

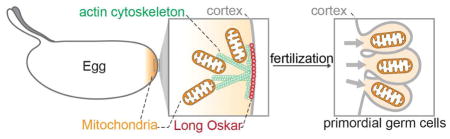
SUMMARY
Inherited mitochondrial DNA (mtDNA) mutations cause severe human disease. In most species, mitochondria are inherited maternally through mechanisms that are poorly understood. Genes that specifically control the inheritance of mitochondria in the germline are unknown. Here, we show that the long isoform of the protein Oskar regulates the maternal inheritance of mitochondria in Drosophila melanogaster. We show that during oogenesis mitochondria accumulate at the oocyte posterior concurrent with the bulk streaming and churning of the oocyte cytoplasm. Long Oskar traps and maintains mitochondria at the posterior at the site of primordial germ cell (PGC) formation through an actin-dependent mechanism. Mutating long oskar strongly reduces the number of mtDNA molecules inherited by PGCs. Therefore, Long Oskar ensures germline transmission of mitochondria to the next generation. These results provide molecular insight into how mitochondria are passed from mother to offspring, and also how they are positioned and asymmetrically partitioned within polarized cells.
eTOC
Mitochondria are inherited maternally through poorly understood mechanisms. Hurd et al. show that the long isoform of Oskar protein specifically controls mitochondrial germline inheritance in Drosophila melanogaster. Through an actin-dependent mechanism, Long Oskar traps mitochondria where the cells that give rise to the next generation form, ensuring efficient mtDNA inheritance.
INTRODUCTION
During cell division, it is essential that functional organelles are properly positioned such that they are correctly and sufficiently apportioned to each daughter cell. Mutations that disrupt the partitioning of functional mitochondria between mother and daughter cells cause premature aging and can influence cell fate (Katajisto et al., 2015; Vevea et al., 2014). Correct partitioning is particularly important for mitochondria because, unlike most other organelles, they contain their own genomes, which cannot be made de novo and must therefore be inherited. In flies and humans, mitochondrial genomes are exclusively inherited through the female germline (Stewart and Chinnery, 2015). Inheritance of defective mitochondrial DNA (mtDNA) is a major cause of human disease (Stewart and Chinnery, 2015). Recent studies have associated germline mtDNA mutations with aging and suggest they may play a causal role in this process (Ross et al., 2013). Studies have also associated commonly inherited mtDNA polymorphisms with several late-onset degenerative disorders (Hudson et al., 2014). As disease severity often correlates with the levels of mtDNA mutations inherited, understanding the mechanisms that underlie mtDNA inheritance is crucial.
Despite its scientific and medical importance, mechanisms of germline mitochondrial inheritance are not well understood on a molecular level. To gain a better understanding of this essential process, we developed imaging approaches that permitted us to follow mitochondria and count mtDNA molecules throughout the lifecycle of the D.melanogaster germline. With these new approaches, we observed a striking accumulation of mitochondria and mtDNA at the posterior of embryos. In D. melanogaster, primordial germ cells (PGCs) form at the posterior pole of the embryo, incorporating germ plasm, a mixture of proteins and RNAs that are deposited there during oogenesis (Eddy, 1975; Mahowald, 1962; Mahowald, 2001). Localization of mitochondria to the posterior occurs independently of germ plasm assembly during mid-oogenesis and is coincident with the microtubule-driven streaming of the bulk oocyte cytoplasm. We found that retention of mitochondria at the posterior depends upon the long isoform of the protein Oskar (Long Oskar), which alters the actin cytoskeleton to trap mitochondria at the site of PGC formation. Without Long Oskar, the number of mitochondrial genomes inherited into PGCs is significantly reduced. Long Oskar thereby plays an essential role in the passage of mitochondria and mtDNA molecules from mother to offspring.
RESULTS
mtDNA copy number increases during oogenesis and drastically decreases upon PGC formation
To understand how mtDNA is transmitted through the female Drosophila germline, we first sought to quantify mtDNA number throughout the various stages of germline development. In order to do this, we developed a quantitative fluorescence in situ hybridization (FISH) methodology. The benefit of this approach is that it allows for measurement of mtDNA spatially and temporally without having to isolate germ cells and disrupt tissue integrity. A mixture of 60 unique DNA probes each 5′ labeled with CAL Fluor® 590 were hybridized along mtDNA and samples were imaged using a confocal microscope. In all samples imaged, distinct foci were observed (Figure 1A and S1A), which strongly associated with a mitochondrial marker, ATP synthase α (Figure S1B), and were insensitive to RNAse A treatment (Figure S1C and S1D), indicating our FISH method specifically recognizes mtDNA molecules in vivo. Using a spot-detection algorithm to quantify the number of mtDNA foci in PGCs, we found that the average number of mtDNA foci per PGCs was 669 (Figure 1B) (Trcek et al., 2015). This finding correlated well with the number of mtDNA molecules in isolated PGCs calculated using a traditional qPCR-based mtDNA assay (Figure 1B), and is in good agreement with estimates in mammalian cells of approximately one to two mtDNA molecules per nucleoid or foci (Kukat et al., 2011). Thus, our mtDNA FISH approach is able to specifically and accurately quantify mtDNA copy number whilst maintaining spatial integrity of the tissue.
Figure 1. mtDNA copy number increases during oogenesis and drastically decreases upon PGC formation in wild type controls.

(A) Distinct mtDNA FISH foci are observed in 0 to 1 hour old syncytial embryos. (B) Quantitative mtDNA FISH can accurately determine the mtDNA copy number in embryonic PGCs in situ. mtDNA was measured in PGCs from 1 to 3 hour old control embryos by mtDNA FISH. mtDNA copy number was determined in FACS-sorted PGCs from 0 to 11 hour old embryos by qPCR. Data are the mean +/− SEM.
(C) Cartoon of D. melanogaster germline development.
(D, E and F) mtDNA in a 2 to 3 hour old embryo (D), a L3 larval gonad (E) and an adult ovariole (F) as visualized using mtDNA FISH. Samples were immunostained with α-Vasa that detects the germline-specific protein Vasa.
(G) mtDNA copy number throughout germline development. mtDNA copy number was determined in PGCs, GSCs, egg chambers and eggs using mtDNA FISH. mtDNA copy number in PGCs from 2 to 4 and 9 to 11 hour old embryos was determined by FACS-sorting and qPCR. Data are the means at least three independent replicates +/− SEM.
See also Figure S1.
Having established a methodology to quantify mtDNA in situ, we used it to assess mtDNA copy number at various stages of germline development (Figure 1C–1G). In D. melanogaster, germline development begins with the formation of PGCs at the posterior of the embryo. PGCs then migrate to and eventually coalesce in the gonad. During the three larval instars, the entire female gonad grows and the number of PGCs increases such that each somatic niche can be occupied with newly specified germline stem cells (GSCs). At the anterior tip of the adult ovaries, GSCs continuously asymmetrically divide to give rise to a chain of differentiating egg chambers that become increasingly more mature as they move posterior, eventually forming a mature egg (Figure 1C).
Using quantitative mtDNA FISH, we were able to detect mtDNA foci at all stages of germline development (Figures 1D–F). Quantification of the number of mtDNA molecules per cell (copy number) indicated that mtDNA copy number remained relatively stable in embryonic PGCs, larval PGCs and adult GSCs (Figure 1G). However, upon differentiation of GSCs into egg chambers, mtDNA copy number substantially increased to over 1000 fold in a mature egg (Figure 1G). This increase in copy number coincides with an increase in germ cell volume. Consequently, as PGCs formed at the posterior of the embryo, there was a far greater than 1000 fold reduction in the mtDNA copy number concomitant with a large reduction in cell volume associated with going from an egg to a PGC. Thus, less than 0.1% of the total mitochondrial genomes produced during oogenesis are partitioned into PGCs and thereby transmitted to the next generation.
Mitochondria are enriched at the embryo posterior
Substantially more sub-sampling of mitochondria occurs during PGC formation than at any other stage of germline development, which raises the question of how mitochondrial genomes are selected for inclusion in PGCs and, thereby, for propagation into the next generation. One strength of our FISH-based approach was the ability to detect differences in the sub-cellular distribution of mtDNA. Interestingly, prior to the formation of PGCs we observed an enrichment of mtDNA at the embryo posterior at the site of PGC formation and we wondered whether this could provide such a mechanism (Figure 1A). It had previously been reported that the mitochondrial large ribosomal RNA (mtlrRNA), but not mitochondria, accumulate at the embryo posterior (Akiyama and Okada, 1992; Amikura et al., 2001; Iida and Kobayashi, 1998; Kobayashi et al., 1993) (Figure 2A). To determine whether mitochondria are in fact enriched at the posterior, we visualized mitochondria with a mitochondrially-targeted EYFP (mito-EYFP). Analysis revealed, consistent with our mtDNA observation, that mitochondria were enriched at the embryo posterior (Figure 2A). The most parsimonious explanation for the very similar overlapping localization patterns of mtDNA, mtlrRNA and mitochondria, is that mtlrRNA at the posterior is present within mitochondria and not outside of them. To test this, we assayed the sub-cellular localization of the mtlrRNA at the posterior using single molecule RNA FISH. Co-localization analysis using the Costes Pearson Correlation Coefficient (PCC) approach (Costes et al., 2004; Trcek et al., 2015) revealed near perfect overlap between the mtlrRNA and mito-EYFP (Figure S2). Our analysis also revealed that mitochondria were positioned proximal to the germ plasm component nos (Figure S2). These results indicate that the majority of mtlrRNA at the embryo posterior is present within mitochondria, and that mitochondria are enriched at the embryo posterior positioning them for entry into PGCs.
Figure 2. Mitochondria are enriched at the embryo posterior and accumulate there during stage 10 of oogenesis. Arrows indicate posteriorly enriched mitochondria.

(A) mtlrRNA and mitochondria were visualized using FISH and mito-EYFP to detect the mtlrRNA and mitochondria, respectively. See also Figure S2.
(B) Mitochondria accumulate posteriorly starting at stage 10 of oogenesis and persist there until embryogenesis. mito-EYFP labeled mitochondria were visualized live using a multi-photon microscope. Mitochondrial accumulation (EYFP fluorescence) is shown using an ICA lookup table which ranges from blue (background) to yellow and white (high accumulation). See also Movie S1.
(C) Percentage of stage 9 to 14 egg chambers and eggs with posteriorly enriched mitochondria.
Mitochondria become posteriorly enriched during mid-oogenesis
Since mitochondria are already enriched at the posterior of 0 to 1hour old embryos, we next wanted to determine when mitochondria first begin to localize there. To do this, we developed a multi-photon live imaging assay to follow EYFP-tagged mitochondria during germline development. Since most germ plasm components are synthesized and deposited at the posterior during oogenesis (Eddy, 1975; Mahowald, 1962; Mahowald, 2001), we first looked at mitochondria in the ovary. Consistent with previous reports, no cortical posterior accumulation of mitochondria was observed during early oogenesis from oocyte specification in the germarium through stage 9 egg chambers (Figure 2B, data not shown) (Cox and Spradling, 2003). However, at stage 10, the posterior accumulation of mitochondria became apparent (Figure 2B, see Movie S1). From stage 10 onward mitochondria appeared to increase in number until stage 13, where enrichment persisted at the posterior into embryogenesis (Figures 2B, 2C, and Movie S1).
Concomitant with the posterior enrichment of mitochondria at stage 10 of oogenesis, the cytoplasm of the oocyte undergoes a microtubule-dependent, unidirectional churning known as cytoplasmic streaming (Ganguly et al., 2012; Gutzeit and Koppa, 1982) (Figure 3A). The appearance of mitochondria at the posterior concurrent with cytoplasmic streaming suggests that this movement might provide the force driving mitochondrial localization. Consistent with this hypothesis, mitochondria underwent unidirectional streaming (Figure 3B) and moved at a similar speed to other particles known to stream (Figure 3E) (Serbus et al., 2005). To determine whether mitochondria require cytoplasmic streaming to localize, we selectively inhibited streaming by culturing stage 10 egg chambers in the presence of the microtubule inhibitor colcemid. Consistent with previous reports, colcemid inhibited streaming, but not subsequent development, as egg chambers initiated nurse cell dumping and progressed through oogenesis normally (Movie S2) (Forrest and Gavis, 2003; Gutzeit, 1986). Inhibition of cytoplasmic streaming strongly reduced the unidirectional movement (Figure 3C), speed (Figure 3E) and accumulation of mitochondria at the posterior (Figures 3D, 3F and Movie S2). This suggests that streaming is necessary for efficient posterior localization. As colcemid blocks all motor-based movements it remains possible that microtubules also contribute more directly to the transport of mitochondria to the posterior. Taken together, these results suggest that mitochondria require cytoplasmic streaming to accumulate at the oocyte posterior, beginning at stage 10b of oogenesis.
Figure 3. Cytoplasmic streaming is required to localize mitochondria to the oocyte posterior.

(A) Cartoon of cytoplasmic streaming.
(B and C) mito-EYFP tagged mitochondria undergo cytoplasmic streaming in stage 10/11 egg chambers but not in those treated with colcemid. Each panel represents time projections acquired over 0, 40 or 80 seconds (s). EYFP, white.
(D) mito-EYFP tagged mitochondria do not accumulate at the posterior of stage 10 egg chambers treated with colcemid (0 min) and aged 100 minutes. See also Movie S2.
(E) The speed of mito-EYFP tagged mitochondria in stage 10/11 egg chambers treated with and without colcemid. Data are the mean +/− SEM.
(F) Percentage of egg chambers treated with or without colcemid at stage 10 and aged 100 minutes that had posteriorly enriched mitochondria.
Mitochondrial localization requires Long Oskar, but not other germ plasm components
Having identified how mitochondria are transported to the posterior of the oocyte, we next sought to identify genes that are required to retain mitochondria at the posterior once they arrive. The germ plasm proteins Oskar, Vasa and Tudor are good candidates for this because they accumulate just prior to the posterior localization of mitochondria (Bardsley et al., 1993; Eddy, 1975; Kim-Ha et al., 1991; Lasko and Ashburner, 1990; Mahowald, 1962; Mahowald, 2001). To determine whether the genes encoding these proteins were necessary to trap mitochondria at the posterior, we removed each and assayed mitochondrial distribution. As germ plasm components are not required for cytoplasmic streaming, we assessed mitochondrial distribution in embryos. Interestingly, removal of oskar, but not vasa or tudor, completely abolished the posterior accumulation of mitochondria (Figures 4A and S3).
Figure 4. Long Oskar and its N-terminal domain are both necessary and sufficient to trap and retain mitochondria at the cortex. Arrows indicate mitochondrial accumulation.
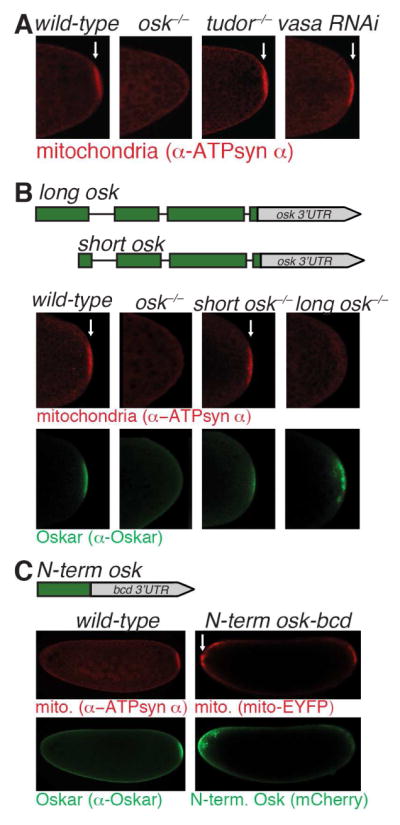
(A) Loss of oskar, but not tudor nor vasa, prevents posterior mitochondrial accumulation. 0 to 1 hour old embryos from wild-type, oskar null mutant (oskA87/Df(3R)p-XT103), tudor null mutant (tudortux46/Df(tudor)), or Vasa knockdown (nosgal4::VP16, mito-EYFP/uas-vasa RNAi) mothers were immunostained with α-ATP synthase α. See also Figure S3.
(B) Loss of long, but not short, oskar prevents posterior mitochondrial accumulation. 0 to 1 hour old embryos from wild-type mothers, oskar null mutant mothers (oskA87/Df(3R)p-XT103), or oskar null mutant mothers transgenically expressing either Long Oskar (short osk−/−; oskM139L (go M2-12)/+; oskA87/Df(3R)p-XT103) or Short Oskar (long osk−/− ;oskM1L(go M1-7)/+;oskA87/Df(3R)p-XT103) were immunostained with α-ATP synthase α and α-Oskar.
(C) The N-terminal domain is sufficient to trap mitochondria at the anterior when it is expressed there. Left, 0 to 1 hour old embryos from wild-type mothers were immunostained with α-ATP synthase α and α-Oskar. Right, 0 to 1 hour old embryos from mothers expressing the N-terminus of Long Oskar fused to mCherry, FLAG, and HA at the anterior using the bcd 3’UTR (N-term osk-bcd 3’UTR: uas-osk N-term::mCherry::3xFLAGHA/+; nosgal4::VP16, mito-EYFP/+) were imaged with EYFP (mitochondria) and mCherry (N-term. Oskar). See also Figure S4.
oskar is necessary and sufficient for specifying functional germ cells (Ephrussi and Lehmann, 1992; Ewen-Campen et al., 2012; Smith et al., 1992). oskar encodes two protein isoforms, termed Short and Long, which are generated by alternative start codon usage (Markussen et al., 1995) (Figure 4B). Short Oskar is necessary for germ plasm assembly and PGC formation (Markussen et al., 1995), while Long Oskar is largely dispensable for these processes, and instead helps to anchor germ plasm to the posterior cortex (Vanzo et al., 2007; Vanzo and Ephrussi, 2002). To determine which isoform of Oskar is necessary for mitochondrial posterior accumulation, we assessed mitochondrial distribution in embryos from oskar null mutants transgenically expressing either the Long or the Short isoforms of Oskar. This analysis revealed that Long, but not Short, Oskar is required for the posterior mitochondrial accumulation (Figure 4B).
The only difference in primary amino acid sequence between the two isoforms of Oskar is the presence of an N-terminal domain in Long Oskar (Figure 4C). That Long Oskar, but not Short Oskar, is required to trap mitochondria at the posterior indicates that the N-terminal domain is necessary for mitochondrial entrapment; however, whether it is also sufficient or if other domains in Oskar are required remained unclear. To test this, we targeted the N-terminal domain of Long Oskar to the anterior of the embryo by fusing its coding sequence to the bicoid (bcd) 3′UTR. The bcd 3′UTR contains an anterior localization signal which restricts RNAs containing it and in some cases the proteins they encode to the anterior of oocytes and embryos (Ephrussi and Lehmann, 1992; Gavis and Lehmann, 1992). In order to visualize the N-terminal domain of Long Oskar we tagged it with mCherry, FLAG and HA. If the N-terminal domain is sufficient to trap mitochondria, then mitochondria should accumulate anteriorly as cytoplasmic streaming occurs throughout the oocyte such that mitochondria swirl past the anterior as well as the posterior. Indeed, we found anterior localization of the N-terminal domain was sufficient to ectopically trap mitochondria at the anterior pole of the embryo (Figure 4C). Consistent with this result, versions of anteriorly targeted Long Oskar lacking various domains outside the N-terminus also accumulated mitochondria at the embryo anterior (Figures S3 and S4). Taken together, our findings show that Long Oskar, through its N-terminal domain, is both necessary and sufficient to trap mitochondria.
Long Oskar traps mitochondria through an actin-dependent mechanism
Long Oskar has not previously been implicated in the entrapment of mitochondria. Instead, it is known to be present on endosomes where it is thought to stimulate endocytosis together with Rab5 and Rbsn-5 (Tanaka and Nakamura, 2011). To determine how Long Oskar traps mitochondria at the posterior, we conducted co-immunoprecipitation experiments to identify Long Oskar interacting proteins by mass spectrometry. To uncover proteins that uniquely and specifically interact with the N-terminus of Long Oskar, we transgenically expressed mCherry-, FLAG- and HA-tagged Long Oskar, Short Oskar, and the N-terminal domain of Long Oskar and immunoprecipitated them from 0 to 2 hour old embryos. For all three co-immunoprecipitations, appreciable and similar amounts of bait were pulled down (Figure 5A, red boxes and Table S1, bait). As expected, several previously identified Short Oskar interacting proteins, including Vasa and Tudor, were among the most abundant proteins present in the Short, but not N-terminal, Oskar pull-downs (Table S1).
Figure 5. Long Oskar interacts with the actin cytoskeleton.

(A) Silver stained polyacrylamide gel of proteins co-immunoprecipitated with Long Oskar, Short Oskar or the N-terminal domain of Long Oskar (N-term). Proteins were co-immunoprecipitated from 0 to 2 hour old embryos from mothers expressing Long Oskar (;uas-long oskar::mCherry::3xFLAG HA/+; nosgal4::VP16, mito-EYFP/+), Short Oskar (;uas-short oskar::mCherry::3xFLAG HA/+; nosgal4::VP16, mito-EYFP/+) or N-term (;uas-osk N-term::mCherry::3xFLAG HA/+; nosgal4::VP16, mito-EYFP/+). Baits are indicated by red boxes as determined by apparent molecular weight (MW).
(B) Ten most abundant proteins enriched in Long and N-term Oskar, but not Short Oskar, co-immunoprecipitations. Protein abundance (Abd.) was normalized to bait abundance. Long/Short and N-term/Short, were calculated by dividing normalized protein abundances. See also Tables S1 and S2.
(C) Gene ontology enrichment analysis of proteins that co-immunoprecipitated with Long Oskar and its N-terminal domain, but not Short Oskar.
(D–G) F-actin in fixed S2R+ cells expressing either Long Oskar (act-gal4 uas-long oskar::mCherry::3xFLAG HA) (D to F) or Short Oskar (act-gal4 uas-short oskar::mCherry::3xFLAG HA) (G). Images are of single sections (D, F and G) or maximum intensity projections (E). F-actin was visualized with Alexa Fluor® 488 phalloidin (red), Oskar with mCherry (green) and DNA with DAPI (blue). The white dashed circles indicate the Oskar expressing cells.
Mass spectrometry analysis revealed 44 unique proteins that interacted specifically with both Long Oskar and its N-terminus but not Short Oskar (Figures 5A, 5B, and Table S2). Gene ontology enrichment analysis of Long Oskar and N-terminal domain interacting proteins revealed a significant enrichment in actin binding proteins (p = 1.33 × e−7) (Figure 5C). In fact, 22 of the most abundant and all of the top ten proteins co-immunoprecipitated were either actin or actin-binding proteins (Figure 5B and Table S2). Most top Short, Long and N-terminal Oskar-specific interactors were reproducibly immunopreciptated (Table S2). Together these results demonstrate that Long Oskar physically interacts with the actin cytoskeleton and/or actin binding proteins. To establish a functional link between Long Oskar and the actin cytoskeleton, we expressed Long Oskar or Short Oskar in S2R+ cells and stained the actin cytoskeleton with phalloidin. Cortical F-actin was greatly reduced in cells expressing high levels of Long Oskar demonstrating that Long Oskar can regulate actin (Figure 5D–G). Together, this suggests that Oskar interacts with the actin cytoskeleton and can influence its dynamics.
Mitochondria have been shown to interact with the actin cytoskeleton in neurons and yeast (Sheng and Cai, 2012; Vevea et al., 2014). Thus, Long Oskar might directly alter the actin cytoskeleton, which in turn traps mitochondria at the posterior during cytoplasmic streaming. To test this possibility, we treated stage 12 egg chambers in which mitochondria had already become enriched at the posterior with the cell-permeable actin polymerization inhibitor cytochalasin D, which has previously been used to disrupt the actin cytoskeleton at this stage of oogenesis (Forrest and Gavis, 2003). Incubation with cytochalasin D caused mitochondria to disassociate from the posterior of the embryo, suggesting that the actin cytosketelon is indeed required to maintain mitochondria at the posterior (Figures 6A, 6C and 6D). In contrast, once localized, mitochondria no longer required microtubules, as the microtubule depolymerizing agent colcemid had no effect on mitochondrial localization (Figures 6A, 6B and 6D). Together, these data demonstrate that the actin cytoskeleton is required to retain mitochondria at the posterior.
Figure 6. Posterior trapped mitochondria require the actin cytoskeleton.

(A to C) Treatment of stage 12 egg chambers with the actin polymerization inhibitor cytochalsin D (cyto. D) (C), but not the microtubule depolymerizing agent colcemid (colc.) (B) or vehicle control (A), caused mito-EYFP mitochondria to detach from the posterior. Images were taken 0, 1 and 2 hours after the addition of drug. White arrows and dashed shapes indicate posteriorly enriched mitochondria.
(D) Percentage of stage 12 colc., cyto. D, or vehicle only (control) treated egg chambers that had discernable posteriorly enriched mitochondria.
(E) In the absence of TmII, anteriorly expressed Oskar is not able to retain mitochondria at the embryo anterior. 0 to 1 hour old embryos from mothers transgenically expressing Oskar at the anterior in the presence (P{ry+, osk-bcd}42/+;TmIIgs1/+) or absence (P{ry+, osk-bcd}42/+;TmIIgs1/ TmIIgs) of TmII were immunostained with α-ATP synthase α and α-Oskar. Arrows indicate anteriorly accumulated mitochondria.
(F) Percentage of 0 to 1 hours old embryos with posteriorly and/or anteriorly enriched mitochondria.
See also Fiugre S5.
To establish a further molecular link between Long Oskar and mitochondria, we sought to identify Long and N-terminal Oskar interacting proteins required for the posterior entrapment of mitochondria. The non-muscle tropomyosin, tropomyosin II (TmII) (also known as Tm1), is known to accumulate at the posterior (Erdélyi et al., 1995) and was one of the most enriched, abundant Long and N-terminal Oskar interacting proteins (Figure 5B and Table S2). We could not assess mitochondrial localization at the posterior in TmII mutants because TmII is required to localize Oskar RNA at an earlier stage of oogenesis (Erdélyi et al., 1995). Instead, we ectopically expressed Oskar at the anterior in an TmII mutant background and assessed mitochondrial distribution in embryos. Mutation of TmII significantly diminished mitochondrial accumulation at the embryo anterior without abolishing Oskar localization (Figures 6E and 6F). In contrast, perturbation of the Long Oskar co-immunoprecipitants Myosin V (Didum) and Myosin VI (Jar) did not abolish mitochondrial accumulation in ovaries and embryos, respectively (Figure S5). This demonstrates that in addition to actin Long Oskar requires TmII to tether mitochondria to the cortex.
Posterior mitochondrial enrichment ensures transmission of mtDNA to the next generation
Having identified the mechanism by which mitochondria localize to the posterior, we are able to assess the consequences of compromised mitochondrial enrichment for the first time. Long Oskar is dispensable for PGC formation, and therefore mitochondrial enrichment is not strictly required for PGCs to form (Markussen et al., 1995;Vanzo et al., 2002). However, the ultimate purpose of PGCs is to pass genetic material on to the next generation. As such, we reasoned that the localization of mitochondria to the posterior is a mechanism to make sure PGCs inherit mtDNA to be transmitted to subsequent generations. If this is correct, we expect the following. First, posteriorly localized mitochondria will be taken into PGCs as they form. To test, we generated a strain in which we targeted a photo-activatable GFP (PAGFP) (Patterson and Lippincott-Schwartz, 2002) to mitochondria. We then photo-activated posteriorly enriched mitochondria and followed them to see if they were taken up by the PGCs. Indeed, mitochondria enriched at the posterior could be observed entering PGCs as they formed (Figure 7A, 7B and Movie S3). Second, depletion of localized mitochondria should decrease the number of mtDNA molecules within PGCs. To this end, we visualized mtDNA using quantitative mtDNA FISH in control and CRISPR generated long oskar null mutant embryos (Figure S6), and observed a significant reduction in the number of mitochondrial genomes in PGCs lacking Long Oskar (Figure 7C to 7E). To verify this result, we measured the number of mtDNA molecules present in PGCs from oskar null maternal mutants transgenically expressing either the wild type or the Short isoform of Oskar by qPCR, and consistently found a significant reduction in the number of mtDNA genomes (Figure S7A and S7B). Furthermore, Long Oskar appeared to be specific for mitochondria as we did not observe enrichment of the Golgi apparatus or the endoplasmic reticulum to the embryo posterior relative to the anterior (Figure S7C and S7D). Taken together, these results demonstrate that Long Oskar-mediated mitochondrial enrichment at the embryo posterior is an active process that ensures incorporation of mitochondria into PGCs and transmission of mtDNA to the next generation.
Figure 7. Most mitochondria require Long Oskar to enter PGCs.

(A) Cartoon of PGC formation.
(B) Mitochondrial targeted PAGFP (mito-PAGFP) just prior to photoactivation (left), immediately after photoactivation (middle) and during PGC formation (right). Dashed red box indicates the area of photoactivation. See also Movie S3.
(C and D) mtDNA FISH of embryos just after PGC formation from control (C) and long oskar mutant (D) mothers. PGCs were stained with α-Vasa and are delineated by the white dashed shape. See also Figure S6.
(E) The number of mtDNA copies per PGC as determined by quantitative mtDNA FISH. Data are the mean +/− SEM. For long osk−/+, 99 PGCs from 5 embryos were counted. For long osk−/−, 70 PGCs from 4 embryos were counted. ** p < 0.01 (independent two-tailed Student’s t-test). See also Figure S7.
(F) The number of PGCs in cycle 14 embryos from control and long oskar null mutant mothers. Data are the mean +/− SEM. 20 embyros were counted for each genotype. ** p < 0.001 (independent two-tailed Student’s t-test).
PGC number is reduced and oogenesis is impaired in long oskar mutants
To further explore the physiological consequences of reduced mitochondrial inheritance, we counted the number of PGCs present in embryos from control and long oskar null mutants. A significant reduction in the number of PGCs was observed in blastoderm stage (nuclear cycle 14) embryos from long oskar mutants (Figure 7F). To verify this result, we measured PGC number in embryos from oskar null mutants transgenically expressing either the wild type or the Short isoform of Oskar, and consistently found a significant reduction in PGC number (Figure S7E). To further explore how this influences adult fertility, we analyzed ovaries from the progeny of oskar null mutants transgenically expressing either wild type or the Short isoform of Oskar. Rudimentary ovaries with no discernable late stage egg chambers were observed at a significantly higher frequency in mutants lacking Long Oskar when compared to controls (Figure S7F). Immunofluorescences analysis using an antibody that detects the germline marker Vasa revealed that these ovaries were often completely lacking germline (Figure S7G and S7H). In summary, these data demonstrate that Long Oskar is required for normal PGC number and subsequent germline development in adults, and suggest that Long Oskar-mediated mitochondrial inheritance may contribute to this.
DISCUSSION
Germ cells are the means by which sexually reproducing organisms transmit genetic material to subsequent generations to ensure the continuance of the species. Consequently, the formation and specification of germ cells is one of the most important events in development. PGC formation can occur either through the cytoplasmic inheritance of maternally deposited determinants, called germ plasm, or through inductive cell signaling events (Evans et al., 2014; Extavour and Akam, 2003). In D. melanogaster, PGCs are formed by the deposition of germ plasm at the posterior of the embryo (Eddy, 1975; Ephrussi and Lehmann, 1992; Mahowald, 1962; Mahowald, 2001). The germ plasm has long been known to be rich in mitochondria (Eddy, 1975; Kloc et al., 2004; Mahowald, 1962; Mahowald, 2001). In fact, in mammals one of the names for germline granules is the intermitochondrial cement (Aravin and Chan, 2011). The reason for this curious association, however, has been unclear until now.
Here, we have shown that mitochondria accumulate in the germ plasm to ensure the transmission of their genomes to the next generation. In D. melanogaster, we find that most mitochondria are transported to the germ plasm during cytoplasmic streaming in developing oocytes and maintained there by an actin-dependent mechanism. Long Oskar controls mitochondrial anchoring at the posterior and is not only necessary but also sufficient to tether mitochondria wherever it is expressed. Mutating long oskar decreases the number of mitochondrial genomes transmitted to the next generation, demonstrating that Long Oskar is important for mtDNA inheritance. Long Oskar mutants also have reduced numbers of PGCs and frequently impaired oogenesis (Vanzo et al., 2002). Our data suggest that a potential cause of this is a failure to enrich mitochondria at the posterior. However, it remains to be determined whether the reduction in the number of mitochondria at the posterior and in PGCs affects PGCs survival, formation or division. Long Oskar-mediated mitochondrial enrichment could also play a role in the formation, biogenesis and/or anchoring of germ plasm to the posterior prior to PGC formation (Ryu and Macdonald et al, 2015; Vanzo et al., 2002). Alternatively, the defects in long oskar mutants could be due to some other function of Long Oskar independent of its role in trapping mitochondria at the posterior.
Previous studies have analyzed mitochondrial distribution during earlier stages of Drosophila oogenesis (Cox and Spradling, 2003; Hill et al., 2014). They show that mitochondria initially enter the oocyte travelling on microtubules and once there coalesce into a single mass resembling a structure called the Balbiani body (Cox and Spradling, 2003). Recent data suggest that selective replication of mtDNA may restrict the transmission of deleterious mtDNA mutations at this time (Hill et al., 2014). Further experiments showed that Balbiani body mitochondria associate with the posterior until stage 7, when the oocyte repolarizes its microtubule network. In our study we analyzed mitochondrial distribution at later stages of oogenesis. We show that the vast majority of mitochondria passed into PGCs accumulate during and after stage 10b, and thus may be predominantly nurse cell derived and Balbiani body-independent. In the absence of Long Oskar a small amount of mitochondria do enter the PGCs however and it is possible that these could constitute a different pool that entered the oocyte at an earlier stage. Direct visualization of mitochondrial populations are needed to determine whether specific sources of mitochondria reach the posterior pole or whether they are randomly selected from the oocyte pool.
Mitochondrial transport is often an active process in which motor proteins and their adapters move mitochondria along the cytoskeleton (Sheng and Cai, 2012). Interestingly, this is not likely the case in D. melanogaster stage 10 oocytes. Instead, we find that mitochondria move apparently passively, caught in the bulk flow of the oocyte cytoplasm, in order to localize to the oocyte posterior. This mode of localization is not unique to mitochondria; germ plasm RNAs, such as nanos, also use it to localize to the embryo posterior (Forrest and Gavis, 2003; Sinsimer et al., 2011; 2013). Cytoplasmic streaming occurs in a wide variety of other contexts, across a range of organisms and developmental stages (Ganguly et al., 2012). Given our findings it will be interesting to investigate whether cytoplasmic streaming is used in other contexts as a means of mitochondrial transport or asymmetric localization.
How Long Oskar uses the actin cytoskeleton to anchor mitochondria remains unclear. Oskar is present in two forms, Short and Long (Markussen et al., 1995). Short Oskar is an integral member of germ plasm and is both necessary and sufficient to form functional PGCs (Ephrussi and Lehmann, 1992; Ewen-Campen et al., 2012; Smith et al., 1992). In stark contrast, Long Oskar is distinctly localized to endocytic membranes and is not required for PGC formation per se (Tanaka and Nakamura, 2011; Vanzo et al., 2007). Long Oskar may instead function to help anchor the germ plasm by promoting yolk endocytosis and remodeling of the actin cytoskeleton (Tanaka and Nakamura, 2011; Vanzo et al., 2007). Unexpectedly, we did not identify any endocytic proteins in our Long Oskar co-immunoprecipitation experiments (Table S2). Instead, the most abundant Long Oskar interacting proteins identified were actin and actin-binding proteins including surprisingly a number of muscle-specific actinomyosin proteins. This leaves open the possibility that Long Oskar, and more specifically its N-terminal domain, nucleates actin directly or regulates proteins that modify actin. If so, Long Oskar would likely represent a new type of actin modifying protein, as its N-terminal domain bears no sequence homology to any actin modifying protein in Drosophila or elsewhere (Ahuja and Extavour, 2014). Overexpression of Long Oskar in S2R+ cells caused gross alteration to the F-actin cytoskeleton, which is also consistent with Long Oskar binding the actin cytoskeleton and possibly competing with other actin cytoskeletal binding proteins (Davidson and Wood, 2016). Further experiments will be required to determine exactly how Long Oskar alters the actin cytoskeleton and whether cytoskeletal-mediated mitochondrial localization requires endosomal components.
The actin cytoskeleton is necessary for mitochondrial retention at the posterior pole of the embryo. Defects in mitochondrial interactions with the cytoskeleton are associated with many neurodegenerative diseases (Sheng, 2014). Interestingly, disruption of F-actin with actin depolymerizing drugs affects mitochondrial retention, but not transport, in Drosophila neurons (Chada and Hollenbeck, 2004). Furthermore, in vertebrate axonal neurons mitochondria have been shown to interact with actin microfilaments (Morris and Hollenbeck, 1995). As both Oskar and TmII are reported to be expressed in Drosophila neurons, it would be interesting to determine whether these two proteins similarly anchor mitochondria in this cell type (Li and Gao, 2003; Xu et al., 2013). Further high-resolution imaging is also required to determine the regulation and dynamics of this potentially general mechanism of mitochondrial retention (Sinsimer et al., 2013).
Long Oskar acts as the main mechanism of mitochondrial inheritance in PGCs. Whether mitochondria that localize to the posterior and represent the majority of those inherited, are chosen at random or are selected based on fitness, health or some other attribute remains to be determined. In yeast, such a ‘fitness’ based mechanism of inheritance has been observed (Vevea et al., 2014). There, bundles of F-actin extend from the bud tip to the mother cell and serve as tracks for mitochondrial movement (Vevea et al., 2014). Far from static, these actin cables are continuously moving away from the bud (Vevea et al., 2014). Therefore, in order for mitochondria to be inherited into daughter cells they must “crawl upstream” against the opposing movement of the actin cables, creating a fitness test such that only the healthiest mitochondria make it and are inherited (Vevea et al., 2014). It is possible that a similar situation also occurs at the posterior of Drosophila oocytes. Though mitochondria appear to be statically anchored at the posterior in the embryo, our analysis does not exclude the possibility that they are undergoing short-range movements on actin filaments. Indeed, purifying selection against deleterious mtDNA mutations have been observed in the Drosophila germline (Hill et al., 2014; Ma et al., 2014). It will be interesting to explore whether the accumulation and inheritance of mitochondria serves as a mechanism to test fitness and/or select against those that carry harmful mutations.
Most organisms inherit mitochondria uniparentally. Why exactly this is remains unclear. Recent evidence suggests that inheritance of paternal mtDNA can be harmful (Zhou et al., 2016). Consistent with this multiple pathways have been described in Drosophila preventing the transmission of paternal mtDNA (DeLuca and O’Farrell, 2012; Politi et al., 2014). Clearly, understanding mechanistically how mitochondria are transmitted and the genes that regulate this process is a key step in ultimately determining why this unusual mode of inheritance is so prevalent in nature.
EXPERIMENTAL PROCEDURES
D. melanogaster strains
D. melanogaster were maintained on cornmeal molasses yeast medium at 25°C using standard procedures. All D. melanogaster strains generated and used in this study are listed in the Supplemental Experimental Procedures. DNA constructs were generated by standard molecular biological methods and injected into D. melanogaster by BestGene Inc.
Embryo, ovary and S2R+ cell immunofluorescence
Immunofluorescence was performed on fixed embryos, ovaries and S2R+ cells using ATP5A, GFP, 1B1, Vasa, Tudor and Oskar antisera following standard procedures. Imaging was conducted using Zeiss 510Meta and LSM780 confocal microscopes. Further detail and antibody information is provided in the Supplemental Experimental Procedures.
Fluorescence in situ hybridization (FISH)
mtlrRNA, single molecule RNA and mtDNA FISH was performed on fixed embryos, larval gonads and ovaries. mtlrRNA FISH was conducted on 0 to 1 hour embryos as previously described (Lécuyer et al., 2008). Single molecule RNA FISH (smFISH) was performed as previously described (Trcek et al., 2015) using Custom Stellaris® RNA smFISH Probes labed with Quasar® 670 and designed against the mtlrRNA, ND1 RNA and a germ plasm RNA nanos. Quantitative mtDNA FISH was adapted from previously described methods (McKim et al., 2009; Trcek et al., 2015) using a mixture of 60 5′ labeled CAL Fluor® 590 DNA oligonucleotides. Mixtures of labeled probes hybridizing along transcripts strongly amplifies the signal-to-noise ratio and therefore enhances detection sensitivity. For quantification of mtDNA, foci were counted in the 3D image stack using a spot detection algorithm (Airlocalize) as previously described (Lionnet et al., 2011; Trcek et al., 2015). Imaging was conducted using a Widefield Epifluorescence microscope for smFISH and Zeiss 510Meta and LSM780 confocal microscopes for mtlrRNA and mtDNA FISH. Further details, and Stellaris® probe and primer sequences used to generate FISH probes are listed in the Supplemental Experimental Procedures.
Live imaging
Live imaging was performed on a Zeiss LSM780 confocal microscope and a Prairie Ultima multiphoton microscope equipped with a pulsed 4W Ti:sapphire Chameleon laser. Dissected egg chambers were incubated with 50 μg/ml colcemid or 20 μg/ml cytochalasin D in Schneider’s medium in Lab-Tek II coverslip dishes. Embryos were dechorionated and transferred to a heptane glue coated coverslip before covering with Halocarbon Oil. Further details are provided in the Supplemental Experimental Procedures.
Co-immunoprecipitation and mass spectrometry
Co-immunoprecipitations were performed on 0 to 2 hour old embryos lysed with NP-40 lysis buffer using anti-FLAG® M2 affinity gel. Proteins were digested with trypsin and analyzed using a LC-MS/MS Orbitrap. Further details are provided in the Supplemental Experimental Procedures.
Protein gel electrophoresis and silver staining
Co-immunoprecipitants were electrophoresed through a NuPAGE® 4–12% Bis-Tris Gel before being stained using SilverQuest™ staining kit according to the maunfacturer’s directions. Further details are provided in the Supplemental Experimental Procedures.
qPCR mtDNA copy number measurements
mtDNA copy number was measured by qPCR as using a LightCycler® 480 instrument as described previously (Hunter et al., 2010). PGCs were isolated by FACS (Figure 1) or manually (Figure S7B). Further details and primer sequences are listed in the Supplemental Experimental Procedures.
S2R+ transfection
DNA was transfected into S2R+ cells using Qiagen’s Effectene reagent according to the manufacturer’s directions. Further details are provided in the Supplemental Experimental Procedures.
Statistical and image analysis
Data are expressed as the mean +/− SEM. Significance was assessed using an independent two-tailed Student’s t-test. Images were analyzed using ImageJ (Fiji).
Supplementary Material
RESEARCH HIGHLIGHTS.
< 0.1% of oocyte mtDNA partitions into primordial germ cells in Drosophila
Mitochondria localize to the posterior of the egg independently of germ plasm
- Binding of the long isform of Oskar to the actin cytoskeleton entraps mitochondria
Without Long Oskar, mtDNA is drastically reduced in primordial germ cells
Acknowledgments
We would like to thank the Anne Ephrussi and Akira Nakamura for very kindly providing strains and reagents. We thank Tatjana Trcek for her assistance with single molecule RNA and mtDNA FISH. We thank NYULMC Proteomics Core for assistance. We thank Alexely Soshnev for help with the schematic in Figure 7A. We thank Allison Blum, Lacy Barton, Katie Kistler, Maija Slaidina, Benjamin Lin and Prashanth Rangan for their critical reading of the manuscript. We acknowledge the Bloomington Stock Center for reagents. T.R.H. was supported by the CIHR and R.L. is an HHMI investigator and is supported by NIH R01/R37HD41900.
Footnotes
AUTHOR CONTRIBUTIONS
T.R.H. and R.L. designed the study. T.R.H., J.Sauerwald and B.H. performed the embryo immunofluorescence. T.R.H and J. Sanny generated the oskar transgenic strains. M.G. performed the single molecule RNA FISH. T.R.H. performed all other experiments. T.R.H. and R.L. wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahuja A, Extavour CG. Patterns of molecular evolution of the germ line specification gene oskar suggest that a novel domain may contribute to functional divergence in Drosophila. Dev Genes Evol. 2014;224:65–77. doi: 10.1007/s00427-013-0463-7. [DOI] [PubMed] [Google Scholar]
- Akiyama T, Okada M. Spatial and developmental changes in the respiratory activity of mitochondria in early Drosophila embryos. Development. 1992;115:1175–1182. doi: 10.1242/dev.115.4.1175. [DOI] [PubMed] [Google Scholar]
- Amikura R, Kashikawa M, Nakamura A, Kobayashi S. Presence of mitochondria-type ribosomes outside mitochondria in germ plasm of Drosophila embryos. Proc Natl Acad Sci USA. 2001;98:9133–9138. doi: 10.1073/pnas.171286998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin AA, Chan DC. piRNAs meet mitochondria. Dev Cell. 2011;20:287–288. doi: 10.1016/j.devcel.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Bardsley A, McDonald K, Boswell RE. Distribution of tudor protein in the Drosophila embryo suggests separation of functions based on site of localization. Development. 1993;119:207–219. doi: 10.1242/dev.119.1.207. [DOI] [PubMed] [Google Scholar]
- Chada SR, Hollenbeck PJ. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr Biol. 2004;14:1272–1276. doi: 10.1016/j.cub.2004.07.027. [DOI] [PubMed] [Google Scholar]
- Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox RT, Spradling AC. A Balbiani body and the fusome mediate mitochondrial inheritance during Drosophila oogenesis. Development. 2003;130:1579–1590. doi: 10.1242/dev.00365. [DOI] [PubMed] [Google Scholar]
- Davidson AJ, Wood W. Unravelling the Actin Cytoskeleton: A New Competitive Edge? Trends Cell Biol. 2016;26:569–576. doi: 10.1016/j.tcb.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca SZ, O’Farrell PH. Barriers to male transmission of mitochondrial DNA in sperm development. Dev Cell. 2012;22:660–668. doi: 10.1016/j.devcel.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy EM. Germ plasm and the differentiation of germ cell line. Int Rev Cytol. 1975;43:229–280. doi: 10.1016/s0074-7696(08)60070-4. [DOI] [PubMed] [Google Scholar]
- Ephrussi A, Lehmann R. Induction of germ cell formation by oskar. Nature. 1992;358:387–392. doi: 10.1038/358387a0. [DOI] [PubMed] [Google Scholar]
- Erdélyi M, Michon AM, Guichet A, Glotzer JB, Ephrussi A. Requirement for Drosophila cytoplasmic tropomyosin in oskar mRNA localization. Nature. 1995;377:524–527. doi: 10.1038/377524a0. [DOI] [PubMed] [Google Scholar]
- Evans T, Wade CM, Chapman FA, Johnson AD, Loose M. Acquisition of germ plasm accelerates vertebrate evolution. Science. 2014;344:200–203. doi: 10.1126/science.1249325. [DOI] [PubMed] [Google Scholar]
- Ewen-Campen B, Srouji JR, Schwager EE, Extavour CG. Oskar predates the evolution of germ plasm in insects. Curr Biol. 2012;22:2278–2283. doi: 10.1016/j.cub.2012.10.019. [DOI] [PubMed] [Google Scholar]
- Extavour CG, Akam M. Mechanisms of germ cell specification across the metazoans: epigenesis and preformation. Development. 2003;130:5869–5884. doi: 10.1242/dev.00804. [DOI] [PubMed] [Google Scholar]
- Forrest KM, Gavis ER. Live imaging of endogenous RNA reveals a diffusion and entrapment mechanism for nanos mRNA localization in Drosophila. Curr Biol. 2003;13:1159–1168. doi: 10.1016/s0960-9822(03)00451-2. [DOI] [PubMed] [Google Scholar]
- Gavis ER, Lehmann R. Localization of nanos RNA controls embryonic polarity. Cell. 1992;71:301–313. doi: 10.1016/0092-8674(92)90358-j. [DOI] [PubMed] [Google Scholar]
- Ganguly S, Williams LS, Palacios IM, Goldstein RE. Cytoplasmic streaming in Drosophila oocytes varies with kinesin activity and correlates with the microtubule cytoskeleton architecture. Proc Natl Acad Sci USA. 2012;109:15109–15114. doi: 10.1073/pnas.1203575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutzeit H, Koppa R. Time-lapse film analysis of cytoplasmic streaming during late oogenesis of Drosophila. Development. 1982;67:101–111. [Google Scholar]
- Gutzeit HO. The role of microfilaments in cytoplasmic streaming in Drosophila follicles. J Cell Sci. 1986;80:159–169. doi: 10.1242/jcs.80.1.159. [DOI] [PubMed] [Google Scholar]
- Hill JH, Chen Z, Xu H. Selective propagation of functional mitochondrial DNA during oogenesis restricts the transmission of a deleterious mitochondrial variant. Nat Genet. 2014;46:389–392. doi: 10.1038/ng.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson G, Gomez-Duran A, Wilson IJ, Chinnery PF. Recent mitochondrial DNA mutations increase the risk of developing common late-onset human diseases. PLoS Genet. 2014;10:e1004369. doi: 10.1371/journal.pgen.1004369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter SE, Jung D, Di Giulio RT, Meyer JN. The QPCR assay for analysis of mitochondrial DNA damage, repair, and relative copy number. Methods. 2010;51:444–451. doi: 10.1016/j.ymeth.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida T, Kobayashi S. Essential role of mitochondrially encoded large rRNA for germ-line formation in Drosophila embryos. Proc Natl Acad Sci USA. 1998;95:11274–11278. doi: 10.1073/pnas.95.19.11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katajisto P, Döhla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, Zoncu R, Chen W, Weinberg RA, Sabatini DM. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science. 2015;348:340–343. doi: 10.1126/science.1260384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloc M, Bilinski S, Etkin LD. The Balbiani body and germ cell determinants: 150 years later. Curr Top Dev Biol. 2004;59:1–36. doi: 10.1016/S0070-2153(04)59001-4. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Amikura R, Okada M. Presence of mitochondrial large ribosomal RNA outside mitochondria in germ plasm of Drosophila melanogaster. Science. 1993;260:1521–1524. doi: 10.1126/science.7684857. [DOI] [PubMed] [Google Scholar]
- Kukat C, Wurm CA, Spåhr H, Falkenberg M, Larsson NG, Jakobs S. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proceedings of the National Academy of Sciences. 2011;108:13534–13539. doi: 10.1073/pnas.1109263108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasko PF, Ashburner M. Posterior localization of vasa protein correlates with, but is not sufficient for, pole cell development. Genes Dev. 1990;4:905–921. doi: 10.1101/gad.4.6.905. [DOI] [PubMed] [Google Scholar]
- Lécuyer E, Necakov AS, Cáceres L, Krause HM. High-resolution fluorescent in situ hybridization of Drosophila embryos and tissues. CSH Protoc. 2008 doi: 10.1101/pdb.prot5019. pdb.prot5019. [DOI] [PubMed] [Google Scholar]
- Li W, Gao F-B. Actin filament-stabilizing protein tropomyosin regulates the size of dendritic fields. J Neurosci. 2003;23:6171–6175. doi: 10.1523/JNEUROSCI.23-15-06171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionnet T, Czaplinski K, Darzacq X, Shav-Tal Y, Wells AL, Chao JA, Park HY, de Turris V, Lopez-Jones M, Singer RH. A transgenic mouse for in vivo detection of endogenous labeled mRNA. Nat Methods. 2011;8:165–170. doi: 10.1038/nmeth.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Xu H, O’Farrell PH. Transmission of mitochondrial mutations and action of purifying selection in Drosophila melanogaster. Nat Genet. 2014;46:393–397. doi: 10.1038/ng.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahowald AP. Fine structure of pole cells and polar granules in Drosophila melanogaster. J Exp Zoo. 1962;151:201–215. [Google Scholar]
- Mahowald AP. Assembly of the Drosophila germ plasm. Int Rev Cytol. 2001;203:187–213. doi: 10.1016/s0074-7696(01)03007-8. [DOI] [PubMed] [Google Scholar]
- Markussen FH, Michon AM, Breitwieser W, Ephrussi A. Translational control of oskar generates short OSK, the isoform that induces pole plasma assembly. Development. 1995;121:3723–3732. doi: 10.1242/dev.121.11.3723. [DOI] [PubMed] [Google Scholar]
- McKim KS, Joyce EF, Jang JK. Cytological analysis of meiosis in fixed Drosophila ovaries. Methods Mol Biol. 2009;558:197–216. doi: 10.1007/978-1-60761-103-5_12. [DOI] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol. 1995;131:1315–1326. doi: 10.1083/jcb.131.5.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- Politi Y, Gal L, Kalifa Y, Ravid L, Elazar Z, Arama E. Paternal mitochondrial destruction after fertilization is mediated by a common endocytic and autophagic pathway in Drosophila. Dev Cell. 2014;29:305–320. doi: 10.1016/j.devcel.2014.04.005. [DOI] [PubMed] [Google Scholar]
- Ross JM, Stewart JB, Hagström E, Brené S, Mourier A, Coppotelli G, Freyer C, Lagouge M, Hoffer BJ, Olson L, Larsson NG. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature. 2013;501:412–415. doi: 10.1038/nature12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu YH, Macdonald PM. RNA sequences required for the noncoding function of oskar RNA also mediate regulation of Oskar protein expression by Bicoid Stability Factor. Dev Biol. 2015;407:211–223. doi: 10.1016/j.ydbio.2015.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbus LR, Cha BJ, Theurkauf WE, Saxton WM. Dynein and the actin cytoskeleton control kinesin-driven cytoplasmic streaming in Drosophila oocytes. Development. 2005;132:3743–3752. doi: 10.1242/dev.01956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Z-H. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J Cell Biol. 2014;204:1087–1098. doi: 10.1083/jcb.201312123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Z-H, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13:77–93. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinsimer KS, Jain RA, Chatterjee S, Gavis ER. A late phase of germ plasm accumulation during Drosophila oogenesis requires lost and rumpelstiltskin. Development. 2011;138:3431–3440. doi: 10.1242/dev.065029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinsimer KS, Lee JJ, Thiberge SY, Gavis ER. Germ Plasm Anchoring Is a Dynamic State that Requires Persistent Trafficking. Cell Rep. 2013;5:1169–1177. doi: 10.1016/j.celrep.2013.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JL, Wilson JE, Macdonald PM. Overexpression of oskar directs ectopic activation of nanos and presumptive pole cell formation in Drosophila embryos. Cell. 1992;70:849–859. doi: 10.1016/0092-8674(92)90318-7. [DOI] [PubMed] [Google Scholar]
- Stewart JB, Chinnery PF. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet. 2015;16:530–542. doi: 10.1038/nrg3966. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Nakamura A. Oskar-induced endocytic activation and actin remodeling for anchorage of the Drosophila germ plasm. Bioarchitecture. 2011;1:122–126. doi: 10.4161/bioa.1.3.17313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trcek T, Grosch M, York A, Shroff H, Lionnet T, Lehmann R. Drosophila germ granules are structured and contain homotypic mRNA clusters. Nat Comms. 2015;6:7962. doi: 10.1038/ncomms8962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanzo N, Oprins A, Xanthakis D, Ephrussi A, Rabouille C. Stimulation of endocytosis and actin dynamics by Oskar polarizes the Drosophila oocyte. Dev Cell. 2007;12:543–555. doi: 10.1016/j.devcel.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Vanzo NF, Ephrussi A. Oskar anchoring restricts pole plasm formation to the posterior of the Drosophila oocyte. Development. 2002;129:3705–3714. doi: 10.1242/dev.129.15.3705. [DOI] [PubMed] [Google Scholar]
- Vevea JD, Swayne TC, Boldogh IR, Pon LA. Inheritance of the fittest mitochondria in yeast. Trends Cell Biol. 2014;24:53–60. doi: 10.1016/j.tcb.2013.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Brechbiel JL, Gavis ER. Dynein-dependent transport of nanos RNA in Drosophila sensory neurons requires Rumpelstiltskin and the germ plasm organizer Oskar. J Neurosci. 2013;33:14791–14800. doi: 10.1523/JNEUROSCI.5864-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Li H, Li H, Nakagawa A, Lin JLJ, Lee ES, Harry BL, Skeen-Gaar RR, Suehiro Y, William D, Mitani S, Yuan HS, Kang BH, Xue D. Mitochondrial endonuclease G mediates breakdown of paternal mitochondria upon fertilization. Science. 2016;353:394–399. doi: 10.1126/science.aaf4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.