

Abstract
MOTIVATION
Among many large-scale proteomic quantification methods, 18O/16O labeling requires neither specific amino acid in peptides nor label incorporation through several cell cycles, as in metabolic labeling; it does not cause significant elution time shifts between heavy- and light-labeled peptides, and its dynamic range of quantification is larger than that of tandem mass spectrometry-based quantification methods. These properties offer 18O/16O labeling the maximum flexibility in application. However, 18O/16O labeling introduces large quantification variations due to varying labeling efficiency. There lacks a processing pipeline that warrants the reliable identification of differentially expressed proteins (DEPs). This motivates us to develop a quantitative proteomic approach based on 18O/16O labeling and apply it on Kaposi sarcoma-associated herpesvirus (KSHV) microRNA (miR) target prediction. KSHV is a human pathogenic γ-herpesvirus strongly associated with the development of B-cell proliferative disorders, including primary effusion lymphoma. Recent studies suggest that miRs have evolved a highly complex network of interactions with the cellular and viral transcriptomes, and relatively few KSHV miR targets have been characterized at the functional level. While the new miR target prediction method, photoactivatable ribonucleoside-enhanced cross-linking and immunoprecipitation (PAR-CLIP), allows the identification of thousands of miR targets, the link between miRs and their targets still cannot be determined. We propose to apply the developed proteomic approach to establish such links.
METHOD
We integrate several 18O/16O data processing algorithms that we published recently and identify the messenger RNAs of downregulated proteins as potential targets in KSHV miR-transfected human embryonic kidney 293T cells. Various statistical tests are employed for picking DEPs, and we select the best test by examining the enrichment of PAR-CLIP-reported targets with seed match to the miRs of interest among top ranked DEPs returned by statistical tests. Subsequently, the list of DEPs picked by the selected statistical test is filtered with the criteria that they must have downregulated gene expressions, must have reported as targets by an miR target prediction algorithm SVMcrio, and must have reported as targets by PAR-CLIP.
RESULT
We test the developed approach in the problem of finding targets of KSHV miR-K1. The RNAs of three DEPs are identified as miR-K1 targets, among which RAB23 and HNRNPU are novel. Results from both Western blotting and Luciferase reporter assays confirm the novel targets. These results show that the developed quantitative approach based on 18O/16O labeling can be combined with genomic, PAR-CLIP, and target prediction algorithms for the confident identification of KSHV miR targets. The developed approach could also be applied in other applications.
Keywords: proteomics, miRNA target prediction, O18 labeling, KSHV
Introduction
Finding differentially expressed proteins (DEPs) between two conditions is a common problem in biological and clinical research. There are several quantification methods for measuring DEPs based on liquid chromatography–mass spectrometry/tandem mass spectrometry (LC-MS/MS). These are either label-free or stable isotope labeling methods.1 Label-free methods offer wider dynamic range and broader proteome coverage, while stable isotope labeling approaches offer higher quantification precision and accuracy.2
Among many labeling methods, chemical isobaric tagging (including Isobaric tag for relative and absolute quantitation [iTRAQ] and Tandem mass tag [TMT]) provides up to 8-plex analysis by quantifying at the tandem MS level. However, it suffers from severe dynamic range compression and reduced quantitative accuracy due to precursor interference when samples are complex.3,4 Considering that a lot of research studies need to deal with complex samples, we have to consider quantification at the LC-MS level.
Among labeling methods at the LC-MS level, stable isotope labeling of amino acid in cell culture (SILAC)5 has high quantification accuracy. However, SILAC requires several cell cycles to incorporate the labels, and in research like microRNA (miR) target prediction, the miR-mediated regulation of proteins with long half-lives may not be detected by measuring steady-state protein levels using SILAC.6 Pulsed SILAC, which only compares the differential expression of newly synthesized proteins at different time points, is a great analytical tool. However, it is relatively expensive, time-consuming, and not practical for analyzing biological samples that cannot be grown in culture, such as tissues or body fluids.6 In addition, most proteomic centers that run LC-MS/MS experiments cannot perform required cell culturing because of licensing issues in handling virus-transfected cells. Laboratories with cells that need LC-MS/MS analysis may not have the resource and time in implementing a complex laboratory protocol, such as SILAC/pulsed SILAC. These practical aspects limit the application of SILAC/pulsed SILAC.
An alternative LC-MS quantification method based on chemical labeling is dimethylation of peptides. However, deuterated peptides show a small but significant retention time difference in reversed phase chromatography compared to their nondeuterated counterparts.7 This complicates data analysis because the relative quantities of two peptides cannot be determined accurately from one spectrum, and it requires integration across the whole chromatographic timescale. Considering that there exists a lot of co-eluting peptides that contaminate elution peaks, integrating across the whole chromatographic timescale becomes impractical.
Isotope-coded affinity tagging is a chemical labeling method that was first described by the Aebersold lab. This method only quantifies cysteine-containing peptides carrying heavy and light isotope tags, which limit its quantification coverage.
Reverse phase protein array8 is another protein quantification method; however, it is limited with the availability of high-quality protein antibody.
18O/16O labeling has relatively low cost and complexity. It does not require specific amino acid in peptides9 and label incorporation through several cell cycles, nor does it cause significant elution time shifts between heavy- and light-labeled peptides. Its dynamic range of quantification is larger than that of tandem MS-based quantification methods. These properties offer 18O/16O labeling the maximum flexibility in application.
In this work, we propose to develop an LC-MS-based quantitative proteomic approach for identifying DEPs based on 18O/16O labeling and apply the approach to the problem of Kaposi sarcoma-associated herpesvirus (KSHV) miR-K1 target prediction. MiRs are short RNAs that regulate target gene expression levels.10,11 Dysregulation of miRs may lead to disease progression and cancer pathogensis,12 but the underlying mechanisms are still not very clear. The understanding of miR function is not possible without the knowledge of target messenger RNAs (mRNAs) of miRs. KSHV is the causative agent of Kaposi sarcoma (KSar), which is associated with primary effusion lymphoma (PEL), and a subset of multicentric Castleman disease.13 KSHV encodes dozens of miRs derived from 12 pre-miRNAs, among which miR-K1 is a very important one. It directly regulates the lκBα protein by targeting the 3′ untranslated region (UTR) of its transcript. The expression of miR-K1 is sufficient to rescue lκBα protein activity and inhibit viral lytic replication, whereas the inhibition of miR-K1 in KSHV-infected PEL cells has the opposite effect.14 We aim to identify miR-K1 targets by identifying DEPs between the human embryonic kidney 293T cells transfected with KSHV miR-K1 and the control group transfected with an empty vector for 48 hours.14 We use tandem MS for peptide identification (not quantification) and LC-MS for quantification. KSHV-transfected sample is digested with trypsin in 18O-water, and the control sample is digested in normal water. Subsequently, after protein digestion, the samples are mixed together. Twenty strong cation exchange (SCX)15 fractions of peptides are collected, and each fraction is further divided into three technical replicates.
In 18O/16O data processing, one needs to properly address the following issues.
We need to perform peptide feature alignment between different technical replicates, so that a peptide can be quantified multiple times to reduce quantification variations.
Due to interactions between peptide ion clouds inside mass spectrometers, smaller ion clouds in Orbitrap instruments can get torn apart by larger ion clouds16 and the abundance measurements of smaller ion clouds are suppressed randomly, as shown in our previous research.17 This suppression effect leads to unknown bias and variation in fold change measurements.
Multiple peptides of a protein could have been quantified in different data fractions with different bias and variation, which cannot be estimated by assuming a simple Gaussian noise model.17 Consequently, it is hard to estimate protein expression levels based on peptide measurements.
We do not know what statistical test is most appropriate for picking DEPs, given the complex structure of LC-MS/MS data, where each peptide has its unique measurement variance.
Although there are several algorithms and software programs published for 18O/16O labeling,18–22 the abovementioned issues have not been addressed properly. To overcome these issues, we first apply an alignment algorithm called statistical corresponding feature identification algorithm (SCFIA)23 to boost the total number of quantifications per peptide per protein. In this way, the majority of tandem MS-identified peptides within an SCX fraction can be quantified three times in three technical replicates. Otherwise, a lot of peptides are only quantified once, as in the widely distributed packages such as Trans-Proteomic Pipeline (TPP)24 and MaxQuant.19 With more measurements per peptide, we can reduce the measurement variance. After alignment, we employ a peptide quantification method that we developed in a previous study25 to partially remove interference and random suppression effects.
After peptide quantification, we need to find downregulated DEPs because miRs regulate proteins through suppression. Thus, we need to estimate the direction of protein expression. We develop a variance estimation method, so that the estimated variance can be used for weighing peptide measurements. We assume that unique and nonunique peptides have uniform variance within their fractions. The uniform variance assumption allows us to weigh unique and nonunique measurements properly.
After we determine the direction of protein expression, various statistical tests are employed for picking DEPs. In genomics, tests are either homoscedastic or heteroscedastic. The former model assumes uniform variance for all protein/gene measurements, and obviously, this does not fit our proteomic data. Heteroscedastic model in genomic data processing assumes gene-by-gene variance26; however, multiple measurements of genes are still assumed to have the same distribution, which does not fit our data either. A recent research27 compares several statistical tests often used in proteomics. However, none of the tests, including the widely used t-test, clearly performs better than other methods with different sample size and variance, and there is no clear guide for selecting the appropriate statistical test.
In this paper, we select statistical tests by examining the enrichment of photoactivatable ribonucleoside-enhanced cross-linking and immunoprecipitation (PAR-CLIP) predicted targets with seed match to miR-K1 among top DEPs returned by different statistical tests. PAR-CLIP uses 4-thiouridine to label mRNAs in vivo combined with ultraviolet cross-linking to improve recovery and to facilitate the identification of the cross-linking site.28 Although PAR-CLIP pulls down the cross-linked mRNAs and miRs, it does not build direct links between them and further validation of individual interactions will be required.29
As seed match is one of the known mechanisms of miR–mRNA binding, a correlation should exist between the PAR-CLIP-predicted targets with seed match to miR-K1 and the DEPs predicted by a good statistical test. On the other hand, the two lists are not expected to overlap completely because: (1) There may exist other binding mechanisms,30 and consequently, PAR-CLIP plus seed match produces a target list that contains false positives. (2) Downregulated DEPs could be attributed to secondary effects of miR transfection.
We developed a statistical test called Kullback–Leiberler31 distance test (KL test), which uses the KL distance as a goodness-of-fit measure for comparing peptide fold changes of a protein to that of a background protein with the same number of peptide measurements. Through the KL test, we obtained a significant enrichment of PAR-CLIP-predicted targets with seed match to miR-K1 (PAR-CLIPSMK1). We also examined the t-test, Kolmogorov–Smirnov (KS) test,32 and their modified versions when considering the number of peptide measurements per protein. None of these tests and MaxQuant and TPP returned DEPs that are significantly enriched with PAR-CLIPSMK1s.
After the statistical test, we had a list of DEPs whose mRNAs are potential miR-K1 targets. However, due to protein–protein interactions, the mRNAs of these DEPs may not be direct miR-K1 targets, and we have to jointly consider miR-K1 targets returned by other prediction methods. Computational methods have been widely used to predict miR targets,33 and most of them rely on sequence complementarity between the 5′UTR end of mature miRs and the 3′UTR of target genes (seed match).34,35 While seed match is an important mechanism for miR target binding, there are other possibilities.36 For example, the SVMicro37 miR target prediction algorithm investigates over 30 features and statistically combines these features for target prediction. However, computational target prediction algorithms suffer from both high false-positive and false-negative rates38 due to the lack of a comprehensive understanding of the binding mechanisms.
Alternatively, we can measure mRNA abundance changes using high-throughput microarray approaches39 for target prediction because miRs cause downregulation at the gene level. However, this method could be problematic if most miRs regulate gene expression by translational inhibition rather than mRNA degradation, which has been shown to be the case in animals.40,41 Even if mRNA degradation is the main gene regulation mechanism,42 there would still be a lot of downregulated genes due to the secondary effects of miR binding that are not direct miR targets.
To improve the true positive rate of identified DEPs as mi-K1 targets, we propose to combine various target prediction methods by further filtering the list of DEPs picked by the KL test using the following criteria: (1) The corresponding mRNAs of DEPs must have downregulation in microarray experiments; (2) the DEPs must have been predicted to be possible targets by SVMicro37; and (3) the DEPs must have been reported as PAR-CLIPSMK1s.
After applying these criteria, the list of DEPs is reduced to three in our experiment, among which RAB23 and HNRNPU are novel. These novel targets have been confirmed by both Western blotting and Luciferase reporter assays, and it shows that the developed quantitative approach based on 18O/16O labeling can be combined with the genomic, PAR-CLIP, and target prediction algorithms to identify KSHV miR targets with high confidence. The developed approach will also have wide applications in other biological and clinical research.
Methods
Data collection
The LC-MS/MS data were pre-separated in 20 fractions, and each fraction consisted of three technical replicates. The samples from human embryonic kidney 293T cells (ATCC) were cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum. One group was transfected with an expression vector expressing miR-K1 of KSHV, while the control group was transfected with a vector for 48 hours.14 These two groups of samples were lysed in 8 M urea and 50 mM ammonium bicarbonate (pH 8.3). The lysates were subjected to centrifugation at 13,000 rpm for 20 minutes, and the supernatants were collected. Then, the two samples were denatured in 8 M urea, reduced using 10 mM dithiothreitol, alkylated with 30 mM iodoacetamide, and digested with trypsin (using an enzyme-to-protein ratio of 1:50) at 37 °C overnight. The samples were desalted with Sep-Pak cartridges, separated into two tubes, and dried in a SpeedVac. The first sample was resuspended in 100 mL 18O-water (purity > 98%), containing 50 mM ammonium bicarbonate, 10 mM calcium chloride, and trypsin (1–50 w/w trypsin:peptide) pH 7.8. The second sample was treated in the same manner except that the 18O-water was replaced with purified 16O-water. After incubation with shaking at 450 rpm for five hours at 37 °C, the labeling reaction was terminated by first boiling the sample for 10 minutes and then adding 5 mL of formic acid to further inhibit any residual trypsin activity. A bicinchoninic acid assay was performed to determine peptide concentration. Two hundred micrograms of equally combined sample was fractionated into 20 fractions, using SCX. Then, the four samples were subjected to reverse phase–reverse phase LC followed by ETD-LTQ-Orbitrap Velos MS.
PAR-CLIP data were downloaded from http://bugs.mimnet.northwestern.edu/labs/gottweinlab/Data.html.
For more information, please refer Ref. 29. Details of gene expression data based on miR-K1 transfection were published in Ref. 37.
LC-MS/MS data processing
Figure 1 shows the overall processing flow chart that consists of four steps: (1) preprocessing, (2) peptide quantification, (3) protein quantification, and (4) identification of DEPs.
Figure 1.

The overall processing flow chart.
Preprocessing
The goal of preprocessing is to obtain a list of tandem MS-identified peptides, based on which, we can quantify these peptides at the LC-MS level. We use TPP for this purpose. Raw LC-MS data collected from Orbitrap are first converted to the mzXML formats and are submitted for tandem MS peptide identification using X! Tandem, which is called by TPP. As mentioned in the previous section, there are 20 SCX fractions and 3 technical replicates within each fraction, which results in 60 LC-MS files. All the 60 files are processed in TPP in a signal run. The protein database used is International Protein Index (IPI) human database version 3.68 (http://www.mmnt.net/db/0/5/ftp.ebi.ac.uk/pub/databases/IPI/old/HUMAN/). For X! Tandem, the parent mass and fragment ions are searched with maximal mass errors of 7 ppm and 0.5 dalton, respectively. Methionine oxidation and n-terminal acetylation are considered as variable modifications, and cysteine carbamidomethylation is selected as the fixed modification. The modification mass of the C is set to 57.021464, and the potential modification mass of M is set to 15.994915. The input of the cleavage C-terminal mass change is set to 21.01. In database search, the minimum length of peptide is set to 6 and the maximum missed cleavage sites is set to 2. The peptide prophet score threshold is set to 0.9 to guarantee high confident identification. Peptides that are identified multiple times are combined, which resulted in a list of 31,268 distinct peptides and 4,740 distinct proteins. For more information about the protein expression, please refer Supplementary File (http://compgenomics.utsa.edu/zgroup/miRTargetprediction/miR-Targets.htm).
MaxQuant processing
MaxQuant is a widely available software package that can process 18O/16O data. In order to compare our approach with that of MaxQuant, we download MaxQuant_1.3.0.5 from the webpage www.maxquant.org. IPI human database version 3.83 is selected as the source of protein sequences. We set the MS1 tolerance to 20 ppm for the first search and 6 ppm for the main search. We set the MS/MS tolerance to 20 ppm, peptide false detection rate (FDR) to 0.01, protein FDR to 0.01, site FDR to 0.01, and heavy labels to 18O. We have selected oxidation (M) and acetyl(protein N-term) as modification sites. In database search, the minimum length of peptide is set to 7 and the maximum missed cleavage sites is set to 2, as these are the default values of the software.
Peptide quantification
Although both TPP and Max-Quant perform peptide quantification, they are not specifically optimized for 18O/16O data. The ASAPRatio algorithm used in TPP is designed for low resolution data, and Max-Quant is shown to have large bias.25
The quantification process is briefly described as the following. We first obtain a union list of tandem MS-identified peptides across all the three technical replicates in each fraction. Then, candidate LC peaks of peptides are identified at the LC-MS level. We employ a special LC peak detection algorithm that is effective in removing interference from co-eluting peptides.25
If a peptide is identified by tandem MS with elution time information, we will pick the LC peak that matches in elution time. If a peptide is identified in other technical replicates, but not in current one, we employ SCFIA developed by Ciu et al.23 to find its LC peak in the current technical replicate.
Once the peptide peaks are identified in all the technical replicates, a linear regression method is used to quantify the heavy light ratios (HLRs) between the labeled and unlabeled peptides.43
Protein quantification
Once peptide level quantification is finished, we need to integrate peptide HLRs to that of proteins. However, peptide HLRs have different measurement variances. For example, nonunique peptides that are shared among several proteins have larger variance than unique peptides. Peptides measured on different fractions have different variances. These variances cannot be estimated for every peptide, and we cannot hope to get accurate protein quantification without knowing the variances. To best approximate the true protein expression level, we have to make some simplifying assumptions so that we can at least estimate the direction of protein expression. For this purpose, we assume that unique or nonunique peptides share the same measurement variance, within an LC-MS dataset, which can be estimated as described in the following sections:
Estimating peptide expression variances. To estimate the variance of measurements within a dataset, we consider two peptides from the same protein. Specifically, suppose two peptides from a protein in an LC-MS/MS dataset are measured to have HLRs as r1 and r2 and their distributions can be approximated as normal: and , their log ratio difference (LRD) can be defined as follows:
| (1) |
Then, we have
| (2) |
As the peptides of the same protein in the same dataset share the same bias in sample preparation and instrument suppression/distortion, the means of the two log ratios can be assumed identical, and we have:
| (3) |
It is easy to show that
| (4) |
As we assume that the variance of LRD of unique peptides (varLRDU) is uniform within an LC-MS run, we can take many samples within an LC-MS file to estimate the varLRDU, which reflects variations from the labeling process and the instrument. The LRD of nonunique peptides (varLRDNU) will reflect the interference from other proteins in addition to that from the labeling process and the instrument. varLRDU and varLRDNU are estimated for each dataset (60 in total). We denote varLRDNU as Vi,0 and varLRDU as Vi,1, where i is the file number. For details of the LRD calculation, please refer Supplementary File (http://compgenomics.utsa.edu/zgroup/miRTargetprediction/suppl/Supplementary.pdf). We find that varLRDUs and varLRDNUs are similar among replicates, but very different among fractions.
Determining protein expression direction using weighted average of peptide HLRs. Before protein quantification, we first need to combine peptide HLRs in three technical replicates within each fraction. We first consider taking the weighted average of three measurements when assuming independent instrument noise. However, this approach results in higher varLRDU and varLRDNU within each fraction. This can be attributed to the fact that distortions caused by the instrument are not completely independent in technical replicates. Therefore, we opt for selecting the replicate with the smallest varLRDU.
After peptide measurements within each fraction have been determined, we further consider the problem of combining different peptide measurements from different fractions. In such cases, the variation can be attributed to operational and experimental sources in different LC-MS runs, which are much larger than instrumental variations and can be assumed as independent.
Suppose a protein is measured for N times on different peptides with [(r1,V1,j),(r2,V2,j),…,(rN,Vn,j)], in which ri represents the ith peptide measurement, and it is annotated with its measurement variance, Vi,j, where j ∈ (0,1) indicates whether the peptide is unique or not. The weighted mean (µ) and variance (V) of the protein are:
| (5) |
and
| (6) |
Of note, the sign of µ does not change if the set of estimated variances, Vi,js, are proportional to the true variance of a peptide. Using this method, we have quantified 4,740 proteins, and in contrast, TPP has quantified 4,665 proteins in total.
Differential expression prediction
The variance of peptides is calculated based on the assumption that all unique/nonunique peptides share the same variance within the same LC-MS run. While this is a reasonable assumption for estimating the direction of protein expressions, it ignores that each peptide has different labeling efficiency and goes through the instrument with its own random distortion.17 We cannot rely on the estimated protein expression level for picking DEPs.
Popular statistical tests, such as the t-test, assume a Gaussian distribution and uniform variance for different peptide measurements, which do not fit our data. The KS test32 is a goodness-of-fit test that does not assume any specific distribution among the samples; however, it can only be applied appropriately for relatively larger sample sizes (>4). However, a lot of proteins only have 1–3 peptide measurements in our case, and it is not a perfect statistical test as well. For this reason, we developed our own statistical test named as the KL test, which is described as follows.
The KL31 divergence (information divergence) is a non-symmetric measure of the difference between two probability distributions, and the KL divergence between two normalized histograms P and Q is D(P∥Q) = Σipilog(pi/qi), where i represents the ith interval on the histogram of P/Q. pi and qi are the portion of samples within the ith interval in P and Q, respectively.
We want to determine if a protein with N peptide measurements with a normalized histogram P is differentially expressed when compared to the background histogram Q. The problem is that there is a great size mismatch: P is usually derived based on few peptide measurements and Q is based on many samples drawn randomly from the pool of all the peptide measurements. P is usually a poor representation of a distribution, and random variations could affect the statistics of the KL test when P is poorly represented.
To avoid such problems, we draw 5,000 random samples of normalized histograms Jks, where k ε {1,…, 5,000}. Each Jk is built by drawing N random peptide measurements from Q, which allow us to calculate KLk = D(Jk∥Q),∀k. Subsequently, we treat the histogram of KLks as the null distribution and use it to score the significance of D(P∥Q). In this way, as we match the number of peptide measurements between Jks and P, we do not have to worry about the effect of great size mismatch between P and Q. In Figure 2, we show the flow diagram of the KL test. We notice that the effective null distributions based on KLks are different for proteins with different sizes and ranking the DEPs based on the P-values is no longer valid. On the other hand, D(P∥Q) is evaluated for different proteins based on the same Q, and therefore, we can sort DEPs based on D(P∥Q) directly.
Figure 2.
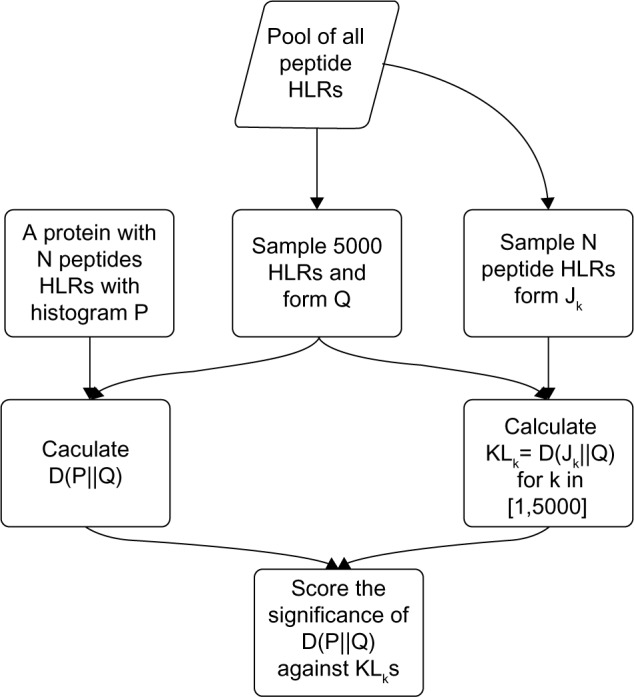
The flowchart of calculating the KL score and significance of each protein.
We have also applied other statistical tests and quantification methods (TPP and MaxQuant) to pick the DEPs. As there is also the problem of significant mismatch in sample size in the t-test and KS test, we have also investigated two modified versions of these tests, which are described as follows.
In these tests, we construct the background peptide distribution Q by randomly picking 5,000 peptide measurements from the pool of all the peptide measurements (31,312 peptides).
In t-test, if a protein has N (N > 2) measurements and forms an empirical distribution R, the t-test between R and Q is first calculated and the resulted P-values are converted to FDRs44 by an MATLAB function mafdr. Down-regulated proteins with FDR < 0.01 are picked as DEPs.
The KS test is applied in a similar way as the t-test.
TPP returns a list of protein measurements and P-values. Downregulated proteins with P-value <0.05 are picked as DEPs.
The modified t-test is very similar to our proposed KL test. The difference is, instead of calculating KLk = D(Jk∥Q), we calculate the t-test statistics between Jk and Q as (Tk(Jk,Q),∀k) using the MATLAB function t-test(). Then, we treat Tk as the null distribution and estimate the P-value of T(P, Q). Finally, we pick proteins with P-value <0.05 as DEPs.
Modified KS test is constructed by replacing the t-test with the KS test in the modified t-test.
MaxQuant is a popular software package that can process 18O/16O data. MaxQuant returns a list of protein measurements. As MaxQuant does not offer P-values, we rank downregulated proteins with their HLRs in ascending order.
Results
We compared and evaluated different test statistics based on the enrichment of PAR-CLIPSMK1 proteins within the top ranked DEPs picked by different methods. The rationale is that if the test statistics reflects the real differential expression, then enriched PAR-CLIPSMK1 proteins should exist among top ranked proteins.
The enrichment rate is calculated by counting the percentage of PAR-CLIPSMK1 targets among all downregulated DEPs at a given rank of the DEPs sorted by the selected test statistics. For comparison, we also calculated the enrichment rate of PAR-CLIP targets without seed match to miR-K1.
The results are summarized in Figure 3. From these figures, we can see that DEPs ranked through the KS test/modified KS test, t-test/modified t-test, TPP, and MaxQuant do not show an enrichment of PAR-CLIPSMK1, especially among the top 50 DEPs. However, clear enrichment of PAR-CLIPSMK1s is shown based on the KL test. Thus, we have selected the DEPs returned by the KL test for further verification.
Figure 3.

Enrichment of PAR-CLIPSMK1 targets in top ranked DEPs using (A) t-test FDR, (B) KS FDR, (C) TPP, (D) KL tests, (E) modified t-test statistics, (F) modified KS test statistics, and (G) MaxQuant protein measurements. X-axis represents the rank of DEPs, and y-axis represents the enrichment rate.
Further filtering of DEPs
Based on the KL test, 331 significantly DEPs are identified. Among them, 13 proteins are found to have overlap with PAR-CLIPSMK1 proteins. The proteins and their corresponding information are listed in Table 1. The enrichment curve is plotted in Figure 3D.
Table 1.
Protein expressed level.
| GENE NAME | PROTEIN INDEX | KL DISTANCE | SVM SCORE | GENE EXPRESSION |
|---|---|---|---|---|
| RAB23 | IPI00008034 | 2.4432 | 0.7998 | −0.0245 |
| GTPBP8 | IPI00107246 | 2.4432 | −0.6822 | 0.1717 |
| CAMK2D | IPI00172636 | 2.4432 | 0.8751; −0.9980; 0.8821 | −0.0281 |
| MRPL1 | IPI00549381 | 2.4432 | −0.9279 | −0.1361 |
| PHF3 | IPI00170770 | 2.4432 | 1.1149 | 0.3065 |
| KPNA1 | IPI00303292 | 2.1908 | −0.9738 | −0.0697 |
| EIF3M | IPI00102069 | 2.1184 | −0.6817 | 0.0242 |
| CPNE3 | IPI00024403 | 2.0685 | −0.9892 | −0.0997 |
| MTDH | IPI00328715 | 2.0463 | 0.7519 | 0.1066 |
| PPP2CA | IPI00008380 | 2.0430 | 0.7073 | −0.1338 |
| RAD21 | IPI00006715 | 1.8131 | −1.0477 | −0.4094 |
| NUP153 | IPI00292059 | 1.7519 | −0.9754 | 0.0567 |
| HNRNPU | IPI00479217 | 1.7326 | 0.3299 | −0.1262 |
We further filtered the 13 proteins by inspecting if their corresponding gene expressions are downregulated and if their mRNAs are predicted as miR-K1 targets using the bioinformatics tool SVMicro.37 This cuts down the list to RAB23, PPP2CA, and HNRNPU. Of note, CAMK2D had three protein isoforms and their returned SVMicro scores are different in all the three cases. We did not include it after filtering.
MiR-K1 target verification
In order to verify that the mRNAs of the three DEPs are identified by the proposed approach as miR-K1 targets, we have further performed Western blotting and Luciferase reporter assay.
Western blotting
For target verification, 293T cells were transfected with synthetic miR-K1 mimic (50 nM). Forty-eight hours after transfection, endogenous protein levels were assessed by Western blotting. Tubulin was used as internal controls. Nitrocellulose (NC) was used as negative control for miR-K1 mimic, which was synthesized by Sigma-Aldrich. Locked Nucleic Acid (LNA) suppressors were chemically synthesized by Exiqon. The sequences are listed in Supplementary Table 1 (http://compgenomics.utsa.edu/zgroup/miRTargetprediction/suppl/Supplementary.pdf). Reverse transfection of RNA oligoribonucleotide(s) was done using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s protocol.
In Western blotting, equal amount of protein samples was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. The blots were blocked with 5% nonfat milk and incubated with primary antibody followed by a horseradish peroxidase- conjugated secondary antibody (Sigma-Aldrich). Specific bands were revealed with chemiluminescence substrates (Roche) and recorded with BioSpectrum Imaging System (UVP Inc.). Antibodies to PPP2CA were obtained from Cell Signaling Technology (CST). Antibodies to RAB23 and HNRNPU were obtained from Abcam.
RNA oligoribonucleotides and cell transfection
miR-K1 mimic was obtained from Sigma-Aldrich. The sequences were listed in Table 2. A scrambled oligonucleotide containing random sequence was used as a control. RNA oligos were transfected using Lipofectamine RNAiMAX (Invitrogen). The transfection of plasmid DNA with RNA oligos was performed using Lipofectamine 2000 (Invitrogen).
Table 2.
Sequences of miRNA mimics and PCR primers.
| NAME | SEQUENCE miRNA MIMICS— |
|---|---|
| miR-K1 | AUUACAGGAAACUGGGUGUAAGC (Sense) |
| UUACACCCAGUUUCCUGUAACUU (Antisense) | |
| Primers for constructing 3′UTR WT and mutant reporters | |
| RAB23 Site 1 WT |
AGTGGTACCTGCAAAATGAGCTTGGGTTT (Forward) |
| AGTCTCGAGTGTGGGACTGACAGCTCTTG (Reverse) | |
| RAB23 Site 1 mutant |
AGTCATTCAGGAGGTGGACAGTAGTGTGGTGATGC (Forward) |
| GCATCACCACACTACTGTCCACCTCCTGAATGACT (Reverse) | |
| RAB23 Site 2 WT |
AGTGGTACCCAAGAGCTGTCAGTCCCACA (Forward) |
| AGTCTCGAGATTTTGCCCCCAAAACCTAT (Reverse) | |
| RAB23 Site 2 mutant |
AGCATTGCAAAATGACATTAAATAACTTTTATT (Forward) |
| AATAAAAGTTATTTAATGTCATTTTGCAATGCT (Reverse) | |
| hnRNPU WT | AGTGGTACCAGGACGAGGAAACAATCGTG (Forward) |
| AGTCTCGAGAGCTTACCCCCTCCACTACC (Reverse) | |
| hnRNPU Mutant | GGTTCTACATTTTATGACATTAATGT-GACTTTTT (Forward) |
| AAAAAGTCACATTAATGTCATAAAATGTAGAACC (Reverse) |
Construction of wild-type and mutant 3′UTR reporters
Wild-type (WT) and mutant 3′UTR reporters were generated as reported in a previous study.45 A WT 3′UTR fragment of the human RAB23 or hnRNPU mRNA containing the putative binding sites for miR-K1 and its 5′ and 3′ flanking regions (271 and 258 bp for RAB23 site 1, 186 and 355 bp for RAB23 site 2, and 344 and 255 bp for hnRNPU, respectively) was polymerase chain reaction (PCR)-amplified and inserted into the Kpn I and Xho I sites, downstream of the stop codon of the firefly luciferase in the pGL3 vector. The mutant 3′UTRs, which carried the mutated sequences in the complementary seed region of miR-K1, were generated using fusion PCR based on the construct using the WT 3′UTR reporters as templates.
Luciferase reporter assays
Reporter assays for the 3′UTR reporters were carried out in 48-well plates as described in a previous study.45 For each well, cells were cotransfected with 10 ng of the luciferase reporter plasmid, 2 ng of pRL-TK (Promega Corporation), and 10 nM of miR mimic. Cells were collected at 48-hour posttransfection and analyzed using the Dual-Luciferase reporter assay system (Promega Corporation). The pRL-TK vector providing the constitutive expression of Renilla luciferase was used as an internal control. Transfection was performed in duplicate, and all experiments were independently repeated at least three times.
Western blotting and reporter assay results
Western blotting results are shown in Figure 4. We can see that Western blotting results confirm all the three targets picked through the proteomic approach. Among the three confirmed targets, RAB23 and HNRNPU are novel. PPP2CA has been reported as an miR-K1 target in a previous publication.29 IkBa14 and p2146 are confirmed miR-K1 targets, but they are not identified in this experiment. We investigated and found that the proteins of IkBa and p21 are not identified by tandem MS before quantification, which randomly samples peptides and has limited coverage, which cannot be controlled by our quantification methods. The reporter assay results of the two novel targets, RAB23 and HNRNPU, are shown in Figures 5 and 6, respectively.
Figure 4.
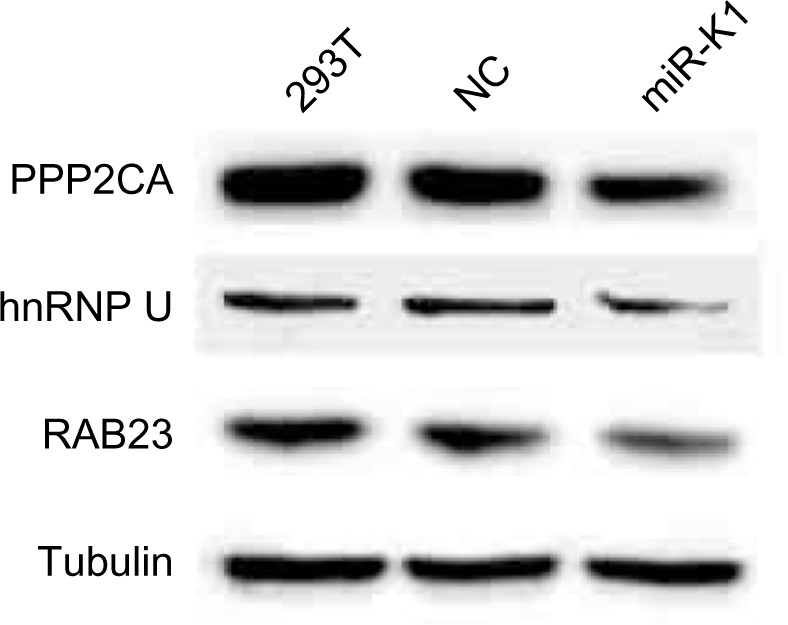
293T cells transfected with synthetic miR-K1 mimic (50 nM). Forty-eight hours after transfection, endogenous protein levels were assessed by Western blotting. Tubulin was used as internal controls. NC was used as negative control for miR-K1 mimic.
Figure 5.

RAB23 is a direct target of miR-K1. (A) Sequence alignment of miR-K1 with the RAB23 3′UTR. (B–C) miR-K1 suppressed the activity of luciferase through its binding site 1 (B) and site 2 (C) in RAB23 3′UCTR. 293T cells were cotransfected with the miR-K1 mimic or the scrambled control, a firefly luciferase reporter containing the WT or mutant 3′UTR reporter, and a Renilla luciferase expressing construct. The firefly luciferase activity of each sample was normalized to the Renilla luciferase activity. The mean of normalized luciferase activity of scrambled control in each experiment was set as 1.
Notes: P < 0.05; P < 0.001, compared with scrambled control.
Figure 6.
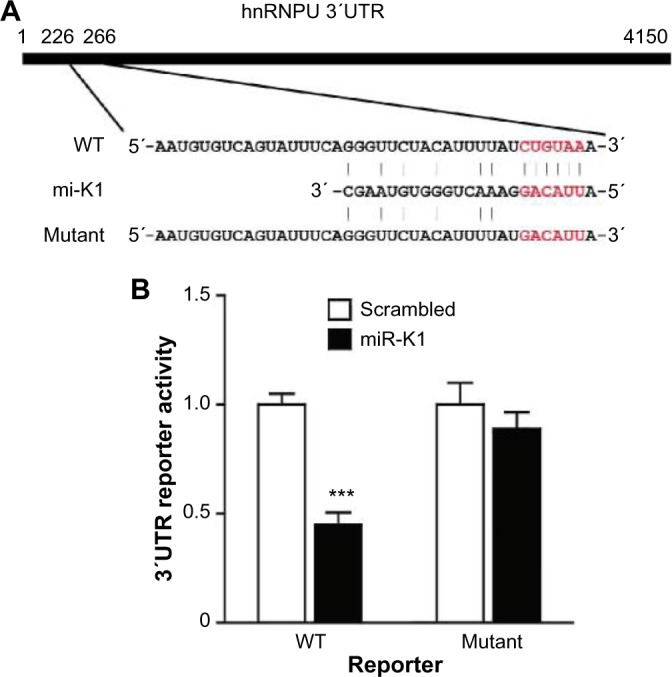
hnRNPU is a direct target of miR-K1. (A) Sequence alignment of miR-K1 with the hnRNPU 3′UTR. (B) miR-K1 suppressed the activity of luciferase through its binding site in hnRNPU 3′UTR. 293T cells were cotransfected with the miR-K1 mimic or the scrambled control, a firefly luciferase reporter containing the WT or mutant 3′UTR reporter, and a Renilla luciferase expressing construct. The firefly luciferase activity of each sample was normalized to the Renilla luciferase activity. The mean of normalized luciferase activity of scrambled control in each experiment was set as 1.
Notes: P < 0.05; P < 0.001, compared with scrambled control.
These results show that our developed method is highly effective in identifying novel biomarkers based on 18O/16O labeling, which can be applied in a wide range of applications.
Function of identified targets
PPP2CA encodes the phosphatase 2A catalytic subunit. Protein phosphatase 2A is one of the four major Ser/Thr phosphatases, and it is implicated in the negative control of cell growth and division, and is involved in breast cancer.47
Among the two novel targets, RAB23 encodes a small GTPase of the Ras superfamily and Rab proteins are involved in the regulation of diverse cellular functions associated with intracellular membrane trafficking, including autophagy and immune response to bacterial infection.48 The encoded protein may play a role in central nervous system development by antagonizing sonic hedgehog signaling.49 Disruption of this gene has been implicated in Carpenter syndrome as well as cancer.50
HNRNPU belongs to the subfamily of ubiquitously expressed heterogeneous nuclear ribonucleoproteins (hnRNPs). The hnRNPs are RNA-binding proteins, and they form complexes with heterogeneous nuclear RNA. These proteins are associated with pre-mRNAs in the nucleus and appear to influence pre-mRNA processing and other aspects of mRNA metabolism and transport.51 It has been shown that hnRNPU directly interacts with WT1 and modulates WT1 transcriptional activation,52 which is the Wilms’ tumor suppressor gene. Other diseases associated with HNRNPU include diffuse gastric cancer.53
Future Work
In this work, the aim is to return miRNA targets with low false-positive rate and we have applied three filtering criteria outlined in the Introduction section on DEPs detected by the proposed algorithm. The results have shown that these three criteria effectively ensured a zero false-positive rate in this case, which greatly reduced the cost of biological validation of the computed targets. However, the need of applying these filters has not been investigated, and there could be real miRNA targets that have been missed, which should be investigated in the future.
Conclusion
In this paper, we developed and applied a proteomic approach for identifying the targets of KSHV miR. The developed method is shown to be effective in finding miRNA targets. The developed method is based on 18O/16O labeling that can be used in many applications. Two novel and one previously identified miR-K1 targets are picked by the proposed method. They are further confirmed based on Western blotting and Luciferase reporter assay.
The core of the proteomic approach is based on the statistical test called the KL distance test, which uses the KL distance as a goodness-of-fit measure for comparing peptide fold changes of a protein to that of a background protein with the same number of peptide measurements. Through the KL test, we obtained a significant enrichment of PAR-CLIP-predicted targets with seed match to miR-K1 (PAR-CLIPSMK1). In comparison, none of the other statistical tests, such as t-test and the KS test, and their modified versions, as well as other proteomic software programs, including MaxQuant and TPP, find any DEPs that are significantly enriched with PAR-CLIPSMK1s.
Although the proposed method has a limitation as DEPs can arise through some unknown mechanism other than miRNA involvement even in miRNA-transfected cells, we have shown that the application of additional filters based on PAR-CLIP and SVMicro can greatly reduce false-positive rate.
Supplementary Material
All supplementary materials cited herein are available at http://compgenomics.utsa.edu/zgroup/miRTargetprediction/miRTargets.htm.
Acknowledgments
We thank the Computational Biology Initiative (UTSA/UTHSCSA) for providing access and training to the analysis software used. We also thank the Center for Proteomics, Translational Genomics Research Institute, for LC-MS data generation. Some aspects of this research were supported by the Virginia G. Piper Charitable Trust.
Footnotes
ACADEMIC EDITOR: J. T. Efird, Editor in Chief
PEER REVIEW: Three peer reviewers contributed to the peer review report. Reviewers’ reports totaled 797 words, excluding any confidential comments to the academic editor.
FUNDING: This project was supported by a grant from the National Institute on Minority Health and Health Disparities (G12MD007591) from the National Institutes of Health, and NIH grant R01CA096512. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE).
Author Contributions
Conceived and designed the experiments: SJG, TT, YZ, JZ. Analyzed the data: XM, JZ. Wrote the first draft of the manuscript: XM, JZ. Contributed to the writing of the manuscript: YZ, SJG. Advised XM regarding the usage of SVMicro software and the design of the comparison experiments: YH. Agree with manuscript results and conclusions: JZ, SJG, YZ, XM. Jointly developed the structure and arguments for the paper: XM, JZ, SJG. Made critical revisions and approved final version: JZ. All authors reviewed and approved of the final manuscript.
REFERENCES
- 1.Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem. 2007;389(4):1017–31. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- 2.Xie F, Liu T, Qian W, Petyuk VA, Smith RD. Liquid chromatography-mass spectrometry-based quantitative proteomics. J Biol Chem. 2011;286(29):25443–9. doi: 10.1074/jbc.R110.199703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ow SY, Salim M, Noirel J, Evans C, Rehman I, Wright PC. iTRAQ underestimation in simple and complex mixtures: the good, the bad and the ugly. J Proteome Res. 2009;8(11):5347–55. doi: 10.1021/pr900634c. [DOI] [PubMed] [Google Scholar]
- 4.Christoforou A, Lilley KS. Taming the isobaric tagging elephant in the room in quantitative proteomics. Nat Methods. 2011;8(11):911–3. doi: 10.1038/nmeth.1736. [DOI] [PubMed] [Google Scholar]
- 5.Ong S-E, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 6.Li C, Xiong Q, Zhang J, Ge F, Bi LJ. Quantitative proteomic strategies for the identification of microRNA targets. Expert Rev Proteomics. 2012;9(5):549–59. doi: 10.1586/epr.12.49. [DOI] [PubMed] [Google Scholar]
- 7.Zhang R, Sioma CS, Wang S, Regnier FE. Fractionation of isotopically labeled peptides in quantitative proteomics. Anal Chem. 2001;73(21):5142–9. doi: 10.1021/ac010583a. [DOI] [PubMed] [Google Scholar]
- 8.Tibes R, Qiu Y, Lu Y, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5(10):2512–21. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- 9.Ye X, Luke B, Johann D, Jr, et al. Optimized method for computing (18)O/(16)O ratios of differentially stable-isotope labeled peptides in the context of postdigestion (18)O exchange/labeling. Anal Chem. 2010;82(13):5878–86. doi: 10.1021/ac101284c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X, Wang X. Systematic identification of microRNA functions by combining target prediction and expression profiling. Nucleic Acids Res. 2006;34(5):1646–52. doi: 10.1093/nar/gkl068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Volinia S, Calin G, Liu C, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103(7):2257–61. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qin Z, Jakymiw A, Findlay V, Parsons C. KSHV-encoded microRNAs: lessons for viral cancer pathogenesis and emerging concepts. Int J Cell Biol. 2012;2012:603961. doi: 10.1155/2012/603961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samols M, Skalsky R, Maldonado A, et al. Identification of cellular genes targeted by KSHV-encoded microRNAs. PLoS Pathog. 2007;3(5):e65. doi: 10.1371/journal.ppat.0030065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lei X, Bai Z, Ye F, et al. Regulation of NF-k B inhibitor Ik Bα and viral replication by a KSHV microRNA. Nat Cell Biol. 2010;12(2):193–9. doi: 10.1038/ncb2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X, Valentine SJ, Plasencia MD, Trimpin S, Naylor S, Clemmer DE. Mapping the human plasma proteome by SCX-LC-IMS-MS. J Am Soc Mass Spectrom. 2007;18(7):1249–64. doi: 10.1016/j.jasms.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bresson JA, Anderson GA, Bruce JE, et al. Improved isotopic abundance measurements for high resolution Fourier transform ion cyclotron resonance mass spectra via time-domain data extraction. J Am Soc Mass Spectrom. 1998;9(8):799–804. [Google Scholar]
- 17.Ma X, Hestilow T, Cui J, Zhang J. Suppression correction and characteristic study in liquid chromatography/Fourier transform mass spectrometry measurements. Rapid Commun Mass Spectrom. 2011;25(4):551–7. doi: 10.1002/rcm.4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, Zhang H, Ranish J, Aebersold R. Automated statistical analysis of protein abundance ratios from data generated by stable-isotope dilution and tandem mass spectrometry. Anal Chem. 2003;75(23):6648–57. doi: 10.1021/ac034633i. [DOI] [PubMed] [Google Scholar]
- 19.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized ppb-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26(12):1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 20.Han DK, Eng J, Zhou H, Aebersold R. Quantitative profiling of differentiation-induced microsomal proteins using isotope-coded affinity tags and mass spectrometry. Nat Biotechnol. 2001;19(10):946–51. doi: 10.1038/nbt1001-946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramos-Fernández A, López-Ferrer D, Vázquez J. Improved method for differential expression proteomics using trypsin-catalyzed 18O labeling with a correction for labeling efficiency. Mol Cell Proteomics. 2007;6(7):1274–86. doi: 10.1074/mcp.T600029-MCP200. [DOI] [PubMed] [Google Scholar]
- 22.Yao X, Afonso C, Fenselau C. Dissection of proteolytic 18O labeling: endoproteasecatalyzed 16O-to-18O exchange of truncated peptide substrates. J Proteome Res. 2003;2(2):147–52. doi: 10.1021/pr025572s. [DOI] [PubMed] [Google Scholar]
- 23.Cui J, Ma X, Chen L, Zhang J. SCFIA: a statistical corresponding feature identification algorithm for LC/MS. BMC Bioinformatics. 2011;12(1):439. doi: 10.1186/1471-2105-12-439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pedrioli PG. Trans-proteomic pipeline: a pipeline for proteomic analysis. In: Hubbard SJ, Jones AR, editors. Proteome Bioinformatics. Berlin: Springer; 2010. pp. 213–38. [DOI] [PubMed] [Google Scholar]
- 25.Cui J, Petritis K, Tegeler T, et al. Accurate LC peak boundary detection for 16O/18O labeled LC-MS data. PLoS One. 2013;8(10):e72951. doi: 10.1371/journal.pone.0072951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jeanmougin M, De Reynies A, Marisa L, Paccard C, Nuel G, Guedj M. Should we abandon the t-test in the analysis of gene expression microarray data: a comparison of variance modeling strategies. PLoS One. 2010;5(9):e12336. doi: 10.1371/journal.pone.0012336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christin C, Hoefsloot HC, Smilde AK, et al. A critical assessment of feature selection methods for biomarker discovery in clinical proteomics. Mol Cell Proteomics. 2013;12(1):263–76. doi: 10.1074/mcp.M112.022566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scheibe M, Butter F, Hafner M, Tuschl T, Mann M. Quantitative mass spectrometry and PAR-CLIP to identify RNA-protein interactions. Nucleic Acids Res. 2012;40(19):9897–902. doi: 10.1093/nar/gks746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gottwein E, Corcoran D, Mukherjee N, et al. Viral microRNA targetome of KSHV-infected primary effusion lymphoma cell lines. Cell Host Microbe. 2011;10(5):515–26. doi: 10.1016/j.chom.2011.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–55. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kullback S, Leibler R. On information and sufficiency. Ann Math Stat. 1951;22(1):79–86. [Google Scholar]
- 32.Wilcox R. Encyclopedia of Biostatistics. Wiley Online Library; 2005. Kolmogorov–Smirnov test. [Google Scholar]
- 33.Rajewsky N. microRNA target predictions in animals. Nat Genet. 2006;38:S8–13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- 34.Grimson A, Farh KK-H, Johnston WK, Garrett-Engele P, Lim LP, Bartel DP. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27(1):91–105. doi: 10.1016/j.molcel.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee I, Ajay SS, Yook JI, et al. New class of microRNA targets containing simultaneous 5-UTR and 3-UTR interaction sites. Genome Res. 2009;19(7):1175–83. doi: 10.1101/gr.089367.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones-Rhoades MW, Bartel DP. Computational identification of plant microRNAs and their targets, including a stress-induced miRNA. Mol Cell. 2004;14(6):787–99. doi: 10.1016/j.molcel.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 37.Liu H, Yue D, Chen Y, Gao SJ, Huang Y. Improving performance of mammalian microRNA target prediction. BMC Bioinformatics. 2010;11(1):476. doi: 10.1186/1471-2105-11-476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin G, Schouest K, Kovvuru P, Spillane C. Prediction and validation of microRNA targets in animal genomes. J Biosci. 2007;32:1049–52. doi: 10.1007/s12038-007-0106-0. [DOI] [PubMed] [Google Scholar]
- 39.Frankel L, Christoffersen N, Jacobsen A, Lindow M, Krogh A, Lund AH. Programmed cell death 4 (PDCD4) is an important functional target of the microRNA miR-21 in breast cancer cells. J Biol Chem. 2008;283(2):1026–33. doi: 10.1074/jbc.M707224200. [DOI] [PubMed] [Google Scholar]
- 40.Bartel D. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116(2):281–97. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 41.Yang Y, Chaerkady R, Beer M, Mendell JT, Pandey A. Identification of miR-21 targets in breast cancer cells using a quantitative proteomic approach. Proteomics. 2009;9(5):1374–84. doi: 10.1002/pmic.200800551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bagga S, Bracht J, Hunter S, et al. Regulation by let-7 and lin-4 miRNAs results in target mRNA degradation. Cell. 2005;122(4):553. doi: 10.1016/j.cell.2005.07.031. [DOI] [PubMed] [Google Scholar]
- 43.Eckel-Passow J, Oberg A, Therneau T, et al. Regression analysis for comparing protein samples with 16O/18O stable-isotope labeled mass spectrometry. Bioinformatics. 2006;22(22):2739–45. doi: 10.1093/bioinformatics/btl464. [DOI] [PubMed] [Google Scholar]
- 44.Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003;100(16):9440–5. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lei X, Zhu Y, Jones T, Bai Z, Huang Y, Gao SJ. A Kaposi’s sarcoma-associated herpesvirus microRNA and its variants target the transforming growth factor β pathway to promote cell survival. J Virol. 2012;86(21):11698–711. doi: 10.1128/JVI.06855-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gottwein E, Cullen BR. A human herpesvirus microRNA inhibits p21 expression and attenuates p21-mediated cell cycle arrest. J Virol. 2010;84(10):5229–37. doi: 10.1128/JVI.00202-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baldacchino S, Saliba C, Petroni V, Fenech AG, Borg N, Grech G. Deregulation of the phosphatase, PP2A is a common event in breast cancer, predicting sensitivity to FTY720. EPMA J. 2014;5:3. doi: 10.1186/1878-5085-5-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nozawa T, Aikawa C, Goda A, Maruyama F, Hamada S, Nakagawa I. The small GTPases Rab9 A and Rab23 function at distinct steps in autophagy during group A streptococcus infection. Cell Microbiol. 2012;14(8):1149–65. doi: 10.1111/j.1462-5822.2012.01792.x. [DOI] [PubMed] [Google Scholar]
- 49.Eggenschwiler JT, Espinoza E, Anderson KV. Rab23 is an essential negative regulator of the mouse Sonic hedgehog signalling pathway. Nature. 2001;412(6843):194–8. doi: 10.1038/35084089. [DOI] [PubMed] [Google Scholar]
- 50.Hou Q, Wu YH, Grabsch H, et al. Integrative genomics identifies RAB23 as an invasion mediator gene in diffuse-type gastric cancer. Cancer Res. 2008;68(12):4623–4630. doi: 10.1158/0008-5472.CAN-07-5870. [DOI] [PubMed] [Google Scholar]
- 51.Kukalev A, Nord Y, Palmberg C, Bergman T, Percipalle P. Actin and hnRNP U cooperate for productive transcription by RNA polymerase II. Nat Struct Mol Biol. 2005;12(3):238–44. doi: 10.1038/nsmb904. [DOI] [PubMed] [Google Scholar]
- 52.Spraggon L, Dudnakova T, Slight J, et al. hnRNP-U directly interacts with WT1 and modulates WT1 transcriptional activation. Oncogene. 2007;26(10):1484–91. doi: 10.1038/sj.onc.1209922. [DOI] [PubMed] [Google Scholar]
- 53.Hope NR, Murray GI. The expression profile of RNA-binding proteins in primary and metastatic colorectal cancer: relationship of heterogeneous nuclear ribonucleoproteins with prognosis. Hum Pathol. 2011;42(3):393–402. doi: 10.1016/j.humpath.2010.08.006. [DOI] [PubMed] [Google Scholar]