

Abstract
Homotetrameric R67 dihydrofolate reductase possesses 222 symmetry and a single active site pore. This situation results in a promiscuous binding site that accommodates either the substrate, dihydrofolate (DHF), or the cofactor, NADPH. NADPH interacts more directly with the protein as it is larger than the substrate. In contrast, the p-aminobenzoyl-glutamate tail of DHF, as monitored by nuclear magnetic resonance and crystallography, is disordered when bound. To explore whether smaller active site volumes (which should decrease the level of tail disorder by confinement effects) alter steady state rates, asymmetric mutations that decreased the half-pore volume by ∼35% were constructed. Only minor effects on kcat were observed. To continue exploring the role of tail disorder in catalysis, 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide-mediated cross-linking between R67 DHFR and folate was performed. A two-folate, one-tetramer complex results in the loss of enzyme activity where two symmetry-related K32 residues in the protein are cross-linked to the carboxylates of two bound folates. The tethered folate could be reduced, although with a ≤30-fold decreased rate, suggesting decreased dynamics and/or suboptimal positioning of the cross-linked folate for catalysis. Computer simulations that restrain the dihydrofolate tail near K32 indicate that cross-linking still allows movement of the p-aminobenzoyl ring, which allows the reaction to occur. Finally, a bis-ethylene-diamine-α,γ-amide folate adduct was synthesized; both negatively charged carboxylates in the glutamate tail were replaced with positively charged amines. The Ki for this adduct was ∼9-fold higher than for folate. These various results indicate a balance between folate tail disorder, which helps the enzyme bind substrate while dynamics facilitates catalysis.
R67 dihydrofolate reductase (DHFR) catalyzes the NADPH-dependent reduction of dihydrofolate (DHF) to tetrahydrofolate (THF). R67 has been proposed to be a primitive enzyme, and it shares no sequence or structural homology with chromosomal DHFR.1Figure 1 shows R67 DHFR is a homotetramer possessing 222 symmetry as well as a single active site pore.2 The symmetry imposed on the single active site results in overlapping binding sites for substrate, DHF, and cofactor, NADPH. The promiscuous surface, coupled with a narrow central constriction, limits ligand binding by R67 DHFR to a total of two ligands: either two NADPH molecules (Kd1 = 2.5 μM, and with negative cooperativity, Kd2 = 95 μM) or two folate/DHF molecules (for folate, Kd1 = 195 μM, and with positive cooperativity, Kd2 = 48 μM) or one NADPH and one folate/DHF molecule.3 The first two complexes are dead-end complexes, while the third is the productive catalytic complex. Because of the symmetry, this generalized binding surface is not optimized for binding of either ligand.
Figure 1.
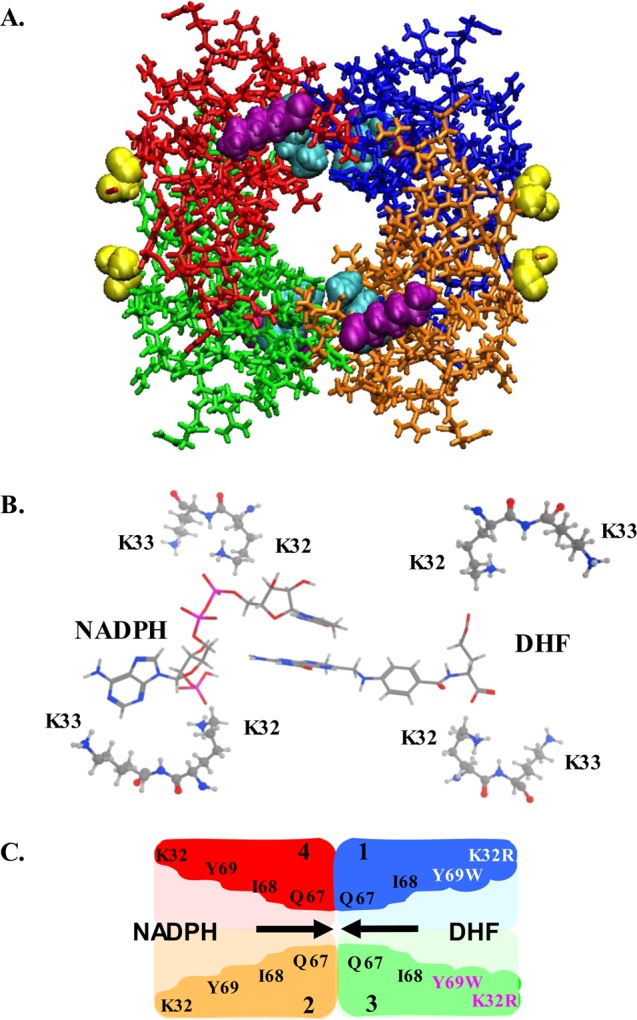
Structure of apo R67 DHFR (PDB entry 2RH2).5 Each different color corresponds to a different monomer. Symmetry-related K32 residues are colored cyan; K33 residues are colored purple, and the first ordered amino acid at the chymotrypsin-truncated N-terminus (residue 22) is colored yellow. The active site pore appears in the middle of the structure. Panel B shows a slice through the active site indicating the relative positions of K32 and K33 with respect to bound DHF and NADPH in the ternary complex structure.5 This image is related to panel A by a 90° rotation around the y-axis. The pABA-glu tail of DHF was modeled as described by Kamath et al.6 The pterin ring of the substrate and the nicotinamide ring of the cofactor stack at the center of the pore. The atoms are colored as follows: white for hydrogen, gray for carbon, red for oxygen, magenta for phosphate, and blue for nitrogen. The cartoon in panel C also depicts a slice through the central pore of R67 DHFR, with the same orientation as panel B and a color code as in panel A. The residues lining the active site pore are labeled as is each monomer (1–4). The pore is depicted by the lighter color. In Quad3, the predicted positions of the mutations are shown. For example, the K32R:1+3 mutations (labeled in white and magenta) lie in the same half-pore (right side) while the Y69W:1+3 mutations (white and magenta) occur further toward the center of the pore. The I68A:3 mutation would occur in monomer 3 (not labeled). NADPH is shown entering the wild-type side of the pore (left), forcing DHF to enter from the right. Note that addition of S59A and H362L mutations locks Quad3 into a single topology (not shown).17
In the R67 DHFR·2 folate structure (PDB entry 1VIF), electron density is observed for the pteridine rings, but not for the p-aminobenzoyl-glutamate (pABA-glu) tail.2 In an NMR study, positive NOEs were seen for the pABA-glu tail of folate bound to R67 DHFR, consistent with disorder.4 No electron density for the pABA-glu tail was seen in a crystal structure of DHF bound in a ternary complex structure with NADP+, again indicating the tail of the bound substrate is disordered.5 Because the distance between the two carboxylates of folate/DHF is not long enough to span the width of the active site pore, there is likely an ensemble of states that participate in direct ionic interactions as well as solvent-separated ion pairs.
Recent molecular dynamics simulations predict the pABA-glu tail translates back and forth between symmetry-related K32 residuesa as well as flips the α- and γ-carboxylate positions.6 This study predicted that movement of the pABA-glu tail, coupled with clamping of the pteridine ring by interactions at the central constriction of the pore, could result in puckering of the pteridine ring, ultimately leading to transition state formation.6 A positive correlation between the radius of gyration of the DHF tail and kcat of wild-type (wt) R67 DHFR and several mutants suggested greater tail movement could lead to a faster catalytic rate. Protein dynamics is also known to impact the mechanism of chromosomal DHFR, where the puckering of the pteridine ring is induced by a conserved phenylalanine interacting with the pABA ring.7 Additionally, the substrate carboxylate groups interact with conserved regions in chromosomal DHFRs, where changes have been shown to have important evolutionary consequences.8
Recent reviews of intrinsically disordered proteins (IDPs) have suggested functional roles for disorder.9,10 Of particular interest is the apparent difficulty in reconciling molecular recognition with disorder in complex formation. Mittag et al. suggest complexes can display a continuum of disorder and that polyelectrostatic interactions can play a role in binding.11 In the polyelectrostatic model, it is the cumulative electrostatic environment that is important, allowing “fuzzy” complex12 formation. For the case of R67 DHFR, it is the ligand tail that possesses disorder, a notion usually considered as antagonistic to catalysis. However, it may be that binding is permitted by polyelectrostatic interactions, while a subset of motions could lead to puckering of the DHF pteridine ring, contributing to catalysis in R67 DHFR.
This study continues to examine the role of disorder associated with the DHF/folate molecule bound by R67 DHFR as well as the relationship between the K32 residue and the glutamate tail of the bound substrate. Exploration of ion pair formation involved examining the effect of 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide (EDC) cross-linking on enzyme activity. To study the effect of altering the radius of gyration on kcat, the folate tail was tethered by a cross-link to K32 and the rate of turnover monitored. In addition, the volume of the active site was decreased by site-directed mutagenesis to determine if there was any change in steady state kinetic parameters associated with an altered radius of gyration. Finally, the relationship between K32 and folate was probed by synthesis of a bis-ethylene-diamine folate-α,γ-amide adduct where the negative charges associated with the glutamate tail were removed and replaced with positive charges.
Materials and Methods
Protein Purification
High yields of R67 DHFR were obtained as previously described.13 Briefly, ammonium sulfate precipitation and ion-exchange column chromatography were used to purify the protein to homogeneity. Purified samples were dialyzed against distilled, deionized water and then lyophilized. Protein concentrations were determined with a BCA (bicinchoninic acid, Pierce Biotechnology) assay.
In some experiments, a His-tagged R67 DHFR was used. This was constructed by cloning a SacI–EcoRI fragment of the synthetic R67 DHFR gene13 into the pRSETB vector from Invitrogen.14 This results in substitution of the N-terminal MIR- sequence with MRGSHHHHHHGMASMTGGQQMGRDLYDDDDKDP-. His-tagged R67 DHFR was purified using a nickel-nitrilotriacetic acid (Ni-NTA) column (Qiagen), followed by elution from a DEAE fractogel column. His-tagged R67 DHFR is almost fully active, with only ∼2-fold increases in both Km values.15
Truncation of 16 residues from the N-terminus of native R67 DHFR (or 47 residues from the His-tagged protein) was performed by incubating the enzyme overnight at room temperature with immobilized chymotrypsin (Sigma-Aldrich) in 10 mM Tris/1 mM EDTA buffer (pH 8.0).13 A single truncated species was obtained no matter which N-terminal sequence was present. Chymotrypsin cleavage after F16 in the wt R67 DHFR or after F47 in the His-tagged construct results in the same 62-amino acid monomer that assembles into an active homotetramer.2 The extent of the reaction was monitored by sodium dodecyl sulfate (SDS) electrophoresis. Immobilized chymotrypsin was removed by an Acrodisc (0.2 μm filter). The truncated tetramer was separated from peptide fragments by gel filtration at pH 8 using G75 Sephadex. Alternatively, if the His-tagged protein was used, a Ni-NTA column allowed separation of the N-terminus from the tetrameric core of the protein. This step was followed by dialysis using a 7 kDa cutoff membrane.
Steady State Kinetics
Steady state rates were obtained using a PerkinElmer λ35 spectrophotometer interfaced with an IBM personal computer as previously described.16 For analysis of site-directed mutants in MTA polybuffer (100 mM Tris, 50 mM MES, and 50 mM acetic acid), the concentration of DHF was held constant while the concentration of NADPH was varied. This process was repeated using several additional DHF concentrations. Concentration ranges utilized were 3–87 μM DHF and 4–150 μM NADPH. The data were then globally fit to an equation describing either the bisubstrate kinetic reaction of DHFR or a bisubstrate reaction showing substrate inhibition.17,18 A nonlinear, global fit using SAS17,18 resulted in best fit values for kcat, both Km values, and a Kd2DHF value describing binding of a second DHF molecule.
DHF was produced by reduction of folate as described by Blakley.19 NADPH was obtained from Alexis Biochemicals. Concentrations of DHF and NADPH were measured using their respective extinction coefficients, 7.75 × 103 and 6.22 × 103 M–1 cm–1 at 340 nm.20 For the reaction, the extinction coefficient was 12.3 × 103 mol–1 cm–1.21
For cross-linking studies, steady state rates were measured at 30 °C by the addition of saturating concentrations of substrate (DHF) and cofactor (NADPH), followed by the addition of enzyme to initiate the reaction.
Reduction of folate was monitored using an extinction coefficient of 18400 M–1 cm–1 at 340 nm22 or 14900 M–1 cm–1 at 360 nm. Care was taken to subtract out any nonenzymatic drift rates by placing matching concentrations of cofactor and folate in the reference cuvette. Initial rates were monitored, and argon kept the solution anaerobic.
Mutagenesis
A tandem array of four R67 DHFR genes (named Quad3) was previously constructed where the genes were linked in-frame.17,23 The introduction of asymmetric mutations into this construct allows breaking of the 222 symmetry of the R67 DHFR protein. Two K32R mutations were placed in gene copies 1 and 3 (K32R:1+3).b Two Y69W mutations were placed in gene copies 1 and 3 of the K32R:1+3 double mutant; this construct was named K32R:1+3 plus Y69W:1+3. C-Terminally His-tagged versions of these genes were synthesized by GenScript and cloned into pUC57. All mutants were verified by DNA sequencing. The resulting proteins were purified using Ni-NTA and DEAE columns.
Models of the mutant structures were constructed using the R67 DHFR crystal structure (PDB entry 2RH2)5 or a model of the reactant complex.6 Asymmetric K32R and Y69W mutations were modeled by the computer program MOE (version 2009.10, Chemical Computing Group, Ltd., Montréal, QC). A range of rotamer conformations in the rotamer explore option were selected, and the active site pore volume was calculated using CASTp (Dundas et al.24 and http://sts.bioe.uic.edu/castp/calculation.php).
EDC Modification
Zero length cross-linking was performed in 50 mM Hepes buffer (pH 7.0) using 7–12 μM R67 DHFR with addition of 5 mM 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide (EDC) and either 0.07–2 mM folate or 640 μM DHF.3 NADPH (1 mM) was added to determine if it could protect the active site from modification. Inactivation rates were calculated using a fit to a single exponential with a linear rate.
The stoichiometry of EDC labeling was determined using samples inactivated to <10% remaining activity. Depending on the folate concentration and the enzyme species used, this could require an overnight incubation. The reaction was quenched by addition of ammonium chloride or Tris buffer. Unreacted folate was removed by passing the solution over a DEAE-fractogel column (Supelco, 11 cm × 1.5 cm) equilibrated in 10 mM Tris and 1 mM EDTA (pH 8.0) or an epifuge spin column loaded with small amounts of this resin. For reactions using a His-tagged R67 DHFR construct, a His Spintrap column (GE Healthcare) was used to separate the labeled protein from unreacted folate. Protein concentrations were monitored by a BCA assay, while folate concentrations were measured by absorbance at 350 nm with an extinction coefficient of 7000 mol–1 cm–1.25
Matrix-Assisted Laser Desorption Ionization Time-of-Flight (MALDI-TOF) Analyses
The molecular masses of cross-linked R67 DHFR species were determined by mass spectrometry using a Bruker MicroFlex MALDI-TOF instrument. MALDI experiments were performed in positive ion mode (delayed extraction/linear mode). A 337 nm nitrogen laser was used for all experiments, and 1000 laser shots per data acquisition round were employed. The accelerating voltage was 20 kV with a grid voltage of 96% and a delayed extraction time of 220 ns. The matrix was prepared by dissolving 10 mg of α-cyano-4-hydroxycinnamic acid in 50% acetone and 50% 2-propanol with 5 mg of added nitrocellulose.26 The matrix was spotted onto a MALDI plate and allowed to dry. Next, the protein sample was diluted 1/10 to 1/100 in 0.1% trifluoroacetic acid (w/v) and 50% methanol and spotted. A multipoint calibration was performed using protein 1 standard (Bruker).
Sedimentation Velocity
Sedimentation velocity experiments were conducted using a Beckman Optima XL-I ultracentrifuge and absorbance optics. Protein samples were dialyzed into 10 mM Tris buffer and 1 mM EDTA (pH 8.0), and the dialysate was used as the optical reference. Protein was loaded into double-sector charcoal-filled Epon centerpieces, and sedimentation velocity analysis was conducted at 50000 rpm and 25 °C using an An50 Ti eight-hole rotor. Sedimentation velocity analysis was performed by direct boundary modeling using solutions of the Lamm equation and the program Sedfit (see Schuck27 and www.analyticalultracentrifugation.com). Partial specific volume, buffer density, and viscosity values were determined using software developed by J. Philo (SEDNTERP; see www.jphilo.mailway.com/download.htm).
Scaled-Up Cross-Linking Reaction
To obtain a larger quantity of cross-linked protein, ∼30 mL of 10 μM K33M His-tagged protein was mixed with a low folate concentration (270 μM) and 5 mM EDC. Cross-linking continued until ∼20% activity remained. Excess folate was removed by passing the solution over a Ni-NTA column. The His-tagged protein was eluted with buffer containing 250 mM imidazole and 300 mM NaCl. To remove any folates cross-linked to the N-terminus or the lysine in the His tag, the protein was truncated by chymotrypsin treatment. [Chymotrypsin should cleave after residue 26 in the His tag sequence introduced by pRSETB and after residue 47, which corresponds to F16 in the wt R67 DHFR sequence (see Figure S1 for a comparison of sequences).] Both fragments possess molecular masses in the range of 2–3 kDa. Passing this mixture over a Ni-NTA column allows removal of any His-tagged fragments or uncut full length protein from the truncated species. The latter no longer binds the column. The truncated K33M tetramer mass is calculated to be 26916 Da. Dialysis (7 kDa molecular mass cutoff) and concentration (Vivaspin with a 10 kDa molecular mass cutoff) were used to remove peptides from the solution. Whether the cross-linked folate associated with the truncated, K33M R67 DHFR (12–25 μM) could be reduced was assessed by addition of NADPH (200 μM). The rate of reduction was obtained by a linear fit of the initial rate. The data were also fit to a single exponential and a linear rate to obtain the amplitude of the observed absorbance change, which describes the concentration of folate that was reduced.
Computational Modeling
This modeling of the hydride transfer step was performed using the empirical valence bond (EVB) method, developed by Warshel and co-workers.28,29 The modeled reaction describes hydride transfer from NADPH to protonated DHF producing NADP+ and tetrahydrofolate. More specifically, a hydride is transferred from the C4N carbon on the cofactor (the donor carbon, CD) to C6 on the protonated substrate DHF (the acceptor carbon, CA). Figure S2 gives the reactive rings of the substrate and cofactor and atom labels. The EVB method, in combination with classical molecular mechanics, was used for sampling of the conformations along the hydride transfer trajectory. The homotetrameric model of R67 with one bound protonated substrate molecule and one bound reduced cofactor, in explicit solvent, was used as previously described.6 Cross-linking of the substrate tail was modeled by applying a weak restraint between K32:3 (the bottom right K32 in Figure 1B) and the γ-carboxylate group of DHF. A distance restraint (req = 3.0 Å, and force constant = 100 kcal mol–1 Å–2) between the C atom of the DHF γ-carboxylate and the Nε atom of the K32:3 side chain was applied. The activation energy barrier for the hydride transfer catalyzed by wild-type R67 was set to 17.6 kcal/mol by appropriate selection of EVB parameters.6 This restraint allowed the carboxylate and amino groups to be held in the proximity of each other and to explore various conformations. A weak restraint instead of a covalent linkage was used for modeling to avoid introduction of an artificial bias on the tail conformation. As demonstrated in a previous computational study, the starting conformation and the flexibility of the DHF tail are important for the progress of the reaction.6 Two alternate but complete sets of EVB simulations were performed for the purpose of comparison. These two sets (arbitrarily labeled as EVB1 and EVB2) differ in the starting conformation of the protein (see ref (6) for details).
Synthesis of Bis[2-N′,N′-(1-aminoethyl)] Folate (α,γ)-Amide (Bis-EDA−folate)
Folate adduct formation followed the general procedure of Wang et al.30,31 with several modifications. Folate (500 mg) was dissolved in 20 mL of dimethyl sulfoxide and activated by treatment with a 2.2-fold molar excess of N-hydroxysuccinimide and a 20-fold molar excess of dicyclohexylcarbodiimide at room temperature for 1 h. This solution was further reacted with a 20-fold molar excess of ethylenediamine (EDA, 1.5 mL) for 24 h. The reaction mixture was acidified, and the folates were precipitated with cold acetone. The pellet was washed with diethyl ether as well as ethanol. The product was initially purified with a silica column. Impurities were eluted with 95% ethanol, while a mixture of folate products was eluted with water (pH 9). The bis-EDA–folate adduct was separated from any monoadducts and unreacted folate by being passed over a 1.5 cm × 15 cm DEAE-fractogel column equilibrated in 10 mM NH4HCO3. The bis-adduct eluted with 50 mM NH4HCO3. Using a gravimetric approach, the extinction coefficient for the folate analogue was calculated as 26000 M–1 cm–1 at 282 nm and 7700 M–1 cm–1 at 340 nm.
The identity of the bis-EDA adduct was confirmed by NMR (see Figure S3). The bis-EDA–folate adduct had limited solubility in many buffers, including MTA polybuffer. It was, however, soluble in 10 mM Tris-HCl buffer (pH 7), thus this buffer with 100 mM NaCl was used for assays. The ability of this compound to inhibit R67 DHFR activity was monitored using a saturating NADPH concentration (51 μM) and varying DHF concentrations (4–56 μM). The bis-EDA-folate concentration range was 0–100 μM. A Ki was calculated using a replot of the slopes from the Lineweaver–Burk plots as described by Segel.32
Results
Mutagenesis
One approach to probing the role of disorder associated with the pABA-glu tail of DHF is to vary the volume of the active site pore. For example, alterations in cavity size in GroEL resulted in changes in folding rates as well as specificity; i.e., “size matters” due to confinement effects.33,34 Confinement will restrict the chain entropy.35,36 In other words, as the pore volume decreases so should the tail disorder. To probe the effect of pore volume on binding and catalysis, we examined the R67 DHFR structure and proposed mutations at K32, I68, and/or Y69. To target DHF binding rather than the binding of both ligands, we mutagenized only half the pore. Because NADPH is the larger ligand and its binding uses more protein contacts as well as a “dry” interface (i.e., no bridging waters),5,37 interactions with the cofactor are more likely to be disrupted by these amino acid substitutions. Also from ITC studies, a preference exists for NADPH to bind first,3 predicting that if the mutations have a strong effect, the cofactor would bind to the wt half of the pore. Substrate would then be forced into the mutant half of the pore, consistent with the tail disorder and use of a “wet” interface.37
Addition of asymmetric mutations can be achieved by using our gene quadruplication construct in which four R67 DHFR gene copies are linked in frame.17 The linker between gene copies encodes the natural N-terminus. The resulting protein, Quad3, possesses 4 times the mass of the R67 monomer and is almost fully active with a 1.6-fold decrease in kcat and a 1.5-fold increase in Km(NADPH). This construct allows creation of asymmetry by adding mutations to specific gene copies. A first mutant gene encoded two K32R mutations in gene copies 1+3; in the resulting mutant protein, the two K32R substitutions occur in only one side of the active site pore (see Figure 1C).
A longer side chain associated with the K32R mutations could decrease the distance across the edge of the pore by up to 6 Å depending on which rotamers are present. A shorter distance introduces the possibility of the α- and γ-carboxylates in the pABA-glu tail being able to span the pore and form two direct ionic interactions with the K32R residues. This possibility, coupled with any decreased tail motion associated with greater confinement, could result in tighter binding of DHF.37,38 A potential countereffect might be a greater desolvation penalty associated with formation of two ion pairs.39
A second set of mutations, Y69W:1+3, was added to the K32R:1+3 mutant. The Y69 position moves partway toward the center of the pore (see Figure 1C). Using the CASTp algorithm,24 a 34–35% decrease in the half-pore volume is estimated in this multimutant.
A last mutant explored the role of the I68 residue. Here, we decreased the amino acid size, introducing a single I68A mutation into Quad3. The volume was decreased as I68 occurs on the side walls of the central constriction, and mutations at this position seemed less likely to alter the glutamate tail location and more likely to alter the pteridine and/or pABA ring positions.
Steady state kinetic analysis of the three mutants mentioned above finds that these changes in volume do not greatly alter the kcat and Km values as noted in Table 1. While the Km for DHF decreases ∼2-fold in the K32R:1+3 mutant compared to that of Quad3, so does the kcat value, suggesting a nonproductive binding effect. Also, the Km for DHF increases ∼2-fold in the K32R:1+3 plus Y69W:1+3 multimutant.
Table 1. Comparison of Steady State Kinetic Parameters at pH 7.0 for Numerous R67 DHFR Constructs.
| enzyme | kcat (s–1) | NADPH Km (μM) | DHF Km (μM) | kcat/Km(NADPH) (s–1 M–1) | kcat/Km(DHF) (s–1 M–1) | any DHF inhibition? |
|---|---|---|---|---|---|---|
| Quad3a | 0.81 ± 0.02 | 4.4 ± 0.4 | 6.7 ± 0.4 | 1.8 × 105 | 1.2 × 105 | no |
| K32R:1+3 | 0.41 ± 0.01 | 6.5 ± 0.6 | 3.9 ± 0.3 | 6.3 × 104 | 1.1 × 105 | no |
| K32R:1+3 plus Y69W:1+3 | 0.80 ± 0.03 | 3.8 ± 0.4 | 12 ± 1 | 2.1 × 105 | 6.7 × 104 | no |
| I68A:3 | 0.56 ± 0.02 | 3.5 ± 0.6 | 5.6 ± 0.6 | 1.6 × 105 | 1.0 × 105 | yes with a Kd2 of 8.6 ± 2.0 μM |
| wt R67 DHFRb | 1.3 ± 0.07 | 3.0 ± 0.06 | 5.8 ± 0.02 | 4.3 × 105 | 2.2 × 105 | no |
One surprising result is the observation of substrate inhibition by DHF in the I68A:3 mutant (see Figure S4). Obviously one role for the I68 side chain is to help discriminate between formation of productive versus nonproductive (e.g., 2DHF) complexes.
Cross-Linking
A cross-linking strategy was utilized to tether the glutamate tail of bound folate (or DHF) to R67 DHFR. The cross-linking results are followed by a description of the reduction rate of the cross-linked folate molecule. pKa values were calculated for the amine groups in R67 DHFR; however, no perturbed pKa values were predicted (see the Supporting Information).
Wild-type (wt) R67 DHFR was incubated with 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide (EDC) in the presence or absence of DHF or folate. EDC cross-links amines to carboxylates.40−42 The available amines in R67 DHFR are the N-terminus as well as the K32 and K33 residues. From the ternary complex crystal structure,5 a model of bound folate places the carboxylate groups close to the K32 residues, so a specific cross-link between bound folate and K32 might occur.6 As shown in Figure 2A, the negative controls show little to no activity loss over the incubation period. However, addition of high concentrations of either folate (2 mM) or DHF (640 μM) leads to >90% loss of activity within several hours. Inactivation rates of 0.018 ± 0.001 and 0.014 ± 0.001 min–1 were obtained for folate and DHF modification, respectively. As folate is more stable than DHF, it was used in subsequent cross-linking experiments. Protection of the active site by addition of 1 mM NADPH was not observed under these conditions. However, when the folate concentration was decreased, a slower inactivation rate was noted and NADPH protection observed (see Figure 2B).
Figure 2.

Inactivation of R67 DHFR via cross-linking to folate. In panel A, 10 μM R67 DHFR was incubated with 5 mM EDC and 2 mM folate (● points). Aliquots were withdrawn at various times, and the remaining activity was monitored. Alternatively, DHFR was incubated with 5 mM EDC and 640 μM DHF (gray circle points). Another sample consisting of 7 μM His-tagged K33M mutant was incubated with the same concentrations of EDC and folate (■ points). Negative controls included R67 DHFR with EDC (△ points), R67 DHFR only (☆ points), His-tagged K33M DHFR with EDC (▽ points), and His-tagged K33M DHFR only (□ points). Exponential fits to the data are shown by the solid (R67 with folate), dashed (R67 with DHF), and dotted–dashed (His-tagged K33M with folate) lines. Panel B shows the protection of R67 DHFR by NADPH from inactivation. Data points describe inactivation of 11 μM R67 DHFR by 5 mM EDC in the presence of 80 μM folate (●, solid line). The protection afforded by addition of 1 mM NADPH is shown by gray square points and the dashed line. Best fits to an exponential process with a linear component are shown.
An EDC reaction was additionally performed with a K33M mutant containing an N-terminal His tag sequence43 (see Figure 2A). This reaction, in the presence of 2 mM folate, also resulted in activity loss, with a slightly slower inactivation rate (0.009 ± 0.001 min–1).
The presence of intermolecular cross-links was assessed using reducing sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE). After cross-linking, a strong monomer band was observed; however, dimer and higher-order oligomer bands also occurred. Details of these analyses are given in the Supporting Information.
As mutations at K32 destabilize the active R67 tetramer and favor the inactive dimer,43 it seemed possible that the activity loss seen in Figure 2 might be due to dimer formation. To test this possibility, sedimentation velocity experiments were performed using a chymotrypsin-truncated R67 DHFR variant that is fully active (new N-termini at V17).13 For the truncated enzyme control, the predominant species has a molecular mass of 28500 Da. For comparison, the calculated tetramer mass from the protein sequence is 26906 Da. As shown in Figure S5, a small peak corresponding to dimer was also noted, indicating the truncated enzyme exists as a mixture of dimer and tetramer. For the truncated protein cross-linked in the presence of 150 μM folate, the major species has a mass of 29900 Da. No dimer was observed, and a shift in mass corresponding to folate adduction occurs.
Mass spectrometry was used to monitor the shift in enzyme mass associated with the cross-linking reactions. The chymotrypsin-truncated R67 DHFR variant was used; its monomer mass was predicted to be 6726.5 Da, and a mass of 6729 Da was observed (see Figure S6). For the EDC only (control) reaction, a large peak for the truncated protein and a second, smaller peak corresponding to an EDC adduct (mass of 6885 Da) were observed (data not shown). While isoacylureas are typically unstable and hydrolyze in aqueous solution,40,41 stable N-acylureas have previously been reported40,44 as well as tyrosine and cysteine adducts.40,45,46
The various species detected by MALDI for the EDC reaction between truncated R67 and folate are shown in Figure S6. Depending on the folate concentration used as well as the time length of the reaction, different combinations of peaks can be observed. In the presence of 72 μM folate and with 25% DHFR activity remaining, a large peak corresponding to truncated enzyme remains. A second large peak corresponds to a folate adduct (mass of 7155 Da). A smaller peak corresponding to addition of two folates per monomer (mass of 7576 Da) can also be observed. At higher folate concentrations (150 μM folate) and with <10% activity remaining, only a small peak remains for unmodified enzyme and the largest peak (mass of 7155 Da) corresponds to the folate adduct. Another prominent peak (mass of 7308 Da) likely corresponds to a species containing both folate and EDC adducts. (For example, an O-acyl isourea adduct between EDC and a tyrosine has been seen by MS in myoglobin,47 and such a species in an R67 DHFR–folate adduct would result in a mass of 7304 Da.) A smaller peak corresponding to adduction of two folates per R67 monomer can additionally be seen.
To probe the labeling stoichiometry, initial experiments used the His-tagged R67 DHFR protein; the protein–folate adduct was isolated using a His Spintrap column. The protein content was measured by a BCA assay and the folate concentration obtained by absorbance at 350 nm. At a high folate concentration (2 mM), a labeling ratio of 4.2 folates per monomer was found, consistent with labeling of the N-terminus, a lysine introduced by the His tag cloning step, K32, and K33. If the K33M mutant in the His-tagged construct was used in the cross-linking reaction, 2.9 folates per monomer were found. If lower folate concentrations (0.1 mM) were used with the wt His-tagged protein, only 0.91 residues per monomer were labeled. These differences in stoichiometry suggest nonspecific labeling occurs at higher folate concentrations, while more specific labeling occurs at lower folate levels. Indeed, previous studies have shown nonspecific labeling of catalase and superoxide dismutase by high concentrations of folate.48
When wt R67 DHFR was used in cross-linking reactions with low folate concentrations, the protein–folate adduct showed 0.48–1.1 folates per monomer (four separate experiments). To determine if there was a mixture of labeling sites (for example, K32 as well as the N-terminus), the N-terminus of the folate–protein adduct was truncated by chymotrypsin treatment, followed by separation of the N-terminal peptide fragment from the β-barrel core of the protein by gel filtration. Following this step, the folate:protein monomer ratio decreased to 0.32–0.46 (three separate experiments). This value correlates strongly with the ligand binding stoichiometry of two folates bound per tetramer.3 Neither collision-induced dissociation (CID) MS analysis by Alphalyse (http://www.alphalyse.com/) nor Edman degradation was able to identify which lysine was cross-linked.
It is not clear which folate carboxylate, α or γ, is involved in the cross-link. Two carbodiimide conjugation studies suggest a preferred reaction occurs with the more basic carboxylate.30,44 Because the pKa for the γ-carboxylate of folate is 4.5 while that for the α-moiety is 2.5, it seems likely that the major cross-linked species involves the γ-carboxylate of folate.30
Can the Covalently Linked Folate Be Reduced?
To obtain sufficient protein to determine whether the cross-linked folate could be reduced, we scaled up our cross-linking reaction. The K33M His-tagged protein was cross-linked using a low folate concentration until ∼20% activity remained. Excess folate was removed using a Ni-NTA column. From a BCA assay and absorbance at 350 nm, the full length, cross-linked species contained an average of 3.7 folates per tetramer. To remove any folates cross-linked to the N-terminus or the lysine in the His-tagged pRSETB sequence, the protein was truncated by chymotrypsin treatment. Passing this mixture over a Ni-NTA column removed the His-tagged N-terminal peptide from the active, truncated species that no longer binds the column. Any other peptides were removed by dialysis and concentration. The resulting species had an average of 1.7 folates per tetramer.
As the K33M lysine has been removed by mutagenesis and the N-termini have been removed by chymotrypsin treatment, the only remaining amine that could be tagged by folate is K32. The pool should contain a mixture of unmodified protein as well as singly and doubly modified species. As any unmodified protein does not contain cross-linked folate, it is not expected to be active as the substrate is not added to the activity assay; i.e., the cross-link provides the folate source. Also, the tetramer containing two folates is predicted not to be active because of blockage of the active site pore. The species of interest should have one cross-linked folate per active site pore. Addition of NADPH leads to cofactor binding in the empty half-pore, resulting in a single turnover of folate to DHF followed by release of NADP+ and rebinding of a second NADPH molecule, followed by the reduction of DHF to tetrahydrofolate. Other possible scenarios are discussed in the Supporting Information.
The kcat value for the control reaction, folate reduction by His-tagged K33M R67 DHFRs (pH 7), is 0.065 ± 0.017 min–1. In contrast, reduction of the cross-linked folate in the truncated K33M mutant gave a rate of 0.002 ± 0.0007 min–1. The initial rate was obtained by a linear fit. The reaction terminated in ∼6–12 h depending on the protein concentration. In comparison to the K33M control rate for folate reduction, the rate for reduction of cross-linked folate is decreased ≤30-fold. The ≤ modifier comes from the amplitude of the absorbance change indicating the fraction of active species. This is ∼40%.
Computational Modeling
The effect of cross-linking K32:3 (bottom right K32 in Figure 1B) and the γ-carboxylate group of bound protonated DHF was computationally analyzed using a weak restraint. This allowed the distance between the K32:3 side chain and the DHF γ-carboxylate group to be maintained at ≤3 Å during the hydride transfer reaction. Two independent trajectories were calculated and compared to each other (see Figure 3 and Figure S7). The EVB1 trajectory showed an activation barrier of 18.6 kcal/mol (error bar of ∼1.0 kcal/mol), corresponding roughly to a rate of ∼0.17 s–1, which would be ∼6-fold slower compared to a wild-type barrier of 17.6 kcal/mol6 with a rate of ∼1 s–1 (see Figure 3A). The second simulation, EVB2, showed an energy barrier of 16.2 kcal/mol, corresponding to a rate of ∼9.7 s–1. However, this trajectory indicated that the substrate adopted an unusual conformation (Figure 3B). Our previous detailed computational study6 indicates that various torsions and angles around the pABA ring and pABA-glu tail impact the reaction barrier. Even though the EVB2 simulation describes a reaction faster than that for the wild-type enzyme with a free tail, it requires the pABA-glu tail to be present in an alternate conformation. Overall, the computational simulations indicate that the cross-linked tail still allows the hydride transfer reaction to occur. Even though the end of the tail is restricted, there appears to be enough freedom around the pABA ring to allow for movement/torsion at the active site center to adopt a suitable conformation for reaction to proceed. The overall rate would be a function of the concentration of the various transition states and their respective rates.
Figure 3.

Computational modeling of the hydride transfer reaction catalyzed by R67 DHFR with a cross-linked DHF tail. Cross-linking was modeled by applying a distance harmonic restraint between K32 and the DHF γ-carboxylate group. (A) EVB modeling indicated activation barrier heights of 18.6 and 16.2 kcal/mol for two independent EVB simulations (arbitrarily labeled EVB1 and EVB2) based on a wild-type (free DHF tail) barrier of 17.6 kcal/mol.6 (B) Structural analysis indicates that even though the glu tail is held in the proximity of the K32 residues, other parts of the pABA-glu tail can still move or rotate, allowing the substrate to adopt alternative conformations that allow the reaction center to achieve a reactive conformation(s). The conformations of bound NADPH, protonated DHF, and K32:3 near the transition state are given for the two trajectories. EVB1 is colored green and EVB2 cyan. The structures at the transition state from two simulations are shown from two different angles (left and right) to provide better views of the substrate and cofactor orientations. The panel at the right shows a change in the N5–C6–C9–C10 dihedral angle and a different placement of the pABA ring.
Bis-EDA Adduct of Folate
Another way to explore the role of the proposed ion pair between symmetry-related K32 residues in R67 DHFR with the glu tail of folate is to alter the substrate. The ability of the bis-EDA–folate adduct to inhibit R67 DHFR was analyzed. Weaker binding was evident as higher concentrations were required to inhibit the enzyme. Figure 4 shows noncompetitive inhibition was observed with respect to DHF. A Ki of 140 ± 15 μM was obtained. As bis-EDA−folate did not display parabolic inhibition under these conditions,32 it does not appear to be forming a 2bis-EDA–folate complex with R67. Rather, the inhibitory complex is DHF·bis-EDA–folate. By contrast, folate is a competitive inhibitor of DHF with a Ki of 16 ± 3 μM [10 mM Tris and 100 mM NaCl buffer (pH 7)].3
Figure 4.

Inhibition of R67 DHFR activity (51 μM NADPH and 4–56 μM DHF) with folate derivatives in 10 mM Tris-HCl, 100 mM NaCl, and 1 mM EDTA (pH 7.0) at 25 °C. (A) Competitive inhibition of R67 by folate yields a Ki of 16 ± 3 μM. Folate concentrations are 0 μM (—), 13.5 μM (−–−), 26 μM (···), and 62 μM (−-−). (B) The Bis-EDA–folate adduct inhibits R67 DHFR in a noncompetitive manner (Ki = 140 ± 15 μM). Bis-EDA–folate adduct concentrations are 0 μM (—), 50 μM (−–−), and 100 μM (···).
Discussion
Analysis of the various R67 DHFR crystal structures2,5 as well as docking49 and MD6 studies has found the pABA-glu tail of the bound substrate to be disordered. The disorder is proposed to be due to the many combinations that are available for direct and solvent-separated ion pairs between symmetry-related K32 residues and the two carboxylates in the glu tail of bound folate. This study further explores the role of disorder by cross-linking experiments, changes in the volume of the active site pore, and modification of folate.
Cross-Linking
At high folate concentrations, we find labeling of all possible amine targets in R67 DHFR, consistent with specific as well as nonspecific labeling. When we use lower folate concentrations, most of the nonspecific labeling is eliminated and NADPH addition provides some protection to the enzyme activity. While neither CID MS by the company Alphalyse nor Edman degradation allowed identification of the cross-linked residue that results in a loss of enzyme activity, we can still analyze the various data sets to make a strong prediction. The N-terminus was eliminated as the cross-linking target that causes activity loss as three different N-terminal sequences were employed (wt N-terminal MIR-, the chymotrypsin-truncated N-terminal V17F18P19S20-, and the His-tagged N-terminal MRGSHHHHHH-), and all species showed inactivation in the presence of EDC and folate. Additionally, when higher folate concentrations were used, the labeled N-terminus of R67 could be removed by chymotrypsin treatment, but DHFR activity was not restored.
Modification of K33 could potentially cause a loss of enzyme activity if the cross-linked folate is able to enter the active site pore due to torsioning of the K33 side chain. However, K33 is unlikely to be the cross-linking target associated with activity loss as a K33M mutant still shows inactivation, with a rate that is only 2-fold slower than that of wt R67 DHFR (Figure 2A).
The remaining amine that could be involved in activity loss is K32. Labeling of this residue seems most likely to result in a loss of activity by proximity considerations as K32 is ∼3–5 Å from the glu tail in a model of bound DHF. Molecular dynamics calculations also propose the glu tail of the substrate switches between direct ionic interactions and solvent-separated ion pairs with K32.5,6 Additionally, folate adduction stabilizes the tetramer as measured by analytical ultracentrifugation, suggesting the covalent linkage provides additional contacts to the complex, consistent with folate being docked in the active site. Third, chymotrypsin treatment of the R67 DHFR–folate adduct yields a labeling stoichiometry of 0.32–0.46 folate per monomer. These values correlate with our previous ITC binding studies in which two folates bind per R67 tetramer (0.5 folate per monomer).3 All these results strongly support the hypothesis that cross-linking of two folates to two symmetry-related K32 residues on opposite sides of the pore blocks access to the active site, leading to loss of activity.
Disordered pABA-glu Tail Effects
While the experiments outlined above describe our present research, we have previously studied the pABA-glu tail disorder using salt effects,43 asymmetric mutants,50,51 and molecular dynamics.6 To provide a comprehensive discussion, we consider these studies, as well.
Because of the 222 symmetry imposed on the active site pore, each R67 binding site must accommodate both ligands. While NADPH binding utilizes many contacts and docks into a single conformation,37 binding of DHF appears to be less optimal, using fewer contacts and displaying a disordered pABA-glu tail. We have previously found different salt effects on kcat and kcat/Km, suggesting that while ion pairs are involved in ligand binding, a productive path to the transition state involves breaking one of these salt bridges.43 This is an unusual result as the ground state in most enzymes typically resembles the transition state and changes in ionic interactions are not usually observed as the reaction progresses.43,52 In R67 DHFR, the ion pair that is proposed to be lost upon transition state formation (based on salt effects on asymmetric K32M mutants)50,51 occurs between K32 and the glu tail of bound DHF.
More recently, using a MD approach, Kamath et al.6 predicted the pABA-glu tail of bound DHF samples many possible conformations by frequent switching of direct ion pairs to solvent-separated ion pairs, indicating the various ion pair combinations are reasonably isoenergetic. Given the wide range of motions, it was difficult to completely deconvolute which motions of the pABA-glu tail might be important. However, Kamath et al. suggested a general correlation between the radius of gyration for the pABA-glu tail and the reaction barrier height for wt and several asymmetric K32M mutants. A proposal was advanced that “tail movement at the edge of the active site, coupled with the fixed position of the pteridine ring in the center of the pore, leads to puckering of the pteridine ring, which can promote transition state formation.” Another necessary event to reach the transition state is concurrent puckering of the nicotinamide ring.
To vary the disorder of the pABA-glu tail of the bound substrate, we utilized two different approaches. The first involved mutations that decreased the half-pore volume by ∼35%. A reduced active site size should decrease the entropy associated with the pABA-glu tail. For example, if a larger radius of gyration does lead to higher kcat values, then a smaller pore volume would be expected to decrease enzyme activity. However, the effects of the K32R:1+3 and Y69W:1+3 mutations were minimal, suggesting R67, in the form of Quad3, had no difficulty in binding DHF and reaching its transition state. It may be that the radius of gyration alone is not well correlated with kcat.
A second experiment for exploring the role of substrate disorder asked if folate cross-linked to the K33M mutant DHFR could be reduced. Here, the glu tail of folate is tethered to the enzyme by the EDC cross-linking reaction and its movement severely restricted. This gambit resulted in a ≤30-fold decrease in folate reduction rates, a ≤2 kcal/mol effect. Because cross-linked folate can still be reduced, albeit at a reduced rate, it seems likely that the tethered folate possesses a suboptimal position for reduction.
While our tethered substrate experiments described folate reduction and our MD calculations used protonated DHF, one computational observation is that the tethered substrate can still sample many different poses. This may provide a solution for how the tethered folate may still act as a substrate. For example, if tethering of the glu tail places folate in a poor position for catalysis and there is sufficient room in the active site pore for the cross-linked folate to move, this may allow an alternate conformer to attain the transition state. This is not the paradigm for catalysis in most enzymes as they typically have many contacts that hold the substrate in place. In fact, well-evolved enzymes are proposed to have active sites that lack gating or fluctuations in the donor·acceptor distance.53,54 In contrast, R67 has been shown to require significant gating to reach its transition state.55
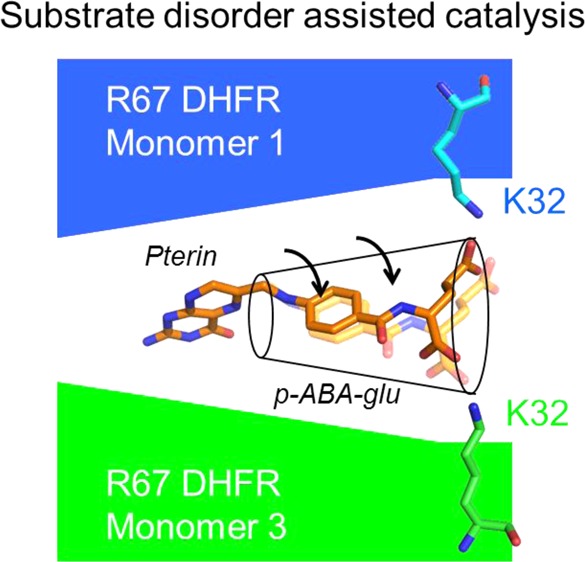
Our combined results suggest the larger space available to bound DHF and the concomitant tail disorder allow R67 to deal with various impediments such as some degree of confinement and/or tethered substrate tails. This view suggests a high adaptability56 associated with this protein, albeit coupled with a low catalytic efficiency. In other words, entropy allows many possible solutions but caps the activity at a low level. “Catch 222” appears to describe R67 DHFR function.57Figure 5 provides a cartoon of our view of how the R67 enzyme functions using a “fuzzy” ground state to bind the substrate, followed by loss of an ion pair, which leads to the transition state.
Figure 5.

Cartoon of folate bound to half the active site pore in R67 DHFR. An asymmetric slice through the protein is shown with the protein colored green and blue (shading as per Figure 1C) and the active site pore colored yellow. The pterin ring of folate is denoted as Pte, and the nicotinamide ring of cofactor is denoted as Nic. The other side of the pore with bound NADPH is not shown for the sake of clarity. Two symmetry-related K32 residues in the right half-pore are shown as blue and green ovals. The α- and γ-carboxylates of folate are shown as orange and cyan ovals, respectively. Water is shown as a “v” with two small white circles attached. Close contact of the ovals indicates ion pair formation. As the folate tail is not long enough to span the pore and interact with both symmetry-related lysines, water mediates one interaction. Many possible solvent-separated ion pairs (SSIPs) are possible. From MD studies, the glu tail can flip and/or translate to provide various pairings.6 From studies of the effects of salt on catalysis, one ion pair is lost to form the transition state.43
Discrimination between Productive and Nonproductive Complex Formation
The I68A:3 mutant in Quad3 resulted in DHF inhibition (see Figure S4). Previous I68L and I68M mutants in R67 DHFR also displayed slight to moderate levels of substrate inhibition.58 Further, the Q67H mutant showed substantial substrate inhibition.59 These various results indicate that Q67 and I68 near the center of the hourglass pore as well as the tight constriction that occurs in this area play a role in discriminating between the productive NADPH–DHF pair or the nonproductive 2DHF complex.
Further, our bis-EDA–folate results find loss of the ion pair between K32 and the glu tail of DHF leads to noncompetitive inhibition, consistent with formation of an R67 DHFR·DHF·bis-EDA–folate complex. This result suggests that another role of the ion pair between K32 and the DHF glu tail is to help partition the reaction path toward the productive ternary complex rather than a nonproductive species. Our bis-EDA–folate results may also correlate with use of NADH as a cofactor, which leads to DHF inhibition.43,60 In this case, loss of an ion pair between K32 and the 2′-phosphate of NADH weakens the preference for the cofactor·substrate pair and allows formation of the nonproductive 2DHF complex. In other words, it appears easy to tip the balance from the NADPH–DHF complex to other bound species by either mutations or use of alternate substrates.
Polyelectrostatic theory applied to the disordered protein Sic1, a CDK inhibitor, shows increasingly tighter binding to Cdc4, a SCF ubiquitin ligase subunit, as up to nine sites in Sic1 can be phosphorylated.61 A plot of ln Kd versus the net charge for the Sic1–Cdc4 pair shows a straight line, consistent with increasing numbers of phosphorylations allowing additional ion pair combinations as well as electrostatic potential effects.62 A change in charge from +2 to −4 results in ∼100-fold tighter binding. A plot of the number of negative charges associated with folate or folate analogues versus their Ki in R67 DHFR is given in Figure 6. A different pattern is observed as folate analogues apparently only need one negative charge to bind reasonably tightly (Ki values of 37 and 28 μM for folate–histidine and folate–ornithine, respectively).63 Addition of another negative charge tightens binding 2-fold (folate Ki of 16 μM), while addition of one additional negative charge and three additional negative charges in polyglutamylated folates do not tighten binding further.63 (From analysis of the crystal structure, the additional glutamates are not expected to contact the protein. Rather, they will dangle out of the pore into solvent.) In contrast, ablation of the −2 charge by the bis-EDA analogue weakens binding by ∼9-fold (Ki of 140 μM).
Figure 6.

(A) Plot of Ki vs the net charge on folate or its analogues. The Ki values are listed in Table S1 of the Supporting Information. PG2 and PG4 are pteroyl-diglutamate and pteroyl-tetraglutamate, respectively. Lines are provided to guide the eye and do not represent a fit. (B) Plot of Km(DHF) for Quad3 and Quad4 and their asymmetric K32M mutants51,64 vs the number of K32 residues in one half-pore. The Km values are listed in Table S1 of the Supporting Information. Lines connecting the data points do not represent a fit. As NADPH prefers to bind first3 and utilize two ion pairs,43 we predict it will dock into the least mutated half-pore that is available. Then DHF will be forced to bind to the other, mutant half-pore. A similar pattern appears in both panels when either the ligand or the protein loses one or two contributors to the ion pair between K32 and the glu tail of folate. Loss of one K32 residue or loss of one carboxylate on the folate tail does not have a large detrimental effect, likely because of switching from the fuzzy complex depicted in Figure 5 to a single ion pair. Loss of both K32 residues in the protein or both carboxylates in the folate ligand has a large effect as no ion pairs or solvent-separated ion pairs remain possible.
Loss of all possible ion pairing with substrate can also be accomplished via site-directed mutagenesis. For example, asymmetric K32M mutants have previously been constructed in a quadruplicated gene product. These constructs result in no, one, or two K32 residues per half-pore.51,64 A brief description of these mutants is provided in the supplement. The K32M:1+2 and K32M:1+4 double mutants that allow only one K32M mutation per half-pore display DHF Km values of 14.4 and 10.5 μM, respectively. In contrast, a K32M:1+3 double mutant with no K32 residues in one half-pore shows a DHF Km value of 165 μM.64Figure 6B plots the DHF Km versus the number of K32 residues in the half-pore. We conclude the K32 residue tightens DHF binding by ∼20–30-fold.
Panels A and B in Figure 6 show a similar pattern in that loss of all possible ion pairs between the glu tail of folate/DHF and K32 residues results in ∼10–30-fold weaker binding. In contrast, loss of one possible ion pair can apparently be offset by switching of the fuzzy complex depicted in Figure 5 to a more traditional ion pair. The EDC cross-linked folate species constructed in this research mimics the more traditional ion pair. However, because of the cross-linking reaction, the ion pair becomes a covalent bond. Fixing the position of the glu tail does not abolish turnover; rather, the less optimal position for catalysis reduces kcat ≤30-fold.
Conclusion
The disorder and/or dynamics associated with bound DHF likely arises due to the 222 symmetry associated with the R67 active site. As the R67 DHFR active site is large, this leads to many compromises in R67’s behavior. For example, the ability of a tethered folate to be reduced most likely arises due to movement of the pterin and pABA rings. As the disorder of the pABA-glu tail plays a role in leading to the transition state, this may be an example of substrate-assisted catalysis.65 Thus, wt R67 DHFR appears to have evolved to a balancing point where the pABA-glu tail disorder helps the enzyme to function. However, when mutants or alternate ligands are used, the disorder associated with the pABA-glu tail can tip the seesaw toward an inability to discriminate between the productive NADPH·DHF complex and the nonproductive 2DHF complex.
Acknowledgments
We thank Nancy Horn for her help in setting up our initial MALDI experiments. We thank Badri Krishnan for initial help with the asymmetric mutants and Kusum Rathore for her initial experiments exploring the stoichiometry of folate labeling of R67 DHFR in the EDC reaction. We thank Russ Dooling and Jordan Grubbs for their help in the cross-linking experiments. We thank Charles Murphy for running several MS experiments and Edman reactions.
Glossary
Abbreviations
- DHF
dihydrofolate
- THF
tetrahydrofolate
- EDA
ethylenediamine
- EDC
1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide
- EVB
empirical valence bond
- MTA buffer
100 mM Tris, 50 mM MES, and 50 mM acetic acid polybuffer
- NADP+ and NADPH
oxidized and reduced nicotinamide adenine dinucleotide phosphate, respectively
- Ni-NTA
nickel-nitrilotriacetic acid
- pABA-glu
p-amino-benzoyl-glutamate
- PG2 and PG4
-
pteroyl-diglutamate and pteroyl-tetraglutamate
, respectively
- SSIP
solvent-separated ion pair
- wt
wild-type
- Quad3
a tandem array of four R67 DHFR genes fused in frame, yielding a monomer containing 312 amino acids and possessing the essential tertiary structure of the wild-type R67 DHFR homotetramer
- R67 DHFR
R67 dihydrofolate reductase
- PDB
Protein Data Bank.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.5b00981.
A comparison of the amino acid sequences of wt and His-tagged R67 DHFR, structures of the pterin and nicotinamide rings computationally modeled in the DHFR reaction, an NMR spectrum of the bis-EDA–folate adduct, a figure with kinetic data for the I68A:3 mutant in Quad3, a sedimentation velocity plot for chymotrypsin-truncated R67 DHFR and cross-linked, truncated DHFR, a mass spectrometry figure of R67 DHFR cross-linked with folate and EDC, figures from MD analysis describing a weak link between K32 and the γ-carboxylate of folate, a discussion of pKa calculations, a discussion of SDS–PAGE experiments, a comment on whether folate reduction is intra- or intermolecular, a cartoon describing the asymmetric K32M mutations, and a table of Ki and Km values for data depicted in Figure 6 (PDF)
Author Present Address
∥ S.C.: Department of Cell Biology and Physiology, University of North Carolina, Chapel Hill, NC 27599-7545.
This work was supported by National Science Foundation Grant MCB-0817827 to E.E.H. and in part by National Institutes of Health Grants GM 110669 (to E.E.H.) and GM105978 (to P.K.A.).
The authors declare no competing financial interest.
Footnotes
The amino acids in the first monomer of R67 DHFR (A) are labeled 1–78. Those in the second monomer (B) are labeled 101–178. Those in the third monomer (C) are labeled 201–278. Those in the fourth monomer (D) are labeled 301–378. For the sake of brevity, when a single residue is mentioned, all four symmetry-related residues are implied. We use this numbering scheme even when a His tag is added to the N-terminus.
The following system is used to name the asymmetric mutants in Quad3. The residue, residue number, and mutation are listed first, followed by a colon. The position of the asymmetric mutation is indicated numerically, where 1 refers to gene copy 1, 2 refers to gene copy 2, etc. For example, the K32R:1+3 double mutant contains two mutations: lysine 32 in gene copy 1 has been mutated to arginine, as has lysine 32 located in gene copy 3. In this mutant, both lysines in one half-pore are mutated to arginine.
Supplementary Material
References
- Howell E. E. (2005) Searching sequence space: two different approaches to dihydrofolate reductase catalysis. ChemBioChem 6, 590–600. 10.1002/cbic.200400237. [DOI] [PubMed] [Google Scholar]
- Narayana N.; Matthews D. A.; Howell E. E.; Xuong N.-h. (1995) A plasmid-encoded dihydrofolate reductase from trimethoprim-resistant bacteria has a novel D2-symmetric active site. Nat. Struct. Biol. 2, 1018–1025. 10.1038/nsb1195-1018. [DOI] [PubMed] [Google Scholar]
- Bradrick T. D.; Beechem J. M.; Howell E. E. (1996) Unusual binding stoichiometries and cooperativity are observed during binary and ternary complex formation in the single active pore of R67 dihydrofolate reductase, a D2 symmetric protein. Biochemistry 35, 11414–11424. 10.1021/bi960205d. [DOI] [PubMed] [Google Scholar]
- Li D.; Levy L. A.; Gabel S. A.; Lebetkin M. S.; DeRose E. F.; Wall M. J.; Howell E. E.; London R. E. (2001) Interligand overhauser effects in type II dihydrofolate reductase. Biochemistry 40, 4242–4252. 10.1021/bi0026425. [DOI] [PubMed] [Google Scholar]
- Krahn J.; Jackson M.; DeRose E. F.; Howell E. E.; London R. E. (2007) Structure of a type II dihydrofolate reductase ternary complex: use of identical binding sites for unrelated ligands. Biochemistry 46, 14878–14888. 10.1021/bi701532r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath G.; Howell E. E.; Agarwal P. K. (2010) The tail wagging the dog: Insights into catalysis in R67 dihydrofolate reductase. Biochemistry 49, 9078–9088. 10.1021/bi1007222. [DOI] [PubMed] [Google Scholar]
- Agarwal P. K.; Billeter S. R.; Rajagopalan P. T.; Benkovic S. J.; Hammes-Schiffer S. (2002) Network of coupled promoting motions in enzyme catalysis. Proc. Natl. Acad. Sci. U. S. A. 99, 2794–2799. 10.1073/pnas.052005999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. T.; Hanoian P.; French J. B.; Pringle T. H.; Hammes-Schiffer S.; Benkovic S. J. (2013) Functional significance of evolving protein sequence in dihydrofolate reductase from bacteria to humans. Proc. Natl. Acad. Sci. U. S. A. 110, 10159–10164. 10.1073/pnas.1307130110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habchi J.; Tompa P.; Longhi S.; Uversky V. N. (2014) Introducing protein intrinsic disorder. Chem. Rev. 114, 6561–6588. 10.1021/cr400514h. [DOI] [PubMed] [Google Scholar]
- Wright P. E.; Dyson H. J. (2014) Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 16, 18–29. 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittag T.; Kay L. E.; Forman-Kay J. D. (2010) Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. 23, 105–116. 10.1002/jmr.961. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M.; Tompa P. (2012) Fuzzy complexes: a more stochastic view of protein function. Advances in experimental medicine and biology 725, 1–14. 10.1007/978-1-4614-0659-4_1. [DOI] [PubMed] [Google Scholar]
- Reece L. J.; Nichols R.; Ogden R. C.; Howell E. E. (1991) Construction of a synthetic gene for an R-plasmid-encoded dihydrofolate reductase and studies on the role of the N-terminus in the protein. Biochemistry 30, 10895–10904. 10.1021/bi00109a013. [DOI] [PubMed] [Google Scholar]
- Park H. (1997) Creation and Characterization of Asymmetric Mutants in R67 Dihydrofolate Reductase. Ph.D. Thesis, Department of Biochemistry & Cellular and Molecular Biology, University of Tennessee, Knoxville, TN. [Google Scholar]
- Strader M. B. (1998) Constructing a Hybrid of R67 Dihydrofolate Reductase to Study Asymmetric Mutations in the Active Site. M.S. Thesis, Department of Biochemistry & Cellular and Molecular Biology, University of Tennessee, Knoxville, TN. [Google Scholar]
- Howell E. E.; Warren M. S.; Booth C. L.; Villafranca J. E.; Kraut J. (1987) Construction of an altered proton donation mechanism in Escherichia coli dihydrofolate reductase. Biochemistry 26, 8591–8598. 10.1021/bi00400a015. [DOI] [PubMed] [Google Scholar]
- Smiley R. D.; Stinnett L. G.; Saxton A. M.; Howell E. E. (2002) Breaking symmetry: mutations engineered into R67 dihydrofolate reductase, a D2 symmetric homotetramer possessing a single active site pore. Biochemistry 41, 15664–15675. 10.1021/bi026676j. [DOI] [PubMed] [Google Scholar]
- Smiley R. D.; Saxton A. M.; Jackson M. J.; Hicks S. N.; Stinnett L. G.; Howell E. E. (2004) Nonlinear fitting of bisubstrate enzyme kinetic models using SAS computer software: application to R67 dihydrofolate reductase. Anal. Biochem. 334, 204–206. 10.1016/j.ab.2004.06.040. [DOI] [PubMed] [Google Scholar]
- Blakley R. L. (1960) Crystalline dihydropteroylglutamic acid. Nature 188, 231–232. 10.1038/188231a0. [DOI] [Google Scholar]
- Horecker B. L.; Kornberg A. (1948) The extinction coefficients of the reduced band of pyridine nucleotides. J. Biol. Chem. 175, 385–390. [PubMed] [Google Scholar]
- Baccanari D.; Phillips A.; Smith S.; Sinski D.; Burchall J. (1975) Purification and properties of Escherichia coli dihydrofolate reductase. Biochemistry 14, 5267–5273. 10.1021/bi00695a006. [DOI] [PubMed] [Google Scholar]
- Hillcoat B. L.; Blakley R. L. (1966) Dihydrofolate reductase of Streptococcus faecalis. I. Purification and some properties of reductase from the wild strain and from strain A. J. Biol. Chem. 241, 2995–3001. [PubMed] [Google Scholar]
- Bradrick T. D.; Shattuck C.; Strader M. B.; Wicker C.; Eisenstein E.; Howell E. E. (1996) Redesigning the quaternary structure of R67 dihydrofolate reductase. Creation of an active monomer from a tetrameric protein by quadruplication of the gene. J. Biol. Chem. 271, 28031–28037. 10.1074/jbc.271.45.28031. [DOI] [PubMed] [Google Scholar]
- Dundas J.; Ouyang Z.; Tseng J.; Binkowski A.; Turpaz Y.; Liang J. (2006) CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 34, W116–118. 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams E. A.; Morrison J. F. (1992) Human dihydrofolate reductase: reduction of alternative substrates, pH effects, and inhibition by deazafolates. Biochemistry 31, 6801–6811. 10.1021/bi00144a022. [DOI] [PubMed] [Google Scholar]
- Zhao X.; Barber-Singh J.; Shippy S. A. (2004) MALDI-TOF MS detection of dilute, volume-limited peptide samples with physiological salt levels. Analyst 129, 817–822. 10.1039/b407322c. [DOI] [PubMed] [Google Scholar]
- Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619. 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A. (1984) Dynamics of Enzymatic-Reactions. Proc. Natl. Acad. Sci. U. S. A. 81, 444–448. 10.1073/pnas.81.2.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A. (1991) Computer Modeling of Chemical Reactions in Enzymes, Wiley-Interscience, New York. [Google Scholar]
- Wang S.; Lee R. J.; Mathias C. J.; Green M. A.; Low P. S. (1996) Synthesis, purification, and tumor cell uptake of 67Ga-deferoxamine-folate, a potential radiopharmaceutical for tumor imaging. Bioconjugate Chem. 7, 56–62. 10.1021/bc9500709. [DOI] [PubMed] [Google Scholar]
- Wang S.; Luo J.; Lantrip D. A.; Waters D. J.; Mathias C. J.; Green M. A.; Fuchs P. L.; Low P. S. (1997) Design and synthesis of [111In]DTPA-folate for use as a tumor-targeted radiopharmaceutical. Bioconjugate Chem. 8, 673–679. 10.1021/bc9701297. [DOI] [PubMed] [Google Scholar]
- Segel I. H. (1975) Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems, John Wiley and Sons, New York. [Google Scholar]
- Tang Y. C.; Chang H. C.; Roeben A.; Wischnewski D.; Wischnewski N.; Kerner M. J.; Hartl F. U.; Hayer-Hartl M. (2006) Structural features of the GroEL-GroES nano-cage required for rapid folding of encapsulated protein. Cell 125, 903–914. 10.1016/j.cell.2006.04.027. [DOI] [PubMed] [Google Scholar]
- Radford S. E. (2006) GroEL: More than Just a folding cage. Cell 125, 831–833. 10.1016/j.cell.2006.05.021. [DOI] [PubMed] [Google Scholar]
- Zhou H. X. (2004) Protein folding and binding in confined spaces and in crowded solutions. J. Mol. Recognit. 17, 368–375. 10.1002/jmr.711. [DOI] [PubMed] [Google Scholar]
- Zhou H. X. (2004) Polymer models of protein stability, folding, and interactions. Biochemistry 43, 2141–2154. 10.1021/bi036269n. [DOI] [PubMed] [Google Scholar]
- Janin J. (1999) Wet and dry interfaces: the role of solvent in protein-protein and protein-DNA recognition. Structure 7, R277–279. 10.1016/S0969-2126(00)88333-1. [DOI] [PubMed] [Google Scholar]
- Chacko S.; Silverton E.; Kam-Morgan L.; Smith-Gill S.; Cohen G.; Davis D. (1995) Structure of an antibody-lysozyme complex unexpected effect of conservative mutation. J. Mol. Biol. 245, 261–274. 10.1006/jmbi.1994.0022. [DOI] [PubMed] [Google Scholar]
- Chong L. T.; Dempster S. E.; Hendsch Z. S.; Lee L. P.; Tidor B. (1998) Computation of electrostatic complements to proteins: a case of charge stabilized binding. Protein Sci. 7, 206–210. 10.1002/pro.5560070122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means G., and Feeney R. (1971) Chemical Modification of Proteins, Holden-Day, Inc., San Francisco. [Google Scholar]
- Gilles M. A.; Hudson A. Q.; Borders C. L. Jr. (1990) Stability of water-soluble carbodiimides in aqueous solution. Anal. Biochem. 184, 244–248. 10.1016/0003-2697(90)90675-Y. [DOI] [PubMed] [Google Scholar]
- Okamura-Ikeda K.; Fujiwara K.; Motokawa Y. (1999) Identification of the folate binding sites on the Escherichia coli T-protein of the glycine cleavage system. J. Biol. Chem. 274, 17471–17477. 10.1074/jbc.274.25.17471. [DOI] [PubMed] [Google Scholar]
- Hicks S. N.; Smiley R. D.; Hamilton J. B.; Howell E. E. (2003) Role of ionic interactions in ligand binding and catalysis of R67 dihydrofolate reductase. Biochemistry 42, 10569–10578. 10.1021/bi034643d. [DOI] [PubMed] [Google Scholar]
- Nakajima N.; Ikada Y. (1995) Mechanism of amide formation by carbodiimide for bioconjugation in aqueous media. Bioconjugate Chem. 6, 123–130. 10.1021/bc00031a015. [DOI] [PubMed] [Google Scholar]
- Carraway K. L.; Koshland D. E. Jr. (1968) Reaction of tyrosine residues in proteins with carbodiimide reagents. Biochim. Biophys. Acta, Protein Struct. 160, 272–274. 10.1016/0005-2795(68)90102-5. [DOI] [PubMed] [Google Scholar]
- Carraway K. L.; Triplett R. B. (1970) Reaction of carbodiimides with protein sulfhydryl groups. Biochim. Biophys. Acta, Protein Struct. 200, 564–566. 10.1016/0005-2795(70)90112-1. [DOI] [PubMed] [Google Scholar]
- Nika H.; Nieves E.; Hawke D. H.; Angeletti R. H. (2013) C-terminal protein characterization by mass spectrometry using combined micro scale liquid and solid-phase derivatization. Journal of biomolecular techniques: JBT 24, 17–31. 10.7171/jbt.13-2401-003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S.; Murthy N. (2007) Targeted delivery of catalase and superoxide dismutase to macrophages using folate. Biochem. Biophys. Res. Commun. 360, 275–279. 10.1016/j.bbrc.2007.06.054. [DOI] [PubMed] [Google Scholar]
- Howell E. E.; Shukla U.; Hicks S. N.; Smiley R. D.; Kuhn L. A.; Zavodszky M. I. (2001) One site fits both: a model for the ternary complex of folate + NADPH in R67 dihydrofolate reductase, a D2 symmetric enzyme. J. Comput.-Aided Mol. Des. 15, 1035–1052. 10.1023/A:1014824725891. [DOI] [PubMed] [Google Scholar]
- Feng J.; Goswami S.; Howell E. E. (2008) R67, the other dihydrofolate reductase: Rational design of an alternate active site configuration. Biochemistry 47, 555–565. 10.1021/bi701455x. [DOI] [PubMed] [Google Scholar]
- Hicks S. N.; Smiley R. D.; Stinnett L. G.; Minor K. H.; Howell E. E. (2004) Role of Lys-32 residues in R67 dihydrofolate reductase probed by asymmetric mutations. J. Biol. Chem. 279, 46995–47002. 10.1074/jbc.M404484200. [DOI] [PubMed] [Google Scholar]
- Park C.; Raines R. T. (2001) Quantitative analysis of the effect of salt concentration on enzymatic catalysis. J. Am. Chem. Soc. 123, 11472–11479. 10.1021/ja0164834. [DOI] [PubMed] [Google Scholar]
- Wang L.; Goodey N. M.; Benkovic S. J.; Kohen A. (2006) Coordinated effects of distal mutations on environmentally coupled tunneling in dihydrofolate reductase. Proc. Natl. Acad. Sci. U. S. A. 103, 15753–15758. 10.1073/pnas.0606976103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh P.; Abeysinghe T.; Kohen A. (2015) Linking protein motion to enzyme catalysis. Molecules 20, 1192–1209. 10.3390/molecules20011192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahashiri A.; Howell E. E.; Kohen A. (2008) Tuning of the H-Transfer Coordinate in Primitive versus Well-Evolved Enzymes. ChemPhysChem 9, 980–982. 10.1002/cphc.200800067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Loughlin T. L.; Patrick W. M.; Matsumura I. (2006) Natural history as a predictor of protein evolvability. Protein Eng., Des. Sel. 19, 439–442. 10.1093/protein/gzl029. [DOI] [PubMed] [Google Scholar]
- Stinnett L. G.; Smiley R. D.; Hicks S. N.; Howell E. E. (2004) ″Catch 222,″ the effects of symmetry on ligand binding and catalysis in R67 dihydrofolate reductase as determined by mutations at Tyr-69. J. Biol. Chem. 279, 47003–47009. 10.1074/jbc.M404485200. [DOI] [PubMed] [Google Scholar]
- Strader M. B.; Smiley R. D.; Stinnett L. G.; VerBerkmoes N. C.; Howell E. E. (2001) Role of S65, Q67, I68, and Y69 residues in homotetrameric R67 dihydrofolate reductase. Biochemistry 40, 11344–11352. 10.1021/bi0110544. [DOI] [PubMed] [Google Scholar]
- Park H.; Bradrick T. D.; Howell E. E. (1997) A glutamine 67 to histidine mutation in homotetrameric R67 dihydrofolate reductase results in four mutations per single active site pore and causes substantial substrate and cofactor inhibition. Protein Eng., Des. Sel. 10, 1415–1424. 10.1093/protein/10.12.1415. [DOI] [PubMed] [Google Scholar]
- Strader M. B.; Chopra S.; Jackson M.; Smiley R. D.; Stinnett L.; Wu J.; Howell E. E. (2004) Defining the binding site of homotetrameric R67 dihydrofolate reductase and correlating binding enthalpy with catalysis. Biochemistry 43, 7403–7412. 10.1021/bi049646k. [DOI] [PubMed] [Google Scholar]
- Mittag T.; Orlicky S.; Choy W. Y.; Tang X.; Lin H.; Sicheri F.; Kay L. E.; Tyers M.; Forman-Kay J. D. (2008) Dynamic equilibrium engagement of a polyvalent ligand with a single-site receptor. Proc. Natl. Acad. Sci. U. S. A. 105, 17772–17777. 10.1073/pnas.0809222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borg M.; Mittag T.; Pawson T.; Tyers M.; Forman-Kay J. D.; Chan H. S. (2007) Polyelectrostatic interactions of disordered ligands suggest a physical basis for ultrasensitivity. Proc. Natl. Acad. Sci. U. S. A. 104, 9650–9655. 10.1073/pnas.0702580104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson M.; Chopra S.; Smiley R. D.; Maynord P. O.; Rosowsky A.; London R. E.; Levy L.; Kalman T. I.; Howell E. E. (2005) Calorimetric studies of ligand binding in R67 dihydrofolate reductase. Biochemistry 44, 12420–12433. 10.1021/bi050881s. [DOI] [PubMed] [Google Scholar]
- Feng J.; Grubbs J.; Dave A.; Goswami S.; Horner C. G.; Howell E. E. (2010) Radical redesign of a tandem array of four R67 dihydrofolate reductase genes yields a functional, folded protein possessing 45 substitutions. Biochemistry 49, 7384–7392. 10.1021/bi1005943. [DOI] [PubMed] [Google Scholar]
- Xu Q.; Guo H.; Wlodawer A.; Guo H. (2006) The importance of dynamics in substrate-assisted catalysis and specificity. J. Am. Chem. Soc. 128, 5994–5995. 10.1021/ja058831y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.