

Abstract
Myofibrillar myopathies (MFM) are a group of disorders associated with mutations in DES, CRYAB, MYOT, ZASP, FLNC, or BAG3 genes and characterized by disintegration of myofibrils and accumulation of degradation products into intracellular inclusions. We retrospectively evaluated 53 MFM patients from 35 Spanish families. Studies included neurologic exam, muscle imaging, light and electron microscopic analysis of muscle biopsy, respiratory function testing and cardiologic work-up. Search for pathogenic mutations was accomplished by sequencing of coding regions of the six genes known to cause MFM. Mutations in MYOT were the predominant cause of MFM in Spain affecting 18 of 35 families, followed by DES in 11 and ZASP in 3; in 3 families the cause of MFM remains undetermined. Comparative analysis of DES, MYOT and ZASP associated phenotypes demonstrates substantial phenotypic distinctions that should be considered in studies of disease pathogenesis, for optimization of subtype-specific treatments and management, and directing molecular analysis.
Keywords: Myofibrillar myopathy, Desmin, Myotilin, Zasp
1. Introduction
Myofibrillar myopathies (MFMs) are a group of genetically heterogenic disorders having in common myopathological features of disintegration of myofibrils and accumulation of degradation products into inclusions containing desmin and other myofibrillar or ectopic proteins [1]. The proteins involved in MFM vary significantly in their structure and function. Desmin is a muscle-specific type III intermediate filament protein interlinking myofibrils at the level of the Z-disc and connecting them to other cellular organelles thus maintaining the structural and functional integrity of the muscle cell [2]. αB-crystallin belongs to the small heat-shock protein family and serves in the muscle as a chaperone for desmin and other proteins assuring their normal folding [3]. Filamin C cross-links actin at the Z-disc level, binds to other Z-disc proteins and interacts with the dystrophin–dystroglycan complex at the sarcolemma [4]. BAG3 participates in antiapoptotic pathways and exerts co-chaperon activity by binding to Hsp70/HSC70 [5].
Myotilin is a sarcomeric Z-disc protein expressed strongly in skeletal and weakly in cardiac muscle [6]. Myotilin plays a significant role in sarcomere assembly, acting together with α-actinin and filamin C to cross-link actin into tightly packed bundles. The resulting structures support the integrity of the contracting muscle cell [7]. Myotilin is encoded by a single copy gene (MYOT, TTID) located on chromosome 5q31 [8,9]. Mutations in MYOT were originally described in autosomal dominant limb girdle muscular dystrophy type 1A (LGMD1A) [10], and subsequently identified in a subgroup of patients with myofibrillar myopathy [11] as well as a family with spheroid body myopathy [12]. Considering pathological similarity of LGMD 1A [13] and spheroid body myopathy to MFM, these disorders are now classified as MFM.
ZASP (Z band alternatively spliced PDZ-containing protein), also known as LIM-domain-binding-3 (LDB3), CYPHER or Oracle, is a Z-disc-associated protein of skeletal and cardiac muscles. The N-terminal PDZ domain interacts with the C terminus of α-actinin-2, the major component of the Z-disc [14,15]. ZASP also interacts with filamin C and calsarcin, both of which in turn bind to myotilin [16,17]. Mutations in human ZASP gene were discovered in patients suffering from dilated cardiomyopathy or left ventricular non-compaction [18], and another set of ZASP mutations was identified in patients with MFM [19]. One of these mutations was also identified in a large family originally reported by Markesbery et al. [20,21].
No systematic studies reflecting on the relative frequency and comparative phenotypic characteristics of various types of MFM in a single population have been conducted. This study is based on a nation-wide ascertainment and clinical, pathological and molecular analysis of MFM cases in Spain.
2. Materials and methods
2.1. Patient identification and evaluation
Patients with suspected MFM were systematically identified, examined and documented by the study participants within a timeframe of more than 10 years. Periodic patient follow-ups were conducted. Each member of an affected family was fully studied; most commonly, the first patient referred for a study was considered as index case. The following investigations were carried out in each patient: pedigree analysis, neurologic exam, including muscle strength assessment according to the Medical Research Council (MRC) grading scale, serum CK level assessment, electrophysiological studies that included nerve conduction tests and concentric needle EMG, respiratory function tests and cardiologic examination with electrocardiography and echocardiography. Muscle Computer tomography (CT) or Magnetic Resonance Images (MRI) were taken at mid-thigh and mid-calf levels. Time elapsed between the disease onset and the muscle imaging examination varied from 2 to 23 years. Diagnostic muscle biopsy was performed in each sporadic case and at least one patient per affected family. Histopathological diagnostic criteria were similar to those suggested earlier: alterations in trichromatically stained muscle sections consisting of intracellular accumulation of amorphous, hyaline, or granular material that represents ectopic protein deposits immunoreacting with desmin and ultrastructural analysis demonstrating myofibrillar degeneration and disintegration of the sarcomere [22,23]. Data on 22 of 53 patients included in the present study have been described or briefly mentioned in previous publications [24–31].
2.2. Mutation screening and genotyping
Each index case was tested for the presence of mutations in the coding regions of DES, CRYAB, MYOT, ZASP, FLNC, and BAG3 genes known to cause MFM. The priorities for testing of individual genes were established based on clinical, muscle imaging and pathological analysis, which was a learning process that led eventually to a point at which the affected gene could be predicted with an almost 100% certainty.
PCR amplification with intronic primers constructed to amplify each exon was carried out by using an optimal procedure designed for each segment. The resulting DNA fragments were sequenced using BigDyeTerminatore™ protocol on an automated 3100 ABI Prism Genetic Analyzer (Applied Biosystems, Foster City, CA). Mutation identification was accomplished by aligning with database sequences (http://www.ncbi.nlm.nih.gov); positive and negative controls were included as part of each test.
Three microsatellite markers (D5S2115, D5S479, D5S476) and four single nucleotide polymorphisms (SNP) located within a 4-centimorgan MYOT chromosomal region were typed in 16 patients and 48 Spanish control individuals. PCR amplification of microsatellite-containing fragments was performed with primers, one of which was fluorescently labeled. Fragment analysis and sequencing for assessing the SNP phase were performed on an automated 3100 ABI Prism Genetic Analyzer.
Clinical, myopathological and molecular genetic studies were conducted under clinical protocols approved by the Institutional Review Boards of each participating institution; genetic testing was performed in a CLIA-certified laboratory at the Clinical Neurogenetics Unit, National Institute of Neurological Disorders and Stroke, Bethesda, MD.
2.3. Muscle imaging
Muscle imaging studies were performed by using Computed Tomography or Magnetic Resonance Imaging at mid-thigh and mid-calf levels in 36 patients (11 desminopathy, 17 myotilinopathy, 5 zaspopathy, and three patients with no mutations in any of the genes known to cause MFM). The degree of muscle involvement was evaluated using a 5-point scale ranging from 0 to 4 as described [29].
2.4. Muscle biopsy
Open muscle biopsy was carried out for diagnostic purposes in each index case. Skeletal muscle tissue was obtained from a clinically affected muscle, or from a muscle showing involvement on muscle imaging studies. The samples were processed for routine histochemical tests and Congo red staining and immunohistochemistry by using commercial antibodies against myotilin, desmin, αB-crystallin, dystrophin, gelsolin and ubiquitin, and a mouse monoclonal anti-filamin C antibody kindly provided by Fürst and coworkers [32]. The source of antibodies we used is provided in Supplementary Table 1. A small sample of biopsy tissue was fixed in 2% glutaraldehyde, postfixed with 1% osmium tetroxide, and embedded in araldite. Ultrathin sections were stained with uranyl acetate and lead citrate and viewed with a JEOL 1011 electron microscope.
3. Results
3.1. Frequency and types of mutations identified in MFM genes
Mutations have been identified in DES, MYOT, or ZASP genes of 49 patients with clinical/pathological diagnosis of definite MFM originating from 32 families (Table 1). Evaluation of the relative role of the set of genes causing MFM shows that mutations in MYOT were the predominant cause of MFM in Spain affecting 18 of 35 studied families, followed by DES in 11 families and ZASP in 3. No cases with mutations in CRYAB, BAG3, or FLNC have so far been found. In 3 families the cause of MFM remains unidentified. Of the MYOT mutations, p.Ser55Phe and p.Ser60Cys were the most common causes of myotilinopathy in this cohort found in 7 families each. DES p.Leu392Pro, p.Leu370Pro, and p.Pro419Ser have been identified in large families. This is the first report of ZASP p.Ala174Thr and p.Ala165Val mutations in Spanish MFM patients that were identified in one and two families, respectively.
Table 1.
Number of families/patients affected with myofibrillar myopathy.
| Gene | No. of affected families | No. of patients |
|---|---|---|
| DESa | 11 | 18 |
| MYOTb | 18 | 24 |
| ZASPc | 3 | 7 |
| No mutations | 3 | 4 |
| Total | 35 | 53 |
DES mutations identified in Spanish families: p.Arg173_Glu179del, p.Asp214_Glu245del, p.Asn366del, p.Ile367Phe, p.Leu370Pro, p.Leu392-Pro, p. Arg406Pro, p.Pro419Ser.
MYOT mutations identified in Spanish families: p.Ser55Phe, p.Ser60-Cys, p.Ser60Phe, p.Lys36Glu.
ZASP mutations identified in Spanish families: p.Ala147Thr, p.Ala165Val.
3.2. Inheritance patterns
The pattern of inheritance was autosomal dominant in 20 of 35 affected families (Table 2). It was noticeable on comparison that a significant number of MYOT patients (9 of 18), especially those carrying the p.Ser55Phe mutation, did not have family history of the disease. Genotyping of two families and 4 sporadic cases carrying the p.Ser55Phe mutation identified a core haplotype consisting of alleles168–125-Phe-G-C-161 that is shared by eight patients. This haplotype was not found in controls. Subsequent inquiry established that all haplotype-sharing patients have arrived in Barcelona area from Murcia, a Southern region of Spain, and presumably are members of a founder population. There was a massive immigration to Catalonia from southern Spain during the second half of the 20th century. Seven patients carrying the second frequent p.Ser60Cys mutation were similarly genotyped but did not show a common haplotype.
Table 2.
Summary of clinical manifestations observed in patients with mutations in DES, MYOT or ZASP, and patients in which no mutation has been identified.
| Mutated gene
|
||||
|---|---|---|---|---|
| DES | MYOT | ZASP | Unknown | |
| Number of families | 11 | 18 | 3 | 3 |
| Inheritance | ||||
| AD | 6 | 9 | 3 | 1 |
| AR | 1 | 0 | 0 | 0 |
| Sporadica | 4 | 9 | 0 | 2 |
| Number of studied patients | 18 | 24 | 7 | 4 |
| Onset age, mean (yrs) | 28 | 58 | 56 | 56 |
| Standard deviation | 10.1 | 8.7 | 7.1 | 12.2 |
| Gender | ||||
| Male | 8 | 13 | 3 | 2 |
| Female | 10 | 11 | 4 | 2 |
| Initial symptoms | ||||
| Distal LL | 8 | 19 | 6 | 3 |
| Proximal LL | 0 | 3 | 1 | 1 |
| Distal and proximal | 2 | 2 | 0 | 0 |
| Cardiopathy | 8 | 0 | 0 | 0 |
| Advanced illness | ||||
| Distal LL alone | 0 | 4 | 0 | 0 |
| Distal and proximal 4 limbs | 18 | 19 | 4 | 4 |
| Neck/trunk weakness | 7 | 1 | 0 | 0 |
| Facial weakness | 6 | 0 | 1 | 0 |
| Dysphagia | 7 | 2 | 0 | 0 |
| Myalgia/stiffness | 1 | 11 | 2 | 0 |
| Muscle atrophy | 11 | 7 | 5 | 2 |
| Muscle hypertrophy | 0 | 7 | 0 | 0 |
| Respiratory weakness | 9 | 3 | 0 | 1 |
| Neuropathy | 0 | 1 | 0 | 0 |
| Cardiopathy | 16 | 1(?) | 1(?) | 0 |
| Muscle imaging | ||||
| Semitendinosus | 11/11 | 2/17 | 0/5 | 0/3 |
| Semimembranosus | 2/11 | 15/17 | 4/5 | 2/3 |
| Sartorius | 11/11 | 3/17 | 0/5 | 1/3 |
| Gracilis | 9/11 | 1/17 | 0/5 | 0/3 |
| Vastus intermedius/medialis | 3/11 | 10/17 | 3/5 | 2/3 |
| Adductor magnus | 2/11 | 15/17 | 4/5 | 2/3 |
| Biceps femoris | 1/11 | 16/17 | 3/5 | 2/3 |
| Peroneus | 11/11 | 11/17 | 2/5 | 1/3 |
| Soleus | 3/11 | 16/17 | 5/5 | 1/3 |
| Anterior tibialis | 5/11 | 14/17 | 1/5 | 0/3 |
| Medial gastrocnemius | 7/11 | 16/17 | 5/5 | 2/3 |
| Lateral gastrocnemius | 5/11 | 10/17 | 3/5 | 1/3 |
| Disease outcome | ||||
| Wheelchair dependency | 8 | 8 | 2 | 2 |
| Pacemaker/defibrillator | 11 | 0 | 0 | 0 |
| Heart transplantation | 2 | 0 | 0 | 0 |
| Sudden death | 3 | 0 | 0 | 0 |
AD, autosomal dominant; AR, autosomal recessive; LL, lower limbs; UL, upper limbs.
The number of studied patients is indicated as denominator if different from the total.
The parents of these patients were not available for study, therefore it is unknown if the mutations have arisen de novo.
3.3. Clinical manifestations
The age of disease onset was significantly earlier in patients with DES mutations (mean, 28 years; 95% confidence interval [CI] 15.9–36.1) as compared to MYOT (mean, 58; 95% CI 49.3–64.7) or ZASP (mean 56; 95% CI 48.9–63.1). This characterizes MYOT- and ZASP-associated myopathy as a late-onset disease.
Bilateral weakness in the distal lower limb muscles manifesting as foot drop was the predominant initial symptom; proximal muscle weakness leading to difficulties with standing up, climbing stairs or rising arms was the initial sign in only a few patients (3 MYOT and 1 ZASP patients). Cardiopathy was the initial clinical manifestation in 8 patients with DES and none of the MYOT or ZASP patients (Table 2).
In the course of illness, most MFM patients of each subtype developed weakness in both distal and proximal leg muscles and in the majority of patients muscle weakness extended to the upper limbs (Table 2). Facial, neck flexor and trunk muscles were involved quite frequently in patients with DES but not MYOT or ZASP mutations (Table 2). Most significantly, 16 of 18 patients with DES mutations developed cardiopathy in the course of illness, while only 1 elderly patient with MYOT and 1 with a ZASP mutation had suspected cardiac involvement, most likely unrelated to MFM. Among patients with DES mutations, atrioventricular conduction blocks were recorded in 16 (89%), episodes of tachyarrhythmia in 12 patients (67%) and 10 patients developed heart failure as a result of dilated, restrictive or hypertrophic cardiomyopathy confirmed by echocardiographic examination. The disease outcome in these patients was most severe: three DES patients died suddenly at an early age and two received a heart transplant (Table 2). Five MYOT patients died between the ages of 82 and 86 years. Nine patients with DES mutations had restrictive ventilatory insufficiency, while only 3 of MYOT and none of ZASP patients showed signs of respiratory weakness.
Muscle imaging disclosed characteristic patterns of muscle involvement (Fig. 1, Table 2). In patients with DES mutations, the earliest and most consistently affected muscle at mid-thigh is semitendinosus (11/11 patients), followed by sartorius and gracilis. The semimembranosus shows minimal damage. On the contrary, the semimembranosus, hip adductors and biceps femoris were initially affected in MYOT (15/17, 15/17 and 16/17, respectively), and ZASP (4/5, 4/5 and 3/5) subtypes, whereas the semitendinosus remained spared. Of the distal muscles, the earliest abnormalities in DES patients occurred in the peroneal muscles (11/11 patients), followed by tibialis anterior and the posterior compartment muscles (soleus, medial, and lateral gastrocnemius), while in MYOT and ZASP cases the first affected was soleus (16/17 and 5/5 patients) and medial gastrocnemius and subsequently the anterior tibialis and the peroneal group.
Fig. 1.
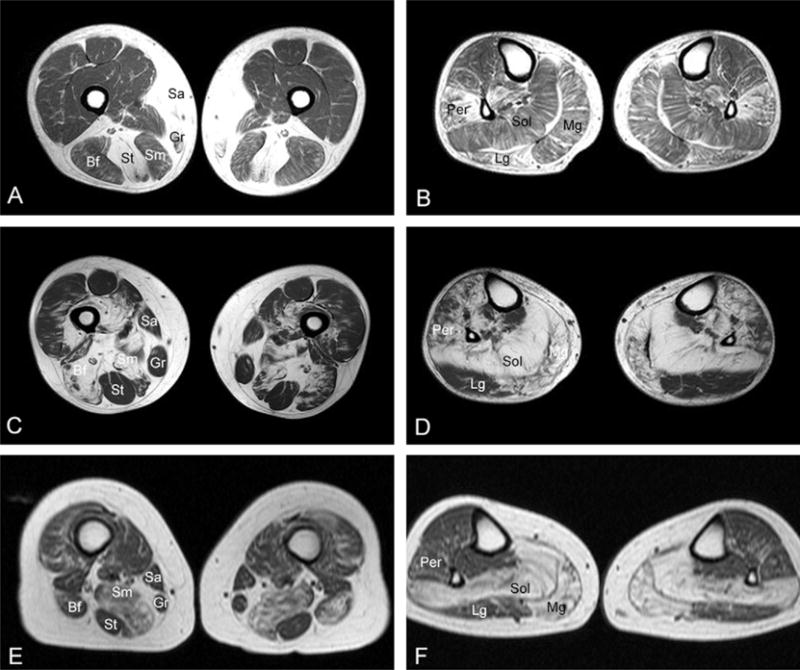
Muscle MRI images in myofibrillar myopathy patients with DES, MYOT, or ZASP mutations. In patients with DES (A, B) mutations, selective involvement of the semitendinosus (St), gracilis (Gr), sartorius (Sa) and peroneal muscles (Per) occur in the early stages of illness. In a patient with a MYOT mutation (C, D), the m. semimembranosus (Sm), biceps femoris (Bf), soleus (Sol), and medial gastrocnemius (Mg), are primarily involved. Less severe abnormalities are seen in the anterior tibialis and peroneal group. The lateral gastrocnemius (Lg) is initially preserved. A similar pattern of involvement is seen in a patient with ZASP mutation (E, F). Note the preservation of semitendinosus, sartorius and gracilis in patients with MYOT and ZASP mutations. A and B: p.Pro419Ser DES; C and D: Ser60Cys MYOT; E and F: p.Ala174Thr ZASP.
Creatine kinase level was normal to moderately elevated and EMG examination shows signs of myopathy with spontaneous activity at rest manifested by positive sharp waves, fibrillation potentials, and high bizarre discharges in most cases. None of the studied patients had peripheral nerve conduction abnormality, except for a single patient with a MYOT mutation.
3.4. Myopathology
Features shared by DES, MYOT and ZASP subtypes include nonspecific myopathic changes ranging from mild variation in the fiber size and increased number of internal nuclei to more advanced degenerative abnormalities comprising muscle fiber atrophy and hypertrophy, fiber splitting, and fibro-fatty tissue proliferation (Table 3, Fig. 2). Nuclear clumps are common in patients with MYOT and ZASP mutations (16/20 and 3/3) but rare in DES patients (3/12) (Table 3, Fig. 2). MYOT subtype is characterized by a more frequent presence of muscle fiber necrosis with phagocytosis (13/20) and in some cases small inflammatory infiltrates (6/20). Collections of cytoplasmic bodies and some nemaline-like bodies appearing as red granules on modified trichrome stain are frequent in both MYOT and ZASP (18/20 and 3/3) but rare in DES patients (3/12) (Table 3, Fig. 2). In addition, spheroid inclusion bodies stained green on modified trichrome were observed in most MYOT and ZASP cases (18/20 and 3/3), but infrequently in DES (2/12). Rimmed vacuoles were present in all studied cases, their number and size being larger in the MYOT and ZASP than DES. Patients with MYOT and ZASP subtypes had in addition a comparatively greater number of non-rimmed vacuoles (Fig. 2).
Table 3.
Myopathological findings in patients with DES, MYOT or ZASP subtype of myofibrillar myopathy.
| Mutated gene
|
|||
|---|---|---|---|
| DES | MYOT | ZASP | |
| Light microscopy | |||
| Number of studied patients | 12 | 20 | 3 |
| Variation of fiber size | 12 | 20 | 3 |
| Fibro-fatty tissue proliferation | 4 | 10 | 3 |
| Vesicular nuclei | 3 | 16 | 3 |
| Necrosis/phagocytosis | 3 | 13 | 2 |
| Inflammatory infiltrates | 1 | 6 | 1 |
| Nemaline-like bodies | 3 | 18 | 3 |
| Core-like lesions | 7 | 20 | 3 |
| Type I fiber predominance | 8 | 18 | 1 |
| Vacuoles | 11 | 17 | 3 |
| Non-hyaline amorphous inclusions | 11 | 10 | 3 |
| Hyaline dense inclusions | 4 | 20 | 3 |
| Spheroid inclusion bodies | 2 | 18 | 3 |
| Rubbed-out fibers | 11 | 6 | 0 |
| Electron microscopy | |||
| Number of studied patients | 9 | 17 | 4 |
| Granulofilamentous material | 9 | 8a | 1a |
| Z-line streaming | 6 | 17 | 4 |
| Tubulofilamentous inclusions | 1 | 14 | 2 |
| Filamentous bundles | 0 | 17 | 4 |
| Autophagic vacuoles | 7 | 17 | 1 |
Biopsied muscle in DES cases: D (n = 4); B (n = 4); AT (n = 1); G (n = 2); Q (n = 1); in MYOT cases: D (n = 6); B (n = 3); AT (n = 1); G (n = 3); Q (n = 6); Sol (n = 1); in ZASP cases: Q (n = 1); not indicated (n = 2), where D, deltoid; B, biceps brachii; AT, anterior tibialis; G, gastrocnemius; Q, quadriceps; Sol, soleus.
Granulofilamentous material in MYOT and ZASP cases was restricted to small foci in a few fibers.
Fig. 2.
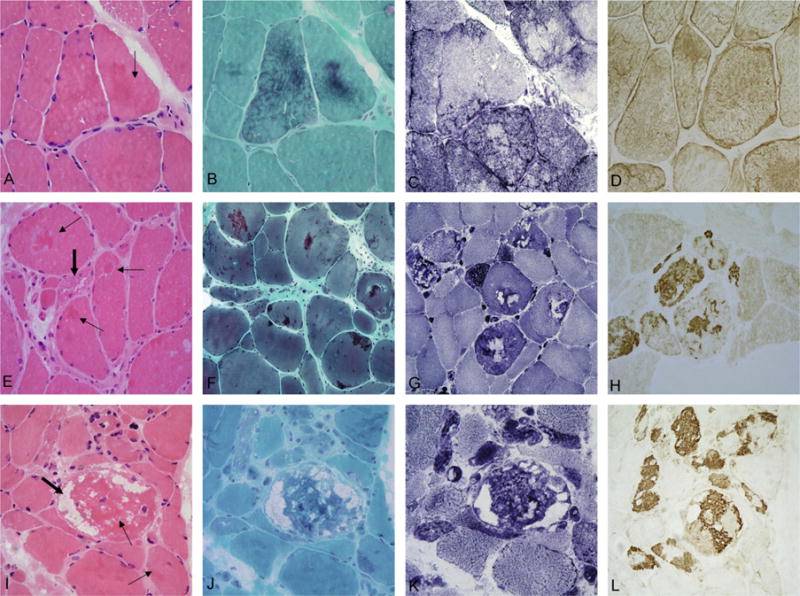
Light microscopy analysis of muscle biopsy samples from myofibrillar myopathy patients with DES, MYOT, or ZASP mutations. The most typical lesions in patients carrying mutations in DES (A–D) are characterized by thin, discrete patches of amorphous eosinophilic (arrow in A) material forming diffuse networks in the cytoplasm. These inclusions are best visualized on trichrome stain (B), are devoid of oxidative enzyme activity causing a “rubbed-out” appearance (C). Increased desmin (D) immunoreactivity is seen under the sarcolemma and within the cytoplasm. In patients with mutations in MYOT (E–H) or ZASP (I–L), dense intensely eosinophilic hyaline inclusions are observed on H&E staining (thin arrows in E and I). Large numbers of fibers contain red to purple inclusions (F) and non-rimmed vacuoles (thick arrows in E and I). On NADH staining (G and K) some abnormal areas lack oxidative enzyme activity whereas in other areas oxidative enzyme activity is increased. Prominent myotilin-immunoreactive aggregates and dense inclusion bodies (H and L) are observed. A–D: p.Ile367Phe DES; E–H: Ser55Phe MYOT; I–L: p.Ala165Val ZASP.
Intracytoplasmic inclusions, the morphological hallmark of MFM, are present in each studied case, but their morphological characteristics vary. In DES cases the inclusions appear as thin patches or spider-web-like formations under the sarcolemma or within the cytoplasm stained eosinophilic and less frequently basophilic on H&E, and green, bluish or red-bluish on modified trichrome stain (Fig. 2). They coexist with well demarcated inclusion bodies in a few cases. Congophilia is lacking or faint. Oxidative enzyme activity is absent in the inclusions, resulting in “rubbed-out” appearance in the affected areas of the muscle fiber (11/12 of DES patients, Table 3 and Fig. 2C). Core-like lesions are seen in the majority of cases (Table 3). Proteinaceous deposits forming sprout networks and less frequently granular or dot-like aggregates strongly react with antibodies against desmin, αB-crystallin, dystrophin and filamin C, and weakly with ubiquitin and myotilin (Fig. 2D).
In patients carrying MYOT and ZASP mutations, muscle fibers contain more dense hyaline material bright pink on H&E and blue/purple on modified trichrome stain (Fig. 2E and I). Round or spheroid inclusion bodies were observed in the majority of patients. Strong congophilia is a prominent feature. Inclusions similar to those seen in DES cases are also present. Irregular core-like lesions, generally surrounded by increased oxidative enzyme activity are encountered in all samples whereas “rubbed-out” fibers are rare (Fig. 2G and K). The inclusions show prominent desmin, αB-crystallin, dystrophin, myotilin (Fig. 2H and L), ubiquitin, and filamin C immunoreactivity.
At EM investigation, initial changes in DES cases feature small electron-dense dots in close proximity to the Z-lines, while larger dappled reticular granulofilamentous structures are seen disrupting the Z-lines in a more advanced stage (Fig. 3A–C). With disease progression, coarse granulofilamentous material covers larger areas centrally and under the plasma membrane. Groups of normally looking mitochondria are positioned between and sometimes within the granulofilamentous masses. Z-line streaming, glycogen granules and autophagic vacuoles containing myelin figures are observed in most DES cases. Studies of an explanted heart of a patient with DES mutation showed lesions identical to those observed in skeletal muscle (Fig. 3D). In a single patient, 15–18 nm tubulofilaments, a filamentous body and dense globoid inclusions were observed in a totaly degenerated fiber. In patients with MYOT and ZASP mutations, early lesions consist of streaming and widened Z-lines. In more severely affected fiber areas, filamentous bundles of Z-disc origin and fine filamentous debris are accumulating within the cytoplasm and subsarcolemmal regions and frequently form globoid inclusions (Fig. 3E, F, I and J). Collections of 15–18 nm tubulofilaments were frequently observed in MYOT (14/17 and 2/2 in Table 3, Fig. 3H). Typical nemaline bodies were observed in one MYOT case and were conspicuous in all ZASP cases. Intranuclear rods were observed in four ZASP samples (data not shown).
Fig. 3.
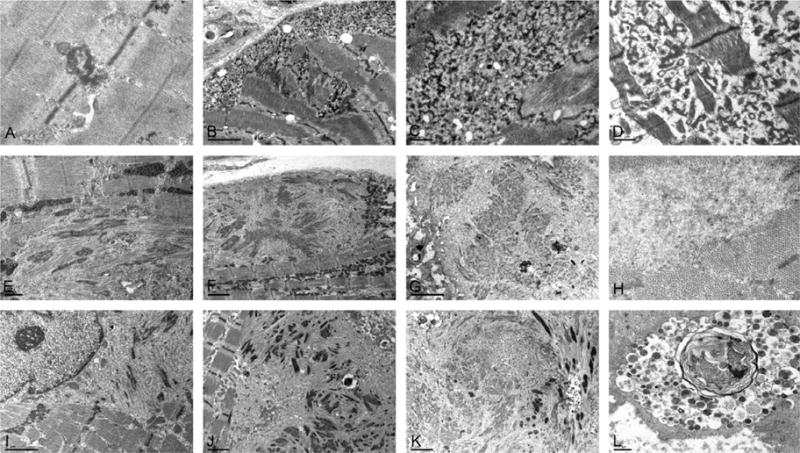
Ultrastructural findings in muscle biopsy samples from myofibrillar myopathy patients with DES, MYOT, or ZASP mutations. DES: early lesions consist of dappled electrondense structures emanating from the Z-discs (A). In more advanced lesions, a thick reticular network of electrondense granulofilamentous material forms in the subsarcolema and intermyofibrillar space (B, C); widening and streaming of Z-discs is also observed (B). Similar accumulation of granulofilamentous material is observed in cardiac muscle (D). MYOT: Dissolved myofibrils with disrupted Z-lines, first focally (E) and later spreading to larger areas and resulting in complete myofibrillar disorganization; multiple filamentous bundles of Z-disc origin and rod-like bodies (E, F), thin filaments and glycogen granules; groups of normal mitochondria surrounding the area of myofibrillar destruction (F); remants of filaments of various electron density accumulating into large inclusions (G); collections of 15–18 nm filaments are seen among normally looking myofibrils (H). ZASP: streaming and widening of the Z-line (I) is followed by sarcomeric disorganization and accumulation of filamentous bundles of Z-disc origin and filamentous debris (J); a spheroid inclusion composed of remants of filaments and rod-like bodies at the periphery (K); degraded material accumulates in autophagic vacuoles (L). A: p.Ile367Phe DES; B and C: p.Asp214_Glu245del DES; D: p.Arg406Pro DES E and F: p.Ser55Phe MYOT; G and H: p.Ser60Cys MYOT; I and J: p.Ala174Thr ZASP; L: p.Ala165Val ZASP. [Note: Figure shown in B is reproduced from Goldfarb L et al. (2010) Desminopathy. In Encyclopedia of Life Sciences. John Wiley & Sons Ltd: Chichester. DOI: 10.1002/9780470015902.a0006173.pub2, with permission from John Wiley & Sons Ltd.]
Mitochondria are absent within the disorganized masses but present at their periphery (Fig. 3F). Autophagic vacuoles (Fig. 3L) and small foci containing granulofilamentous material similar to those described in the DES cases are also observed in some MYOT and ZASP patients.
3.5. Patients with no mutations
Four patients from three families were negative for mutations in any of the known MFM causative genes. The age of onset in these patients varied from 40 to 69 years. Three presented with distal weakness of lower limbs and the remaining one had proximal weakness at onset. One patient developed respiratory insufficiency but none had cardiopathy or peripheral neuropathy. Muscle imaging performed in three cases showed a pattern of muscle involvement similar to that observed in patients carrying mutations in MYOT or ZASP genes. Muscle biopsies of three patients showed features similar to those described for MYOT and ZASP mutations. However, the numbers of muscle fibers containing myofibrillar inclusions were less numerous than in MYOT or ZASP cases.
4. Discussion
MFMs are an expanding and increasingly recognized group of muscular disorders caused by mutations in DES, CRYAB, MYOT, ZASP, FLNC, or BAG3 genes. A comprehensive study of 53 patients from 35 Spanish families molecularly identified as MFM and representing three gene-associated MFM subtypes allows to identify subtype-specific clinical, myopathological and genetic features. Evaluation of the relative frequency of these three subtypes in Spain shows that mutations in MYOT were the predominant cause of MFM, followed by DES and ZASP. This ratio indicates that the late-onset types of MFM with mutations in MYOT and ZASP are a relatively frequent cause of MFM in Spain which correlates with a previous report on the subtype distribution in the largest known series of 63 MFM patients studied in the Mayo Clinic [23]: DES 6%, MYOT 10%, CRYAB 3%, ZASP 15%, FLNC 3%, and mutations not found in 62%. Data presented here suggest that a MYOT p.Ser55Phe mutation may have spread in the founder population of Murcia of South-Eastern Spain and resulted in a relatively increased frequency of the MYOT associated subtype. A similar explanation has previously been suggested for the increased prevalence of C-filaminopathy in Germany [33] and desminopathy in Poland [25].
Our analysis identifies profound clinical differences between the groups of patients with relatively early-onset MFM caused by DES mutations and late-onset subtypes associated with mutations in MYOT and ZASP. Patients with DES mutations show a much wider distribution of muscle weakness spreading to the facial, bulbar, neck and trunk muscles in the advanced illness; these groups of muscles are rarely involved in MYOT and ZASP. Muscle imaging studies show a selective involvement of peroneus, semitendinosus and sartorius muscles at the disease onset in patients with DES mutations, while soleus and semimembranosus are seen the first to be involved in MYOT and ZASP cases. We thus confirm the conclusions regarding the differential topography of muscle involvement made previously [29,34]. Cardiopathy and respiratory weakness are frequently present in the early phase of desminopathy and lead to incapacity and death at young age, while these complications are rare in the late-onset MYOT and ZASP forms. Peripheral neuropathy reported in some cases of myotilinopathy and ZASPopathy [11,19] is not a consistent feature in our patients. A later age of disease onset predicts a more favorable disease outcome. Indeed, the disease progression in both MYOT and ZASP patients was very slow, they remained ambulatory until very late age.
Clinical analysis is corroborated by myopathological findings demonstrating that even though there are similarities between the subtypes, especially in muscle fibers with advanced destructive lesions, there are important distinguishing features. Characteristic hyaline inclusions corresponding at the ultrastructural level to fragmented and compacted filaments in MYOT and ZASP specimens are distinct from non-hyaline inclusions seen at sites occupied by granulofilamentous material in DES-associated cases. Vacuolar changes are most prominent in MYOT and ZASP patients. “Rubbed-out” fibers were regularly observed in DES cases and rarely seen in MYOT and ZASP specimens. These conclusions extend the results of an independent myopathological study based on a smaller number of patients [35].
Although differentiation between the MFM subtypes on the basis of clinical/pathological characteristics may sometimes be difficult, the awareness of clinical, imaging and pathological differences based on studies of representative groups of patients is important for further development of diagnostic criteria and subtype-specific treatments. This study refines clinical features, muscle imaging and pathological data of three subtypes of MFM and shows close similarity between patients with MYOT and ZASP mutations and a significant distinction between the late- and the earlier-onset myofibrillar myopathy caused by mutations in DES.
The inability to identify mutations in a small number of our patients may reflect the fact that there are other unknown genes also causing the MFM phenotype. Of other possible drawbacks, no attempt was made to sequence regulatory gene regions or explore a possibility that rare polymorphisms residing in the known MFM genes could in some circumstances become pathogenic.
MFM pathogenesis has been under intense investigation, including studies of transgenic mice and transfected cell cultures, as well as studies dealing with post-translational protein modifications, ectopic protein expression, abnormal protein aggregation and mechanisms of aberrant protein clearance (reviewed in [36]). However, the answers to many questions regarding the differences in disease phenotypes that may potentially influence subtype-specific treatments are still lacking. The use of new upcoming technologies will help solve these problems and speed up the development of novel therapies.
Supplementary Material
Acknowledgments
We would like to thank the patients and families for collaboration. We also wish to thank D. Moreno, C. Gimenez, and S. Arnedo for their excellent technical assistance. This work was supported in part by a FIS grant PI08-574 and the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, National Institutes of Health.
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.nmd.2011. 05.002.
References
- 1.Nakano S, Engel AG, Waclawik AJ, Emslie-Smith AM, Busis NA. Myofibrillar myopathy with abnormal foci of desmin positivity. 1. Light and electron microscopy analysis of 10 cases. J Neuropathol Exp Neurol. 1996;55:549–62. doi: 10.1097/00005072-199605000-00008. [DOI] [PubMed] [Google Scholar]
- 2.Herrmann H, Aebi U. Intermediate filaments: molecular structure, assembly mechanism, and integration into functionally distinct intracellular scaffolds. Annu Rev Biochem. 2004;73:749–89. doi: 10.1146/annurev.biochem.73.011303.073823. [DOI] [PubMed] [Google Scholar]
- 3.Vicart P, Caron A, Guicheney P, et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nature Genet. 1998;20:92–5. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- 4.Thompson TG, Chan YM, Hack AA, et al. Filamin 2 (FLN2): a muscle-specific sarcoglycan interacting protein. J Cell Biol. 2000;148:115–26. doi: 10.1083/jcb.148.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selcen D, Muntoni F, Burton BK, et al. Mutation in BAG3 causes severe dominant childhood muscular dystrophy. Ann Neurol. 2009;65:83–9. doi: 10.1002/ana.21553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salmikangas P, van der Ven P, Lalowski M, et al. Myotilin, the limb-girdle muscular dystrophy 1A (LGMD 1A) protein, cross-links actin filaments and controls sarcomere assembly. Hum Mol Genet. 2003;12:189–203. doi: 10.1093/hmg/ddg020. [DOI] [PubMed] [Google Scholar]
- 7.van der Ven P, Wiesner S, Salmikangas P, et al. Indications for a novel muscular dystrophy pathway: g-filamin, the muscle-specific filamin isoform, interacts with myotilin. J Cell Biol. 2000;151:235–48. doi: 10.1083/jcb.151.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Salmikangas P, Mykkänen O-M, Grönholm M, Heiska L, Kere J, Carpén O. Myotilin, a novel sarcomeric protein with two Ig-like domains, is encoded by a candidate gene for limb-girdle muscular dystrophy. Hum Mol Genet. 1999;8:1329–36. doi: 10.1093/hmg/8.7.1329. [DOI] [PubMed] [Google Scholar]
- 9.Yamaoka LH, Westbrook CA, Speer MC, et al. Development of a microsatellite genetic map spanning 5q31–q33 andsubsequent placement of the LGMD1A locus between D5S178 and IL9. Neuromuscul Disord. 1994;4:471–5. doi: 10.1016/0960-8966(94)90086-8. [DOI] [PubMed] [Google Scholar]
- 10.Hauser MA, Horrigan SK, Salmikangas P, et al. Myotilin is mutated in limb girdle muscular dystrophy 1 A. Hum Mol Genet. 2000;14:2141–7. doi: 10.1093/hmg/9.14.2141. [DOI] [PubMed] [Google Scholar]
- 11.Selcen D, Engel AG. Mutations in myotilin cause myofibrillar myopathy. Neurology. 2004;62:1363–71. doi: 10.1212/01.wnl.0000123576.74801.75. [DOI] [PubMed] [Google Scholar]
- 12.Foroud T, Pankratz N, Batchman AP, et al. A mutation in myotilin causes spheroid body myopathy. Neurology. 2005;65:1936–40. doi: 10.1212/01.wnl.0000188872.28149.9a. [DOI] [PubMed] [Google Scholar]
- 13.Garvey SM, Miller SE, Claflin DR, Faulkner JA, Hauser MA. Transgenic mice expressing the myotilin T57I mutation unite the pathology associated with LGMD1A and MFM. Hum Mol Genet. 2006;15:2348–62. doi: 10.1093/hmg/ddl160. [DOI] [PubMed] [Google Scholar]
- 14.Faulkner G, Pallavicini A, Formentin E, et al. ZASP: a new Z-band alternatively spliced PDZ-motif protein. J Cell Biol. 1999;146:465–75. doi: 10.1083/jcb.146.2.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou Q, Ruiz-Lozano P, Martone ME, Chen J. Cypher, a striated muscle-restricted PDZ and LIM domain-containing protein, binds to alpha-actinin-2 and protein kinase C. J Biol Chem. 1999;274:19807–13. doi: 10.1074/jbc.274.28.19807. [DOI] [PubMed] [Google Scholar]
- 16.Frey N, Olson EN. Calsarcin-3, a novel skeletal muscle-specific member of the calsarcin family, interacts with multiple Z-disc proteins. J Biol Chem. 2002;277:13998–4004. doi: 10.1074/jbc.M200712200. [DOI] [PubMed] [Google Scholar]
- 17.Klaavuniemi T, Ylänne J. Zasp/Cypher internal ZM-motif containing fragments are sufficient to co-localize with alpha-actinin–analysis of patient mutations. Exp Cell Res. 2006;312:1299–12311. doi: 10.1016/j.yexcr.2005.12.036. [DOI] [PubMed] [Google Scholar]
- 18.Vatta M, Mohapatra B, Jimenez S, et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non-compaction. J Am Coll Cardiol. 2003;42:2014–27. doi: 10.1016/j.jacc.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 19.Selcen D, Engel AG. Mutations in ZASP define a novel form of muscular dystrophy in humans. Ann Neurol. 2005;57:269–76. doi: 10.1002/ana.20376. [DOI] [PubMed] [Google Scholar]
- 20.Markesbery WR, Griggs RC, Leach RP, Lapham LW. Late onset hereditary distal myopathy. Neurology. 1974;24:127–34. doi: 10.1212/wnl.24.2.127. [DOI] [PubMed] [Google Scholar]
- 21.Griggs R, Vihola A, Hackman P, et al. Zaspopathy in a large classic late-onset distal myopathy family. Brain. 2007;130:1477–84. doi: 10.1093/brain/awm006. [DOI] [PubMed] [Google Scholar]
- 22.Dalakas MC, Park K-Y, Semino-Mora LeeHS, Sivakumar K, Goldfarb LG. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med. 2000;342:770–80. doi: 10.1056/NEJM200003163421104. [DOI] [PubMed] [Google Scholar]
- 23.Selcen D, Engel AG. Myofibrillar myopathy. GeneReviews. 2010 http://www.genereviews.org/
- 24.Dagvadorj A, Olivé M, Urtizberea JA, et al. A West European cluster of severe cardiac and skeletal myopathy associated with a de novo R406W mutation in desmin. J Neurol. 2004;251:143–9. doi: 10.1007/s00415-004-0289-3. [DOI] [PubMed] [Google Scholar]
- 25.Kaminska A, Strelkov SV, Goudeau B, et al. Small deletions disturb desmin architecture leading to breakdown of muscle cells and development of skeletal or cardioskeletal myopathy. Hum Genet. 2004;114:306–13. doi: 10.1007/s00439-003-1057-7. [DOI] [PubMed] [Google Scholar]
- 26.Olivé M, Goldfarb L, Shatunov A, Fischer D, Ferrer I. Myotilinopathy: refining the clinical and myopathological phenotype. Brain. 2005;128:2315–26. doi: 10.1093/brain/awh576. [DOI] [PubMed] [Google Scholar]
- 27.Arias M, Pardo J, Blanco-Arias P, et al. Distinct phenotypic features and gender-specific disease manifestations in a Spanish family with desmin L370P mutation. Neuromuscul Disord. 2006;16:498–503. doi: 10.1016/j.nmd.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 28.Olive M, Armstrong J, Miralles F, et al. Phenotypic patterns of desminopathy associated with three novel mutations in the desmin gene. Neuromuscul Disord. 2007;17:443–50. doi: 10.1016/j.nmd.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fischer D, Kley RA, Strach K, et al. Distinct muscle imaging patterns in myofibrillar myopathies. Neurology. 2008;71:758–65. doi: 10.1212/01.wnl.0000324927.28817.9b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gamez J, Armstrong J, Shatunov A, et al. Generalized muscle pseudo-hypertrophy and stiffness associated with the myotilin Ser55Phe mutation: A novel myotilinopathy phenotype? J Neurol Sci. 2009;277:167–71. doi: 10.1016/j.jns.2008.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piñol-Ripoll G, Shatunov A, Cabello A, et al. Severe infantile-onset cardiomyopathy associated with a homozygous deletion in desmin. Neuromuscul Disord. 2009;19:418–22. doi: 10.1016/j.nmd.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Ven PFM, Obermann WMJ, Lemke B, Gautel M, Weber K, Fürst DO. The characterization of mouse filamin isoforms suggest a possible role of gamma-filamin/ABP-L in sarcomeric Z-disc formation. Cell Motil Cytoskelet. 2000;45:149–62. doi: 10.1002/(SICI)1097-0169(200002)45:2<149::AID-CM6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 33.Kley RA, Hellenbroich Y, van der Ven PF, et al. Clinical and morphological phenotype of the filamin myopathy: a study of 31 German patients. Brain. 2007;130:3250–64. doi: 10.1093/brain/awm271. [DOI] [PubMed] [Google Scholar]
- 34.Schramm N, Born C, Weckbach S, Reilich P, Walter MC, Reiser MF. Involvement patterns in myotilinopathy and desminopathy detected by a novel neuromuscular whole-body MRI protocol. Eur Radiol. 2008;18:2922–36. doi: 10.1007/s00330-008-1071-1. [DOI] [PubMed] [Google Scholar]
- 35.Claeys KG, Fardeau M, Schröder R, et al. Electron microscopy in myofibrillar myopathies reveal clues to the mutated gene. Neuromuscul Disord. 2008;18:656–66. doi: 10.1016/j.nmd.2008.06.367. [DOI] [PubMed] [Google Scholar]
- 36.Ferrer I, Olivé M. Molecular pathology of myofibrillar myopathies. Expert Rev Mol Med. 2008;10:e25. doi: 10.1017/S1462399408000793. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.