was used at 50 nM (4 µg/ml), unless otherwise noted.
Phospho-RTK arrays
Phospho-RTK antibody array membranes (R&D Systems) were incubated with cell lysates and processed per the manufacturer's protocol using a phospho-Tyr–specific antibody. In brief, ∼3 × 107 T24 cells were serum-starved overnight and treated with progranulin (240 nM) for 10 min. After incubation, cells were washed with ice-cold DPBS and lysed with a buffer containing 1% NP-40, 20 mM Tris-HCl, pH 8.0, 137 mM NaCl, 10% glycerol, 2 mM EDTA, 1 mM Na3VO4, and aprotinin/leupeptin (10 µg/ml each) for 30 min. A protein assay (Bio-Rad Laboratories) was performed before incubating the RTK membranes.
Solid-phase binding assays
ELISAs were performed according to a standard protocol. Progranulin or EphA2-Fc (Sigma-Aldrich; 100 ng/well) was allowed to adhere to the wells overnight at RT in the presence of carbonate buffer, pH 9.6. Plates were washed with PBS and incubated for 2 h with serial dilutions of EphA2-Fc or progranulin. After incubation, plates were extensively washed with PBS, blocked with 1% BSA/PBS, and incubated for 1 h with an antibody raised against EphA2 (H-77; Santa Cruz Biotechnologies, Inc.) or progranulin (Abcam). An anti-rabbit HRP-conjugated secondary antibody (EMD Millipore) was incubated for 1 h. Signal was developed using Sigma-Fast tablets and read at 450 nm OD.
Binding of progranulin or EphrinA1 to EphA2 via microscale thermophoresis
Protein–protein interactions between purified progranulin and EphA2-Fc or EphrinA1-Fc and EphA2-Fc (R&D Systems) were evaluated by changes in the thermophoretic mobility (Wienken et al., 2010) of progranulin or EphrinA1 labeled with the fluorophore NT-647 (Monolith NT Protein Labeling kit; NanoTemper Technologies). A titration series of EphA2-Fc (1 µM to 0.4 nM) was used, whereas the concentrations of NT-647–labeled progranulin or EphrinA1-Fc remained constant (20 nM). Labeled progranulin (20 nM) with unlabeled BSA served as a negative control. The microscale thermophoretic assay was performed and quantified as previously described (Merline et al., 2011).
Immunoprecipitation
Approximately 8 × 106 HUVECs were serum-starved for 1 h, incubated with progranulin for 30 min, and lysed in 1% NP-40 buffer. Lysates were cleared at 14,000 rpm for 10 min and subjected to immunoprecipitation with an anti–C terminus EphA2 antibody (C-20, Santa Cruz Biotechnologies, Inc.). Samples were separated via 8% SDS-PAGE and immunoblotted for EphA2 and phospho-Tyr. Protein A–conjugated Sepharose beads (GE Healthcare) were rotated with 12 µg EphA2-Fc chimera (Sigma-Aldrich) for 8 h at 4°C. The beads were pelleted by centrifugation, resuspended, and allowed to rotate for 16 h at 4°C with a nonspecific mouse IgG that served to block any unbound protein A. The beads were pelleted again; resuspended in DMEM; and split into aliquots, each receiving 100 nM progranulin, increasing amounts of mouse EphrinA1, or both; and incubated at 37°C for 2 h with occasional tapping. The beads were spun down, washed with PBS three times, resuspended in reducing buffer, and heat-denatured at 100°C. The samples were analyzed on an 8% SDS-PAGE gel. After transfer, blots were blocked with 5% BSA in Tris-buffered saline for 1 h and probed overnight with mouse monoclonal anti His5 (5-Prime) and rabbit polyclonal anti-progranulin (EMD Millipore). The blots were washed four times in Tris-buffered saline/0.1% Tween-20 for 10 min before incubation with IR800-labeled goat anti-rabbit and IR680-labeled goat anti-mouse secondary antibodies (LI-COR Biosciences). Images were obtained using Odyssey Imager V3.0 (LI-COR Biosciences).
In vitro transcription/translation of full-length EphA2
Native, full-length Myc-tagged EphA2 was synthesized using the TnT Quick Coupled Transcription/Translation System (L1170; Promega) with a TrueORF Gold expression plasmid purchased from Origene according to the manufacturer’s instructions. In brief, 1 µg of purified EPHA2 plasmid was combined with TnT quick master mix and 1 mM methionine in the presence of nuclease-free water. After gentle mixing, the reaction was allowed to dwell at 30°C for 90 min, and the products of the in vitro reaction were verified by SDS-PAGE. After identification of the correct product, the lysates were used for immunoprecipitation in the presence of His6-tagged human recombinant progranulin as described earlier.
Capillary morphogenesis assays
Four-well slides (Thermo Fisher Scientific) were coated with ∼200 µl of Matrigel (BD) containing VEGFA (20 ng/ml) and heparin (1 µg/ml) in the presence or absence of progranulin (100 nM). The slides were left in the incubator at 37°C for 30 min to let the Matrigel form a homogeneous solid layer. Approximately 104 HUVECs were then seeded in each well on top of the Matrigel in medium supplemented with VEGFA/heparin as described earlier, with or without progranulin. After transient siRNA transfection, live cell images of the tubes were acquired using a DMIL LED microscope (Leica Biosystems) equipped with a D-LUX3 camera (Leica Biosystems) and processed for further analysis. All data were analyzed with Systat Software in SigmaPlot 12.0.
Preparation of IR800–progranulin and in-cell binding assays
Purified progranulin was labeled with the IR800 dye via the IRDye 800CW labeling kit (LI-COR Biosciences) according to the manufacturer’s instructions. In brief, progranulin was mixed with the dye in a molar ratio of 1:1 and kept for 2 h at RT while protecting the vial from light with aluminum foil. Chemically, IRDye 800CW dye bears a reactive NHS ester group that couples to aliphatic amines, especially those found in lysine residues that form stable conjugates with the purified protein. The free, unconjugated dye was removed from the labeled progranulin by using 0.5-ml Pierce Zeba desalting spin columns. For the in-cell binding assays, confluent PC3 cells were incubated with IR800-progranulin in 0.1% BSA/DMEM in the presence or absence of LCA or after transient siRNA transfection for 1 h, covered, on ice. The cells were washed extensively, fixed in 10% formaldehyde, and scanned with the Odyssey Image system at 800 nm. The cells were washed again and incubated with the far-red fluorescent genomic DNA dye, DRAQ5 (1:10,000; BioStatus) in 0.1% BSA/PBS. After three more washes with PBS, the cells were scanned at 700 nM, and these values were used to normalize the IR-800 signal intensity data.
Transient RNAi-mediated silencing
We transiently transfected both HUVECs and PC3 cells using Lipofectamine RNAiMAX (Thermo Fisher Scientific) mixed with a cocktail of three different and validated siRNA duplexes (e.g., sense and antisense) directed against different regions of Homo sapiens EPHA2, SORT1, or GRN mRNAs (sc-29304, sc-42119, and sc-39261, respectively; Santa Cruz Biotechnologies, Inc.). Sequences for the targeting siRNA oligos used can be found in Table S1. Medium was changed 24 h post-transfection, and biological assays were performed at 48 h, as appropriate. Scramble siRNA (sc-37007; Santa Cruz Biotechnologies, Inc.) served as a control for all siRNA experiments presented herein. This protocol was used subsequent to protein (RIPA) or RNA (TRIzol Reagent; Thermo Fisher Scientific) isolation from samples for further analysis. Verification of siRNA-mediated knockdown of the target proteins was determined via immunoblotting or quantitative PCR as per experimental condition.
Immunofluorescence, confocal laser microscopy, and Pearson’s coefficient of colocalization
Typically, ∼5 × 104 HUVECs or PC3 cells were plated on 0.2% gelatin-coated four-well chamber slides (Nunc, Thermo Fisher Scientific) and grown to full confluence. Cells were treated at the indicated time points with progranulin or LCA (100 µM). After treatment, cells were rinsed twice with cold DPBS (Thermo Fisher Scientific) and fixed in 4% (wt/vol) paraformaldehyde for 30 min. After washing, the slides were incubated with Alexa Fluor 488– or 564–conjugated secondary antibodies for 1 h at RT. Nuclei were visualized with DAPI (Vector Laboratories). Slides were then extensively washed and coverslips mounted with Vectashield (Vector Laboratories). Confocal analysis was performed using a 63×, 1.3 oil-immersion objective of an LSM-780 confocal laser-scanning microscope (ZEISS) with filters set at 488/594 nm for dual-channel imaging. To determine colocalization of the two proteins, z-stack series were acquired maintaining the same number of slices (n = 30). All images were then analyzed in ImageJ and Photoshop CS6 (Adobe Systems). Line scanning plots were generated using SigmaPlot software (Systat Software). For GFP, images were acquired with a 63×, 1.3 oil-immersion objective installed on an DM5500B microscope (Leica Biosystems) with Leica Application Suite v1.8 software.
We calculated the weighted Pearson’s coefficient of colocalization using the on-board colocalization function in the LSM-780 Zen software package. We started by establishing threshold fluorescence levels for each channel (excluding DAPI). The threshold represents the fully saturated population of pixels that each channel intrinsically contains. After the thresholds were determined, all images for that particular experiment were taken without changing that value. Thus, we recorded the percentage of pixels involved in the colocalization separately and combined (e.g., red and green channels). The more intense the colocalization appeared, the higher the proportion of pixels (individual and merged) involved in the image. The tabulated values reflect the mean of three independent experiments (n = 15 images per experiment) and are presented as box plots. Importantly, this quantification was done for all colocalized pixels and does not reflect measurements of a specific cellular or subcellular compartment.
Affinity chromatography
Progranulin (20 µg) was bound overnight at 4°C with rotation to 200 µl of HisPur Ni-NTA beads (Thermo Fisher Scientific) in PBS, 0.3 M NaCl, and 20 mM imidazole, pH 7.4. The beads were allowed to settle in a chromatography column and, once the column was formed, RIPA buffer diluted 1:15 (vol/vol) with binding buffer was passed through it to equilibrate the beads. Approximately 2 × 107 PC3 cells were extracted in RIPA buffer diluted 1:15 (vol/vol) with binding buffer. After extensive washes, elution buffer containing PBS and 500 mM imidazole, pH 7.4, was added to the column, and several fractions were collected and subjected to electrophoresis on 10% SDS/PAGE. Proteins were then transferred to nitrocellulose membranes (Bio-Rad), probed with indicated antibodies, developed with enhanced chemiluminescence (Thermo Fisher Scientific), and detected using ImageQuant LAS-4000 (GE Healthcare).
Quantitative real-time PCR
Expression analysis by quantitative real-time PCR was performed on subconfluent six-well plates seeded with ∼2 × 105 HUVECs or PC3 cells. Cells were treated with progranulin in growth factor–supplemented serum (2%) Basal Endothelial Medium (LifeCell Technology), or Ham’s F12K-1X medium supplemented with 10% FBS, respectively. After incubation, cells were lysed directly in 1 ml of TRIzol reagent to extract total RNA. After quantification, 1 µg of total RNA was annealed with oligo (dT18–20) primers (Thermo Fisher Scientific), and cDNA was synthesized with the aid of SuperScript Reverse transcription II (Thermo Fisher Scientific). Amplicons representing target genes (GRN, SORT1, EPHA2, and GFP) and the endogenous housekeeping gene, ACTB, were amplified in quadruplicate, independent reactions using the Brilliant SYBR Green Master Mix II (Agilent Technologies). All samples were run on the Roche LightCycler 480-II Real Time PCR platform (Roche), and the cycle number (Ct) was recorded for each independent reaction. Fold change determinations were made using the comparative Ct method for expression analysis. ΔCt values represent gene expression levels normalized to ACTB for each reaction. ΔΔCt values then represent the experimental cDNA minus the corresponding gene levels of the calibrator sample. Fold changes were calculated using the double ΔCt method (2−ΔΔCT) ± SEM. Data derive from three to five independent trials run in quadruplicate for each gene of interest.
Promoter luciferase reporter assays
PC3 cells were transiently transfected with the GRN-Luc reporter plasmid containing a 1.8-kb genomic fragment encompassing the human GRN promoter cloned upstream of firefly luciferase. Cells were cotransfected with the Renilla luciferase plasmid for normalization and to determine transfection efficiency. After 48 h, cells were treated individually or in combination with 50 nM progranulin or 100 µM LCA for 6 h. Luciferase activity was assayed using firefly luciferase assay kit and Renilla luciferase assay kit from Biotium. The data from three independent experiments were normalized on Renilla luciferase.
Quantification and statistical analysis
Immunoblots were quantified by scanning densitometry using Scion Image software (National Institutes of Health). Graphs were generated using Sigma Stat 3.10. Significance of the differences was evaluated by Student’s t test with significance at P < 0.05. All data presented herein were collected from a minimum of three independent experiments. Experiments with more than three treatment groups, dose curves, or time courses were subjected to one-way analysis of variance (ANOVA) followed by a Bonferroni post hoc test. Differences among the conditions were considered significant at two-sided P < 0.05.
Online supplemental material
Fig. S1 verifies human recombinant progranulin purity, activation of EphA2 receptor upon binding, dependence of EphA2 in receptor cross talk, and complementary binding methods. Fig. S2 reveals that exogenous progranulin localizes with EphA2 and promotes EphA2 phosphorylation in PC3 cells, triggers EphA2 internalization, and does not associate with TNFR1 or sortilin. Fig. S3 shows phosphorylation of MAPK and Akt over time and reliance on EphA2 for Akt and MAPK activation. Fig. S4 contains bright-field images of a type I collagen capillary morphogenesis assay and ensuing effects of EphA2 loss with quantification as well as images depicting effects of LCA on capillary morphogenesis with quantification. Also, loss of sortilin increases progranulin levels in PC3-conditioned media. Fig. S5 illustrates that exogenous progranulin drives GRN-GFP activity downstream of EphA2. Table S1 shows siRNA sequences.
Supplementary Material
Acknowledgments
We thank S. Goldoni and A. Nyström for their help in the initial stages of this work.
This work was supported in part by National Institutes of Health grants RO1 CA39481 and RO1 CA47282 (to R.V. Iozzo) and RO1 CA164462 (to A. Morrione and R.V. Iozzo), the Benjamin Perkins Bladder Cancer Fund, the Schorsch Family Fund for Innovative Medical Research and Care (to A. Morrione), and the German Research Council (SFB 815, project A5, SFB 1039, project B2, Excellence Cluster ECCPS, to L. Schaefer). T. Neill was supported in part by National Institutes of Health training grant T32 AR060715-04.
The authors declare no competing financial interests.
Footnotes
Abbreviations used:
- ANOVA
- analysis of variance
- EGFR
- epidermal growth factor receptor
- HUVEC
- human umbilical vein endothelial cell
- IC50
- half-maximal inhibitory concentration
- LCA
- lithocholic acid
- RTK
- receptor tyrosine kinase
- TNFR
- TNF receptor
References
- Ahmed Z., Sheng H., Xu Y.F., Lin W.L., Innes A.E., Gass J., Yu X., Wuertzer C.A., Hou H., Chiba S., et al. 2010. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am. J. Pathol. 177:311–324. 10.2353/ajpath.2010.090915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Ayadhi L.Y., and Mostafa G.A.. 2011. Low plasma progranulin levels in children with autism. J. Neuroinflammation. 8:111 10.1186/1742-2094-8-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M., Mackenzie I.R., Pickering-Brown S.M., Gass J., Rademakers R., Lindholm C., Snowden J., Adamson J., Sadovnick A.D., Rollinson S., et al. 2006. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 442:916–919. 10.1038/nature05016 [DOI] [PubMed] [Google Scholar]
- Bateman A., and Bennett H.P.. 2009. The granulin gene family: From cancer to dementia. BioEssays. 31:1245–1254. 10.1002/bies.200900086 [DOI] [PubMed] [Google Scholar]
- Bhandari V., Palfree R.G.E., and Bateman A.. 1992. Isolation and sequence of the granulin precursor cDNA from human bone marrow reveals tandem cysteine-rich granulin domains. Proc. Natl. Acad. Sci. USA. 89:1715–1719. 10.1073/pnas.89.5.1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandari V., Daniel R., Lim P.S., and Bateman A.. 1996. Structural and functional analysis of a promoter of the human granulin/epithelin gene. Biochem. J. 319:441–447. 10.1042/bj3190441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogan C., Chen J., O’Sullivan M.G., and Cormier R.T.. 2009. Loss of EphA2 receptor tyrosine kinase reduces ApcMin/+ tumorigenesis. Int. J. Cancer. 124:1366–1371. 10.1002/ijc.24083 [DOI] [PubMed] [Google Scholar]
- Brantley D.M., Cheng N., Thompson E.J., Lin Q., Brekken R.A., Thorpe P.E., Muraoka R.S., Cerretti D.P., Pozzi A., Jackson D., et al. 2002. Soluble Eph A receptors inhibit tumor angiogenesis and progression in vivo. Oncogene. 21:7011–7026. 10.1038/sj.onc.1205679 [DOI] [PubMed] [Google Scholar]
- Brantley-Sieders D.M., and Chen J.. 2004. Eph receptor tyrosine kinases in angiogenesis: From development to disease. Angiogenesis. 7:17–28. 10.1023/B:AGEN.0000037340.33788.87 [DOI] [PubMed] [Google Scholar]
- Brantley-Sieders D.M., Caughron J., Hicks D., Pozzi A., Ruiz J.C., and Chen J.. 2004. EphA2 receptor tyrosine kinase regulates endothelial cell migration and vascular assembly through phosphoinositide 3-kinase-mediated Rac1 GTPase activation. J. Cell Sci. 117:2037–2049. 10.1242/jcs.01061 [DOI] [PubMed] [Google Scholar]
- Brantley-Sieders D.M., Fang W.B., Hicks D.J., Zhuang G., Shyr Y., and Chen J.. 2005. Impaired tumor microenvironment in EphA2-deficient mice inhibits tumor angiogenesis and metastatic progression. FASEB J. 19:1884–1886. 10.1096/fj.05-4038fje [DOI] [PubMed] [Google Scholar]
- Brantley-Sieders D.M., Zhuang G., Hicks D., Fang W.B., Hwang Y., Cates J.M.M., Coffman K., Jackson D., Bruckheimer E., Muraoka-Cook R.S., and Chen J.. 2008. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J. Clin. Invest. 118:64–78. 10.1172/JCI33154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buraschi S., Xu S.Q., Stefanello M., Moskalev I., Morcavallo A., Genua M., Tanimoto R., Birbe R., Peiper S.C., Gomella L.G., et al. 2016. Suppression of progranulin expression inhibits bladder cancer growth and sensitizes cancer cells to cisplatin. Oncotarget. 7:39980–39995. 10.18632/oncotarget.9556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasquillo M.M., Nicholson A.M., Finch N., Gibbs J.R., Baker M., Rutherford N.J., Hunter T.A., DeJesus-Hernandez M., Bisceglio G.D., Mackenzie I.R., et al. 2010. Genome-wide screen identifies rs646776 near sortilin as a regulator of progranulin levels in human plasma. Am. J. Hum. Genet. 87:890–897. 10.1016/j.ajhg.2010.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenik B., Sephton C.F., Kutluk Cenik B., Herz J., and Yu G.. 2012. Progranulin: A proteolytically processed protein at the crossroads of inflammation and neurodegeneration. J. Biol. Chem. 287:32298–32306. 10.1074/jbc.R112.399170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan B., and Sukhatme V.P.. 2009. Receptor tyrosine kinase EphA2 mediates thrombin-induced upregulation of ICAM-1 in endothelial cells in vitro. Thromb. Res. 123:745–752. 10.1016/j.thromres.2008.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Chang J., Deng Q., Xu J., Nguyen T.A., Martens L.H., Cenik B., Taylor G., Hudson K.F., Chung J., et al. 2013. Progranulin does not bind tumor necrosis factor (TNF) receptors and is not a direct regulator of TNF-dependent signaling or bioactivity in immune or neuronal cells. J. Neurosci. 33:9202–9213. 10.1523/JNEUROSCI.5336-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C., Ansari M.M., Cooper J.A., and Gong X.. 2013. EphA2 and Src regulate equatorial cell morphogenesis during lens development. Development. 140:4237–4245. 10.1242/dev.100727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M., and Van Broeckhoven C.. 2008. Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet. 24:186–194. 10.1016/j.tig.2008.01.004 [DOI] [PubMed] [Google Scholar]
- Cruts M., Gijselinck I., van der Zee J., Engelborghs S., Wils H., Pirici D., Rademakers R., Vandenberghe R., Dermaut B., Martin J.-J., et al. 2006. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 442:920–924. 10.1038/nature05017 [DOI] [PubMed] [Google Scholar]
- Culouscou J.-M., Carlton G.W., and Shoyab M.. 1993. Biochemical analysis of the epithelin receptor. J. Biol. Chem. 268:10458–10462. [PubMed] [Google Scholar]
- Dave A., Martin S., Kumar R., Craig J.E., Burdon K.P., and Sharma S.. 2016. EPHA2 mutations contribute to congenital cataract through diverse mechanisms. Mol. Vis. 22:18–30. [PMC free article] [PubMed] [Google Scholar]
- Dunn K.W., Kamocka M.M., and McDonald J.H.. 2011. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 300:C723–C742. 10.1152/ajpcell.00462.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duxbury M.S., Ito H., Zinner M.J., Ashley S.W., and Whang E.E.. 2004. EphA2: A determinant of malignant cellular behavior and a potential therapeutic target in pancreatic adenocarcinoma. Oncogene. 23:1448–1456. 10.1038/sj.onc.1207247 [DOI] [PubMed] [Google Scholar]
- Farach-Carson M.C., Warren C.R., Harrington D.A., and Carson D.D.. 2014. Border patrol: Insights into the unique role of perlecan/heparan sulfate proteoglycan 2 at cell and tissue borders. Matrix Biol. 34:64–79. 10.1016/j.matbio.2013.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferluga S., Hantgan R., Goldgur Y., Himanen J.P., Nikolov D.B., and Debinski W.. 2013. Biological and structural characterization of glycosylation on ephrin-A1, a preferred ligand for EphA2 receptor tyrosine kinase. J. Biol. Chem. 288:18448–18457. 10.1074/jbc.M113.464008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass J., Lee W.C., Cook C., Finch N., Stetler C., Jansen-West K., Lewis J., Link C.D., Rademakers R., Nykjær A., and Petrucelli L.. 2012. Progranulin regulates neuronal outgrowth independent of sortilin. Mol. Neurodegener. 7:33 10.1186/1750-1326-7-33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghidoni R., Benussi L., Glionna M., Franzoni M., and Binetti G.. 2008. Low plasma progranulin levels predict progranulin mutations in frontotemporal lobar degeneration. Neurology. 71:1235–1239. 10.1212/01.wnl.0000325058.10218.fc [DOI] [PubMed] [Google Scholar]
- Gijselinck I., Van Broeckhoven C., and Cruts M.. 2008. Granulin mutations associated with frontotemporal lobar degeneration and related disorders: An update. Hum. Mutat. 29:1373–1386. 10.1002/humu.20785 [DOI] [PubMed] [Google Scholar]
- Giorgio C., Hassan Mohamed I., Flammini L., Barocelli E., Incerti M., Lodola A., and Tognolini M.. 2011. Lithocholic acid is an Eph-ephrin ligand interfering with Eph-kinase activation. PLoS One. 6:e18128 10.1371/journal.pone.0018128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldoni S., Humphries A., Nyström A., Sattar S., Owens R.T., McQuillan D.J., Ireton K., and Iozzo R.V.. 2009. Decorin is a novel antagonistic ligand of the Met receptor. J. Cell Biol. 185:743–754. 10.1083/jcb.200901129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E.M., Mongiat M., Slater S.J., Baffa R., and Iozzo R.V.. 2003. A novel interaction between perlecan protein core and progranulin: Potential effects on tumor growth. J. Biol. Chem. 278:38113–38116. 10.1074/jbc.C300310200 [DOI] [PubMed] [Google Scholar]
- Grindel B.J., Martinez J.R., Pennington C.L., Muldoon M., Stave J., Chung L.W., and Farach-Carson M.C.. 2014. Matrilysin/matrix metalloproteinase-7 (MMP7) cleavage of perlecan/HSPG2 creates a molecular switch to alter prostate cancer cell behavior. Matrix Biol. 36:64–76. 10.1016/j.matbio.2014.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z., and Bateman A.. 2003. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J. Mol. Med. 81:600–612. 10.1007/s00109-003-0474-3 [DOI] [PubMed] [Google Scholar]
- He Z., Ong C.H.P., Halper J., and Bateman A.. 2003. Progranulin is a mediator of the wound response. Nat. Med. 9:225–229. 10.1038/nm816 [DOI] [PubMed] [Google Scholar]
- Hess A.R., Seftor E.A., Gardner L.M.G., Carles-Kinch K., Schneider G.B., Seftor R.E.B., Kinch M.S., and Hendrix M.J.C.. 2001. Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: Role of epithelial cell kinase (Eck/EphA2). Cancer Res. 61:3250–3255. [PubMed] [Google Scholar]
- Hu F., Padukkavidana T., Vægter C.B., Brady O.A., Zheng Y., Mackenzie I.R., Feldman H.H., Nykjaer A., and Strittmatter S.M.. 2010. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 68:654–667. 10.1016/j.neuron.2010.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Xiao D., Li G., Ma J., Chen P., Yuan W., Hou F., Ge J., Zhong M., Tang Y., et al. 2014. EphA2 promotes epithelial-mesenchymal transition through the Wnt/β-catenin pathway in gastric cancer cells. Oncogene. 33:2737–2747. 10.1038/onc.2013.238 [DOI] [PubMed] [Google Scholar]
- Iozzo R.V. 2005. Basement membrane proteoglycans: From cellar to ceiling. Nat. Rev. Mol. Cell Biol. 6:646–656. 10.1038/nrm1702 [DOI] [PubMed] [Google Scholar]
- Iozzo R.V., and Schaefer L.. 2015. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 42:11–55. 10.1016/j.matbio.2015.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun G., Guo H., Klein B.E., Klein R., Wang J.J., Mitchell P., Miao H., Lee K.E., Joshi T., Buck M., et al. 2009. EPHA2 is associated with age-related cortical cataract in mice and humans. PLoS Genet. 5:e1000584 10.1371/journal.pgen.1000584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessenbrock K., Wang C.-Y., and Werb Z.. 2015. Matrix metalloproteinases in stem cell regulation and cancer. Matrix Biol. 44-46:184–190. 10.1016/j.matbio.2015.01.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlschütter A., and Schulz A.. 2009. Towards understanding the neuronal ceroid lipofuscinoses. Brain Dev. 31:499–502. 10.1016/j.braindev.2008.12.008 [DOI] [PubMed] [Google Scholar]
- Kullander K., and Klein R.. 2002. Mechanisms and functions of Eph and ephrin signalling. Nat. Rev. Mol. Cell Biol. 3:475–486. 10.1038/nrm856 [DOI] [PubMed] [Google Scholar]
- Lema Tomé C.M., Palma E., Ferluga S., Lowther W.T., Hantgan R., Wykosky J., and Debinski W.. 2012. Structural and functional characterization of monomeric EphrinA1 binding site to EphA2 receptor. J. Biol. Chem. 287:14012–14022. 10.1074/jbc.M111.311670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon M.A., and Schlessinger J.. 2010. Cell signaling by receptor tyrosine kinases. Cell. 141:1117–1134. 10.1016/j.cell.2010.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord M.S., Chuang C.Y., Melrose J., Davies M.J., Iozzo R.V., and Whitelock J.M.. 2014a The role of vascular-derived perlecan in modulating cell adhesion, proliferation and growth factor signaling. Matrix Biol. 35:112–122. 10.1016/j.matbio.2014.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord M.S., Jung M., Cheng B., and Whitelock J.M.. 2014b Transcriptional complexity of the HSPG2 gene in the human mast cell line, HMC-1. Matrix Biol. 35:123–131. 10.1016/j.matbio.2013.12.005 [DOI] [PubMed] [Google Scholar]
- Lovat F., Bitto A., Xu S.-Q., Fassan M., Goldoni S., Metalli D., Wubah V., McCue P., Serrero G., Gomella L.G., et al. 2009. Proepithelin is an autocrine growth factor for bladder cancer. Carcinogenesis. 30:861–868. 10.1093/carcin/bgp050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margaryan N.V., Strizzi L., Abbott D.E., Seftor E.A., Rao M.S., Hendrix M.J.C., and Hess A.R.. 2009. EphA2 as a promoter of melanoma tumorigenicity. Cancer Biol. Ther. 8:279–288. 10.4161/cbt.8.3.7485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara T., Mita A., Minami K., Hosooka T., Kitazawa S., Takahashi K., Tamori Y., Yokoi N., Watanabe M., Matsuo E., et al. 2012. PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metab. 15:38–50. 10.1016/j.cmet.2011.12.002 [DOI] [PubMed] [Google Scholar]
- Merline R., Moreth K., Beckmann J., Nastase M.V., Zeng-Brouwers J., Tralhão J.G., Lemarchand P., Pfeilschifter J., Schaefer R.M., Iozzo R.V., and Schaefer L.. 2011. Signaling by the matrix proteoglycan decorin controls inflammation and cancer through PDCD4 and MicroRNA-21. Sci. Signal. 4:ra75 10.1126/scisignal.2001868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao H., Burnett E., Kinch M., Simon E., and Wang B.. 2000. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat. Cell Biol. 2:62–69. 10.1038/35000008 [DOI] [PubMed] [Google Scholar]
- Minami S.S., Min S.W., Krabbe G., Wang C., Zhou Y., Asgarov R., Li Y., Martens L.H., Elia L.P., Ward M.E., et al. 2014. Progranulin protects against amyloid β deposition and toxicity in Alzheimer’s disease mouse models. Nat. Med. 20:1157–1164. 10.1038/nm.3672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misaki R., Morimatsu M., Uemura T., Waguri S., Miyoshi E., Taniguchi N., Matsuda M., and Taguchi T.. 2010. Palmitoylated Ras proteins traffic through recycling endosomes to the plasma membrane during exocytosis. J. Cell Biol. 191:23–29. 10.1083/jcb.200911143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monami G., Gonzalez E.M., Hellman M., Gomella L.G., Baffa R., Iozzo R.V., and Morrione A.. 2006. Proepithelin promotes migration and invasion of 5637 bladder cancer cells through the activation of ERK1/2 and the formation of a paxillin/FAK/ERK complex. Cancer Res. 66:7103–7110. 10.1158/0008-5472.CAN-06-0633 [DOI] [PubMed] [Google Scholar]
- Monami G., Emiliozzi V., Bitto A., Lovat F., Xu S.-Q., Goldoni S., Fassan M., Serrero G., Gomella L.G., Baffa R., et al. 2009. Proepithelin regulates prostate cancer cell biology by promoting cell growth, migration, and anchorage-independent growth. Am. J. Pathol. 174:1037–1047. 10.2353/ajpath.2009.080735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noberini R., Koolpe M., Peddibhotla S., Dahl R., Su Y., Cosford N.D.P., Roth G.P., and Pasquale E.B.. 2008. Small molecules can selectively inhibit ephrin binding to the EphA4 and EphA2 receptors. J. Biol. Chem. 283:29461–29472. 10.1074/jbc.M804103200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nykjaer A., and Willnow T.E.. 2012. Sortilin: A receptor to regulate neuronal viability and function. Trends Neurosci. 35:261–270. 10.1016/j.tins.2012.01.003 [DOI] [PubMed] [Google Scholar]
- Ogawa K., Pasqualini R., Lindberg R.A., Kain R., Freeman A.L., and Pasquale E.B.. 2000. The ephrin-A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene. 19:6043–6052. 10.1038/sj.onc.1204004 [DOI] [PubMed] [Google Scholar]
- Park J.E., Son A.I., Hua R., Wang L., Zhang X., and Zhou R.. 2012. Human cataract mutations in EPHA2 SAM domain alter receptor stability and function. PLoS One. 7:e36564 10.1371/journal.pone.0036564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parri M., Taddei M.L., Bianchini F., Calorini L., and Chiarugi P.. 2009. EphA2 reexpression prompts invasion of melanoma cells shifting from mesenchymal to amoeboid-like motility style. Cancer Res. 69:2072–2081. 10.1158/0008-5472.CAN-08-1845 [DOI] [PubMed] [Google Scholar]
- Petty A., Myshkin E., Qin H., Guo H., Miao H., Tochtrop G.P., Hsieh J.T., Page P., Liu L., Lindner D.J., et al. 2012. A small molecule agonist of EphA2 receptor tyrosine kinase inhibits tumor cell migration in vitro and prostate cancer metastasis in vivo. PLoS One. 7:e42120 10.1371/journal.pone.0042120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prudencio M., Jansen-West K.R., Lee W.C., Gendron T.F., Zhang Y.-J., Xu Y.-F., Gass J., Stuani C., Stetler C., Rademakers R., et al. 2012. Misregulation of human sortilin splicing leads to the generation of a nonfunctional progranulin receptor. Proc. Natl. Acad. Sci. USA. 109:21510–21515. 10.1073/pnas.1211577110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanello M., Piatkowska E., Antoniali G., Cesaratto L., Vascotto C., Iozzo R.V., Delneri D., and Brancia F.L.. 2014. Osteoblastic cell secretome: A novel role for progranulin during risedronate treatment. Bone. 58:81–91. 10.1016/j.bone.2013.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrero G., and Mills D.. 1991. Physiological role of epidermal growth factor on adipose tissue development in vivo. Proc. Natl. Acad. Sci. USA. 88:3912–3916. 10.1073/pnas.88.9.3912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleegers K., Brouwers N., Maurer-Stroh S., van Es M.A., Van Damme P., van Vught P.W.J., van der Zee J., Serneels S., De Pooter T., Van den Broeck M., et al. 2008. Progranulin genetic variability contributes to amyotrophic lateral sclerosis. Neurology. 71:253–259. 10.1212/01.wnl.0000289191.54852.75 [DOI] [PubMed] [Google Scholar]
- Smith K.R., Damiano J., Franceschetti S., Carpenter S., Canafoglia L., Morbin M., Rossi G., Pareyson D., Mole S.E., Staropoli J.F., et al. 2012. Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am. J. Hum. Genet. 90:1102–1107. 10.1016/j.ajhg.2012.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W., Ma Y., Wang J., Brantley-Sieders D., and Chen J.. 2014. JNK signaling mediates EPHA2-dependent tumor cell proliferation, motility, and cancer stem cell-like properties in non-small cell lung cancer. Cancer Res. 74:2444–2454. 10.1158/0008-5472.CAN-13-2136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddei M.L., Parri M., Angelucci A., Onnis B., Bianchini F., Giannoni E., Raugei G., Calorini L., Rucci N., Teti A., et al. 2009. Kinase-dependent and -independent roles of EphA2 in the regulation of prostate cancer invasion and metastasis. Am. J. Pathol. 174:1492–1503. 10.2353/ajpath.2009.080473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W., Lu Y., Tian Q.Y., Zhang Y., Guo F.-J., Liu G.-Y., Syed N.M., Lai Y., Lin E.A., Kong L., et al. 2011. The growth factor progranulin binds to TNF receptors and is therapeutic against inflammatory arthritis in mice. Science. 332:478–484. 10.1126/science.1199214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tangkeangsirisin W., and Serrero G.. 2004. PC cell-derived growth factor (PCDGF/GP88, progranulin) stimulates migration, invasiveness and VEGF expression in breast cancer cells. Carcinogenesis. 25:1587–1592. 10.1093/carcin/bgh171 [DOI] [PubMed] [Google Scholar]
- Tanimoto R., Morcavallo A., Terracciano M., Xu S.Q., Stefanello M., Buraschi S., Lu K.G., Bagley D.H., Gomella L.G., Scotlandi K., et al. 2015. Sortilin regulates progranulin action in castration-resistant prostate cancer cells. Endocrinology. 156:58–70. 10.1210/en.2014-1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto R., Lu K.G., Xu S.Q., Buraschi S., Belfiore A., Iozzo R.V., and Morrione A.. 2016. Mechanisms of progranulin action and regulation in genitourinary cancers. Front. Endocrinol. (Lausanne). 7:100 10.3389/fendo.2016.00100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh H., Chitramuthu B.P., Bennett H.P.J., and Bateman A.. 2011. Structure, function, and mechanism of progranulin; the brain and beyond. J. Mol. Neurosci. 45:538–548. 10.1007/s12031-011-9569-4 [DOI] [PubMed] [Google Scholar]
- Toh H., Cao M., Daniels E., and Bateman A.. 2013. Expression of the growth factor progranulin in endothelial cells influences growth and development of blood vessels: A novel mouse model. PLoS One. 8:e64989 10.1371/journal.pone.0064989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolkatchev D., Malik S., Vinogradova A., Wang P., Chen Z., Xu P., Bennett H.P.J., Bateman A., and Ni F.. 2008. Structure dissection of human progranulin identifies well-folded granulin/epithelin modules with unique functional activities. Protein Sci. 17:711–724. 10.1110/ps.073295308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme P., Van Hoecke A., Lambrechts D., Vanacker P., Bogaert E., van Swieten J., Carmeliet P., Van Den Bosch L., and Robberecht W.. 2008. Progranulin functions as a neurotrophic factor to regulate neurite outgrowth and enhance neuronal survival. J. Cell Biol. 181:37–41. 10.1083/jcb.200712039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kampen J.M., Baranowski D., and Kay D.G.. 2014. Progranulin gene delivery protects dopaminergic neurons in a mouse model of Parkinson’s disease. PLoS One. 9:e97032 10.1371/journal.pone.0097032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.C., Liu H., Talwar A., and Jian J.. 2015. New discovery rarely runs smooth: An update on progranulin/TNFR interactions. Protein Cell. 6:792–803. 10.1007/s13238-015-0213-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesa A.K., Herrem C.J., Mandic M., Taylor J.L., Vasquez C., Kawabe M., Tatsumi T., Leibowitz M.S., Finke J.H., Bukowski R.M., et al. 2008. Enhancement in specific CD8+ T cell recognition of EphA2+ tumors in vitro and in vivo after treatment with ligand agonists. J. Immunol. 181:7721–7727. 10.4049/jimmunol.181.11.7721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitelock J.M., Melrose J., and Iozzo R.V.. 2008. Diverse cell signaling events modulated by perlecan. Biochemistry. 47:11174–11183. 10.1021/bi8013938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienken C.J., Baaske P., Rothbauer U., Braun D., and Duhr S.. 2010. Protein-binding assays in biological liquids using microscale thermophoresis. Nat. Commun. 1:100 10.1038/ncomms1093 [DOI] [PubMed] [Google Scholar]
- Wilusz R.E., Sanchez-Adams J., and Guilak F.. 2014. The structure and function of the pericellular matrix of articular cartilage. Matrix Biol. 39:25–32. 10.1016/j.matbio.2014.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykosky J., and Debinski W.. 2008. The EphA2 receptor and ephrinA1 ligand in solid tumors: Function and therapeutic targeting. Mol. Cancer Res. 6:1795–1806. 10.1158/1541-7786.MCR-08-0244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykosky J., Palma E., Gibo D.M., Ringler S., Turner C.P., and Debinski W.. 2008. Soluble monomeric EphrinA1 is released from tumor cells and is a functional ligand for the EphA2 receptor. Oncogene. 27:7260–7273. 10.1038/onc.2008.328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X., and Serrero G.. 1998. Identification of cell surface binding sites for PC-cell-derived growth factor, PCDGF, (epithelin/granulin precursor) on epithelial cells and fibroblasts. Biochem. Biophys. Res. Commun. 245:539–543. 10.1006/bbrc.1998.8498 [DOI] [PubMed] [Google Scholar]
- Xu K., Zhang Y., Ilalov K., Carlson C.S., Feng J.Q., Di Cesare P.E., and Liu C.J.. 2007. Cartilage oligomeric matrix protein associates with granulin-epithelin precursor (GEP) and potentiates GEP-stimulated chondrocyte proliferation. J. Biol. Chem. 282:11347–11355. 10.1074/jbc.M608744200 [DOI] [PubMed] [Google Scholar]
- Xu S.Q., Buraschi S., Morcavallo A., Genua M., Shirao T., Peiper S.C., Gomella L.G., Birbe R., Belfiore A., Iozzo R.V., and Morrione A.. 2015. A novel role for drebrin in regulating progranulin bioactivity in bladder cancer. Oncotarget. 6:10825–10839. 10.18632/oncotarget.3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y., and Pasquale E.B.. 2004. Eph receptors in the adult brain. Curr. Opin. Neurobiol. 14:288–296. 10.1016/j.conb.2004.04.003 [DOI] [PubMed] [Google Scholar]
- Yang N.Y., Fernandez C., Richter M., Xiao Z., Valencia F., Tice D.A., and Pasquale E.B.. 2011. Crosstalk of the EphA2 receptor with a serine/threonine phosphatase suppresses the Akt-mTORC1 pathway in cancer cells. Cell. Signal. 23:201–212. 10.1016/j.cellsig.2010.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanocco-Marani T., Bateman A., Romano G., Valentinis B., He Z.-H., and Baserga R.. 1999. Biological activities and signaling pathways of the granulin/epithelin precursor. Cancer Res. 59:5331–5340. [PubMed] [Google Scholar]
- Zinchuk V., Wu Y., Grossenbacher-Zinchuk O., and Stefani E.. 2011. Quantifying spatial correlations of fluorescent markers using enhanced background reduction with protein proximity index and correlation coefficient estimations. Nat. Protoc. 6:1554–1567. 10.1038/nprot.2011.384 [DOI] [PubMed] [Google Scholar]
- Zoeller J.J., McQuillan A., Whitelock J., Ho S.-Y., and Iozzo R.V.. 2008. A central function for perlecan in skeletal muscle and cardiovascular development. J. Cell Biol. 181:381–394. 10.1083/jcb.200708022 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.