

Abstract
Adenosine kinase (ADK) deficiency in human patients (OMIM:614300) disrupts the methionine cycle and triggers hypermethioninemia, hepatic encephalopathy, cognitive impairment, and seizures. To identify whether this neurological phenotype is intrinsically based on ADK deficiency in the brain or if it is secondary to liver dysfunction, we generated a mouse model with a brain-wide deletion of ADK by introducing a Nestin-Cre transgene into a line of conditional ADK deficient Adkfl/fl mice. These AdkΔbrain mice developed a progressive stress-induced seizure phenotype associated with spontaneous convulsive seizures and profound deficits in hippocampus-dependent learning and memory. Pharmacological, biochemical, and electrophysiological studies suggest enhanced adenosine levels around synapses resulting in an enhanced adenosine A1 receptor (A1R)-dependent protective tone despite lower expression levels of the receptor. Theta-burst-induced LTP was enhanced in the mutants and this was dependent on adenosine A2A receptor (A2AR) and tropomyosin-related kinase B signaling, suggesting increased activation of these receptors in synaptic plasticity phenomena. Accordingly, reducing adenosine A2A receptor activity in AdkΔbrain mice restored normal associative learning and contextual memory and attenuated seizure risk. We conclude that ADK deficiency in the brain triggers neuronal adaptation processes that lead to dysregulated synaptic plasticity, cognitive deficits, and increased seizure risk. Therefore, ADK mutations have an intrinsic effect on brain physiology and may present a genetic risk factor for the development of seizures and learning impairments. Furthermore, our data show that blocking A2AR activity therapeutically can attenuate neurological symptoms in ADK deficiency.
SIGNIFICANCE STATEMENT A novel human genetic condition (OMIM #614300) that is based on mutations in the adenosine kinase (Adk) gene has been discovered recently. Affected patients develop hepatic encephalopathy, seizures, and severe cognitive impairment. To model and understand the neurological phenotype of the human mutation, we generated a new conditional knock-out mouse with a brain-specific deletion of Adk (AdkΔbrain). Similar to ADK-deficient patients, AdkΔbrain mice develop seizures and cognitive deficits. We identified increased basal synaptic transmission and enhanced adenosine A2A receptor (A2AR)-dependent synaptic plasticity as the underlying mechanisms that govern these phenotypes. Our data show that neurological phenotypes in ADK-deficient patients are intrinsic to ADK deficiency in the brain and that blocking A2AR activity therapeutically can attenuate neurological symptoms in ADK deficiency.
Keywords: adenosine kinase, epilepsy, gene mutation, human genetic disorder, learning and memory, mouse model
Introduction
Adenosine kinase (ADK) is the key metabolic regulator of the purine ribonucleoside adenosine. In the adult brain, ADK is primarily expressed in astrocytes and determines the availability of adenosine in the synaptic cleft (Boison, 2013). Overexpression of ADK in the brain has been associated with the development of epilepsy and cognitive impairment (Li et al., 2008; Boison et al., 2012), whereas therapeutic adenosine augmentation is considered a promising therapeutic strategy for the treatment of epilepsy (Boison et al., 2002a; Pritchard et al., 2010; Boison, 2013). Recently, the first human mutations in the Adk gene have been described (OMIM:614300) (Bjursell et al., 2011; Staufner et al., 2016). Consistent with a prominent role of ADK for the maintenance of transmethylation reactions in the liver (Boison et al., 2002b), six patients from three unrelated families displayed a hepatic phenotype composed of disruption of the transmethylation cycle, dysregulation of hepatic metabolites, and hepatic encephalopathy (Bjursell et al., 2011). Affected individuals presented with global psychomotor delay and convulsive seizures commencing between the first and third year of life (Bjursell et al., 2011). Subsequently, 11 additional patients with ADK deficiency from eight families were identified; microvesicular hepatic steatosis and global developmental delay were prominent and most patients developed seizures and cognitive impairment (Staufner et al., 2016). Given the neuroprotective and anticonvulsive properties of adenosine, the neurological phenotype of patients with inborn ADK deficiency is somewhat surprising.
In the brain, adenosine modulates neurotransmission primarily through binding to its two high-affinity G-protein-coupled receptors: the inhibitory adenosine A1 receptor (A1R) and the stimulatory A2A receptor (A2AR) (Chen et al., 2013). The predominant action of adenosine is the inhibition of synaptic transmission via A1R-mediated signaling and, in rodent models of epilepsy, this signaling pathway is impaired, accounting for increased excitability and susceptibility to seizures (Rebola et al., 2003). Despite lower expression levels of the A2AR in the hippocampus, activation of A2ARs influences the release and uptake of neurotransmitters and also facilitates excitatory tropomyosin-related kinase B (TrkB)-mediated BDNF actions (Diógenes et al., 2004; Fontinha et al., 2008). To determine whether the neurological phenotype in ADK-deficient patients is secondary to hepatic encephalopathy or if it is intrinsic to ADK deficiency in the brain, we generated mice with a brain-wide deletion of ADK. This was achieved by breeding conditional Adk-flox (Adkfl/fl) with Nestin-Cre mice (Burns et al., 2007) to yield Nestin-Cre+/−:Adkfl/fl (AdkΔbrain) mice. These mutants completely lacked ADK in the brain and developed progressive stress-induced seizures and deficits in learning and memory. We identified a novel mechanism in which the complete lack of ADK in the brain drives neuronal adaptation processes that lead to enhanced basal synaptic transmission.
Materials and Methods
Transgenic mice.
The Adk gene targeting vector to produce global ADK knock-out mice (Adktm1bois) has been fully described previously (Boison et al., 2002b; Fedele et al., 2004). Briefly, the targeting construct was reengineered and exon 7 of the Adk gene was flanked with loxP sites. Through homologous recombination in embryonic stem cells, an Adkfl/− allele was created, which was used to generate a line of Adkfl/fl mice. Adkfl/fl mice were crossbred with constitutive Nestin-Cre mice (Tronche et al., 1999) (The Jackson Laboratory, RRID: IMSR_JAX:003771) to generate Nestin-Cre+/−:Adkfl/fl mice. Nestin-Cre mice express the Cre driver as early as embryonic day 11, resulting in Cre activation in neuronal and astroglial lineages (Tronche et al., 1999), a strategy chosen to achieve a brain-wide deletion of ADK, which is predominantly expressed in astrocytes of the adult brain (Studer et al., 2006). To exclude any Cre related experimental confounds, additional Nestin-Cre+/− mice without Adk-flox alleles were included in our in vivo studies. Nestin-Cre+/−:Adkfl/fl:A1R−/− mice and Nestin-Cre+/−:Adkfl/fl:A2AR−/− were generated by breeding Nestin-Cre+/−:Adkfl/fl mice with global A1R−/− (Johansson et al., 2001) or A2AR−/− mice (Day et al., 2003), respectively. Offspring were then backcrossed until Nestin-Cre+/−:Adkfl/fl:A1R−/− and Nestin-Cre+/−:Adkfl/fl:A2AR−/− mice were generated. Breeding of the experimental animals followed either a Nestin-Cre+/−:Adkfl/fl mice × Nestin-Cre−/−:Adkfl/fl, Nestin-Cre+/−:Adkfl/fl:A1R−/− × Nestin-Cre−/−:Adkfl/fl:A1R−/− or Nestin-Cre+/−:Adkfl/fl:A2AR−/− × Nestin-Cre−/−:Adkfl/fl:A2AR−/− mating protocol, which generated ADK-deficient and normal mice in a 1:1 ratio as littermates. All mice were generated and propagated on an identical C57BL/6 background and were genotyped at weaning by PCR. If not indicated otherwise, male subjects were used. All animals were social housed under standardized conditions of light, temperature, humidity, and environmental enrichment and had ad libitum access to food and water. Experimental animals used for this study were taken between 2 and 8 months of age. In vivo studies were conducted in an Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC)-accredited facility in accordance with protocols approved by the Legacy Institutional Animal Care and Use Committee. Ex vivo assays were performed according to procedures approved by the Portuguese authorities (DL 113/2013) and European Community Guidelines for Animal Care (European Communities Council Directive 2010/63/EU).
PCR.
Tissue from mice was obtained by tail clipping or from specific organs. Genomic DNA was prepared following standard procedures. For genotyping, Cre DNA sequence amplification with primers 5′-GGACATGTTCAGGGATCGCCAGGCG-3′ and 5′-GGACATGTTCAGGGATCGCCAGGCG-3′ was performed. For genomic Adk, the primer sequences were as follows: 5′-CCTCTATGAGTTGAGATCCTGTCTCC-3′ and 5′-ATTTATTAACTTTACATAGATTCAGACAG-3′. The Cre and Adk PCRs were paired with a genomic internal positive control using primers oIMR7338 (5′-CTAGGCCACAGAATTGAAAGATCT-3′) and oIMR7339 (5′-GTAGGTGGAAATTCTAGCATCATCC-3′) (The Jackson Laboratory). PCR products were loaded in a 2% agarose gel dyed with ethidium bromide for band visualization. For qRT-PCR, cortical samples were homogenization in QiAzol Lysis Reagent (Qiagen) and RNA was extracted, using the RNeasy Lipid Tissue Mini Kit (Qiagen). For first-strand cDNA synthesis, 1.5 μg of total RNA was applied in each reaction according to the manufacturer's protocol (SuperScript III Reverse Transcriptase; Invitrogen Life Technologies). For cDNA amplification, 2 μl of 1:10 diluted cDNA was added to 12.5 μl of 2× Power SYBR Green PCR Master Mix (Life Technologies) and 1 μl of each primer (5 μm) in a reaction volume of 25 μl. All reactions were performed in duplicate. The qRT-PCRs were performed on an RT-PCR Rotor Gene 6000 device (Corbett Life Science). Melting curves were analyzed to confirm primer specificity and the comparative Ct (threshold cycle) method was for quantification according to the following formula: Ct = Ct(target gene) − Ct(reference gene). The genes used to normalize the expression of the target sequences were PPIA peptidylprolyl isomerase A (cyclophilin A, CypA) and ribosomal protein L13A (RpL13A). Primers used were as follows: 5′-TATCTGCACTGCCAAGACTGAGTG-3′ and 5′-CTTCTTGCTGGTCTTGCCATTCC-3′ for CypA; 5′-GGATCCCTCCACCCTATGACA-3′ and 5′-CTGGTACTTCCACCCGACCTC-3′ for RpL13A; and 5′-TCGGCTGGCTACCACCCCTTG-3′ and 5′- CCAGCACCCAAGGTCACACCAAAGC-3′ for A1R.
Analysis and quantification of induced seizures.
Stress-induced seizures were evoked in AdkΔbrain mice by placing animals into a novel environment and characterized by tonic–clonic seizure activity followed by rearing and falling. Seizures were scored for occurrence and duration. Please see Movie 1 for a representative stress-induced seizure. AdkΔbrain mice used for the quantification of stress-induced seizures were naive to any additional treatments. During this period, the mice were exclusively handled by the same investigators. The probability of an evoked seizure was calculated for each mouse as the number of evoked seizures relative to the total number of trials within a month and then reported as the group average. Additional animals were equipped with a bipolar electrode surgically implanted into the hippocampus (AP: −1.94; ML: −1.25, DV: −1.5, relative to bregma) with a surface cortical monopolar screw electrode and cerebellum reference screw electrode while under general anesthesia (2% isofluorane, 100% O2). Video EEG recordings occurred at least 1 week after surgery. Mice were tethered for the EEG recordings. Electrical brain activity was amplified and digitized using a Nervus EEG recording system. Pharmacologically induced seizures were evoked by intraperitoneal injection of the A1R antagonist DPCPX (up to 3.5 mg/kg; Sigma-Aldrich). For the DPCPX threshold test, mice received 0.5 mg/kg DPCPX intraperitoneally every 10 min until a convulsive tonic–clonic seizure was induced up to a maximum cumulative dose of 3.5 mg/kg DPCPX. For the high-dose DPCPX experiment, a single dose of 3.0 mg/kg intraperitoneal DPCPX was administered and the latency to tonic–clonic seizure and mortality were indexed. The A2AR antagonist SCH58261 or the TrkB antagonist Ana-12 (0.5 mg/kg, 5% DMSO, i.p.; Sigma-Aldrich) were administered either 30 min or 4 h before DPCPX, respectively.
Stress-induced tonic convulsive seizure in an AdkΔbrain mouse. The movie is of a representative stress-induced seizure that is triggered by placing the mouse in a novel environment. The seizure is characterized by multiple bouts of tonic-clonic, rearing, and falling activity.
Assessment of baseline EEG activity and spontaneous seizures.
Spontaneous seizures were assessed by video EEG in 6-month-old AdkΔbrain mice (n = 6) and nestin-Cre+/− mice (n = 4) for 7 d. Additional baseline EEG recordings were conducted in all three lines of control mice (wild-type, nestin-Cre+/−, and Adkfl/fl) plus AdkΔbrain mice for 3 d (n = 5/genotype). Animals were equipped with EEG recording electrodes as described above. Electrical brain activity was monitored using a Nervus EEG recording system connected with a Nervus Magnus 32/8 Amplifier and filtered (high-pass filter 50 Hz cutoff, low-pass 1 Hz). The digital EEG signal was recorded, stored, and visualized using a NicoletOne-System (Viasys Healthcare). Videos were acquired with Lorex cameras and a DVR. EEG recordings were scored in their entirety for seizure activity. EEG seizure activity was defined as high-amplitude rhythmic discharges that clearly represented a new pattern of tracing lasting for >5 s. EEG seizures were confirmed to have a tonic–clonic behavioral correlated with the corresponding time-matched video.
Behavioral assessment of mice.
Conditioned learning and contextual memory were conducted using a classic fear-conditioning paradigm (Wehner and Radcliffe, 2004) with a Med Associates Fear Conditioning chamber and Video Freeze software (RRID: SCR_014574). Testing consisted of a 2 d paradigm. Day 1 was conditioned learning, including a baseline activity trial (3 min in the arena) and then 4 trials of conditioning stimuli (CS, 90 dB white noise, 30 s) immediately followed by aversive foot shock (0.3 mA, 1 s) with an intertrial interval (ITI) of 3 min. Freezing was measured during the ITI. Day 2 was contextual memory testing, in which mice were returned to the same arena and freezing was measured during 8 consecutive 1 min bins. Data are represented as the group average for percentage time freezing. The fear-conditioning paradigm was performed in animals that did not respond to seizure induction or in animals that were in their refractory period 2 h after a preceding seizure. There were no significant differences between the two groups of animals (associative learning, p = 0.58; contextual memory, p = 0.79). Animals that had a stress-induced seizure during the fear-conditioning paradigm were excluded from the analysis.
Brain dissection for electrophysiology.
All mice were quickly anesthetized with isofluorane before decapitation to minimize stress. No signs of seizures were detected in AdkΔbrain mice at the time of euthanasia. The brain was quickly removed and hippocampi and cortices were dissected in ice-cold aCSF containing the following (in mm): NaCl 124, KCl 3, NaH2PO4 1.25, NaHCO3 26, MgSO4 1, CaCl2 2, and glucose 10, pH 7.4. While the other brain areas were frozen at −80°C until further use, one of the hippocampi was sliced (400 μm) perpendicularly to its longitudinal axis using a McIlwain tissue chopper. Slices were then immediately transferred to a resting chamber filled with the same solution at room temperature and allowed to recover for at least 1 h before use in extracellular recordings. For each of the experimental paradigms described below, we used at least one slice per mouse; all n values stated are based on the group size of mice with their respective genotype.
Extracellular recordings.
Slices were transferred to a recording chamber and submerged with oxygenated aCSF solution at 32°C, continuously superfused at a flow rate of 3 ml/min. When indicated, drugs were added to the superfused solution. Recordings were obtained with an Axoclamp 2B amplifier and digitized (Molecular Devices). Evoked field EPSPs (fEPSPs) were recorded extracellularly through a microelectrode filled with 4 m NaCl (2–6 MΩ resistance) placed in the stratum radiatum of the CA1 area. A concentric electrode was placed on the Schaffer collateral–commissural fibers in stratum radiatum near the CA3–CA1 border and used to deliver the stimulation (rectangular 0.1 ms pulses once every 15 s). Individual responses were monitored and averages of eight (basal synaptic transmission) or six (LTP induction and input–output curve) consecutive responses were continuously stored on a personal computer with the WinLTP software (RRID: SCR_008590).
Drugs used for electrophysiological studies.
DPCPX (1,3-dipropyl-8-cyclopentylxanthine, A1R antagonist) was obtained from Ascent Scientific (Bristol, UK). CGS21680 (2-[p-(2-carboxyethyl)phenethylamino]-5-N-ethyl-carboxamido adenosine, A2AR agonist) was purchased from Sigma (St Louis, MO). K252a (Tyrosine kinase inhibitor) and SCH58261 (2-(2-Furanyl)-7-(2-phenylethyl)-7H-pyrazolo[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-amine, A2AR antagonist were purchased from Tocris Bioscience Cookson (Ballwin, MO). DPCPX, SCH58261 and CGS21680 were prepared in a 5 mm and K252a in a 1 mm stock solution, all in DMSO. The percentage of DMSO in each experiment did not exceed 0.001%.
Basal synaptic transmission.
The intensity of stimulus (80–200 μA) was initially adjusted to obtain a submaximal fEPSP slope (near 0.5 mV/ms) with a minimum population spike contamination. Alteration in synaptic transmission induced by drugs was evaluated as the percentage change in the average slope of the fEPSP in the presence of the drug in relation to the average slope of the fEPSP measured during the 10 min that preceded its addition.
Input–output curve.
Once obtaining a stable baseline for at least 15 min, the stimulus delivered to the slice was decreased to 60 μA, with fEPSPs disappearance. The stimulus was delivered to the slice in successively increased steps of 20 μA until a supramaximal stimulation of 340 μA. For each stimulation condition, data from three consecutive average fEPSP were stored. The Input-Output curve was plotted as the relationship of fEPSP slope (ms) vs stimulus intensity (μA).
LTP induction.
After obtaining a 14 min stable baseline of fEPSP slope by 0.5 ms, LTP was induced by a theta-burst protocol as described previously (Diógenes et al., 2011). This protocol consisted of three trains of three stimuli delivered at 100 Hz, each separated by 200 ms; the intensity of the stimulus was kept constant before, during, and after the induction protocol. LTP was quantified as the percentage change in the average slope of fEPSP taken from 52–60 min after LTP induction in relation to the average slope of the fEPSP measured during the baseline period (10 min before LTP induction). While assessing LTP in the presence of drugs, LTP was only induced no less than 30 min after starting the drug perfusion and only after fEPSP slope values had stabilized.
Western blot and saturation binding assays.
Dissected brain samples were homogenized in chilled 0.32 m sucrose solution with 50 mm Tris, pH 7.6, plus protease inhibitors (Hoffmann LaRoche) and then centrifuged. Protein was quantified using a Bio-Rad Protein assay kit. Next, 25–200 μg of aqueous protein extracts from tissue were loaded and separated on 10% SDS-PAGE gels and transferred onto PVDF membranes (Millipore). The blots were probed overnight at 4°C with primary antibody anti-ADK (1:4000, RRID pending; Gouder et al., 2004) and anti-GAPDH (1:500, catalog #sc-47724 RRID:AB_627678; Santa Cruz Biotechnology) as internal standards. The specificity of the ADK antibody has been validated previously in knock-out samples (Fedele et al., 2004; Gouder et al., 2004). The membranes were incubated with secondary antibody anti-mouse (1:2000, catalog #A-10677 RRID:AB_2534060; Thermo Fisher Scientific) or anti-rabbit (1:10,000, catalog #G-21234 RRID:AB_2536530; Thermo Fisher Scientific) conjugated with horseradish peroxidase for 1 h at room temperature. Chemoluminescence detection was performed with an ECL-PLUS Western blot detection reagent (GE Healthcare) using X-ray films (Fujifilm). The saturation binding experiments were adapted from a previous protocol (Batalha et al., 2013). [3H]DPCPX (radiolabeled A1R antagonist, specific activity 120 Ci/mmol) was from GE Healthcare. [3H]DPCPX (0–42 nm) (specific activity 120 Ci/mmol) was incubated for 2 h at room temperature with 39–77 μg of protein homogenate and 4 U/ml adenosine deaminase in a solution containing 50 mm Tris, 2 mm MgCl2 · 6H2O, pH 7.4, with a final volume of 300 μl. Each reaction was performed in duplicate. Nonspecific binding was measured in the presence of 2 μm XAC and normalized for protein concentration. Binding reactions were stopped by vacuum filtration with a Skatron semiautomatic cell harvester using chilled incubation solution. Filtermats 1.5 μm (Molecular Devices) were used and placed in scintillation vials to which 3 ml of scintillation mixture (OptiPhase HiSafe 2; PerkinElmer) was added. Radioactivity bound to the filters was determined after 12 h with an efficiency of 55–60% for 2 min.
Immunohistochemistry.
ADK and Nissl staining was performed on 4% paraformaldehyde-fixed coronal brain sections (40 μm) using standard protocols (Studer et al., 2006). High-resolution digital images were acquired using equivalent settings with a Leica DM1000 bright-field microscope equipped with a DCF295 camera and LAS AF Image Acquisition Software (RRID: SCR_013673; Leica Microsystems).
Statistical analysis.
Analyses were conducted with Prism 7 software (RRID: SCR_002798; GraphPad) and statistical significance was assumed at p < 0.05. Repeated-measures two-way ANOVA followed by Tukey's, Dunnett's, or Sidak's multiple-comparison tests; two-way ANOVA followed by Tukey's multiple-comparison test; nonlinear regression fit test; or two-tailed unpaired t tests were used as appropriate. The two-tailed Mann–Whitney test or Kruskal–Wallis test followed by Dunn's multiple-comparisons test were used for nonparametric analysis where appropriate. Kaplan–Meier survival curves were analyzed by log–rank (Mantel–Cox) tests.
Results
Conditional deletion of the Adk gene causes brain-wide ADK deficiency
To provide mechanistic evidence that ADK deficiency in the brain could be a primary cause for neurological symptoms in ADK-deficient patients, we engineered a novel line of mice with brain-wide disruption of ADK. First, our Adk gene-targeting construct (Boison et al., 2002b) was modified to flank exon 7 of the Adk gene with loxP sites (Fig. 1A). This construct was used to generate Adkfl/fl mice, which were then crossed with Nestin-Cre mice (Burns et al., 2007) to generate mutant AdkΔbrain mice and normal Adkfl/fl littermates. PCR analysis validated selective deletion of the Adk allele in representative brain regions of AdkΔbrain mice, whereas the intact Adk gene was maintained in peripheral organs of AdkΔbrain mice and in all tissues from Adkfl/fl mice (Fig. 1B). Consistent with intact Adk gene expression in the liver of AdkΔbrain mice, the gross liver morphology was normal and did not show any signs of lipid accumulation with oil-red-O staining (data not shown). In addition, AdkΔbrain mice did not show any appreciable differences in reproduction and life span. Western blot analysis (Fig. 1C) and ADK immunohistochemistry (Fig. 1D,E) corroborated a complete lack of ADK expression throughout the brains of the AdkΔbrain mice without affecting gross morphology (Fig. 1F,G).
Figure 1.

Brain-specific deletion of ADK in AdkΔbrain mice. A, Transgenic strategy: exon 7 of the Adk allele is flanked with loxP sites in Adkfl/fl mice. B, Adk PCR on genomic DNA extracts from the cortex (C), striatum (S), hippocampus (Hp), cerebellum (Cb), heart (Ht), lung (L), liver (Lv), and spleen (Sp) from Adkfl/fl and AdkΔbrain mice. Tail DNA from an Adkfl/fl (T) and wild-type mouse (WT) were included as positive controls. Water (−) was included as a no-template control. Adk forward and reverse primer (P1 and P2) sites are depicted in A. C, ADK (40 kDa) Western blots on cortical, striatal, and hippocampal protein extracts from Adkfl/fl and AdkΔbrain mice; n = 2/genotype are used as representatives. ADK Western blots were reprobed with GAPDH (37 kDa) as a loading control. D, E, ADK immunohistochemistry of cortical brain tissue from Adkfl/fl (D) and AdkΔbrain (E) mice. F, G, Nissl stain of hippocampal formation from Adkfl/fl (F) and AdkΔbrain (G) mice.
Brain-wide disruption of ADK results in increased susceptibility to seizure induction
AdkΔbrain mice initially developed normally. However, from 2 months on, the animals developed increased susceptibility to stress-induced seizures that were reliably induced by placing animals into a novel environment (i.e., a clean observation area) and characterized by tonic–clonic convulsions (see a representative seizure in Movie 1). These evoked seizures lasted an average of 2.48 ± 0.18 min (n = 7) and a marked progression of this seizure phenotype with age was found. By 3 months of age, 44% of all AdkΔbrain mice reacted with a seizure upon being placed into a novel environment; by 4 months of age, the incidence of seizure response within the same AdkΔbrain population was at 89% and continued to 100% by 6 months of age. Likewise, the probability of an evoked seizure response in the AdkΔbrain mice rose from 7.4% at 2 months of age to 77% at 6 months. Because pharmacological blockade of ADK is a very effective therapeutic strategy to suppress seizures in clinically relevant animal models of epilepsy (Gouder et al., 2004; McGaraughty et al., 2005; Boison, 2016a, 2016b), the emergence of the stress-induced seizure phenotype in AdkΔbrain mice was an unexpected finding. To identify the underlying mechanisms of seizure induction in AdkΔbrain mice, we next sought to replicate seizure induction in AdkΔbrain mice with well controlled pharmacological tools.
Because adenosine A1Rs link to potent anticonvulsant mechanisms (Gouder et al., 2003; Fedele et al., 2006; Kochanek et al., 2006; Gomes et al., 2011; Chen et al., 2013), A1R function in AdkΔbrain mice was tested. Injection of the A1R antagonist DPCPX (1.0 - 2.0 mg/kg, i.p.) at doses that do not trigger seizures in control mice (Masino et al., 2011; Fig. 2B) triggered a convulsive seizure in 5 of 6 AdkΔbrain mice (Fig. 2A,C, Movie 2). Furthermore, Adkfl/fl control mice (n = 5) were unaffected by higher doses up 3.5 mg/kg (Fig. 2B,C). Therefore, pharmacological blockade of the A1R could be used to induce seizures reliably in lieu of stress in AdkΔbrain mice (Mantel–Cox test, χ(1)2 = 6.73, **p = 0.0095; Fig. 2C). Next, it was determined that a single injection with a higher dose of the A1R antagonist DPCPX (3 mg/kg i.p.) consistently triggered lethal status epilepticus in AdkΔbrain mice (10 of 10 mice), but not in Adkfl/fl mice (0 of 10 mice; Fig. 2D,E). Together, these data demonstrate that, despite increased susceptibility for inducible seizures in AdkΔbrain mice, the A1R maintains an inhibitory and protective function.
Figure 2.

Loss of brain ADK results in increased susceptibility to seizure induction. A, Representative cortical EEG trace that includes a complete DPCPX- induced seizure (110 s) in an AdkΔbrain mouse that received a total cumulative dose of 2.0 mg/kg DPCPX intraperitoneally. High-resolution portions (15 s) of the DPCPX-induced seizure are depicted in cortical/hippocampal EEG1 and EEG2. These EEG traces correspond to boxes demarcated with 1 and 2 in upper cortical trace. See Movie 2 for a matching behavioral seizure. B, DPCPX does not trigger seizures in Adkfl/fl control mice. Shown is a representative section of a cortical EEG trace (110 s) recorded after a cumulative dose of 3.5 mg/kg DPCPX intraperitoneally. C, DPCPX dose response (0.5 mg/kg, i.p., every 10 min) in AdkΔbrain mice (n = 6) and Adkfl/fl mice (n = 5). D, E, DPCPX (3 mg/kg, i.p.) administered to AdkΔbrain mice (n = 10) causes status epilepticus (D, ****p < 0.0001) and increased mortality (E, ****p < 0.0001) compared with DPCPX-treated Adkfl/fl mice (n = 10), vehicle-treated Adkfl/fl mice (n = 6), and vehicle-treated AdkΔbrain mice (n = 6). Statistical analysis: log–rank (Mantel–Cox) test.
DPCPX-induced tonic convulsive seizure in an AdkΔbrain mouse. The movie is of a representative DPCPX-induced seizure that was triggered with a final cumulative dose of 2.0 mg/kg DPCPX. The seizure is the behavioral correlate of the cortical and hippocampal EEG traces shown in Figure 2A. The seizure is characterized by bouts of tonic-clonic, rearing, falling, running, and jumping activity.
Brain-wide disruption of ADK results in spontaneous seizures
To rule out that the Nestin-Cre expression or the Adk flox mutation affects the EEG baseline, 72 h blocks of hippocampal EEGs obtained from C57BL/6 (WT), Adkfl/fl, and Nestin-Cre+/− mice without floxed Adk alleles were compared with those from AdkΔbrain mice (n = 5, each) All control lines had normal hippocampal baseline EEGs, similar to those recorded from AdkΔbrain mice. Neither the AdkΔbrain mutation nor the Nestin-Cre expression affected baseline EEG activity (Fig. 3A). During those initial recordings, spontaneous seizures were found only in the AdkΔbrain mice, not in the controls. Therefore, an additional 7 d of vEEG recording blocks in a new set of AdkΔbrain mice (n = 6) and Nestin-Cre+/− mice (n = 4; Fig. 3B) were performed. Five of six AdkΔbrain mice had spontaneous seizures, whereas none of the controls was affected (χ2 test of contingency, χ(1)2 = 6.67, z = 2.58, **p = 0.0098). The spontaneous seizures were identified first on EEG and defined as high-amplitude rhythmic discharges that clearly represented a new pattern of tracing lasting for >5 s. The seizures identified by EEG always had a tonic–clonic behavioral correlate (Movie 3). The average spontaneous seizure rate assessed by vEEG was estimated to be 0.85 seizures per day with the average duration of each seizure being 43.6 ± 7.4 s.
Figure 3.

Loss of brain ADK results in spontaneous seizures. A, Thirty seconds of representative hippocampal EEG traces from AdkΔbrain mice and C57BL6 (WT), Nestin-Cre+/−:Adk+/+ (Nes-Cre+/−), and Adkfl/fl control mice demonstrate comparable baseline seizure activity. B, Representative cortical EEG recording of a complete spontaneous convulsive seizure (55 s) from an AdkΔbrain mouse (top trace). Bottom traces are high-resolution cortical and hippocampal EEG recordings of a 20 s portion of seizure demarcated by the box in top trace. See Movie 3 for a matching behavioral seizure.
Spontaneous tonic convulsive seizure in an AdkΔbrain mouse. The movie is of a representative spontaneous seizure characterized by tonic–clonic activity. The seizure is the behavioral correlate of the cortical and hippocampal EEG traces shown in Figure 3B.
Brain-wide disruption of ADK results in learning and memory impairment
Patients with ADK deficiency have severe cognitive impairments (Bjursell et al., 2011; Staufner et al., 2016); therefore, we next assessed learning and memory in our AdkΔbrain mice. AdkΔbrain and Adkfl/fl controls were subjected to a classic fear-conditioning paradigm in which a cue (CS) is paired with a mild electric foot shock. AdkΔbrain mice had severe cognitive impairments reflected by deficits in both associative learning and contextual memory compared with Adkfl/fl mice (Fig. 4A,B). Associative learning was indexed by an increase in percentage time freezing during the CS, with CS1 being the first baseline tone response. AdkΔbrain mice had a significant decrease in percentage time freezing during CS2 compared with Adkfl/fl mice (repeated-measures ANOVA, genotype effect F(3,48) = 6.62; AdkΔbrain vs Adkfl/fl ####p < 0.0001; Fig. 4A). Even though AdkΔbrain mice had an associative learning deficit, they showed the general ability to learn, with a significant increase in freezing during CS3 and CS4, compared with CS1 (trial effect F(3,144) = 60.99; CS3 vs CS1 *p = 0.0163, CS4 vs CS1 ****p < 0.0001; Fig. 4B). Therefore, learning was considerably delayed in AdkΔbrain mice, compared with Adkfl/fl mice. Furthermore, AdkΔbrain mice had a profound deficit in contextual memory as demonstrated by freezing rates comparable to baseline, which was significantly different from those observed in Adkfl/fl mice (repeated-measures ANOVA, genotype effect F(3,48) = 13.41; AdkΔbrain vs Adkfl/fl **p = 0.0024; Fig. 4B). To rule out genotype-related confounds, we conducted fear conditioning between three control lines (Adkfl/fl, Nestin-Cre+/−, and WT C57BL/6) and found that learning and memory deficits were not affected by either Cre or the floxed Adk allele (Fig. 4A,B). To rule out sensory-related confounds, we performed a basic behavioral screen and found no differences in the elevated plus maze, acoustic startle response, or spatial working memory tests in AdkΔbrain versus Adkfl/fl mice (data not shown). Furthermore, the response to foot shock, indexed by average (p = 0.51) and maximum motion index (p = 0.38), was comparable between the AdkΔbrain mice and all controls (data not shown). Together, these data demonstrate that brain ADK deficiency is associated with hippocampus-dependent cognitive impairment.
Figure 4.
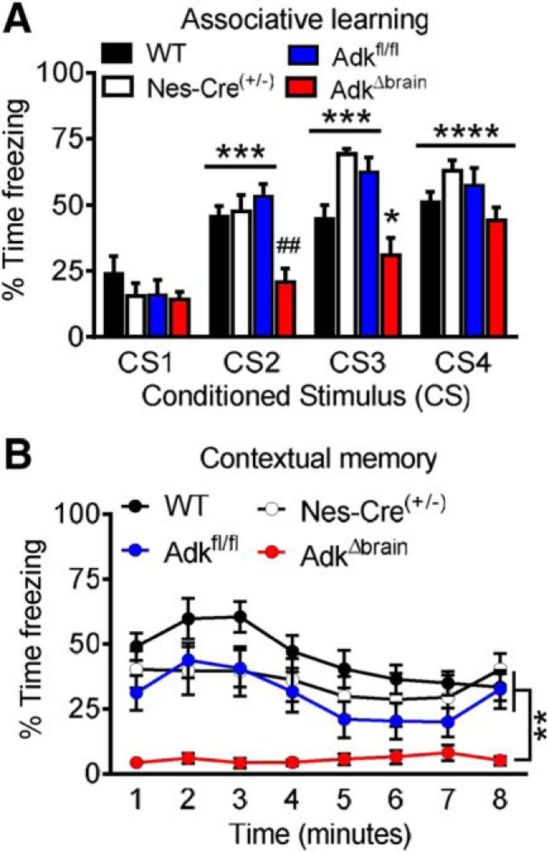
ADK deficiency in the brain results in cognitive impairment. A, Associative learning in the conditioned freezing paradigm is significantly decreased during conditioned stimulus 2 (CS2) in AdkΔbrain (n = 13) mice, compared with WT (n = 15), Nestin-Cre+/− (n = 10), and Adkfl/fl (n = 14) controls. Within AdkΔbrain mice, the percentage time freezing during CS3 and CS4 is significantly increased compared with baseline CS1 freezing. B, Contextual freezing is significantly impaired in AdkΔbrain mice (n = 13), compared with WT (n = 15), Nestin-Cre+/− (n = 10), and Adkfl/fl (n = 14) controls. Data are represented as the mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ##p < 0.01.
AdkΔbrain mice have increased levels of synaptic adenosine
To provide a mechanistic basis for the AdkΔbrain phenotype described above, we performed a detailed biochemical, pharmacological, and electrophysiological analysis of hippocampal brain slices derived from 4-month-old animals. Because ADK is the key metabolic route for adenosine clearance, we first tested for increased levels of brain adenosine. Due to the short half-life of adenosine and the need to gauge the magnitude of changes related to the synaptic pool of adenosine, we pursued a functional assay for synaptic adenosine by testing the influence of the A1R antagonist DPCPX on fEPSPs. We used a hippocampal slice preparation with stimulating electrodes placed in the CA3/CA1 border and recording electrodes placed in CA1 to study excitatory glutamatergic inputs to CA1 pyramidal neurons. We used a supramaximal concentration of DPCPX (50 nm) corresponding to ∼100× the Ki value for A1Rs (Lohse et al., 1988; Sebastião et al., 1990). In hippocampal slices from epileptic AdkΔbrain mice, DPCPX induced a significant increase in the fEPSP slope of 118 ± 20.1% compared with the predrug baseline (repeated-measures ANOVA, time effect F(22,176) = 23.23; 10 min vs 0 min #p = 0.022, 12–38 min vs 0 min ####p < 0.0001; Fig. 5A). This value contrasts with that obtained from age-matched Adkfl/fl mice, in which DPCPX increased the fEPSP slope by only 22 ± 7.8% (genotype effect F(1,8) = 16.25; AdkΔbrain vs Adkfl/fl **p = 0.0042, Fig. 5A; unpaired t test, t(8) = 4.43, **p = 0.0022, Fig. 5B). The higher disinhibition of synaptic transmission caused by the A1R antagonist in AdkΔbrain mice suggests an enhanced A1R-mediated inhibitory tonus. Using calculations described previously (Dunwiddie and Diao, 1994), we estimated the extracellular adenosine levels in the AdkΔbrain brains to be in the 1 μm range, whereas control levels of extracellular adenosine in Adkfl/fl mice were maintained at <200 nm. To assess A1R levels and function, we first quantified A1R mRNA and protein from cortical lysates. qRT-PCR demonstrated significantly decreased A1R mRNA in AdkΔbrain mice compared with controls (Mann–Whitney test, U = 0, **p = 0.0095; Fig. 5C). A1R protein was assessed by saturation binding experiments using [3H]DPCPX, which allow quantification of the maximal number of binding sites (Bmax) as well as the affinity (dissociation constant, Kd) of the receptor for the ligand. Both parameters were obtained by nonlinear regression analysis. The Bmax value indicated a significant reduction of functional A1Rs in AdkΔbrain mice (unpaired t test, t(6) = 8.02, ***p = 0.0002; Fig. 5D). Based on the Kd values, there were no significant differences (p > 0.05) in the affinity of the receptor for its agonist between Adkfl/fl and AdkΔbrain mice (Kd: 4.8 ± 0.9 vs 3.4 ± 1.1 nm). These data suggest decreased A1R density, likely an adaptation to an increased synaptic adenosine tone in the AdkΔbrain mice. Finally, to assess basal synaptic transmission in AdkΔbrain mice, we performed input–output curve analysis by recording the fEPSP responses as a function of increased stimulation intensities delivered to hippocampal slices. The maximum fEPSP slope obtained from AdkΔbrain slices was significantly higher than that from control slices (nonlinear regression fit; top values, F(1,108) = 5.79; AdkΔbrain vs Adkfl/fl, *p = 0.018; Fig. 5F). Together, the data demonstrate increased facilitatory action of DPCPX (Fig. 5A) in the AdkΔbrain mice, a finding that supports our in vivo data (Fig. 2).
Figure 5.

AdkΔbrain mice have increased synaptic adenosine. A, Disinhibition of synaptic transmission by the A1R antagonist DPCPX (50 nm) is facilitated in AdkΔbrain vs Adkfl/fl slices (n = 5 mice each). DPCPX increases the fEPSP slope in AdkΔbrain slices versus baseline. B, Percentage change in fEPSP slope during the last 10 min of DPCPX. C, Decreased A1R mRNA levels of AdkΔbrain (n = 6) versus Adkfl/fl (n = 4 mice) cortex. D, Binding curve for [3H]DPCPX is reduced the AdkΔbrain versus Adkfl/fl cortex (n = 4 mice each). E, Average Bmax values from the binding curves of AdkΔbrain versus Adkfl/fl mice (Bmax: 73 ± 6.5 vs 239 ± 13.4 fmol/mg protein). Data are presented as the mean ± SEM. 100% corresponds to the averaged fEPSP recorded 10 min before drug perfusion (A, B, F, G). F, Input–output curves were obtained from hippocampal slices to address changes in synaptic transmission level. The input–output curves correspond to responses generated by increasing stimulation intensities (60–340 mA) in Adkfl/fl (n = 5) and AdkΔbrain (n = 5) mice. Results are shown as the mean ± SEM and statistical analysis was performed using an F test (*p < 0.05). *p < 0.05, **p < 0.01, ***p < 0.001, ##p < 0.01, ####p < 0.0001.
AdkΔbrain mice have increased A2A receptor-mediated synaptic plasticity
Because synaptic levels of adenosine were found to be elevated in AdkΔbrain samples (> 1 μm), we anticipated increased A2AR activation, although adenosine has a slightly lower potency at A2ARs (EC50 = 0.7 μm) compared with A1Rs (EC50 = 0.3 μm; Fredholm et al., 2001). To test whether baseline A2AR signaling was affected in AdkΔbrain mice, we assessed fEPSPs recorded from hippocampal slices in the presence of a selective A2AR agonist or antagonist. Neither the A2AR agonist CGS21680 nor the antagonist SCH58261 had any effects on fEPSPs recorded from hippocampal AdkΔbrain or Adkfl/fl slices (repeated-measures ANOVA; AdkΔbrain vs Adkfl/fl (A) F(1,6) = 2.58, p = 0.16, Fig. 6A; F(4,108) = 0.029, p = 0.87, Fig. 6B). Next, we tested whether the ADK deletion would affect the tonic influence of A2ARs on LTP. Consistent with previous reports using mild theta-burst LTP induction (Costenla et al., 2011), A2AR blockade was virtually devoid of effect on LTP magnitude, the fEPSP slope 60 min after LTP induction being 122 ± 4.0% in control compared with 124 ± 4.5 in SCH58261 (repeated-measures ANOVA, treatment effect F(1,8) = 0.036, control vs SCH58261, p = 0.85; Fig. 6C). When comparing LTP magnitude in the absence of any drug in Adkfl/fl versus AdkΔbrain slices, a significant increase in LTP was detected (fEPSP: 122 ± 4.0% vs 152 ± 6.9%; two-way ANOVA, interaction F(1,16) = 14.36, Adkfl/fl vs AdkΔbrain, **p = 0.0053; Fig. 6E). Remarkably, the presence of SCH58261 in AdkΔbrain slices reversed the LTP magnitude (fEPSP: 113 ± 5.6%) toward values close to the Adkfl/fl mice (repeated-measures ANOVA, treatment effect F(1,8) = 17.0, control vs SCH58261, **p = 0.0033, Fig. 6E; AdkΔbrain: control vs SCH58261, ***p = 0.0005; Fig. 6F), suggesting that AdkΔbrain mice have enhanced tonic A2AR activation leading to increased synaptic plasticity.
Figure 6.
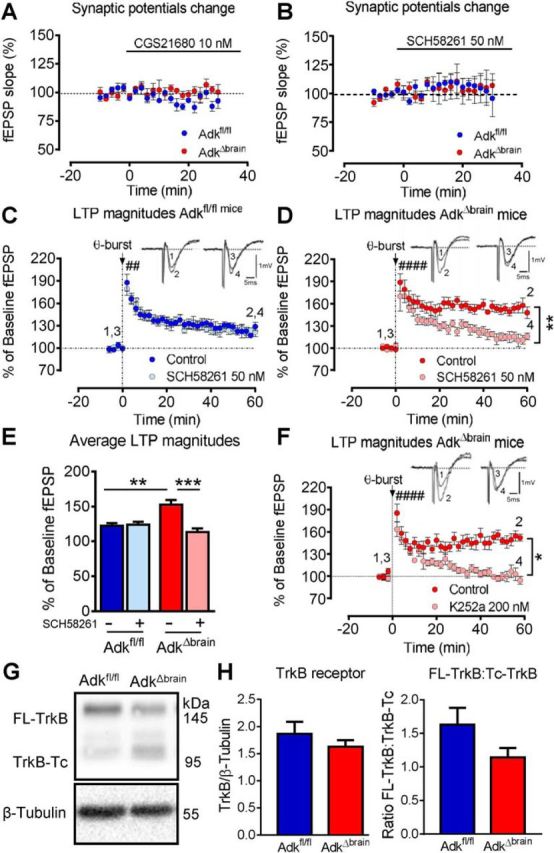
AdkΔbrain mice show enhanced A2A and TrkB receptor- dependent LTP. A, B, The A2AR-selective agonist CGS21680 (10 nm) and antagonist SCH58261 (50 nm) does not affect the fEPSP slope in Adkfl/fl and AdkΔbrain slices (n = 4 mice/genotype/drug). C–E, SCH58261 restores normal theta-burst (3 × 3)-induced LTP in AdkΔbrain mice. C, D, Average change in fEPSP slopes in the absence versus presence of SCH58261 in Adkfl/fl (C) and AdkΔbrain (D) slices (n = 5 mice/genotype/drug). E, Percentage of change in the fEPSP slopes recorded at 52–60 min after LTP induction. F, The TrkB inhibitor K252a (200 nm) restores normal theta-burst (3 × 3)-induced LTP in AdkΔbrain slices (absence: 150 ± 6.7% vs presence: 101 ± 7.0%, n = 5 mice each,). Insets in C, D, and F are representative traces of six consecutive responses composed of the stimulus artifact, presynaptic volley, and fEPSP obtained before (1, 3) and after (2, 4) the theta-burst stimuli in the absence (1, 2) and presence (3, 4) of drug. LTP was significant (####) after theta-burst stimuli versus baseline (C, D, F). G, Representative blot of full-length TrkB receptor (FL-TrkB, 145 kDa), truncated TrkB receptor (TrkB-Tc, 95 kDa), and β-tubulin (55 kDa, loading control) is shown. H, Averaged immunodensities of TrkB receptor normalized to β-tubulin and the ratio of FL-TrkB to TrkB-Tc (n = 8 mice/genotype). Results are presented as mean ± SEM. 100% corresponds to the averaged fEPSP slope recorded 10 min before LTP induction. *p < 0.05, **p < 0.01, ####p < 0.0001.
BDNF signaling is increased in AdkΔbrain mice
High-frequency stimulation triggers the release of BDNF and ATP, the major metabolic precursor of adenosine, therefore favoring the activation of A2ARs, which in turn gate TrkB receptor-mediated facilitatory actions of BDNF upon LTP (Fontinha et al., 2008; Dias et al., 2013). Taking into account the increased LTP magnitude in AdkΔbrain animals and the prevention of this increase on A2AR blockade, we next assessed whether the LTP increase in AdkΔbrain mice could result from enhanced BDNF signaling. To test this hypothesis, the tyrosine kinase inhibitor K252a (200 nm) was added to the perfusion solution and LTP was induced as before. K252a restored normal theta-burst-induced LTP in AdkΔbrain slices (absence: 150 ± 6.7% vs presence: 101 ± 7.0%, n = 5 mice). Therefore, in AdkΔbrain mice, the magnitude of LTP in slices in the presence of K252a was abolished (repeated-measures ANOVA, treatment effect, F(1,4) = 19.82, control vs K252a, *p = 0.012; Fig. 6F). K252a had virtually no effect on LTP magnitude in Adkfl/fl mice (data not shown), in agreement with data reported for wild-type rats (Fontinha et al., 2008). These results suggest a higher level of TrkB receptor activation accounting for a larger LTP in the AdkΔbrain mice. Therefore, our results indicate that the enhanced LTP in AdkΔbrain mice can be attributed to a higher influence of endogenous BDNF on LTP. Finally, we determined that AdkΔbrain mice do not compensate for increased BDNF signaling with changes in TrkB receptor expression (n = 8/genotype, unpaired t test, t(14) = 0.96, p = 0.36; Fig. 6H). However, we observed a modest, but insignificant, decrease in the ratio of full-length TrkB receptor to truncated TrkB receptor (unpaired t test, t(14) = 1.68, p = 0.11; Fig. 6H).
A2AR blockade ameliorates the inducible seizure phenotype and cognitive impairment in AdkΔbrain mice
Increased A2AR activation, as identified here, may promote seizures in AdkΔbrain mice through a BDNF-mediated mechanism. In this case, A2AR antagonists or TrkB antagonists might be of therapeutic value. Furthermore, the balance between inhibitory A1Rs and stimulatory A2ARs is one of the factors that determine the susceptibility to seizures (Sebastião and Ribeiro, 2009; Gomes et al., 2011; Chen et al., 2013). We therefore tested whether A2ARs play a role in determining seizure thresholds in AdkΔbrain mice by first administering a single injection of the A2AR agonist 5′-(N-cyclopropyl)carboxamidoadenosine (CPCA, 0.5 mg/kg). CPCA was ineffective at inducing a seizure (data not shown), which may be indicative of a ceiling effect for A2AR activation in AdkΔbrain mice. To circumvent this, we next sought to focus on the effect of blocking A2AR activity. Pretreatment with antagonists to both A2ARs (SCH58261) and TrkB (Ana-12) significantly extended the survival time after DPCPX-induced seizures in AdkΔbrain mice, compared with vehicle-treated controls (Mantel–Cox test, χ(2)2 = 12.51, **p < 0.0019; Fig. 7A). AdkΔbrain mice that received vehicle injections in lieu of DPCPX and Adkfl/fl mice that received vehicle, SCH58261, or Ana-12 ± DPCPX did not have seizures (data not shown). Next, we generated Nestin-Cre:Adkfl/fl:A1R−/− (=AdkΔbrainA1R−/−) and Nestin-Cre:Adkfl/fl:A2AR−/− (=AdkΔbrainA2AR−/−) triple mutants. Importantly, AdkΔbrainA1R−/− mice maintained their stress-induced seizure phenotype while exhibiting increased mortality, whereas AdkΔbrainA2AR−/− mice showed a marked resistance to stress-induced seizures and normal life expectancy (χ2 test of contingency, χ(2)2 = 18.26, ***p = 0.0001, Fig. 7C; Mantel–Cox test, χ(2)2 = 26.51, ****p < 0.0001, Fig. 7B). Together, these data suggest that AdkΔbrain mice have an enhanced A1R-mediated protective tonus and that additional factors such as stress, A1R blockade, and increased A2AR activation are needed to overcome this hurdle and allow the emergence of induced seizures.
Figure 7.

Blockade of A2AR activity ameliorates the inducible seizure phenotype and cognitive impairment in AdkΔbrain mice. A, Increased survival time of AdkΔbrain mice treated with SCH 58261 (0.5 mg/kg, i.p., 30 min in advance, n = 6) or Ana-12 (0.5 mg/kg, i.p., 4 h in advance, n = 6) before the DPCPX (3 mg/kg, i.p.) challenge compared with DPCPX controls (n = 10). B, Contingency analysis indicates a significant decrease in the percentage of AdkΔbrain:A2AR−/− mice (3 of 10) that develop the inducible seizure phenotype compared with AdkΔbrain (12 of 12) and AdkΔbrain:A1R−/− (8 of 8) mice. C, Kaplan–Meier survival curve indicating increased AdkΔbrain:A1R−/− (n = 18) mortality compared with AdkΔbrain (n = 18) and AdkΔbrain:A2AR−/− (n = 10). D, Associative learning indexed as total percentage time freezing during the CS 2–4 is restored to Adkfl/fl (n = 13) control levels in AdkΔbrain:A2AR−/− (n = 12) mice and is significantly increased compared with AdkΔbrain (n = 13) mice. E, Contextual memory indexed as total percentage time freezing during the 8 min contextual freezing trial is significantly increased in AdkΔbrain:A2AR−/− (n = 12) and Adkfl/fl (n = 13) mice, compared with AdkΔbrain (n = 13) mice. Data are represented as the mean ± SEM; **p < 0.01, ***p < 0.001, ****p < 0.0001.
To establish whether the A2AR is a target for the treatment of cognitive impairment in ADK deficiency, we tested AdkΔbrain:A2AR−/− mice in the fear-conditioning paradigm. Remarkably, the deletion of A2ARs from AdkΔbrain mice restored hippocampus-dependent associative learning (ANOVA, F(2,35) = 15.94; AdkΔbrain vs AdkΔbrain:A2AR−/−, ***p = 0.0008, Adkfl/fl vs AdkΔbrain:A2AR−/−, p = 0.43; Fig. 7D) and contextual memory (Kruskal–Wallis statistic = 27.54; AdkΔbrain vs AdkΔbrain:A2AR−/−, ****p < 0.0001, Adkfl/fl vs AdkΔbrain:A2AR−/−, p = 0.11; Fig. 7E) to Adkfl/fl control levels. Together, our findings suggest that the A2AR might be a therapeutic target for the treatment of the neurological phenotypes associated with ADK deficiency.
Discussion
We generated a novel mouse line, AdkΔbrain, to study specifically the neurological symptoms associated with global ADK deficiency such as those observed in human patients with mutations in the Adk gene (Bjursell et al., 2011; Staufner et al., 2016). We describe a novel mechanism by which ADK deficiency in the brain leads to neuronal adaptation processes that trigger enhanced A2AR- and BDNF-dependent synaptic plasticity. Together, those adaptive processes cause a phenotype characterized by increased propensity to induced and spontaneous seizures and impairment in hippocampus-specific cognitive domains. Therefore, our findings suggest that global ADK deficiency has direct implications on neurological outcome parameters, which might be amenable to treatment. Importantly, we demonstrate that the genetic and pharmacological blockade of A2ARs ameliorates the seizure phenotype and cognitive impairment in AdkΔbrain mice. Several aspects of our study warrant further discussion.
Loss of ADK function
Human patients with global ADK deficiency due to homozygosity of point mutations located in the coding sequence of the ADK gene are rare, with only 17 patients from 11 independent families identified to date (Bjursell et al., 2011; Staufner et al., 2016). Human ADK deficiency shares certain characteristics with a global deletion of ADK in the mouse (Boison et al., 2002b). Both conditions are characterized by disruption of the transmethylation pathway resulting in major physiological aberrations of liver metabolism. Liver is the organ in which 85% of all transmethylation reactions in the body take place and it is also the organ with the highest expression levels of ADK. In the mouse, homozygous deletion of the Adk gene leads to microvesicular hepatic steatosis, whereas the human condition is characterized by hepatic encephalopathy, developmental delay, cognitive impairment, and seizures (Boison et al., 2002b; Bjursell et al., 2011; Staufner et al., 2016). Because global ADK knock-out mice do not survive into adulthood, it was not possible to investigate direct consequences of ADK deficiency on brain physiology. Because Adk expression in the liver of AdkΔbrain mice is normal and heterozygous Adk knock-out (Adk+/−) have a normal liver despite a 50% reduction in liver ADK expression (Boison et al., 2002b), the increased brain adenosine levels in AdkΔbrain mice are not likely to translate into increased peripheral levels of adenosine that affect liver physiology. Our present study with AdkΔbrain mice, in which development is not compromised by liver pathology, reveals a novel mechanism by which brain-wide ADK deficiency leads to neuronal adaptation processes resulting in increased A2AR-dependent synaptic plasticity. We propose that similar mechanisms might play a role for seizure generation and the complex behavioral phenotype of human patients with inborn ADK deficiency. Given the enormous size of the ADK gene (546 kb in humans), it is surprising that only few patients with mutations this gene have been described so far. High evolutionary conservation of the Adk cDNA (>80% identical between man and rodents) suggests that mutations are rarely tolerated. Given the robust neurological phenotype described in homozygous human ADK deficiency (Bjursell et al., 2011; Staufner et al., 2016) and in mice with a homozygous deletion of the Adk gene in the brain, as described here, it remains to be determined whether heterozygosity of Adk gene mutations might affect the development of epilepsy or psychiatric disorders in the human population.
Gain vs loss of ADK function
Although no human gain-of-function mutations for the Adk gene have been described, ADK expression is subject to dynamic regulation during the course of epileptogenesis (Boison, 2008). Astrocytic ADK expression is significantly increased in the epileptogenic hippocampus of patients with epilepsy, as well as in rodent models of epilepsy (Boison, 2013), and is thought to be intrinsically linked to the development of epilepsy (Williams-Karnesky et al., 2013). Both transgenic overexpression of ADK (Li et al., 2008) and adeno-associated virus-mediated overexpression of ADK in astrocytes (Shen et al., 2014) are sufficient to induce spontaneous recurrent seizures in mice. This ADK gain-of-function-related seizure phenotype has been linked to adenosine deficiency and insufficient activation of adenosine A1Rs (Gouder et al., 2004; Fedele et al., 2006). Seizure susceptibility is controlled, not only by the availability of adenosine (i.e., the synaptic adenosine pool), but also by the balance of inhibitory A1R activation versus stimulatory A2AR activation, different distribution patterns of the receptors in the brain, and different potencies of adenosine on the two receptor types (Sebastião and Ribeiro, 2009; Gomes et al., 2011; Chen et al., 2013; Boison, 2016a). Effects mediated by the two receptors are discussed in more detail below.
A1R-dependent effects
Evidence for an inverse relation between ADK expression and adenosine levels has been provided previously (Shen et al., 2011). The synaptic potential recordings reported here (Fig. 5) also revealed a higher synaptic adenosine tonus in epileptic AdkΔbrain mice compared with aged-matched controls because the A1R antagonist DPCPX caused a more pronounced disinhibition of synaptic transmission in AdkΔbrain mice despite the lower expression levels of the A1R and the lower inhibitory effect of A1R agonists on synaptic transmission. Consistent with those findings, we show that a low dose of DPCPX can induce seizures in AdkΔbrain mice that are remarkably similar to the stress-induced seizure phenotype (cf. Movies 1, 2).
A2AR-dependent effects
Brain-specific deletion of ADK increased A2AR function. Interestingly, AdkΔbrain mice expressed a significantly larger LTP magnitude, which was prevented in the presence of an A2AR antagonist, suggesting a higher tonic influence of this receptor in synaptic plasticity modulation. A2AR antagonists do not affect basal synaptic transmission in hippocampal slices (Sebastião and Ribeiro, 1992; Cunha et al., 1997), which suggests only a minor role of the A2AR as a regulator of hippocampal synaptic transmission under low-frequency neuronal firing. However, the release of purines is more pronounced during physiologically relevant patterns of neuronal activity, namely those that induce hippocampal LTP (Wieraszko and Seyfried, 1989). Adenosine formed from released ATP activates A2ARs preferentially (Cunha et al., 1996). All of these factors favor an influence of A2AR activation on synaptic plasticity. Our data show that these effects, which also result from increased A2AR/BDNF signaling (see below), are markedly exacerbated in AdkΔbrain mice.
Our findings of cognitive impairment in AdkΔbrain mice are consistent with hippocampus-dependent memory deficits in transgenic rats with overexpression of the A2AR in the hippocampus and cortical brain structures (Giménez-Llort et al., 2007). Remarkably, enhanced LTP, together with enhanced A2AR and BDNF signaling, is associated with impaired learning and memory (Diógenes et al., 2011). Here, we show that the genetic ablation of A2ARs restores cognitive function in AdkΔbrain mice. These findings demonstrate directly that increased A2AR activity in AdkΔbrain mice may precipitate the impaired learning and memory phenotype. Increased A2AR function is also consistent with previous findings in the kindling model of epilepsy, in which a long-term increased density of A2ARs was observed (Rebola et al., 2003). Consistent with the anti-epileptic effects of an A2AR antagonist (Etherington and Frenguelli, 2004), A2AR knock-out mice are partially protected from convulsive activity in some experimental models of epilepsy (El Yacoubi et al., 2009). Therefore, enhanced A2AR signaling (Fig. 6) in addition to inhibitory A1R activation (Fig. 5) resulting in an imbalance of A1R/A2AR signaling is a plausible mechanism to permit a “breakthrough” of seizures in an otherwise protected brain environment in AdkΔbrain mice.
BDNF-dependent effects
Our findings of enhanced BDNF-dependent plasticity in AdkΔbrain mice are related to those from BDNF-overexpressing mice (BDNF-tg mice), which likewise develop stress-induced seizures triggered by placing the animals into a new cage. This seizure phenotype only develops later in life, at 5–6 months after birth (Papaleo et al., 2011). In our mechanistic study, we demonstrate that the enhanced A2AR activity in AdkΔbrain mice leads to increased tonic activation of TrkB receptors by BDNF, thereby inducing enhanced synaptic plasticity. Adenosine, through A2AR activation, is an upstream regulator of BDNF-mediated synaptic plasticity (Fontinha et al., 2008; Sebastião and Ribeiro, 2015). Through activation of TrkB, BDNF has facilitatory actions on neuronal activity and synaptic plasticity by operating a cascade of events that lead to imbalanced excitatory transmission (Leal et al., 2015).
Therapeutic implications
Our findings strongly suggest that blocking the A2AR might be of therapeutic value for the treatment of neurological symptoms in ADK-deficient patients. We show here that the pharmacological and genetic ablation of A2AR function ameliorates the seizure phenotype of AdkΔbrain mice. Importantly, the genetic deletion of the A2AR restores hippocampus-dependent learning and memory functions in AdkΔbrain mice. This is a significant finding of translational significance because A2AR inhibitors are in clinical development for the treatment of motor symptoms in Parkinson's disease (Schwarzschild, 2007). We show that those agents might also have procognitive effects under conditions of enhanced adenosine levels in the brain.
Footnotes
This work was supported by the National Institutes of Health (Grants MH083973, NS088024, and HL09556) and the Fundação para a Ciência e Tecnologia (FTC Grant EXPL/BIM-MEC/0009/2013). M.C.-O. was funded by FCT Fellowship SFRH/BD/73276/2010.
The authors declare no competing financial interests.
References
- Batalha VL, Pego JM, Fontinha BM, Costenla AR, Valadas JS, Baqi Y, Radjainia H, Müller CE, Sebastião AM, Lopes LV. Adenosine A(2A) receptor blockade reverts hippocampal stress-induced deficits and restores corticosterone circadian oscillation. Mol Psychiatry. 2013;18:320–331. doi: 10.1038/mp.2012.8. [DOI] [PubMed] [Google Scholar]
- Bjursell MK, Blom HJ, Cayuela JA, Engvall ML, Lesko N, Balasubramaniam S, Brandberg G, Halldin M, Falkenberg M, Jakobs C, Smith D, Struys E, von Döbeln U, Gustafsson CM, Lundeberg J, Wedell A. Adenosine kinase deficiency disrupts the methionine cycle and causes hypermethioninemia, encephalopathy, and abnormal liver function. Am J Hum Genet. 2011;89:507–515. doi: 10.1016/j.ajhg.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. The adenosine kinase hypothesis of epileptogenesis. Prog Neurobiol. 2008;84:249–262. doi: 10.1016/j.pneurobio.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. Adenosine kinase: exploitation for therapeutic gain. Pharmacol Rev. 2013;65:906–943. doi: 10.1124/pr.112.006361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. Adenosinergic signaling in epilepsy. Neuropharmacology. 2016a;104:131–139. doi: 10.1016/j.neuropharm.2015.08.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. The biochemistry and epigenetics of epilepsy: focus on adenosine and glycine. Front Mol Neurosci. 2016b;9:26. doi: 10.3389/fnmol.2016.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Huber A, Padrun V, Déglon N, Aebischer P, Möhler H. Seizure suppression by adenosine-releasing cells is independent of seizure frequency. Epilepsia. 2002a;43:788–796. doi: 10.1046/j.1528-1157.2002.33001.x. [DOI] [PubMed] [Google Scholar]
- Boison D, Scheurer L, Zumsteg V, Rülicke T, Litynski P, Fowler B, Brandner S, Möhler H. Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc Natl Acad Sci U S A. 2002b;99:6985–6990. doi: 10.1073/pnas.092642899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D, Singer P, Shen HY, Feldon J, Yee BK. Adenosine hypothesis of schizophrenia: opportunities for pharmacotherapy. Neuropharmacology. 2012;62:1527–1543. doi: 10.1016/j.neuropharm.2011.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns KA, Ayoub AE, Breunig JJ, Adhami F, Weng WL, Colbert MC, Rakic P, Kuan CY. Nestin-CreER mice reveal DNA synthesis by nonapoptotic neurons following cerebral ischemia hypoxia. Cereb Cortex. 2007;17:2585–2592. doi: 10.1093/cercor/bhl164. [DOI] [PubMed] [Google Scholar]
- Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets-what are the challenges? Nat Rev Drug Discov. 2013;12:265–286. doi: 10.1038/nrd3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costenla AR, Diógenes MJ, Canas PM, Rodrigues RJ, Nogueira C, Maroco J, Agostinho PM, Ribeiro JA, Cunha RA, de Mendonça A. Enhanced role of adenosine A(2A) receptors in the modulation of LTP in the rat hippocampus upon ageing. Eur J Neurosci. 2011;34:12–21. doi: 10.1111/j.1460-9568.2011.07719.x. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Correia-de-Sá P, Sebastião AM, Ribeiro JA. Preferential activation of excitatory adenosine receptors at rat hippocampal and neuromuscular synapses by adenosine formed from released adenine nucleotides. Br J Pharmacol. 1996;119:253–260. doi: 10.1111/j.1476-5381.1996.tb15979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA, Constantino MD, Ribeiro JA. ZM241385 is an antagonist of the facilitatory responses produced by the A2A adenosine receptor agonists CGS21680 and HENECA in the rat hippocampus. Br J Pharmacol. 1997;122:1279–1284. doi: 10.1038/sj.bjp.0701507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day YJ, Huang L, McDuffie MJ, Rosin DL, Ye H, Chen JF, Schwarzschild MA, Fink JS, Linden J, Okusa MD. Renal protection from ischemia mediated by A2A adenosine receptors on bone marrow-derived cells. J Clin Invest. 2003;112:883–891. doi: 10.1172/JCI15483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias RB, Rombo DM, Ribeiro JA, Henley JM, Sebastião AM. Adenosine: setting the stage for plasticity. Trends Neurosci. 2013;36:248–257. doi: 10.1016/j.tins.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Diógenes MJ, Fernandes CC, Sebastião AM, Ribeiro JA. Activation of adenosine A2A receptor facilitates brain-derived neurotrophic factor modulation of synaptic transmission in hippocampal slices. J Neurosci. 2004;24:2905–2913. doi: 10.1523/JNEUROSCI.4454-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diógenes MJ, Costenla AR, Lopes LV, Jerónimo-Santos A, Sousa VC, Fontinha BM, Ribeiro JA, Sebastião AM. Enhancement of LTP in aged rats is dependent on endogenous BDNF. Neuropsychopharmacology. 2011;36:1823–1836. doi: 10.1038/npp.2011.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwiddie TV, Diao L. Extracellular adenosine concentrations in hippocampal brain slices and the tonic inhibitory modulation of evoked excitatory responses. J Pharmacol Exp Ther. 1994;268:537–545. [PubMed] [Google Scholar]
- El Yacoubi M, Ledent C, Parmentier M, Costentin J, Vaugeois JM. Adenosine A2A receptor deficient mice are partially resistant to limbic seizures. Naunyn Schmiedebergs Arch Pharmacol. 2009;380:223–232. doi: 10.1007/s00210-009-0426-8. [DOI] [PubMed] [Google Scholar]
- Etherington LA, Frenguelli BG. Endogenous adenosine modulates epileptiform activity in rat hippocampus in a receptor subtype-dependent manner. Eur J Neurosci. 2004;19:2539–2550. doi: 10.1111/j.0953-816X.2004.03355.x. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Koch P, Brüstle O, Scheurer L, Simpson EM, Möhler H, Boison D. Engineering embryonic stem cell derived glia for adenosine delivery. Neurosci Lett. 2004;370:160–165. doi: 10.1016/j.neulet.2004.08.031. [DOI] [PubMed] [Google Scholar]
- Fedele DE, Li T, Lan JQ, Fredholm BB, Boison D. Adenosine A1 receptors are crucial in keeping an epileptic focus localized. Exp Neurol. 2006;200:184–190. doi: 10.1016/j.expneurol.2006.02.133. [DOI] [PubMed] [Google Scholar]
- Fontinha BM, Diógenes MJ, Ribeiro JA, Sebastião AM. Enhancement of long-term potentiation by brain-derived neurotrophic factor requires adenosine A2A receptor activation by endogenous adenosine. Neuropharmacology. 2008;54:924–933. doi: 10.1016/j.neuropharm.2008.01.011. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Irenius E, Kull B, Schulte G. Comparison of the potency of adenosine as an agonist at human adenosine receptors expressed in Chinese hamster ovary cells. Biochem Pharmacol. 2001;61:443–448. doi: 10.1016/S0006-2952(00)00570-0. [DOI] [PubMed] [Google Scholar]
- Giménez-Llort L, Schiffmann SN, Shmidt T, Canela L, Camón L, Wassholm M, Canals M, Terasmaa A, Fernández-Teruel A, Tobeña A, Popova E, Ferré S, Agnati L, Ciruela F, Martínez E, Scheel-Kruger J, Lluis C, Franco R, Fuxe K, Bader M. Working memory deficits in transgenic rats overexpressing human adenosine A2A receptors in the brain. Neurobiol Learn Mem. 2007;87:42–56. doi: 10.1016/j.nlm.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Gomes CV, Kaster MP, Tomé AR, Agostinho PM, Cunha RA. Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochim Biophys Acta. 2011;1808:1380–1399. doi: 10.1016/j.bbamem.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Gouder N, Fritschy JM, Boison D. Seizure suppression by adenosine A1 receptor activation in a mouse model of pharmacoresistant epilepsy. Epilepsia. 2003;44:877–885. doi: 10.1046/j.1528-1157.2003.03603.x. [DOI] [PubMed] [Google Scholar]
- Gouder N, Scheurer L, Fritschy JM, Boison D. Overexpression of adenosine kinase in epileptic hippocampus contributes to epileptogenesis. J Neurosci. 2004;24:692–701. doi: 10.1523/JNEUROSCI.4781-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Giménez-Llort L, Escorihuela RM, Fernández-Teruel A, Wiesenfeld-Hallin Z, Xu XJ, Hårdemark A, Betsholtz C, Herlenius E, Fredholm BB. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci U S A. 2001;98:9407–9412. doi: 10.1073/pnas.161292398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochanek PM, Vagni VA, Janesko KL, Washington CB, Crumrine PK, Garman RH, Jenkins LW, Clark RS, Homanics GE, Dixon CE, Schnermann J, Jackson EK. Adenosine A1 receptor knockout mice develop lethal status epilepticus after experimental traumatic brain injury. J Cereb Blood Flow Metab. 2006;26:565–575. doi: 10.1038/sj.jcbfm.9600218. [DOI] [PubMed] [Google Scholar]
- Leal G, Afonso PM, Salazar IL, Duarte CB. Regulation of hippocampal synaptic plasticity by BDNF. Brain Res. 2015;1621:82–101. doi: 10.1016/j.brainres.2014.10.019. [DOI] [PubMed] [Google Scholar]
- Li T, Ren G, Lusardi T, Wilz A, Lan JQ, Iwasato T, Itohara S, Simon RP, Boison D. Adenosine kinase is a target for the prediction and prevention of epileptogenesis in mice. J Clin Invest. 2008;118:571–582. doi: 10.1172/JCI33737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ, Klotz KN, Schwabe U, Cristalli G, Vittori S, Grifantini M. 2-Chloro-N6-cyclopentyladenosine: a highly selective agonist at A1 adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol. 1988;337:687–689. doi: 10.1007/BF00175797. [DOI] [PubMed] [Google Scholar]
- Masino SA, Li T, Theofilas P, Sandau US, Ruskin DN, Fredholm BB, Geiger JD, Aronica E, Boison D. A ketogenic diet suppresses seizures in mice through adenosine A1 receptors. J Clin Invest. 2011;121:2679–2683. doi: 10.1172/JCI57813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaraughty S, Cowart M, Jarvis MF, Berman RF. Anticonvulsant and antinociceptive actions of novel adenosine kinase inhibitors. Curr Top Med Chem. 2005;5:43–58. doi: 10.2174/1568026053386845. [DOI] [PubMed] [Google Scholar]
- Papaleo F, Silverman JL, Aney J, Tian Q, Barkan CL, Chadman KK, Crawley JN. Working memory deficits, increased anxiety-like traits, and seizure susceptibility in BDNF overexpressing mice. Learn Mem. 2011;18:534–544. doi: 10.1101/lm.2213711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard EM, Szybala C, Boison D, Kaplan DL. Silk fibroin encapsulated powder reservoirs for sustained release of adenosine. J Control Release. 2010;144:159–167. doi: 10.1016/j.jconrel.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebola N, Coelho JE, Costenla AR, Lopes LV, Parada A, Oliveira CR, Soares-da-Silva P, de Mendonça A, Cunha RA. Decrease of adenosine A1 receptor density and of adenosine neuromodulation in the hippocampus of kindled rats. Eur J Neurosci. 2003;18:820–828. doi: 10.1046/j.1460-9568.2003.02815.x. [DOI] [PubMed] [Google Scholar]
- Schwarzschild MA. Adenosine A2A antagonists as neurotherapeutics: crossing the bridge. Prog Neurobiol. 2007;83:261–262. doi: 10.1016/j.pneurobio.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastião AM, Ribeiro JA. Evidence for the presence of excitatory A2 adenosine receptors in the rat hippocampus. Neurosci Lett. 1992;138:41–44. doi: 10.1016/0304-3940(92)90467-L. [DOI] [PubMed] [Google Scholar]
- Sebastião AM, Ribeiro JA. Adenosine receptors and the central nervous system. Handb Exp Pharmacol. 2009;193:471–534. doi: 10.1007/978-3-540-89615-9_16. [DOI] [PubMed] [Google Scholar]
- Sebastião AM, Ribeiro JA. Neuromodulation and metamodulation by adenosine: Impact and subtleties upon synaptic plasticity regulation. Brain Res. 2015;1621:102–113. doi: 10.1016/j.brainres.2014.11.008. [DOI] [PubMed] [Google Scholar]
- Sebastião AM, Stone TW, Ribeiro JA. The inhibitory adenosine receptor at the neuromuscular junction and hippocampus of the rat: antagonism by 1,3,8-substituted xanthines. Br J Pharmacol. 1990;101:453–459. doi: 10.1111/j.1476-5381.1990.tb12729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HY, Lusardi TA, Williams-Karnesky RL, Lan JQ, Poulsen DJ, Boison D. Adenosine kinase determines the degree of brain injury after ischemic stroke in mice. J Cereb Blood Flow Metab. 2011;31:1648–1659. doi: 10.1038/jcbfm.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HY, Sun H, Hanthorn MM, Zhi Z, Lan JQ, Poulsen DJ, Wang RK, Boison D. Overexpression of adenosine kinase in cortical astrocytes generates focal neocortical epilepsy in mice. J Neurosurg. 2014;120:628–638. doi: 10.3171/2013.10.JNS13918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staufner C, Lindner M, Dionisi-Vici C, Freisinger P, Dobbelaere D, Douillard C, Makhseed N, Straub BK, Kahrizi K, Ballhausen D, la Marca G, Kölker S, Haas D, Hoffmann GF, Grünert SC, Blom HJ. Adenosine kinase deficiency: expanding the clinical spectrum and evaluating therapeutic options. J Inherit Metab Dis. 2016;39:273–283. doi: 10.1007/s10545-015-9904-y. [DOI] [PubMed] [Google Scholar]
- Studer FE, Fedele DE, Marowsky A, Schwerdel C, Wernli K, Vogt K, Fritschy JM, Boison D. Shift of adenosine kinase expression from neurons to astrocytes during postnatal development suggests dual functionality of the enzyme. Neuroscience. 2006;142:125–137. doi: 10.1016/j.neuroscience.2006.06.016. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Wehner JM, Radcliffe RA. Cued and contextual fear conditioning in mice. Curr Protoc Neurosci Chapter. 2004;8 doi: 10.1002/0471142301.ns0805cs27. Unit 8.5C. [DOI] [PubMed] [Google Scholar]
- Wieraszko A, Seyfried TN. Increased amount of extracellular ATP in stimulated hippocampal slices of seizure prone mice. Neurosci Lett. 1989;106:287–293. doi: 10.1016/0304-3940(89)90178-X. [DOI] [PubMed] [Google Scholar]
- Williams-Karnesky RL, Sandau US, Lusardi TA, Lytle NK, Farrell JM, Pritchard EM, Kaplan DL, Boison D. Epigenetic changes induced by adenosine augmentation therapy prevent epileptogenesis. J Clin Invest. 2013;123:3552–3563. doi: 10.1172/JCI65636. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Stress-induced tonic convulsive seizure in an AdkΔbrain mouse. The movie is of a representative stress-induced seizure that is triggered by placing the mouse in a novel environment. The seizure is characterized by multiple bouts of tonic-clonic, rearing, and falling activity.
DPCPX-induced tonic convulsive seizure in an AdkΔbrain mouse. The movie is of a representative DPCPX-induced seizure that was triggered with a final cumulative dose of 2.0 mg/kg DPCPX. The seizure is the behavioral correlate of the cortical and hippocampal EEG traces shown in Figure 2A. The seizure is characterized by bouts of tonic-clonic, rearing, falling, running, and jumping activity.
Spontaneous tonic convulsive seizure in an AdkΔbrain mouse. The movie is of a representative spontaneous seizure characterized by tonic–clonic activity. The seizure is the behavioral correlate of the cortical and hippocampal EEG traces shown in Figure 3B.