

Abstract
BACKGROUND
Cortical motor neurons, also known as upper motor neurons, are large projection neurons whose axons convey signals to lower motor neurons to control the muscle movements. Degeneration of cortical motor neuron axons is implicated in several debilitating disorders, including hereditary spastic paraplegia (HSP) and amyotrophic lateral sclerosis (ALS). Since the discovery of the first HSP gene, SPAST that encodes spastin, over 70 distinct genetic loci associated with HSP have been identified. How the mutations of these functionally diverse genes result in axonal degeneration and why certain axons are affected in HSP remains largely unknown. The development of induced pluripotent stem cell (iPSC) technology has provided researchers an excellent resource to generate patient-specific human neurons to model human neuropathologic processes including axonal defects.
METHODS
In this article, we will frst review the pathology and pathways affected in the common forms of HSP subtypes by searching the PubMed database. We will then summurize the findings and insights gained from studies using iPSC-based models, and discuss the challenges and future directions.
RESULTS
HSPs, a heterogeneous group of genetic neurodegenerative disorders, are characterized by lower extremity weakness and spasticity that result from retrograde axonal degeneration of cortical motor neurons. Recently, iPSCs have been generated from several common forms of HSP including SPG4, SPG3A, and SPG11 patients. Neurons derived from HSP iPSCs exhibit disease-relevant axonal defects, such as impaired neurite outgrowth, increased axonal swellings, and reduced axonal transport.
CONCLUSION
These patient-derived neurons offer unique tools to study the pathogenic mechanisms and explore the treatments for rescuing axonal defects in HSP, as well as other diseases involving axonopathy.
Keywords: HSP, axonal degeneration, pluripotent stem cells, spastin, atlastin-1
1. Introduction
Hereditary spastic paraplegias (HSPs) are a heterogeneous group of genetic disorders that result in progressive lower limb spasticity (Crosby and Proukakis, 2002, Reid, 2003, Fink, 2006, Soderblom and Blackstone, 2006, Salinas et al., 2008). The symptoms are caused by a length-dependent degeneration of axons, most severely affecting corticospinal motor neurons (CSMNs) and those of the dorsal columns. Affected neurons display “dying back” axonopathy, particularly in the spinal cord, resulting in lost lower motor neuron innervations that produce lower limb spasticity and weakness. Over the past decade much work has succeeded in increasing the understanding of the genetic causes of the disorder and the function of the genes involved. To date, there have been over 71 different gene loci linked, denoted SPG 1-71, of which over 54 genes have been shown to lead to HSP when mutated (Hazan et al., 1999, Blackstone et al., 2011). These studies have provided insight into several common pathways whose disruption predominantly affects highly polarized projection neurons, revealing weaknesses in these cells due to their impressive morphology. CSMNs, the main neurons affected in HSP, can have axons that reach up to 1 meter in length, with axoplasm making up over 99% of the total cells volume in some cases (Blackstone et al., 2011). This striking polarity makes these cells sensitive to perturbations to many cellular processes, including axonal transport, cytoskeletal dynamics, endoplasmic reticulum morphogenesis, and endocytic recycling.
HSP can be classified based on its mode of inheritance, with all possible forms, dominant, recessive and X-linked reported (Harding, 1983, 1993, Fink, 2003). On the base of the presence or absence of additional symptoms other than lower limb spasticity and weakness, HSP is classified as “pure” when spasticity is the only clinical observation, or “complex” when it coincides with other clinical features including peripheral neuropathy, cataracts, cognitive impairment, ataxia, and thinning of the corpus callosum (Fink, 1993, Salinas et al., 2008). Most postmortem studies have focused on pure HSP, and the major observation has been distal axon degeneration of the longest descending motor fibers, mainly the corticospinal tract (Deluca et al., 2004). Demyelination was observed in regions of axon degeneration, but it is generally believed that this is a secondary effect that follows axonal problems in most forms of HSP. Thinning of the thoracic spinal cord can be observed in HSP patients by MRI (Hedera et al., 2005). While there is significant axon loss, neuron death is not easily detected in HSP patients, although there are some reports of decreased Betz cells (pyramidal neurons). Breakthroughs in basic science research into the genetic causes and pathogenic mechanisms of HSP have dramatically accelerated in the past decade (Supplemental Table 1). Work in this field has uncovered many cellular processes that are affected by multiple forms of HSP, which have greatly aided in understanding the importance of axonal maintenance in human disease (Fig. 1). Recently, human induced pluripotent stem cells (iPSCs) were successfully established from HSP patients, which allows researchers to study axonal defects in these stem cell-derived neurons directly. Through the rest of this review, some highlights of the recent work and common pathways of HSP will be covered, with a focus on two common forms, SPG4 and SPG3A. Next, we will summarize the findings, implications, and future directions for studying HSP using human pluripotent stem cells (hPSCs).
Figure 1. Major pathways affected in HSP.
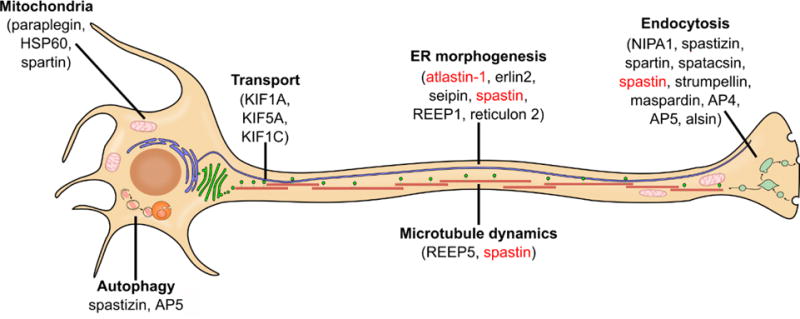
Proteins involved in HSP have a wide range of cellular functions, however many of them cluster into several common cellular pathways. Spastin (for SPG4, the most common form of HSP) and atlastin-1 (for SPG3A, the most common early-onset form of HSP) are shown in red and discussed in detail. Modified from (Blackstone, 2012) with permission from the Annual Review of Neuroscience, Volume 35 by Annual Reviews.
2. Pathology and common pathways of HSP
SPG4 (Spastin)
Mutations to the SPAST (SPG4) gene were found to result in pure autosomal dominant HSP in 1999 and are by far the most common form of HSP, accounting for 40% of all dominant HSP cases (Hazan et al., 1999). The SPAST gene encodes the ubiquitously microtubule-severing protein spastin. All types of possible mutations in SPG4 have been identified, with 28% missense, 15% nonsense, 26.5% splice-site point, 23% deletions and 7.5% insertion mutations (Fonknechten et al., 2000). Spastin is a member of the ATPase associated with diverse cellular activities (AAA) family that also includes the microtubule-severing protein p60 katanin. Four main isoforms for spastin have been identified from the 17 exon protein, generated through combinations of alternative splicing, different promoters and different translational initiation sites (Claudiani et al., 2005, Mancuso and Rugarli, 2008). The two major protein products are a larger, full-length protein, termed M1 spastin, which is 616 amino acids in length, and the shorter 530 amino acid M87 protein that lacks 86 amino acids on its N-terminus (Fig. 2). Two less abundant isoforms exist, which are variations of M1 and M87, that lack exon 4 and the 32 amino acids it encodes (Claudiani et al., 2005). Currently the effects of the presence or absence of this region are not understood, since the role of these 32 amino acids are unknown. Interestingly, M87 spastin is more abundant in all cell types compared to M1, although M1 is more abundant in the brain and spinal cord compared to other locations (Claudiani et al., 2005, Solowska et al., 2008). This observation suggests that M1 spastin may be responsible for HSP development, since the central nervous system is mainly affected. More compelling evidence for the role of M1 spastin in HSP is the observation that two other HSP causative proteins, atlastin-1 and NA14, interact with a region on the N-terminus of spastin, which is only present in the full-length M1 isoform. In rats, it was found that M1 spastin was absent in developing neurons and increased in abundance upon maturation (Solowska et al., 2008). Of particular note was the abundance of M1 spastin in the adult spinal cord that is the location of degenerating axons in HSP patients. The importance of M1 spastin was recently called into question when a report examining SPG4 iPSC-derived neurons found that overexpressing either M1 or M87 spastin could rescue neurite outgrowth defects (Havlicek et al., 2014). This data suggests that both M1 and M87 spastin are important for neurite outgrowth, but it remains unclear if both isoforms contribute equally to axonal maintenance in vivo.
Figure 2. Spastin isoforms and domains.
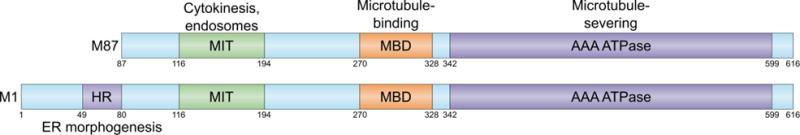
The N-terminus of the protein contains two domains important for protein-protein interactions, the hydrophobic region (HR) and the microtubule interacting and targeting (MIT) domains. The C-terminus contains a microtubule binding domain (MBD) and an AAA ATPase domain, which allows spastin to interact and sever microtubules. Modified from (Blackstone et al., 2011) by permission from Macmillan Publishers Ltd: Nature Reviews Neuroscience, copyright 2011.
A number of studies have shown that spastin is capable of severing microtubules in vitro (Errico et al., 2002). Cell culture studies were originally utilized to determine the function of spastin. When an ATPase-defective form of spastin (K388R) was overexpressed in Cos-7 cells, researchers noticed that the mutated protein localized with microtubules, suggesting that spastin could interact with microtubules, but without the ability to hydrolyze ATP could not dissociate from them (Errico et al., 2002). Also, when wild-type spastin was overexpressed, microtubule disassembly was increased, providing the first evidence for the microtubule-severing activity of spastin. Based on structural studies and comparison with other AAA ATPase proteins, spastin is thought to interact with microtubules through its MTD, allowing the AAA ATPase domain to interact with tubulin. ATP hydrolysis provides energy needed for a conformational change that results in the destabilization of the microtubules. It has been shown that spastin can be present in both monomeric and hexameric form, with the hexamer relying on the N-terminus for assembly (Pantakani et al., 2008). The large variety of different mutations present in the spastin gene of HSP patients (>250) has led to different hypotheses for the mechanism of these mutations. The majority of mutations are nonsense, deletions, or splice site mutations that are believed to reduce the amount of spastin present in a cell, causing disease through a haploinsufficiency mechanism (Lumb et al., 2012). Recently, the observation that almost all of the missense mutations appeared in the AAA domain suggests that some of these mutations could function through a dominant negative mechanism, since the protein forms oligomers. In 2008, it was shown that at least one missense mutation, E442Q, acted in a dominant negative fashion (Pantakani et al., 2008). Using co-localization studies, the researchers were able to show that the mutant spastin was able to interact with wild-type spastin, and perturb its localization.
Perturbed spastin could affect several pathways including endocytosis, endoplasmic reticulum (ER) morphogenesis, and axonal transport (Fig. 1), resulting in axonal defects in SPG4 patients. The regulation of axonal transport fits with the role spastin plays in microtubule severing, as microtubule arrays are present through the entire length of axons and provide structural support as well as serving as the railways for organelle transport. Axonal transport deficits also neatly match the observation that only the longest projection neurons are affected, since they would put the largest strain on transport systems to deliver cellular contents to the most distal portions of the cell. If proper materials are not delivered to the distal regions, it could cause a dying back of the axon, which is seen in HSP. Some of the best lines of evidence linking spastin and transport come from two different HSP mouse models that possess different spastin mutations. The first generated strain had a deletion in spastin that resulted in a premature stop codon to study the effects of reduced levels of spastin (Tarrade et al., 2006). These mutant mice displayed minor gait alterations compared to wild-type at 22 months. At as early as 4 months the mutant mice showed defects in axons located in the spinal cord, with axonal swellings evident in both ascending and descending tracts. In affected axons of the corticospinal tract, dramatic disorganization of cytoskeletal components was observed. These abnormities were restricted to axons, and the neuronal cell bodies remained unaffected. By culturing cortical neurons from these mice, the researchers found that retrograde axonal transport was impaired. This was made evident by the observation that the volume of the axon distal to a region of swelling was significantly larger than the proximal region of the axon. Both mitochondria and peroxisomes accumulated in the distal portion axon swellings, and not in other regions of the cell. In a second HSP mouse model from 2009 containing a splice site mutation that is expected to generate a premature stop codon, many of the similar abnormalities of the first mouse model were also shown (Kasher et al., 2009). Gait abnormalities and axonal swellings were again present in these mice, but what made this study unique was the quantification of both anterograde and retrograde axonal transport. To do this, time-lapse microscopy was used to follow labeled mitochondria and APP as a marker for membrane bound organelles. Contrary to what was suggested in the first HSP mouse model, these researchers found anterograde, not retrograde, axonal transport to be diminished. Also, swellings appeared to be in random locations along the axon, where in the previous mouse model, the swellings were more distally localized. These inconsistencies between the two mouse models suggest that different types of mutations could function in different ways even if both result in decreased spastin levels, and it remains to be seen if this is indeed the case in human neurons.
In addition to regulating axonal transport, spastin plays an important role in membrane trafficking. As mentioned earlier, M1 spastin localizes to the early secretory pathway in cells, interacting with these membranes through the hydrophobic domain at the N-terminus. The HD is predicted to form a hairpin loop capable of inserting itself into the outer leaflet of a lipid bilayer through hydrophobic wedging (Park et al., 2010). Three classes of proteins that display hydrophobic wedging have been shown to also insert themselves into membranes as a mechanism for generating the extreme membrane curvature that is seen in the tubular endoplasmic reticulum, including the reticulons, REEPs and atlastins (Park et al., 2010). Interestingly, members of all three classes are associated with additional forms of autosomal dominant pure HSP, suggesting a similar mechanism.
SPG3A (Atlastin-1)
SPG3A is caused by mutations to the protein atlastin-1 and accounts for approximately 10% of HSP cases, making it the second most common cause of HSP (Zhao et al., 2001). Atlastin-1 mutations result in early onset pure HSP that displays autosomal dominant inheritance. Because deficits are seen at such a young age in SPG3A patients, it has been suggested that this form of HSP is neurodevelopmental, while other late onset forms are neurodegenerative. Atlastin-1 is the founding member of a small group of proteins (atlastin1-3) in the superfamily of dynamin-related GTPases. It is a 558 amino acid protein that possesses a large GTPase domain at the N-terminus, followed by a middle domain that serves an unknown function, and two transmembrane domains at the C-terminus (Zhu et al., 2003) (Fig. 3). Atlastin-1 localizes to the Golgi apparatus and also to particular regions of the tubular endoplasmic reticulum network (Zhu et al., 2003, Hu et al., 2009). The localization of atlastin-1 to the ER is due to the presence of an ER localization signal at the C-terminus. Early work into the function of atlastin-1 has relied on in vitro cellular models. In 2008, a group found that when a mutant or dominant negative atlastin-1 protein was overexpressed in HeLa cells, the endoplasmic reticulum was severely affected. Much less branching was present in the tubular endoplasmic reticulum, while there was an increase in the endoplasmic reticular sheets, associated with the rough ER (Zhu et al., 2003). This observation, along with subsequent studies, has led to the prevailing model that implicates atlastin-1 in the formation of the smooth endoplasmic reticulum network that is present throughout the entirety of all cells, including axons and dendrites of neurons. The tubular ER is important for lipid and cholesterol metabolism and serves as a store for calcium inside of the cell, which is important for synaptic transmission and plasticity (Renvoise and Blackstone, 2010). The ER is a very dynamic organelle with two main subdivisions: the sheets where protein synthesis occurs and the vast network of tubular ER (Moss et al., 2011). The tubular ER is present throughout the entirety of the cell and moves along microtubule tracts, allowing it to localize in even the most distal portions of a neuron’s axon. Atlastin-1 controls ER network morphogenesis by driving the fusion of individual ER tubules, allowing the formation of three-way junctions. The reduction of atlastin-1 in rat cortical neurons resulted in decreased axonal length and the presence of cells that completely lacked an axon (Zhu et al., 2003). In vivo studies of atlastin-1 function have focused on model organisms such as Drosophila and zebrafish Danio rerio. When the atlastin-1 homologue was knocked-down in zebrafish, researchers observed abnormal organization of spinal motor axons that reduced larval movement (Fassier et al., 2010). These abnormalities were credited to alterations in bone morphogenetic protein (BMP) signaling. The observation that atlastin-1 can be partially localized to endosomes suggests that it may alter endocytosis of BMP receptors. There were no observed abnormalities in ER organization, which suggests a potential difference in the function of atlastin-1 between zebrafish and mammals, and further studies are needed to clarify these issues.
Figure 3. Atlasin-1 domains.

Atlastin-1 consists of three main domains: the large GTPase domain, the middle linker domain, and two trans-membrane domains (TMDs). Each TMD partially inserts into ER lipid bilayers through hydrophobic wedging. At the C-terminus is a KDEL ER retention (ERR) signal.
In addition to regulation of ER morphogenesis, atlastin-1 has been shown to regulate lipid droplet production. Lipid droplets consist of lipid ester cores that are surrounded by phospholipid monolayers and serve as the major fat storage organelles in eukaryotic cells (Walther and Farese, 2012). The ER plays an important role in lipid droplet synthesis, as the two organelles are closely associated with one another, and lipid droplet growth requires interaction with the ER (Wilfling et al., 2013). The role of atlastin family members in lipid droplet regulation was first identified in C. elegans, where mutations not only altered ER morphology, but also lipid droplet size (Klemm et al., 2013). In contrast, overexpression of atlastin increased lipid droplet size. These observations were later confirmed in HeLa cells (Falk et al., 2014). This work suggests that atlastin-mediated ER membrane fusion is required for lipid droplet expansion. In the future, it will be interesting to determine if alterations to lipid droplet size have any pathological consequences in SPG3A patients.
ER morphogen complex and common pathway of HSP
Despite the diverse functions of HSP proteins, these proteins are grouped into several common pathways including ER morphogenesis, microtubule dynamics and endocytosis. Atlastin-1 and spastin, as well as two other HSP-related proteins including the receptor expression enhancing protein 1 (REEP1) (Park et al., 2010), were found to localize to the endoplasmic reticulum. REEP1 is encoded by the SPG31 gene whose mutations underlie the third most common cause of HSP. Humans and other mammals have 6 different REEP family members (REEP1-6), which are highly conserved membrane proteins. Through phylogenetic analysis, REEP1 was found to be closely related to the DP1/Yop1p proteins that are involved in shaping tubular ER. In fact, REEP1 predominantly localized with tubular ER, and similar to what was seen in atlastin-1 mutant expressing cells, mutant REEP1 expression impairs tubular ER network formation. In cultured rat cortical neurons, REEP1 colocalizes with both M1 spastin and atlastin-1 through its hydrophobic domains. The ER shaping protein reticulon-2 was identified as the causative gene in the autosomal dominant SPG12 (Montenegro et al., 2012). Like REEP1, reticulon-2 also is a direct binding partner with M1 spastin.
This determination that four HSP proteins localize to the ER and interact with each other has led to the proposal that defects to the so-called “ER morphogen complex” are central to the pathogenesis of over 60% of HSP. The “ER morphogen complex” describes a complex formed by interacting proteins, including atlastin-1, spastin, REEP1 and reticulons, which functions to regulate the formation and localization of the endoplasmic reticulum. Atlastin-1 is needed for the formation of the tubule ER network, by regulating the fusion of tubules to form three-way junctions. REEP1 and reticulons are critical proteins involved in shaping the ER, and can generate the extreme curvature displayed in the tubular ER. Lastly, spastin is involved in linking the ER to cells’ cytoskeleton through microtubule interaction. This is believed to provide structural support for the ER, and also allows transport of the ER throughout the cell. If any one of these components of the “ER morphogen complex” are mutated, the resulting abnormality in ER morphology could negatively affect axons of long motor neurons, leading to loss of synaptic connection and lower limb spasticity. The detailed mechanisms by which impaired ER morphogenesis results in axonal degeneration require further investigation. It is also important to dissect the interplay between ER morphogenesis and other cellular pathways (e.g. microtubule dynamics, bone morphogenetic protein signaling, endocytosis) to better understand the pathogenic mechanisms underlying the axonal degeneration in HSP.
3. Modeling HSP with human pluripotent stem cells
Pluripotent stem cells
Embryonic stem cells (ESC) are a type of pluripotent cells that have two important characteristics. First, they have the ability to proliferate indefinitely, and they can generate any cell type in an adult body from all three germ layers. In 1981, the first mouse embryonic stem cells were isolated from the inner cell mass of preimplantation blastocysts (Evans and Kaufman, 1981, Martin, 1981). These cells could be cultured indefinitely, formed multiple cell types in vitro, and could generate teratocarcinomas when injected into mice. Mouse ESCs have proven to be an invaluable tool for studying mammalian development, and have led to the field of gene targeting (Ben-David et al., 2012). In 1998, the first human embryonic stem cells were generated by Dr. James Thomson (Thomson et al., 1998). This reignited interest in stem cell biology with major implications for regenerative medicine. However, ethical issues concerning the destruction of embryos led to the search for additional sources of pluripotent cells.
As more was understood about the mechanism of pluripotency in mammalian cells, Dr. Shinya Yamanaka’s group sought to reprogram mouse somatic cells using defined factors (Takahashi and Yamanaka, 2006). In a groundbreaking report, researchers chose to force somatic cells to express various combinations of 24 transcription factors that are upregulated in ESCs and found that just OCT3/4, KLF4, SOX2 and c-MYC could reprogram cells back to an ESC-like state. The generated cells are the other type of pluripotent stem cells, termed induced pluripotent stem cells (iPSCs). They were shown to display many similar properties of ESC, including self-renewal and the ability to form cells of all three germ layers. A year later, this feat was accomplished in human cells by two independent labs, with Yamanaka’s group using the same four factors from the mouse study (Yamanaka’s factors) (Takahashi et al., 2007), and the Thomson lab showing similar results with OCT3/4, SOX2, LIN28 and NANOG (Thomson factors) (Yu et al., 2007). Shortly after these reports, some potential benefits became clear to the scientific community, including the ability to generate iPSC lines from patients with genetic disorders to model diseases, and the reduced risk of immune rejection following transplantation of tissues derived from iPSCs that came from the same patient (Guha et al., 2013).
In the past ten years since the first generation of iPSCs, enormous progress has been made to increase the efficiency in generating these cells using much safer reprogramming methods. An important focus of many groups has been to design reprogramming methodologies that are integration free, as numerous reports have shown that integrated transgenes can result in increased tumorigenicity and decreased differentiation potential (Okita et al., 2007). Over the past several decades, a large amount of effort has been directed towards developing reproducible directed differentiation protocols that allow the generation of clinically relevant cell populations from pluripotent stem cells (Murry and Keller, 2008). This work has provided researchers with access to large quantities of human neurons for the first time, including spinal motor neurons (Li et al., 2005), midbrain dopaminergic neurons (Park et al., 2004, Perrier et al., 2004), telencephalic glutamatergic neurons (Reubinoff et al., 2001, Zhang et al., 2001), and medium spiny neurons (Ma et al., 2012). The ability to generate defined populations of neuronal subtypes fueled the excitement over the ability to replace affected cells in various neurodegenerative diseases (Kiskinis and Eggan, 2010). While progress towards this ambitious goal has progressed slowly, work towards another application for stem cell-derived neurons, “in vitro disease modeling”, has rapidly taken off. The ability to generate patient specific neurons allows researchers to examine processes that are not easily observed in postmortem tissues. The combination of directed neuronal differentiation and iPSCs has allowed the examination of patient-derived neurons that harbor pathogenic mutations. This strategy has successfully recapitulated phenotypes from ALS (Dimos et al., 2008, Bilican et al., 2012), spinal muscular atrophy (Ebert et al., 2009), Huntington’s disease (Park et al., 2008, Zhang et al., 2010), Parkinson’s disease (Park et al., 2008, Nguyen et al., 2011), and Alzheimer’s disease (Yagi et al., 2011, Kondo et al., 2013). In the past two years, iPSC-based models have also been generated for several forms of HSP, including SPG4, SPG3A and SPG11.
hPSC models of SPG4
HSP patient-specific iPSC lines were first generated from SPG4, the most common form of HSP. By introducing the pluripotent factors into patient’s fibroblast cells using either lentiviral infection or the non-integrating episomal method, our group successfully established iPSC lines from a SPG4 patient with an intron 4 splice acceptor mutation (c. 683-1, G>T) (Fig. 4A, B) (Denton et al., 2014). To examine the disease-relevant phenotypes, we then differentiated these stem cells into telencephalic glutamatergic neurons (cortical projection neurons), among which some are cortical motor neurons (Fig. 4C). The SPG4 iPSC-derived neurons showed a significant increase in the formation of axonal swellings (Fig. 4D), a common pathological hallmark observed in postmortem patients’ samples and animal models. Increased axonal swellings were also observed in neurons derived from a second SPG4 patient with an amber mutation in the SPAST gene (Fig. 4E,F), suggesting that increased axonal swellings could be a reliable phenotype in hPSC-based models of SPG4. It has been shown that increased formation of axonal swellings can be associated with the accumulation of transported cargos, suggesting impaired axonal transport. To confirm the axonal transport deficits in live HSP neurons, we examined the mitochondrial fast axonal transport in the axons of SPG4 neurons (Fig. 4G). Our data revealed a significant reduction in the percentage and frequency of motile mitochondria in SPG4 neuron axons (Fig. 4H, I). To our knowledge, this is the first evidence of axonal transport deficits in human HSP neurons. By reprogramming fibroblast cells from two SPG4 patients (all with c.1684C>T nonsense mutation) using retroviral infection method, Dr. Winner’s group also successfully established SPG4 patient-specific iPSC lines (Havlicek et al., 2014). The SPG4 iPSC-derived neurons exhibited reduced neurite complexity, increased axonal swellings and impaired axonal transport. These studies together suggest that neurons derived from SPG4 iPSCs recapitulate disease-relevant phenotypes, providing unique models to study axonal defects in HSP.
Figure 4. Establishment of iPSC-based SPG4 and SPG3A models that recapitulate disease-specific axonal phenotypes.
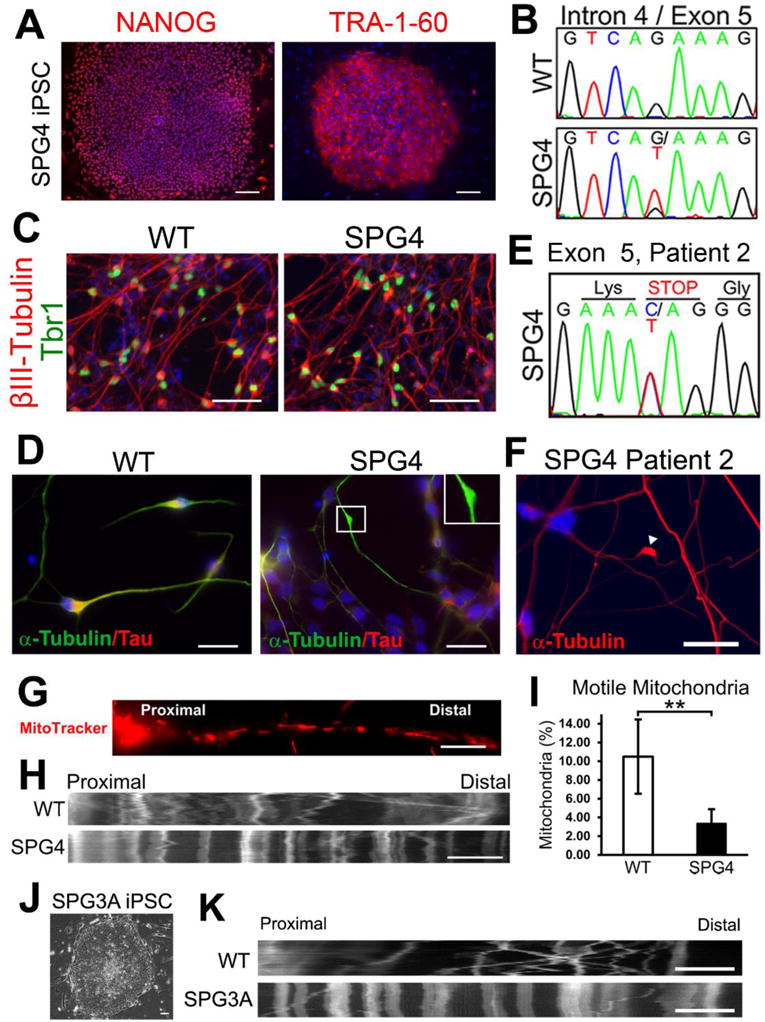
(A, B) Immunostaining showing the expression of pluripotent protein NANOG and TRA-1-60 (A) by the iPSCs derived from a patient with intron 4 splice acceptor mutation (c.683-1G>T; panel B). (C) At 6 weeks after differentiation, telencephalic glutamatergic neurons (Tbr1+/βIII-tubulin+) were efficiently generated from WT (control) and SPG4 iPSCs. (D) Neurons derived from SPG4 iPSCs displayed swellings in Tau+ axons, while control neuron axons were mostly smooth with no swellings. (E, F) Increased formation of axonal swellings was also observed from telencephalic neurons derived from iPSCs of another patient with a C>T transition located in Exon 5 of the SPAST gene (amber mutation, E). (G) To examine fast axonal transport of mitochondria, cells were stained with MitoTracker Red CMXRos (Invitrogen). (H) Representative distance versus time kymographs over a 5 minute recording. (I) Quantification of motile mitochondria in week 8 telencephalic neurons showed a significant decrease of motile mitochondria in SPG4 neurons compared to control neurons. Data presented as mean ± SD. **P < 0.01. (J) SPG3A fibroblast cells were successfully programmed to iPSCs that have typical ESC morphology. (K) As shown by the representative distance versus time kymographs, reduction of motile mitochondria was also observed in SPG3A iPSC-derived telencephalic neurons. Blue indicates Hoechst stained nuclei. Bars, 100 (A), 50 (C), 20 (D,F), 10 (G, H), and 5 (K) μm. Modified from references (Denton et al., 2014; Zhu et al., 2014).
How SPAST mutations result in axonal degeneration in SPG4 patients is largely unclear. Sequencing analysis on SPG4 locus has revealed a wide spectrum of SPAST gene alterations and many SPAST mutations can result in loss of spastin function through haploinsufficiency or a dominant-negative mechanism (Hazan et al., 1999, Lindsey et al., 2000). To examine whether patient-specific neurons have reduced spastin levels, we compared the protein expression levels between forebrain neurons derived from SPG4 iPSCs or control iPSCs. Our data showed that the iPSC-derived forebrain neurons (with the intron 4 splice acceptor mutation) have low levels of spastin protein (~50% of control neurons), suggesting reduced spastin function. To further confirm that loss of spastin function underlies the axonal defects of human SPG4 neurons, we established spastin-knockdown hESCs and differentiated these cells into telencephalic glutamatergic neurons. As in SPG4 neurons, the spastin-knockdown neurons exhibited increased axonal swellings, revealing that the axonal phenotypes in human SPG4 neurons can be caused by loss of spastin function. By introducing spastin into SPG4 neurons, Dr. Winner’s group performed the rescue experiments. Overexpression of spastin in SPG4 neurons restored the neurite complexity and rescued the accumulation of axonal swellings, further confirming that loss of spastin function could result in axonal phenotypes (Havlicek et al., 2014). Interestingly, their study also showed that the protein expression levels of both M1 and M87 spastin isoforms were reduced in SPG4 neurons. Moreover, overexpression of either M1 or M87 isoform can rescue the phenotypes, suggesting the contribution of both isoforms to the pathogenesis of SPG4.
An important advantage for patient-specific iPSC-derived neurons is their potential utilization for future drug screenings. As a proof of principle, we examined the effects of microtubule-targeting drug vinblastine on SPG4 neurons. Our data showed that treatment of nanomolar concentration of the vinblastine can rescue the axonal defects in SPG4 neurons, suggesting that microtubules can be a potential therapeutic target for HSP in human neurons. The beneficial effect of targeting microtubule dynamics in SPG4 was confirmed in olfactory neurosphere-derived cells from SPG4 patients (Fan et al., 2014). Here, researchers found that very low doses of four microtubule-targeting drugs, vinblastine, taxol, epothilone, and noscapine, could rescue peroxisome trafficking deficit in SPG4 cells. With the successful establishment of patient-specific iPSCs with the capacity to efficiently generate telencephalic glutamatergic neurons (the cell types that are affected in patients) these stem cells provide unique sources of patient-specific neurons to screen therapeutic agents to rescue the axonal degeneration in HSP.
iPSC models for SPG3A
SPG3A, the most common early onset form of HSP, is caused by mutations in the atlastin-1 (ATL-1) gene (Zhao et al., 2001, Namekawa et al., 2006). In collaboration with Dr. Craig Blackstone, we generated iPSC lines from fibroblast cells of a SPG3A patient with a novel p.Pro342Ser mutation using episomal transduction method (Zhu et al., 2014). This mutation is in the linker region of atlastin GTPases, which results in both a conformational switch of atlastin and a modest reduction in GTPase activity. Human iPSCs derived from SPG3A patients and normal individuals (control) were differentiated into telencephalic glutamatergic neurons to examine how this mutation affects human neurons. Our data revealed a significant reduction of axon outgrowth and axonal transport (Fig. 4J, K) in SPG3A neurons. Moreover, microtubule-targeting drugs (e.g. vinblastine) could recue the axon outgrowth deficits in SPG3A neurons (Fig. 5). Thus, we demonstrate the successful establishment of SPG3A iPSC models and identify common axonal defects in iPSC models of two common forms of HSP, SPG4 and SPG3A (Fig. 5).
Figure 5. Summary of major phenotypes observed in SPG4 and SPG3A iPSC-derived neurons.
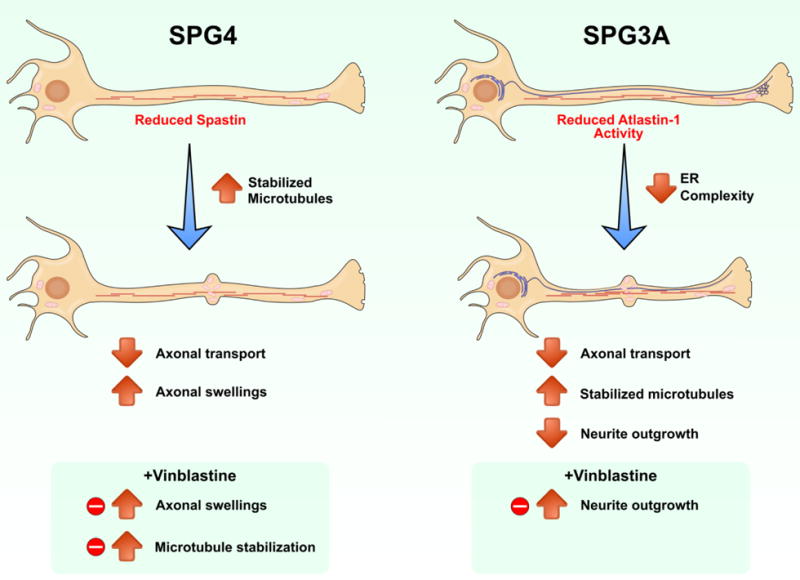
We observed length-dependent axonal swellings in SPG4 neurons, and neurite outgrowth abnormalities in SPG3A neurons. Reduced axonal transport was observed in both SPG4 and SPG3A neurons. These phenotypes were rescued following treatment with the microtubule-targeting drug vinblastine, linking alterations to microtubule dynamics in these forms of autosomal dominant HSP.
The onset of SPG3A is much earlier than other forms of HSP, which coincides with the axonal outgrowth and elongation defects observed in atlastin-1 knockdown rat cortical neurons. Using SPG3A patient-specific iPSCs, our data showed that SPG3A neurons exhibited reduced axonal length and branches, confirming an early axon outgrowth deficit in SPG3A neurons. Interestingly, though both SPG4 and SPG3A neurons have axonal transport deficits, fast axonal transport at the retrograde direction is more affected in SPG4 neurons while SPG3A neurons showed a significant reduction at anterograde direction. This difference could be caused by the differences between cultures or the intrinsic differences between these two HSP proteins. Thus, by generating iPSC lines from different forms of HSP, we could dissect the differences between different forms of HSP and identify the common mechanisms underlying axonal defects in HSP.
Modeling recessive forms of HSP
Besides the more common autosomal dominant forms of HSP, there is a long list of recessive mutations that can lead to complex forms of HSP. SPG11 and SPG15 are the most common recessive forms of HSP, and are clinically almost identical (Stevanin et al., 2007, Hanein et al., 2008). These two forms account for 25% of the autosomal recessive subtypes (Nadar et al., 2008). Both forms present with cognitive impairment, dementia, ataxia, and thinning of the corpus callosum, in addition to lower limb spasticity. SPG11 and SPG15 encode spatacsin and spastizin respectively, both of which are direct binding partners (Murmu et al., 2011, Hirst et al., 2013). These two proteins can also bind to the adaptor protein 5 (AP5), the subunits of which are mutated in the complex SPG48 (Hirst et al., 2013). Another interesting aspect of SPG11, SPG15, and SPG48 is that they can present with prominent juvenile parkinsonism, which has shown improvement following dopaminergic therapy (Guidubaldi et al., 2011, Schicks et al., 2011). It is particularly intriguing that both muscle spasticity, caused by cortical spinal motor neuron degeneration, and parkinsonism, caused by dopaminergic defects, occur following the loss of function of one gene.
By reprogramming fibroblast cells of two SPG11 patients with heterozygous nonsense mutations of spatacsin, Dr. Winner’s group successfully established iPSC lines for SPG11 (Perez-Branguli et al., 2014). To study the role of spatacsin in human neurons, they first examined the expression level and pattern of spatacsin during neural differentiation from human iPSCs. Spatacsin protein is expressed in iPSC-derived neurons, as well as in mouse cortical neurons at multiple stages. Interestingly, spatacsin protein is enriched in axons and dendrites of neurons, and disturbed spatacsin function in SPG11 neurons results in reduced neurite length and complexity. SPG11 neurons also exhibited impaired axonal transport as indicated by a significant reduction of the synaptic vesicle trafficking in the anterograde direction. Similarly, knockdown of spatacsin in mouse cortical neurons led to impaired neurite outgrowth and reduced synaptic vesicle trafficking, revealing the important role of spatacsin in axonal maintenance and transport. Using these patient iPSC-derived neurons, Dr. Winner’s group further investigated the molecular mechanisms underlying axonal defects in SPG11. They observed DNA, mRNA and protein changes related to cell cycle regulation in SPG11 neural progenitors as well as spatacsin knockout neural progenitors that produced a substantial decrease in neuronal proliferation (Mishra et al., 2016). Among the molecular changes they observed in SPG11 neurons, one discovery of particular importance is the GSK3ß/ß-Catenin pathway dysregulation in SPG11 neural precursors that results in a significant reduction of neural cells. Moreover, regulation of the GSK3 pathway can rescue the decreased proliferation of SPG11 neural precursors, implying the GSK3ß/ß-Catenin dysregulation in SPG11 (Mishra et al., 2016). This rescue effect demonstrates the potential role iPSC studies in HSP models can play in discovering novel early therapeutic interventions for these diseases. Although the precise mechanisms underlying spatacsin’s effects on these pathways have not yet been elucidated, these data together provide insights into the physiological role of spatacsin during development and the pathogenic mechanisms in SPG11.
Considering that both dominant (SPG4, SPG3A) and recessive (SPG11) forms of HSP can exhibit similar axonal phenotypes, establishment of iPSC models for multiple forms of HSP would be extremely valuable to dissect the common mechanisms underlying axonal defects in HSP. Furthermore, these iPSC models would also provide unique paradigms to screen therapeutic agents to identify effective therapeutics that may rescue axonal defects in multiple form of HSP, as well as in other diseases involving axonopathy.
4. Challenges and Future Directions
HSP is a group of genetically diverse diseases characterized by the axonopathy of CSMNs. To date, only 13 out of the 71 HSP-linked loci have been studied in zebrafish or drosophila, while 12 have been examined in mice. The stem cell technology allows researchers to examine the role of HSP genes in human neurons with in vitro stem cells models of HSP. The availability of stem cell models for every HSP subtype would greatly enhance the field’s understanding of the pathways necessary for maintenance of long axons. There are some challenges and questions that need to be addressed to better study the axonal degeneration in HSP and other diseases involving axonopathy.
Establishing accurate models of various forms of HSP
One of the roadblocks to modeling all the HSP subtypes is the limited availability of HSP patient fibroblasts, making large cohorts of cell lines difficult to employ. The best way to get around this limitation would be to use newer gene targeting techniques, such as the TALEN (Hockemeyer et al., 2009, Miller et al., 2011, Niu et al., 2014) or CRISPR/Cas systems (Jinek et al., 2012, Mali et al., 2013, Wang et al., 2013a), to introduce mutations identified in patients to every HSP gene in a control cell line, such as H9 hESCs or normal iPSCs. If all of the lines were generated from the same original control line, they would be in effect isogenic, dramatically reducing the potential variability between control and mutant lines that occurs when comparing lines from individuals with different genetic backgrounds. Stem cell based models also allow the identification of events that precede disease onset, since early born neurons are examined (Liu et al., 2012). Though for many neurodegenerative diseases, clinical symptoms appear at adult stage (late-onset), pathological changes in neurons at early developmental stages have been observed. For example, embryonic cortical neurons isolated from SPG4 mouse models (Tarrade et al., 2006, Kasher et al., 2009) exhibited characteristic axonal defects (e.g. accumulation of axonal swellings and impaired axonal transport) which have been recapitulated in our SPG4 iPSC-based disease model (Denton et al., 2014). Late-onset diseases (e.g. Parkinson’s disease) could also be modeled through progerin-induced aging of iPSC-derived neurons (Miller et al., 2013). The ability to model early pathological changes in late-onset diseases is particularly valuable when attempting to identify therapeutic compounds that can prevent disease progression. If a pathogenic event can be detected prior to neurodegeneration, studies can be designed to test for the inhibition of that event. Therapies that prevent disease onset and progression will be more valuable as genetic sequencing becomes cheaper and more widespread. Children who are at risk of inheriting HSP can be sequenced at an early age, allowing preventative measures to be taken.
A significant challenge in modeling human disease with patient-specific iPSCs is the difficulty of studying the circuitry of in vitro cultures. Neocortex is a very specialized structure with different layers and functional areas, each of which has unique cytoarchitecture, chemoarchitecture, input and output (Polleux et al., 2001, O’Leary and Nakagawa, 2002, Grove and Fukuchi-Shimogori, 2003). Considering that cortical motor neuron diseases are developed under complex circuitry in vivo, it is critical to build the circuitry of in vitro cultures for successfully modeling these motor neuron diseases, including HSP. Strategies to address this challenge include building three-dimensional (3D) neural tissues models and co-culturing cortical motor neurons with their target, spinal motor neurons. The accurate model of the in vivo circuitry in hPSC-based models would significantly advance the understanding of the pathogenic mechanisms underlying the axonal defects of cortical motor neurons. It would also be informative to transplant HSP iPSC-derived neurons into animals to test whether or not the phenotypes observed in vitro are seen in vivo. The conditions that cells are exposed to in vitro are much harsher than those in vivo, potentially leading to misleading phenotypes (Halliwell, 2014). Transplantation of cells would also allow for testing the effects of therapeutic agents in a more physiologically relevant environment. This would also allow neurons to be examined after longer periods of time in vivo than would be possible in vitro. With current culturing techniques, stem cell-derived neurons do not survive in vitro indefinitely, but transplanted neurons have been shown to survive at least 2 years in vivo (Hallett et al., 2015).
Overcoming cellular heterogeneity
One of the difficulties when working with stem cell-derived neural cultures is the heterogeneity of cells generated after differentiation. Following neural differentiation, many studies will use immunohistochemistry or transcriptional profiling to determine whether the cell-type of interest is present in the culture, and to what extent. However, studies often fail to fully characterize the other cell-types that are present in the culture, providing a potential significant source of variation, as a number of neurodegenerative diseases result from non-cell autonomous mechanisms. If a culture only contains a small percentage of neurons of interest, this could make it difficult to observe subtle phenotypes, particularly if the other cells in the culture are unaffected in the disease. One way to get around this challenge is to use a purification technique, such as fluorescence-activated cell sorting (FACS), to obtain pure populations of the neuronal subtype of interest. This purification strategy, however, requires a specific marker for the particular neuronal subtype, which is not currently available for all clinically relevant cell populations, such as CSMNs. This strategy has worked well for studies examining diseases affecting spinal motor neurons (Kiskinis et al., 2014) or midbrain dopaminergic neurons (Miller et al., 2013), as specific markers are available to purify these cells. In the case of HSP, using generic telencephalic glutamatergic neurons is appropriate for some autosomal recessive subtypes, such as SPG15 and SPG48, where widespread abnormalities are seen in neurons whose axons pass the corpus callosum. For autosomal pure HSP subtypes, such as SPG3A and SPG4, CSMNs are specifically affected. Currently, a method to specifically generate CSMNs from hPSCs is still not available. In the future, the ability to efficiently generate and/or purify CSMNs will enhance our ability to identify why these neurons are specifically susceptible to degeneration.
Examining the role of HSP proteins
To date, almost no work has been done to examine the physiological importance of HSP proteins in human neurons. The majority of studies have looked at morphological defects of cells when levels of HSP proteins are perturbed. This lack of information needs to be resolved, as a number of HSP proteins are involved in pathways that have direct effect on synaptic transmission and electrophysiology. In addition, mitochondria play a crucial role to presynaptic function, as these regions rely on high levels of ATP to maintain ionic gradients and membrane potential, and to reload synaptic vesicles (Hollenbeck, 2005, Knott et al., 2008). Mitochondria within presynaptic terminals also serve to sequester cytosolic Ca2+ during neurotransmission (Ly and Verstreken, 2006). The involvement of mitochondrial health and transport in multiple forms of HSP suggest it would be interesting to examine whether electrophysiological properties are altered in these HSP neurons.
An important question in examining the role of HSP proteins using patient iPSC-derived neurons is whether the physiological alterations in these patient-derived neurons are directly caused by the mutations/perturbations of the HSP proteins. One strategy to address this question is to perform rescue experiments by correcting the gene mutations and then examining whether the impaired physiological functions can be rescued in these isogenic lines. Similar mutations could also be introduced to normal hESCs for identifying the effects of these mutations on physiological functions by comparing normal and HSP neurons. Since most HSP mutations result in loss of function in these proteins, another strategy is to generate hPSC lines with the knockdown of these HSP genes. If these HSP-knockdown neurons exhibit similar phenotypes as the patient iPSC-derived neurons, it will confirm the direct link between the impaired physiological functions and the HSP protein deficiency. Furthermore, considering that iPSCs have the capacity to generate different neuronal subtypes (Boulting et al., Perrier et al., 2004, Li et al., 2005, Singh Roy et al., 2005, Yan et al., 2005, Roy et al., 2006, Zhang, 2006, Lee et al., 2007, Zeng et al., 2010), the HSP patient-specific iPSCs also provide a unique paradigm to examine the role of HSP proteins in different neuronal subtypes, which will provide important insights into understanding the pathogenic mechanisms underlying axonal degeneration.
Implications for other neurodegenerative disorders
One of the aspects that makes studying HSP relevant to neurodegenerative disorders as a whole is the extensive genetic characterization of the different HSP subtypes (Supplemental Table 1). The fact that mutations to 71 genes and counting all lead to the common phenotype of degeneration of the longest axons can provide insight into the pathways that are needed for axonal maintenance in general and in other neurodegenerative disorders. An understanding of the genes and pathways that result in HSP is also beneficial to understanding the mechanisms of other neurodegenerative disorders, as it seems likely that there are pathways affected in multiple diseases, such as axonal transport (Parkinson’s, Alzheimer’s, ALS, Huntington’s, HSP) (De Vos et al., 2008), ER stress (Huntington’s disease (Vidal et al., 2011), ALS (Kanekura et al., 2009), SBMA (Montague et al., 2014)), RNA processing (SMA (Lunn and Wang, 2008), ALS (Ling et al., 2013)), mitochondrial quality control (Parkinson’s (Valente et al., 2004), Alzheimer’s (Piaceri et al., 2012), ALS (Manfredi and Xu, 2005), HSP (Blackstone et al., 2011)). A recent study performed exome sequencing on 55 families with autosomal recessive HSP, but lacked a genetic diagnosis (Novarino et al., 2014). They identified 18 unknown HSP genes, and also performing pathway analysis using protein interaction databases to show that HSP genes are more highly connected within the network than expected by chance.
There was also a significant overlap between HSP genes and those implicated in Alzheimer’s disease, ALS, and Parkinson’s disease. Interestingly, we have observed the axonal transporter defects in spinal muscular atrophy (SMA) iPSC-derived motor neurons (Xu et al., 2016). Spinal muscular atrophy (SMA), the leading genetic cause of death in infants and toddlers, is caused by homologous deletion or mutations of the survival of motor neuron 1 (SMN1) gene (Lefebvre et al., 1995). Spinal motor neurons are specifically degenerated in SMA, yet the underlying mechanisms are not known. Using human pluripotent stem cell-based SMA cell models, we observed early mitochondrial defects in spinal motor neurons (Wang et al., 2013b, Xu et al., 2016), which is implicated in the specific degeneration of spinal motor neurons in SMA. Interestingly, mitochondrial dysfunction has also been as a major pathological process in many other neurodegenerative diseases, such as ALS, Parkinson’s disease, and Huntington’s disease (Chen and Chan, 2009, Magrane et al., 2014). Together, these findings suggest that common pathways are disrupted in different neurodegenerative disorders, yet different cell types are affected in each. This question of why particular neuronal subtypes are affected in different neurodegenerative diseases has interested researchers for decades. Perhaps in the future, instead of studying each disease separately, combining analysis through bioinformatic approaches of multiple disorders at once may yield new insights.
Developing therapeutics for rescuing axonal degeneration
Axonal degeneration underlies many debilitating disorders, yet there are no effective drugs to prevent, stop, or reverse the axonal degeneration. This is due, at least partially, to the lack of human neuronal systems to screen for therapeutic drugs. It has been shown that human cells may have very different responses to drugs compared to other species, and only about 8% of candidate drugs have been shown to be clinically effective during clinical trials (Kola and Landis, 2004). A recent study utilized both mouse spinal motor neurons and human stem cell-derived spinal motor neurons to screen for small molecules that could improve the motor neuron survival (Yang et al., 2013). This study showed that mouse and human motor neurons responded differentially to certain drugs, indicating the importance of human neurons for testing therapeutic agents. Using iPSC-models of HSP, my group was able to show that low doses of the microtubule-targeting drug vinblastine rescues the axonal swelling phenotype in SPG4 cells and the neurite outgrowth phenotype in SPG3A.
While the studies presented here took advantage of stem cell-derived neurons to test for therapeutic compounds, it was on a very small scale. In the future, these systems can be used to screen many more compounds to identify those with the greatest effect and lowest toxicity. To accomplish this, high content screening systems can be employed, such as the PerkinElmer Opera automated confocal microscope. For the impaired axonal outgrowth in HSP, the measurement of neurite outgrowth parameters can be automatically quantified using existing software (Wang et al., 2010). The automated identification of axonal swellings is a little more difficult because no software is currently available to do so. Future development of methods to quantify axonal swellings would allow large-scale screening of chemical libraries for compounds that can reduce the number of swellings, while not affecting neuron survival or neurite outgrowth. It would also be valuable to develop reporters (e.g. reporters of factors that are specifically enriched in axonal swellings) will serve as for accurately measuring axonal defects in a high-throughput setting. The successful establishment of these methods and reporters would enable high-throughput/high-content drug screenings to identify potential therapeutic agents for HSP.
Future perspectives
HSP comprises a genetically and clinically diverse group of inherited disorders characterized by the dying back axonopathy of cortical motor neurons. How mutations of diverged HSP genes result in axonal degeneration of cortical motor neurons remains largely unclear. Establishing patient-specific hPSC-based HSP models allows the investigation of this important question in patient-derived neurons directly. Future challenges for modeling HSP include the availability of patients’ fibroblast cells, generation of accurate controls (e.g. isogenic controls), cellular heterogeneity of differentiated cells, and recapitulation of circuitry defects in cultures. Successful establishment of accurate models of various forms of HSP in the future will enable the identification of common pathogenic mechanisms that underlie axonal degeneration in multiple forms of HSP. Furthermore, these patient-specific neurons provide a unique source for screening compound library to identify therapeutic agents for rescuing axonal defects in HSP, as well as other devastating diseases involving axonopathy.
Supplementary Material
Acknowledgments
This work has been supported by the Blazer Foundation and a NIH grant (R21NS089042) to X.J.L.
Footnotes
Competing Interests: The authors declare no competing financial interest.
References
- Ben-David U, Kopper O, Benvenisty N. Expanding the boundaries of embryonic stem cells. Cell Stem Cell. 2012;10:666–677. doi: 10.1016/j.stem.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Bilican B, Serio A, Barmada SJ, Nishimura AL, Sullivan GJ, Carrasco M, Phatnani HP, Puddifoot CA, Story D, Fletcher J, Park IH, Friedman BA, Daley GQ, Wyllie DJ, Hardingham GE, Wilmut I, Finkbeiner S, Maniatis T, Shaw CE, Chandran S. Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc Natl Acad Sci U S A. 2012;109:5803–5808. doi: 10.1073/pnas.1202922109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstone C. Cellular pathways of hereditary spastic paraplegia. Annu Rev Neurosci. 2012;35:25–47. doi: 10.1146/annurev-neuro-062111-150400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackstone C, O’Kane CJ, Reid E. Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat Rev Neurosci. 2011;12:31–42. doi: 10.1038/nrn2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulting GL, Kiskinis E, Croft GF, Amoroso MW, Oakley DH, Wainger BJ, Williams DJ, Kahler DJ, Yamaki M, Davidow L, Rodolfa CT, Dimos JT, Mikkilineni S, MacDermott AB, Woolf CJ, Henderson CE, Wichterle H, Eggan K. A functionally characterized test set of human induced pluripotent stem cells. Nat Biotechnol. 29:279–286. doi: 10.1038/nbt.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chan DC. Mitochondrial dynamics–fusion, fission, movement, and mitophagy–in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–176. doi: 10.1093/hmg/ddp326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudiani P, Riano E, Errico A, Andolfi G, Rugarli EI. Spastin subcellular localization is regulated through usage of different translation start sites and active export from the nucleus. Exp Cell Res. 2005;309:358–369. doi: 10.1016/j.yexcr.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Crosby AH, Proukakis C. Is the transportation highway the right road for hereditary spastic paraplegia? Am J Hum Genet. 2002;71:1009–1016. doi: 10.1086/344206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vos KJ, Grierson AJ, Ackerley S, Miller CC. Role of axonal transport in neurodegenerative diseases. Annual review of neuroscience. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- Deluca GC, Ebers GC, Esiri MM. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol Appl Neurobiol. 2004;30:576–584. doi: 10.1111/j.1365-2990.2004.00587.x. [DOI] [PubMed] [Google Scholar]
- Denton KR, Lei L, Grenier J, Rodionov V, Blackstone C, Li XJ. Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells. 2014;32:414–423. doi: 10.1002/stem.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321:1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- Ebert AD, Yu J, Rose FF, Jr, Mattis VB, Lorson CL, Thomson JA, Svendsen CN. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457:277–280. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Errico A, Ballabio A, Rugarli EI. Spastin, the protein mutated in autosomal dominant hereditary spastic paraplegia, is involved in microtubule dynamics. Hum Mol Genet. 2002;11:153–163. doi: 10.1093/hmg/11.2.153. [DOI] [PubMed] [Google Scholar]
- Evans MJ, Kaufman MH. Establishment in culture of pluripotential cells from mouse embryos. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- Falk J, Rohde M, Bekhite MM, Neugebauer S, Hemmerich P, Kiehntopf M, Deufel T, Hubner CA, Beetz C. Functional mutation analysis provides evidence for a role of REEP1 in lipid droplet biology. Hum Mutat. 2014;35:497–504. doi: 10.1002/humu.22521. [DOI] [PubMed] [Google Scholar]
- Fan Y, Wali G, Sutharsan R, Bellette B, Crane DI, Sue CM, Mackay-Sim A. Low dose tubulin-binding drugs rescue peroxisome trafficking deficit in patient-derived stem cells in Hereditary Spastic Paraplegia. Biology open. 2014;3:494–502. doi: 10.1242/bio.20147641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassier C, Hutt JA, Scholpp S, Lumsden A, Giros B, Nothias F, Schneider-Maunoury S, Houart C, Hazan J. Zebrafish atlastin controls motility and spinal motor axon architecture via inhibition of the BMP pathway. Nat Neurosci. 2010;13:1380–1387. doi: 10.1038/nn.2662. [DOI] [PubMed] [Google Scholar]
- Fink JK. Hereditary Spastic Paraplegia Overview 1993 [Google Scholar]
- Fink JK. Advances in the hereditary spastic paraplegias. Exp Neurol. 2003;184(Suppl 1):S106–110. doi: 10.1016/j.expneurol.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Fink JK. Hereditary spastic paraplegia. Curr Neurol Neurosci Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- Fonknechten N, Mavel D, Byrne P, Davoine CS, Cruaud C, Bonsch D, Samson D, Coutinho P, Hutchinson M, McMonagle P, Burgunder JM, Tartaglione A, Heinzlef O, Feki I, Deufel T, Parfrey N, Brice A, Fontaine B, Prud’homme JF, Weissenbach J, Durr A, Hazan J. Spectrum of SPG4 mutations in autosomal dominant spastic paraplegia. Hum Mol Genet. 2000;9:637–644. doi: 10.1093/hmg/9.4.637. [DOI] [PubMed] [Google Scholar]
- Grove EA, Fukuchi-Shimogori T. Generating the cerebral cortical area map. Annu Rev Neurosci. 2003;26:355–380. doi: 10.1146/annurev.neuro.26.041002.131137. [DOI] [PubMed] [Google Scholar]
- Guha P, Morgan JW, Mostoslavsky G, Rodrigues NP, Boyd AS. Lack of immune response to differentiated cells derived from syngeneic induced pluripotent stem cells. Cell Stem Cell. 2013;12:407–412. doi: 10.1016/j.stem.2013.01.006. [DOI] [PubMed] [Google Scholar]
- Guidubaldi A, Piano C, Santorelli FM, Silvestri G, Petracca M, Tessa A, Bentivoglio AR. Novel mutations in SPG11 cause hereditary spastic paraplegia associated with early-onset levodopa-responsive Parkinsonism. Mov Disord. 2011;26:553–556. doi: 10.1002/mds.23552. [DOI] [PubMed] [Google Scholar]
- Hallett PJ, Deleidi M, Astradsson A, Smith GA, Cooper O, Osborn TM, Sundberg M, Moore MA, Perez-Torres E, Brownell AL, Schumacher JM, Spealman RD, Isacson O. Successful Function of Autologous iPSC-Derived Dopamine Neurons following Transplantation in a Non-Human Primate Model of Parkinson’s Disease. Cell Stem Cell. 2015;16:269–274. doi: 10.1016/j.stem.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell B. Cell culture, oxidative stress, and antioxidants: avoiding pitfalls. Biomedical journal. 2014;37:99–105. doi: 10.4103/2319-4170.128725. [DOI] [PubMed] [Google Scholar]
- Hanein S, Martin E, Boukhris A, Byrne P, Goizet C, Hamri A, Benomar A, Lossos A, Denora P, Fernandez J, Elleuch N, Forlani S, Durr A, Feki I, Hutchinson M, Santorelli FM, Mhiri C, Brice A, Stevanin G. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am J Hum Genet. 2008;82:992–1002. doi: 10.1016/j.ajhg.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding AE. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. doi: 10.1016/s0140-6736(83)92879-9. [DOI] [PubMed] [Google Scholar]
- Harding AE. Hereditary spastic paraplegias. Semin Neurol. 1993;13:333–336. doi: 10.1055/s-2008-1041143. [DOI] [PubMed] [Google Scholar]
- Havlicek S, Kohl Z, Mishra HK, Prots I, Eberhardt E, Denguir N, Wend H, Plotz S, Boyer L, Marchetto MC, Aigner S, Sticht H, Groemer TW, Hehr U, Lampert A, Schlotzer-Schrehardt U, Winkler J, Gage FH, Winner B. Gene dosage-dependent rescue of HSP neurite defects in SPG4 patients’ neurons. Hum Mol Genet. 2014;23:2527–2541. doi: 10.1093/hmg/ddt644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazan J, Fonknechten N, Mavel D, Paternotte C, Samson D, Artiguenave F, Davoine CS, Cruaud C, Durr A, Wincker P, Brottier P, Cattolico L, Barbe V, Burgunder JM, Prud’homme JF, Brice A, Fontaine B, Heilig B, Weissenbach J. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat Genet. 1999;23:296–303. doi: 10.1038/15472. [DOI] [PubMed] [Google Scholar]
- Hedera P, Eldevik OP, Maly P, Rainier S, Fink JK. Spinal cord magnetic resonance imaging in autosomal dominant hereditary spastic paraplegia. Neuroradiology. 2005;47:730–734. doi: 10.1007/s00234-005-1415-3. [DOI] [PubMed] [Google Scholar]
- Hirst J, Borner GH, Edgar J, Hein MY, Mann M, Buchholz F, Antrobus R, Robinson MS. Interaction between AP-5 and the hereditary spastic paraplegia proteins SPG11 and SPG15. Mol Biol Cell. 2013;24:2558–2569. doi: 10.1091/mbc.E13-03-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP, Cost GJ, Zhang L, Santiago Y, Miller JC, Zeitler B, Cherone JM, Meng X, Hinkley SJ, Rebar EJ, Gregory PD, Urnov FD, Jaenisch R. Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol. 2009;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ. Mitochondria and neurotransmission: evacuating the synapse. Neuron. 2005;47:331–333. doi: 10.1016/j.neuron.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Shibata Y, Zhu PP, Voss C, Rismanchi N, Prinz WA, Rapoport TA, Blackstone C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–561. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanekura K, Suzuki H, Aiso S, Matsuoka M. ER stress and unfolded protein response in amyotrophic lateral sclerosis. Molecular neurobiology. 2009;39:81–89. doi: 10.1007/s12035-009-8054-3. [DOI] [PubMed] [Google Scholar]
- Kasher PR, De Vos KJ, Wharton SB, Manser C, Bennett EJ, Bingley M, Wood JD, Milner R, McDermott CJ, Miller CC, Shaw PJ, Grierson AJ. Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J Neurochem. 2009;110:34–44. doi: 10.1111/j.1471-4159.2009.06104.x. [DOI] [PubMed] [Google Scholar]
- Kiskinis E, Eggan K. Progress toward the clinical application of patient-specific pluripotent stem cells. J Clin Invest. 2010;120:51–59. doi: 10.1172/JCI40553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiskinis E, Sandoe J, Williams LA, Boulting GL, Moccia R, Wainger BJ, Han S, Peng T, Thams S, Mikkilineni S, Mellin C, Merkle FT, Davis-Dusenbery BN, Ziller M, Oakley D, Ichida J, Di Costanzo S, Atwater N, Maeder ML, Goodwin MJ, Nemesh J, Handsaker RE, Paull D, Noggle S, McCarroll SA, Joung JK, Woolf CJ, Brown RH, Eggan K. Pathways disrupted in human ALS motor neurons identified through genetic correction of mutant SOD1. Cell Stem Cell. 2014;14:781–795. doi: 10.1016/j.stem.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm RW, Norton JP, Cole RA, Li CS, Park SH, Crane MM, Li L, Jin D, Boye-Doe A, Liu TY, Shibata Y, Lu H, Rapoport TA, Farese RV, Jr, Blackstone C, Guo Y, Mak HY. A conserved role for atlastin GTPases in regulating lipid droplet size. Cell Rep. 2013;3:1465–1475. doi: 10.1016/j.celrep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott AB, Perkins G, Schwarzenbacher R, Bossy-Wetzel E. Mitochondrial fragmentation in neurodegeneration. Nat Rev Neurosci. 2008;9:505–518. doi: 10.1038/nrn2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kola I, Landis J. Can the pharmaceutical industry reduce attrition rates? Nat Rev Drug Discov. 2004;3:711–715. doi: 10.1038/nrd1470. [DOI] [PubMed] [Google Scholar]
- Kondo T, Asai M, Tsukita K, Kutoku Y, Ohsawa Y, Sunada Y, Imamura K, Egawa N, Yahata N, Okita K, Takahashi K, Asaka I, Aoi T, Watanabe A, Watanabe K, Kadoya C, Nakano R, Watanabe D, Maruyama K, Hori O, Hibino S, Choshi T, Nakahata T, Hioki H, Kaneko T, Naitoh M, Yoshikawa K, Yamawaki S, Suzuki S, Hata R, Ueno S, Seki T, Kobayashi K, Toda T, Murakami K, Irie K, Klein WL, Mori H, Asada T, Takahashi R, Iwata N, Yamanaka S, Inoue H. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell. 2013;12:487–496. doi: 10.1016/j.stem.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Lee H, Shamy GA, Elkabetz Y, Schofield CM, Harrsion NL, Panagiotakos G, Socci ND, Tabar V, Studer L. Directed differentiation and transplantation of human embryonic stem cell-derived motoneurons. Stem Cells. 2007;25:1931–1939. doi: 10.1634/stemcells.2007-0097. [DOI] [PubMed] [Google Scholar]
- Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Li XJ, Du ZW, Zarnowska ED, Pankratz M, Hansen LO, Pearce RA, Zhang SC. Specification of motoneurons from human embryonic stem cells. Nat Biotechnol. 2005;23:215–221. doi: 10.1038/nbt1063. [DOI] [PubMed] [Google Scholar]
- Lindsey JC, Lusher ME, McDermott CJ, White KD, Reid E, Rubinsztein DC, Bashir R, Hazan J, Shaw PJ, Bushby KM. Mutation analysis of the spastin gene (SPG4) in patients with hereditary spastic paraparesis. J Med Genet. 2000;37:759–765. doi: 10.1136/jmg.37.10.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling SC, Polymenidou M, Cleveland DW. Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron. 2013;79:416–438. doi: 10.1016/j.neuron.2013.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu GH, Qu J, Suzuki K, Nivet E, Li M, Montserrat N, Yi F, Xu X, Ruiz S, Zhang W, Wagner U, Kim A, Ren B, Li Y, Goebl A, Kim J, Soligalla RD, Dubova I, Thompson J, Yates J, 3rd, Esteban CR, Sancho-Martinez I, Izpisua Belmonte JC. Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature. 2012;491:603–607. doi: 10.1038/nature11557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumb JH, Connell JW, Allison R, Reid E. The AAA ATPase spastin links microtubule severing to membrane modelling. Biochim Biophys Acta. 2012;1823:192–197. doi: 10.1016/j.bbamcr.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Lunn MR, Wang CH. Spinal muscular atrophy. Lancet. 2008;371:2120–2133. doi: 10.1016/S0140-6736(08)60921-6. [DOI] [PubMed] [Google Scholar]
- Ly CV, Verstreken P. Mitochondria at the synapse. The Neuroscientist: a review journal bringing neurobiology, neurology and psychiatry. 2006;12:291–299. doi: 10.1177/1073858406287661. [DOI] [PubMed] [Google Scholar]
- Ma L, Hu B, Liu Y, Vermilyea SC, Liu H, Gao L, Sun Y, Zhang X, Zhang SC. Human embryonic stem cell-derived GABA neurons correct locomotion deficits in quinolinic acid-lesioned mice. Cell Stem Cell. 2012;10:455–464. doi: 10.1016/j.stem.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet. 2014;23:1413–1424. doi: 10.1093/hmg/ddt528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso G, Rugarli EI. A cryptic promoter in the first exon of the SPG4 gene directs the synthesis of the 60-kDa spastin isoform. BMC Biol. 2008;6:31. doi: 10.1186/1741-7007-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi G, Xu Z. Mitochondrial dysfunction and its role in motor neuron degeneration in ALS. Mitochondrion. 2005;5:77–87. doi: 10.1016/j.mito.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci U S A. 1981;78:7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, Dulay GP, Hua KL, Ankoudinova I, Cost GJ, Urnov FD, Zhang HS, Holmes MC, Zhang L, Gregory PD, Rebar EJ. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, Mandal PK, Vera E, Shim JW, Kriks S, Taldone T, Fusaki N, Tomishima MJ, Krainc D, Milner TA, Rossi DJ, Studer L. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell. 2013;13:691–705. doi: 10.1016/j.stem.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra HK, Prots I, Havlicek S, Kohl Z, Perez-Branguli F, Boerstler T, Anneser L, Minakaki G, Wend H, Hampl M, Leone M, Bruckner M, Klucken J, Reis A, Boyer L, Schuierer G, Behrens J, Lampert A, Engel FB, Gage FH, Winkler J, Winner B. GSK3ss-dependent dysregulation of neurodevelopment in SPG11-patient iPSC model. Ann Neurol. 2016 doi: 10.1002/ana.24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montague K, Malik B, Gray AL, La Spada AR, Hanna MG, Szabadkai G, Greensmith L. Endoplasmic reticulum stress in spinal and bulbar muscular atrophy: a potential target for therapy. Brain. 2014;137:1894–1906. doi: 10.1093/brain/awu114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montenegro G, Rebelo AP, Connell J, Allison R, Babalini C, D’Aloia M, Montieri P, Schule R, Ishiura H, Price J, Strickland A, Gonzalez MA, Baumbach-Reardon L, Deconinck T, Huang J, Bernardi G, Vance JM, Rogers MT, Tsuji S, De Jonghe P, Pericak-Vance MA, Schols L, Orlacchio A, Reid E, Zuchner S. Mutations in the ER-shaping protein reticulon 2 cause the axon-degenerative disorder hereditary spastic paraplegia type 12. J Clin Invest. 2012;122:538–544. doi: 10.1172/JCI60560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss TJ, Daga A, McNew JA. Fusing a lasting relationship between ER tubules. Trends Cell Biol. 2011;21:416–423. doi: 10.1016/j.tcb.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murmu RP, Martin E, Rastetter A, Esteves T, Muriel MP, El Hachimi KH, Denora PS, Dauphin A, Fernandez JC, Duyckaerts C, Brice A, Darios F, Stevanin G. Cellular distribution and subcellular localization of spatacsin and spastizin, two proteins involved in hereditary spastic paraplegia. Mol Cell Neurosci. 2011;47:191–202. doi: 10.1016/j.mcn.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Murry CE, Keller G. Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell. 2008;132:661–680. doi: 10.1016/j.cell.2008.02.008. [DOI] [PubMed] [Google Scholar]
- Nadar VC, Ketschek A, Myers KA, Gallo G, Baas PW. Kinesin-5 is essential for growth-cone turning. Curr Biol. 2008;18:1972–1977. doi: 10.1016/j.cub.2008.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekawa M, Ribai P, Nelson I, Forlani S, Fellmann F, Goizet C, Depienne C, Stevanin G, Ruberg M, Durr A, Brice A. SPG3A is the most frequent cause of hereditary spastic paraplegia with onset before age 10 years. Neurology. 2006;66:112–114. doi: 10.1212/01.wnl.0000191390.20564.8e. [DOI] [PubMed] [Google Scholar]
- Nguyen HN, Byers B, Cord B, Shcheglovitov A, Byrne J, Gujar P, Kee K, Schule B, Dolmetsch RE, Langston W, Palmer TD, Pera RR. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 2011;8:267–280. doi: 10.1016/j.stem.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu J, Zhang B, Chen H. Applications of TALENs and CRISPR/Cas9 in Human Cells and Their Potentials for Gene Therapy. Mol Biotechnol. 2014 doi: 10.1007/s12033-014-9771-z. [DOI] [PubMed] [Google Scholar]
- Novarino G, Fenstermaker AG, Zaki MS, Hofree M, Silhavy JL, Heiberg AD, Abdellateef M, Rosti B, Scott E, Mansour L, Masri A, Kayserili H, Al-Aama JY, Abdel-Salam GM, Karminejad A, Kara M, Kara B, Bozorgmehri B, Ben-Omran T, Mojahedi F, Mahmoud IG, Bouslam N, Bouhouche A, Benomar A, Hanein S, Raymond L, Forlani S, Mascaro M, Selim L, Shehata N, Al-Allawi N, Bindu PS, Azam M, Gunel M, Caglayan A, Bilguvar K, Tolun A, Issa MY, Schroth J, Spencer EG, Rosti RO, Akizu N, Vaux KK, Johansen A, Koh AA, Megahed H, Durr A, Brice A, Stevanin G, Gabriel SB, Ideker T, Gleeson JG. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014;343:506–511. doi: 10.1126/science.1247363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary DD, Nakagawa Y. Patterning centers, regulatory genes and extrinsic mechanisms controlling arealization of the neocortex. Curr Opin Neurobiol. 2002;12:14–25. doi: 10.1016/s0959-4388(02)00285-4. [DOI] [PubMed] [Google Scholar]
- Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313–317. doi: 10.1038/nature05934. [DOI] [PubMed] [Google Scholar]
- Pantakani DV, Swapna LS, Srinivasan N, Mannan AU. Spastin oligomerizes into a hexamer and the mutant spastin (E442Q) redistribute the wild-type spastin into filamentous microtubule. J Neurochem. 2008;106:613–624. doi: 10.1111/j.1471-4159.2008.05414.x. [DOI] [PubMed] [Google Scholar]
- Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. Cell. 2008;134:877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Lee KS, Lee YJ, Shin HA, Cho HY, Wang KC, Kim YS, Lee HT, Chung KS, Kim EY, Lim J. Generation of dopaminergic neurons in vitro from human embryonic stem cells treated with neurotrophic factors. Neurosci Lett. 2004;359:99–103. doi: 10.1016/j.neulet.2004.01.073. [DOI] [PubMed] [Google Scholar]
- Park SH, Zhu PP, Parker RL, Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J Clin Invest. 2010;120:1097–1110. doi: 10.1172/JCI40979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Branguli F, Mishra HK, Prots I, Havlicek S, Kohl Z, Saul D, Rummel C, Dorca-Arevalo J, Regensburger M, Graef D, Sock E, Blasi J, Groemer TW, Schlotzer-Schrehardt U, Winkler J, Winner B. Dysfunction of spatacsin leads to axonal pathology in SPG11-linked hereditary spastic paraplegia. Hum Mol Genet. 2014;23:4859–4874. doi: 10.1093/hmg/ddu200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrier AL, Tabar V, Barberi T, Rubio ME, Bruses J, Topf N, Harrison NL, Studer L. Derivation of midbrain dopamine neurons from human embryonic stem cells. Proc Natl Acad Sci U S A. 2004;101:12543–12548. doi: 10.1073/pnas.0404700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piaceri I, Rinnoci V, Bagnoli S, Failli Y, Sorbi S. Mitochondria and Alzheimer’s disease. Journal of the neurological sciences. 2012;322:31–34. doi: 10.1016/j.jns.2012.05.033. [DOI] [PubMed] [Google Scholar]
- Polleux F, Dehay C, Goffinet A, Kennedy H. Pre- and post-mitotic events contribute to the progressive acquisition of area-specific connectional fate in the neocortex. Cereb Cortex. 2001;11:1027–1039. doi: 10.1093/cercor/11.11.1027. [DOI] [PubMed] [Google Scholar]
- Reid E. Science in motion: common molecular pathological themes emerge in the hereditary spastic paraplegias. J Med Genet. 2003;40:81–86. doi: 10.1136/jmg.40.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renvoise B, Blackstone C. Emerging themes of ER organization in the development and maintenance of axons. Curr Opin Neurobiol. 2010;20:531–537. doi: 10.1016/j.conb.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reubinoff BE, Itsykson P, Turetsky T, Pera MF, Reinhartz E, Itzik A, Ben-Hur T. Neural progenitors from human embryonic stem cells. Nat Biotechnol. 2001;19:1134–1140. doi: 10.1038/nbt1201-1134. [DOI] [PubMed] [Google Scholar]
- Roy NS, Cleren C, Singh SK, Yang L, Beal MF, Goldman SA. Functional engraftment of human ES cell-derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat Med. 2006;12:1259–1268. doi: 10.1038/nm1495. [DOI] [PubMed] [Google Scholar]
- Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. doi: 10.1016/S1474-4422(08)70258-8. [DOI] [PubMed] [Google Scholar]
- Schicks J, Synofzik M, Petursson H, Huttenlocher J, Reimold M, Schols L, Bauer P. Atypical juvenile parkinsonism in a consanguineous SPG15 family. Mov Disord. 2011;26:564–566. doi: 10.1002/mds.23472. [DOI] [PubMed] [Google Scholar]
- Singh Roy N, Nakano T, Xuing L, Kang J, Nedergaard M, Goldman SA. Enhancer-specified GFP-based FACS purification of human spinal motor neurons from embryonic stem cells. Exp Neurol. 2005;196:224–234. doi: 10.1016/j.expneurol.2005.06.021. [DOI] [PubMed] [Google Scholar]
- Soderblom C, Blackstone C. Traffic accidents: molecular genetic insights into the pathogenesis of the hereditary spastic paraplegias. Pharmacol Ther. 2006;109:42–56. doi: 10.1016/j.pharmthera.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Solowska JM, Morfini G, Falnikar A, Himes BT, Brady ST, Huang D, Baas PW. Quantitative and functional analyses of spastin in the nervous system: implications for hereditary spastic paraplegia. J Neurosci. 2008;28:2147–2157. doi: 10.1523/JNEUROSCI.3159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevanin G, Santorelli FM, Azzedine H, Coutinho P, Chomilier J, Denora PS, Martin E, Ouvrard-Hernandez AM, Tessa A, Bouslam N, Lossos A, Charles P, Loureiro JL, Elleuch N, Confavreux C, Cruz VT, Ruberg M, Leguern E, Grid D, Tazir M, Fontaine B, Filla A, Bertini E, Durr A, Brice A. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet. 2007;39:366–372. doi: 10.1038/ng1980. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tarrade A, Fassier C, Courageot S, Charvin D, Vitte J, Peris L, Thorel A, Mouisel E, Fonknechten N, Roblot N, Seilhean D, Dierich A, Hauw JJ, Melki J. A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum Mol Genet. 2006;15:3544–3558. doi: 10.1093/hmg/ddl431. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Vidal R, Caballero B, Couve A, Hetz C. Converging pathways in the occurrence of endoplasmic reticulum (ER) stress in Huntington’s disease. Curr Mol Med. 2011;11:1–12. doi: 10.2174/156652411794474419. [DOI] [PubMed] [Google Scholar]
- Walther TC, Farese RV., Jr Lipid droplets and cellular lipid metabolism. Annual review of biochemistry. 2012;81:687–714. doi: 10.1146/annurev-biochem-061009-102430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Lagerstrom R, Sun C, Bishof L, Valotton P, Gotte M. HCA-vision: Automated neurite outgrowth analysis. Journal of biomolecular screening. 2010;15:1165–1170. doi: 10.1177/1087057110382894. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013a;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZB, Zhang X, Li XJ. Recapitulation of spinal motor neuron-specific disease phenotypes in a human cell model of spinal muscular atrophy. Cell Res. 2013b;23:378–393. doi: 10.1038/cr.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilfling F, Wang H, Haas JT, Krahmer N, Gould TJ, Uchida A, Cheng JX, Graham M, Christiano R, Frohlich F, Liu X, Buhman KK, Coleman RA, Bewersdorf J, Farese RV, Jr, Walther TC. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Developmental cell. 2013;24:384–399. doi: 10.1016/j.devcel.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu CC, Denton KR, Wang ZB, Zhang X, Li XJ. Abnormal mitochondrial transport and morphology as early pathological changes in human models of spinal muscular atrophy. Dis Model Mech. 2016;9:39–49. doi: 10.1242/dmm.021766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, Yamanaka S, Okano H, Suzuki N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20:4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- Yan Y, Yang D, Zarnowska ED, Du Z, Werbel B, Valliere C, Pearce RA, Thomson JA, Zhang SC. Directed differentiation of dopaminergic neuronal subtypes from human embryonic stem cells. Stem Cells. 2005;23:781–790. doi: 10.1634/stemcells.2004-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YM, Gupta SK, Kim KJ, Powers BE, Cerqueira A, Wainger BJ, Ngo HD, Rosowski KA, Schein PA, Ackeifi CA, Arvanites AC, Davidow LS, Woolf CJ, Rubin LL. A small molecule screen in stem-cell-derived motor neurons identifies a kinase inhibitor as a candidate therapeutic for ALS. Cell Stem Cell. 2013;12:713–726. doi: 10.1016/j.stem.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, Slukvin II, Thomson JA. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Zeng H, Guo M, Martins-Taylor K, Wang X, Zhang Z, Park JW, Zhan S, Kronenberg MS, Lichtler A, Liu HX, Chen FP, Yue L, Li XJ, Xu RH. Specification of region-specific neurons including forebrain glutamatergic neurons from human induced pluripotent stem cells. PLoS One. 2010;5:e11853. doi: 10.1371/journal.pone.0011853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, An MC, Montoro D, Ellerby LM. Characterization of Human Huntington’s Disease Cell Model from Induced Pluripotent Stem Cells. PLoS Curr. 2010;2:RRN1193. doi: 10.1371/currents.RRN1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SC. Neural subtype specification from embryonic stem cells. Brain Pathol. 2006;16:132–142. doi: 10.1111/j.1750-3639.2006.00008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SC, Wernig M, Duncan ID, Brustle O, Thomson JA. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol. 2001;19:1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- Zhao X, Alvarado D, Rainier S, Lemons R, Hedera P, Weber CH, Tukel T, Apak M, Heiman-Patterson T, Ming L, Bui M, Fink JK. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat Genet. 2001;29:326–331. doi: 10.1038/ng758. [DOI] [PubMed] [Google Scholar]
- Zhu PP, Denton KR, Pierson TM, Li XJ, Blackstone C. Pharmacologic rescue of axon growth defects in a human iPSC model of hereditary spastic paraplegia SPG3A. Hum Mol Genet. 2014;23:5638–5648. doi: 10.1093/hmg/ddu280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PP, Patterson A, Lavoie B, Stadler J, Shoeb M, Patel R, Blackstone C. Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J Biol Chem. 2003;278:49063–49071. doi: 10.1074/jbc.M306702200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.