

Abstract
Stress influences memory, an adaptive process crucial for survival. During stress, hippocampal synapses are bathed in a mixture of stress-released molecules, yet it is unknown whether or how these interact to mediate the effects of stress on memory. Here, we demonstrate novel synergistic actions of corticosterone and corticotropin-releasing hormone (CRH) on synaptic physiology and dendritic spine structure that mediate the profound effects of acute concurrent stresses on memory. Spatial memory in mice was impaired enduringly after acute concurrent stresses resulting from loss of synaptic potentiation associated with disrupted structure of synapse-bearing dendritic spines. Combined application of the stress hormones corticosterone and CRH recapitulated the physiological and structural defects provoked by acute stresses. Mechanistically, corticosterone and CRH, via their cognate receptors, acted synergistically on the spine-actin regulator RhoA, promoting its deactivation and degradation, respectively, and destabilizing spines. Accordingly, blocking the receptors of both hormones, but not each alone, rescued memory. Therefore, the synergistic actions of corticosterone and CRH at hippocampal synapses underlie memory impairments after concurrent and perhaps also single, severe acute stresses, with potential implications to spatial memory dysfunction in, for example, posttraumatic stress disorder.
SIGNIFICANCE STATEMENT Stress influences memory, an adaptive process crucial for survival. During stress, adrenal corticosterone and hippocampal corticotropin-releasing hormone (CRH) permeate memory-forming hippocampal synapses, yet it is unknown whether (and how) these hormones interact to mediate effects of stress. Here, we demonstrate novel synergistic actions of corticosterone and CRH on hippocampal synaptic plasticity and spine structure that mediate the memory-disrupting effects of stress. Combined application of both hormones provoked synaptic function collapse and spine disruption. Mechanistically, corticosterone and CRH synergized at the spine-actin regulator RhoA, promoting its deactivation and degradation, respectively, and destabilizing spines. Notably, blocking both hormones, but not each alone, prevented the enduring memory problems after acute concurrent stresses. Therefore, synergistic actions of corticosterone and CRH underlie enduring memory impairments after concurrent acute stresses, which might be relevant to spatial memory deficits described in posttraumatic stress disorder.
Keywords: acute stress, dendritic spine, hippocampus, long-term potentiation, memory, RhoGTPase
Introduction
The ability of the mammalian brain to modulate learning and memory in response to stress—to learn from stressful events and to forget severely traumatic experiences—is crucial for survival and for emotional health (Lupien et al., 2009). Stress, signaling a potential or actual threat, is prevalent and unavoidable (Lupien et al., 2009; Maras and Baram, 2012) and exerts variable influences on memory. These influences depend on the context, nature, and duration of the stress (Shors et al., 2001; Joëls and Baram, 2009; Lupien et al., 2009; Conboy and Sandi, 2010; Wagner et al., 2013). In general, an acute stress lasting minutes facilitates memory, whereas chronic stress worsens it. Unexpectedly, we found recently that concurrent acute stresses impaired spatial memory (Maras et al., 2014), yet little is known about the longevity of these surprising effects and the role of specific stress hormones in their mechanisms. This information is important because humans are exposed to concurrent acute stresses in daily life. The emergence of memory disorders in firemen and other “rapid responders” after even a few episodes of short calamities (Corneil et al., 1999; Adler et al., 2013) suggests that multiple simultaneous stresses might have enduring and as yet undiscovered effects on memory processes.
Stress influences memory by modulating the integrity and plasticity of synapses that are fundamental for memory processes (Kim and Diamond, 2002; Maggio and Segal, 2009; Popoli et al., 2011; Wang et al., 2013). During stress, hippocampal synapses are concurrently exposed to a mixture of stress-mediators, including adrenal corticosterone (CORT) and locally released corticotropin-releasing hormone (CRH), as well as neurotransmitters (Chen et al., 2001; Joëls and Baram, 2009; Krugers et al., 2010; Popoli et al., 2011; Wang et al., 2011). Much is known about the individual effects of CORT on hippocampus-dependent functions and these effects are time and dose dependent (Lupien et al., 2009; Salehi et al., 2010; Joëls et al., 2011; Liston and Gan, 2011). CRH also influences memory functions in a time-dependent manner (Reyes et al., 2008; Sheng et al., 2008; Ivy et al., 2010; Refojo et al., 2011; Chen et al., 2012; Thoeringer et al., 2012; Wang et al., 2013). Surprisingly, little is known about potential interactions of these stress mediators and their role in the consequences of stress on synaptic integrity, plasticity, and memory (Joëls and Baram, 2009; Maggio and Segal, 2009; Luksys and Sandi, 2011).
Materials and Methods
Animals.
C57BL/6J (http://jaxmice.jax.org/strain/013636.html) and B6.Cg-Tg Thy1-yellow fluorescent protein (YFP) (https://www.jax.org/strain/003782) male mice (2–4 months) were group housed in a quiet, uncrowded facility on a 12/12 h light/dark schedule (lights on at 7:00 A.M.), with ad libitum access to food and water. All experiments were conducted according to the National Institutes of Health's guidelines for the care of experimental animals, with approval by the Institutional Animal Care and Use Committee of the University of California–Irvine.
Multiple simultaneous acute stresses.
The multiple simultaneous acute stresses (MAS) consisted of restraint, bright light, unpredictable loud noise, physical jostling and awareness of peer discomfort (Chen et al., 2008, 2010; Maras et al., 2014). Briefly, mice were put in a restrainer fashioned from a 50 ml plastic tube (Corning), placed five per cage (social stress) on a laboratory shaker, and jostled in a brightly lit room bathed in loud rap music (dB level = 95) for 1–2 h. Initial studies used a 2 h stress to enable exclusion of the genomic effects of glucocorticoid receptor (GR) and, guided by the electrophysiology, we then imposed the paradigm for 90 min and 1 h. The results were superimposable, so the groups were all included here. After MAS, mice were returned to their home cages. Control animals remained in the animal facility. Mice were trained for object location memory a minimum of 2 h after the end of the MAS, based on experiments showing that serum corticosterone levels normalized and exploration behavior was as robust as in controls (see Fig. 1B,C).
Figure 1.

MAS impairs hippocampus-dependent spatial memory. A, During an object location test, mice exposed to simultaneous stresses lasting 60–90 min failed to spend more time exploring the novel location compared with the familiar, as indicated by the discrimination ratio (novel vs familiar; n = 5 mice/group; t(12) = 3.23, *p < 0.01). B, Plasma CORT levels at the time of the training session, 2 h after the end of the stresses, were comparable between controls and stressed mice (n = 4 mice/group, p = 0.15). C, Comparable exploration durations between control and stressed mice during the training (10 min) and testing (5 min) phases of a novel location task (n = 8 mice/group, p > 0.05). D–F, Memory deficits provoked by multiple acute stresses persisted 1 d and 1 week later. Another cohort of stressed mice was allowed to recover for 24 h or 1 week, which was followed by training (10 min) and testing (5 min). Controls explored the object in a novel location more than the object left in the familiar location, whereas stressed mice trained 1 d or 1 week after the MAS and tested 6 h later spent almost equal time exploring either (D) (main effect of stress, F(1, 28) = 5.86, *p < 0.05). Exploration times during the training and testing phases did not distinguish between the controls and stressed animals that recovered for 24 h (E) or 1 week (F) (n = 8 mice per group; p > 0.05).
Object location memory task.
C57BL/6 male mice (3 months old) were used. MAS mice and controls were placed in a quiet dimly lit room for 2 h. Preparatory experiments demonstrated that this recovery period suffices for return of CORT levels of MAS mice to baseline and for equal performance for MAS and control groups in an object location training session. Specifically, object exploration durations and overall activity of MAS mice were not appreciably different from those of controls. The object location tests were performed as described previously (Vogel-Ciernia et al., 2013; Maras et al., 2014). Before training, mice were handled 2 min for 6 d and then habituated to the experimental apparatus for 10 min per day for 5–6 d in the absence of objects. In the training session, two identical objects were presented for exploration for 10 min. Training sessions took place 2 h, 24 h, or 1 week after the MAS. In the testing session, 6 or 24 h later, object exploration was scored for 5 min. Object exploration was scored when a mouse's head was oriented toward the object within a distance of 1 cm or when the mouse sniffed the object. Exploration times of object in familiar and novel locations were recorded and expressed as a ratio (novel/familiar). Total exploration times were calculated and the novel/familiar ratio was compared between controls and MAS mice. Mice that explored both objects for <20 s in total during either training or testing were removed from further analysis. Mice that demonstrated an object preference during training (ratio <0.5 or >2.0) were also excluded. Statistical outliers were excluded based on Tukey's range test. Both training and testing phases were video recorded and scored without knowledge of treatment group.
Surgical procedures and receptor blockade.
To investigate the role of CRH and GR in stress-induced memory deficit, spine loss, and synaptic plasticity, we blocked each one separately as well as together. We conducted dose-finding experiments to identify doses for each receptor blocker that do not prevent the peripheral release of stress hormones, yet are known to be effective in vivo and in vitro (Chen et al., 2006; Joëls et al., 2011; Liston and Gan, 2011). Therefore, the selective GR blocker RU38486 (750 ng in 1 μl; catalog #M8046; Sigma-Aldrich) and the selective CRFR1 receptor blocker NBI 30775 (5 μg in 1 μl; provided by Dr. DE Grigoriadis, Neurocrine Biosciences), alone or together (in 1 μl) were infused intracerebroventricularly to groups of control mice and 30 min before multiple concurrent acute stresses (MAS) in stressed mice. This was accomplished via cannulae implanted 6–7 d earlier as described previously (Chen et al., 2006, 2010). Experimental groups included the following: (1) stress-free (nonstressed mice with or without cannula insertion; these two subgroups were combined because the cannula did not alter novel object location testing scores and spine density); (2) stress-free given RU38486, NBI 30775, or both; (3) MAS (with or without vehicle administration; the vehicle infusion did not alter object location memory or spine density so that these two subgroups were combined); and (4) MAS mice receiving RU38486, NBI 30775, or both NBI 30775 and RU38486. Subsequent to memory tests (see above), mice were deeply anesthetized with sodium pentobarbital (100 mg/kg, i.p.) and perfused intracardially with fresh 4% paraformaldehyde (PFA) in 0.1 m sodium phosphate buffer (PB; pH 7.4, 4°C). Brains were cryoprotected and stored (Chen et al., 2001).
Electrophysiology studies.
Mice were decapitated and the brain was quickly submerged in oxygenated, ice-cold, high-magnesium artificial CSF (aCSF) containing the following (in mm): 124 NaCl, 3 KCl, 1.25 KH2PO4, 5 MgSO4, 26 NaHCO3, and 10 dextrose. Mid-dorsal hippocampal slices were prepared and transferred to an interface recording chamber (31 ± 1°C; 95% O2/5% CO2) as described previously (Chen et al., 2010, 2013). Slices were continuously perfused with preheated oxygenated aCSF at a rate of 60–70 ml/h. Recordings began ≥1.5 h after transfer to the chamber.
Field EPSPs (fEPSPs) were recorded in the apical dendritic field by positioning a glass electrode (2 m NaCl, 2–3 MΩ) in CA1b stratum radiatum while stimulating (twisted nichrome wire, 65 μm) in CA1c as described previously (Trieu et al., 2015). Input–output curves were generated using stimulation currents from 10 to 100 μA. To evaluate paired-pulse facilitation (PPF), two stimulation pulses were delivered at intervals of 40, 60, 100, and 200 ms and the initial slope of the responses was measured. Long-term potentiation (LTP) was induced by delivering a 2 s theta-burst train (TBS) consisting of 10 bursts separated by 200 ms (bursts of four pulses at 100 Hz) after a minimum of 20 min of stable baseline recording. In all instances, initial slopes and amplitudes were measured from digitized fEPSPs (NACGather 2.0; Theta Burst). Facilitation of responses to theta pulse trains was analyzed using an in-house MATLAB script (The MathWorks) and normalized to the amplitude of the first pulse within the train.
Acute hippocampal slices and in vitro hormone studies.
Transverse hippocampal slices (350 μm) were prepared from 3-month-old male mice as described previously (Chen et al., 2010, 2013). Slices were cut into ice-cold aCSF containing the following (in mm): 124 NaCl, 3 KCl, 1.25 KH2PO4, 2.5 CaCl2, 1.5 MgSO4, 26 NaHCO3, and 10 dextrose, pH 7.4, distributed across two interface recording chambers and maintained at 31 ± 1°C with the upper surface exposed to humidified 95% O2/5% CO2. aCSF was perfused constantly at a rate of 60 ml/h. Slices from dorsal hippocampus equilibrated in the chamber for 1.5–2 h before treatment. CORT, CRH, or the combination was added to the bath via a second perfusion line to attain a final 100 nm concentration, whereas control slices received vehicle–ACSF. Slices were collected at 120 min for electrophysiology and spine and synapses analysis and at 30 min for RhoA localization and activation. Immediately after treatment, slices were collected into cold 4% PFA and fixed for 15 min for RhoA detection at 4°C or 60 min for YFP detection before sectioning (20 μm) and processing for single/double immunolabeling.
Single and dual immunocytochemistry.
Animals were perfused transcardially with saline for 2 min followed by 4% PFA for 25 min (Chen et al., 2001). Brains were sectioned coronally or transversely (20 μm) using a cryostat. Single or double immunocytochemistry was performed on free-floating sections (Chen et al., 2001, 2013). Antibodies included mouse anti-GFP (1:8000; catalog #G6539; Sigma-Aldrich) and anti-postsynaptic density 95 (anti-PSD-95) antibody (1:2000; catalog #MA1-25629, RRID: AB_795156; Thermo Fisher Scientific). Antibody binding was visualized with anti-mouse IgG conjugated to Alexa Fluor 488 (1:400; catalog #A-11001; Invitrogen). Single secondary-labeled sections were also checked for secondary antibody cross-reactivity.
Acute slices were briefly fixed in 4% PFA in 0.1 m phosphate buffer, pH 7.4, and immediately cryoprotected and subsectioned (25 μm). Single or double immunocytochemistry was performed on sections mounted on gelatin-coated slides. Antibodies included rabbit anti-total RhoA (1:1000; NewEast Biosciences), mouse anti-active RhoA (1:1000; NewEast Biosciences), and goat anti-PSD-95 (1:1000; catalog #ab12093, RRID: AB_298846; Abcam). Antibody binding was visualized with anti-mouse IgG conjugated to Alexa Fluor 594 or Alexa Fluor 594 anti-rabbit IgG in combination with anti-goat IgG Alexa Fluor 488 (1:1000; Invitrogen).
Golgi procedures.
Mice were perfused transcardially with saline for 2 min, followed by a 15 min perfusion with a fixative provided in the Golgi kit (Bioenno sliceGolgi Kit; Bioenno Tech). Fixed brains were cut at 200 μm (easiSlicer; Ted Pella) and sections including the dorsal hippocampus collected in 0.1 m PB, pH 7.4. Free-floating sections were impregnated in the dark for 3–4 d (22°C), followed by staining (2 min) and poststaining (30 s). Sections were mounted on gelatin-coated slides, cleared with xylene, and coverslipped with Permount mounting medium.
Imaging and quantification of dendritic spines.
All analyses were performed without knowledge of treatment group. For in vivo studies, neurons were chosen using systematic unbiased sampling from the anterior third of the anterior/posterior extent of the hippocampus (8–12 neurons per mouse) (Chen et al., 2001). For in vitro acute slices using Golgi impregnated neurons, 10–14 neurons per group were analyzed. CA1 pyramidal neurons were selected for analyses to include equal representation of long- and short-shaft populations. Golgi-stained dendritic branches and spines were captured using a Nikon E400 equipped with a Nikon digital sight camera system (version 3). Image Z-stacks of second-fourth dendritic branches of CA1 pyramidal neurons were collected under 100×/1.3 oil lens and reconstructed using ImageJ (RRID: SCR_002285) and Adobe Photoshop (version 6, RRID:SCR_014199).
For YFP-expressing neurons from mice, Z-stack images were captured, reconstructed, and drawn using a Zeiss 510 confocal microscope with 63× objective, Imaris (version 7.1.0, RRID: SCR_007370; Bitplane) and Adobe Photoshop (version 6, RRID:SCR_014199). The second-fourth apical dendritic branches of CA1 pyramidal neurons were collected at 0.2 μm focal steps through the entire depth of each dendrite. A total of 5–6 branches per animal and 5 animals per group were evaluated.
The number of spines (spine density) was quantified comparing dendritic branches of the same order. Reconstructed spines were identified and characterized (Chen et al., 2013). Mushroom-type and thin spines were compared and filopodia were excluded. Spine density was expressed as the number of spines per 10 or 20 μm of dendrite length. The diameter (vertical to the long-axis of spine) and area of heads of mushroom-type spines were further measured and calculated using ImageJ and ImageTool (version 3.01). No correction factors were applied to the spine counts because high-magnification neuronal reconstruction permitted all spines of a given dendritic segment to be visualized.
Imaging and quantification of excitatory synapses.
A separate cohort of MAS and control mice (n = 5 mice per group) were used to assess the effects of concurrent acute stresses on the number and size of dorsal hippocampal synapses, using PSD-95 as a reliable marker for mature synapses (Chen et al., 2013). Sections from treatment and control groups were processed concurrently and analyzed without knowledge of treatment group. Wide-field 3D image stacks were deconvolved for 10 iterations using AutoQuant (version 2.10, RRID: SCR_002465; MediaCybernetics) and analyzed as described previously (Chen et al., 2013; Maras et al., 2014).
Methods and quantification of total and activated RhoA within synapses.
Two independent experimental cohort, driven by feasibility issues with the rapid processing of the slices for activated RhoA, were run. Each cohort contained all four groups, attaining group sizes of: vehicle = 12; CORT = 7; CRH = 8; CORT+CRH (combo) = 8. Wide-field 3D image stacks were collected in CA1 stratum radiatum (105 × 136 × 2 μm sample field per stack) at 0.2 μm steps, deconvolved (Volocity 4.1, RRID: SCR_002668; PerkinElmer), and numbers of both single- and double-labeled PSD-95-positive and active RhoA-positive puncta were quantified as described previously (Seese et al., 2013). In addition, the levels of colocalization of total RhoA with PSD-95 were quantified for all four groups (n = 3–5 slices/group). These measures were then used to determine whether, in any given slice, the treatment led to a reduced amount of activated RhoA at synapses, which should promote shifts between double- and single-labeled profiles Therefore, we expressed the data as a ratio of double-labeled to single-labeled PSDs. Notably, such a normalization obviates any potential issue of slight differences in total PSD number per slice or staining condition.
Radioimmunoassay.
Plasma corticosterone levels were determined by radioimmunoassay (Maras et al., 2014) using a commercially available kit (MP Biomedicals). Corticosterone assay sensitivity was 7.7 ng/ml and interassay variability, determined by 2 dilutions of adult mouse serum, averaged 10%.
Quantitative and statistical analyses.
All analyses were performed without knowledge of treatment group. Data were analyzed using Prism 5 software (RRID: SCR_002798; GraphPad). Tests included one-way or two-way ANOVA as indicated in the text, with Bonferroni's or Newman–Keuls post hoc test. A one-sample t test was also used to distinguish whether exploration times of objects were different from those predicted by chance. Significance was set at 95% confidence and values are presented as mean ± SEM.
Results
Enduring memory deficits arise after acute concurrent stresses
We subjected mice to acute MAS lasting 1–2 h and then trained them in a spatial memory task. After their recovery, previously stressed mice failed to encode object location, a behavior dependent upon hippocampus (Broadbent et al., 2009; Buzsáki and Moser, 2013). Specifically, control mice explored a novel-location object preferentially, relative to a familiar-location object (one-sample t test, t(4) = 2.86, p < 0.05), when tested 6 h after training. In contrast, stressed mice treated the objects equally (one-sample t test, t(3) = 0.87, p = 0.45; unpaired t test, control vs MAS, t(12) = 3.23, p < 0.01; Fig. 1A). The reduced ability to remember the location of an object was not a result of residual effects of the stress on general features of behavior: total object-exploration times during training and testing did not distinguish the stressed and control groups (p > 0.05; Fig. 1C). Moreover, plasma levels of CORT, a typical index of stress, were comparable between groups at the time of training (p > 0.05; Fig. 1B). Therefore, it appeared that the stress experience interfered with the ability of the mice to learn and remember the location of objects and these impairments seemed to require concurrent stresses because individually restraint or noise stresses of the same intensity and lasting as long as 5 h did not disrupt memory (Maras et al., 2014).
To evaluate the stability of the spatial memory impairment and to further exclude potentially subtle residual effects of the short stresses, we allowed a cohort of MAS mice to recover for 24 h and then trained them for object location memory. Upon testing, the memory deficits persisted (novel/familiar ratio = 1.67 in controls, 1.19 in stressed mice). Remarkably, the mice failed to learn object location even 1 week after the acute stresses (novel/familiar ratio = 2.0 in controls, 1.29 in mice stressed a week earlier) (main effect of stress, F(1,28) = 5.86, p < 0.05; Fig. 1D). Total object exploration times again did not distinguish the groups (Fig. 1E,F). To generate such persistent defects, the acute stresses must have led to a robust and fundamental disruption of processes involved in spatial memory. We then sought to examine these cellular and molecular mechanisms.
Electrophysiological mechanisms of memory impairments provoked by short combined stresses
Conventional hippocampal slices were used to test the effects of acute concurrent stresses on fEPSPs elicited in hippocampal field CA1 by stimulation of the Schaffer-commissural projections. Input/output curves produced by different stimulation intensities were not detectably different in slices prepared from control versus stressed animals (Fig. 2A,B). PPF was also comparable between groups, indicating that simultaneous short stresses did not affect transmitter release characteristics significantly (Fig. 2C). Evaluating responses generated by a 2 s TBS train, we found no group differences in the area of the composite response to the first burst in the train nor in the facilitation of the response to the second burst (Fig. 2D). In marked contrast, the LTP produced by TBS was greatly reduced in slices derived from MAS mice. Potentiation recorded immediately after the theta train was not measurably different, but LTP failed to stabilize in the subsequent 10 min and decayed steadily (Fig. 2E). These results strongly suggest that concurrent acute stresses did not affect the complex machinery involved in the induction of LTP, but instead had a surprisingly profound influence on the synaptic events responsible for maintaining synapses in the potentiated state.
Figure 2.

Multiple concurrent acute stresses impair LTP at area CA1 Schaffer–commissural synapses. A, Graph comparing the fEPSP and fiber-volley amplitudes in response to increasing intense stimulation in slices prepared from control (brown) and stressed (green) animals. Note that no difference was detected in slices prepared from the two groups (n = 5–6 mice per group). B, Representative fEPSPs recorded from slices from control (brown) and stressed (green) mice in response to increasing stimulation intensity. Scale bars: y = 1 mV, x = 20 ms. C, Graph illustrating PPF associated with different interpulse intervals in control (brown) and stressed (green) mouse slices demonstrates no group differences. D, Bar graph summarizing the effect of burst number on response size in control (brown) and stressed (green) mouse slices. E, LTP after delivery of TBS was attenuated in slices from stressed (green) mice compared with controls (brown) within 10 min of induction. Representative traces from baseline (black lines) and 30 min after induction (colored lines) are shown. All slices in the stress group were prepared from animals <1 h after the concurrent acute stresses (*p < 0.01 for minutes 30–80). Results are shown as means ± SEM. Values in parentheses are the numbers of slices from n = 5–6 mice.
Disrupted dendritic spines and synapses underlie the loss of synaptic plasticity
The loss of LTP suggested a disruption of basic synaptic integrity. Therefore, we visualized synapses directly in field CA1, the location of the electrophysiological deficits, using the same slices used for electrophysiology. In regions of the apical dendritic trees corresponding to the affected Schaffer–commissural synaptic connections, the density of dendritic spines was significantly lower in slices from stressed mice (16.91 ± 0.55 and 13.94 ± 0.55 spines/10 μm in control and stress, respectively; t(57) = 3.78, p < 0.01; Fig. 3A,B). This was accounted for primarily by a reduction of the density of thin spines (11.82 ± 0.59 and 9.26 ± 0.54 spines/10 μm, control and stress, respectively; t(57) = 3.15, p < 0.01; Fig. 3A, arrows); the density of mature, mushroom-type spines did not differ between groups (4.49 ± 0.23 and 4.30 ± 0.33 spines/10 μm, control and stress, respectively; t(57) = 0.47, p = 0.64; Fig. 3A, arrowheads). A second anatomical change was an apparent increase in the diameter of mature, mushroom-type spines in stressed versus control hippocampi (0.62 ± 0.010 μm and 0.54 ± 0.006 μm, t(8) = 5.21, p < 0.01; Fig. 3A, arrowheads, C). Therefore, multiple acute stresses shifted the size distribution of mature spine diameter toward larger values (Fig. 3C, left). These data were unexpected because individual acute stresses are not reported to provoke a loss of synapses or a change in the size of dendritic spine heads. Therefore, we conducted the same analyses using an independent method. Studies using wide-field imaging and 3D fluorescence deconvolution tomography (FDT) of PSD-95-immunolabeled spine synapses (Kramár et al., 2009; Rex et al., 2009) revealed the same effects of concurrent acute stresses (reduced synaptic density, t(58) = 1.92, *p = 0.05; shifted size distribution, t(29) = 2.30, p < 0.05; Fig. 3D–F). Together, these findings identified a novel consequence of acute stresses on synapse and spine structure, leading to a search for the responsible mediators.
Figure 3.

Concurrent acute stresses (MAS) reduce the density of thin dendritic spines and increase the size of mushroom-type spines. A, Representative CA1 apical dendritic segments from stress-free controls versus mice experiencing MAS. Arrows indicate thin spines; arrowheads, mushroom-type spines; note augmented size of spine heads with stress. Scale bar, 5 μm. B, With stress, spine density is reduced on apical dendritic trees in CA1 stratum radiatum, corresponding to the Schaffer–commissural terminal field. (t(57) = 3.78, *p < 0.01) and this is primarily a result of reduced numbers of thin spines in stressed versus control mice (t(57) = 3.15, *p < 0.01). C, Mean diameter of mushroom-type spine heads is larger in stressed versus control mice (t(8) = 5.21, *p < 0.01) and the size–frequency distribution of mushroom-type spines is “shifted to the right” in stressed mice. D, Representative wide-field microscopy followed by FDT demonstrates fewer and larger PSD-95-positive synapses in stressed mice. Scale bar, 10 μm. E, Reduced PSD-95 positive synapse density (t(58) = 1.92, *p = 0.05). F, Shift in the size–frequency distributions of PSD-95 puncta are apparent using the independent method (n = 20 slices and 5 mice per group). The number of large synapses is augmented in MAS mice. (t(29) = 2.30, *p < 0.05).
Corticosterone and CRH together mediate structural and functional hippocampal deficits
To identify molecules mediating the effects of acute concurrent stresses, we tested whether acute exposure to either CORT or CRH at levels likely to be present in hippocampus during stress influences synaptic physiology in the region in which short multiple stresses produced the above described changes. Hippocampal slices from adult mice were treated with CORT at 100 nm (Joëls and Baram, 2009; van Gemert et al., 2009): Synaptic responses (fEPSP slope) in field CA1 did not change appreciably after CORT application throughout the 2 h duration of recording. Treatment with CRH at 100 nm (Chen et al., 2012) did not change the fEPSP slope during the first 75 min of infusion (Fig. 4). Responses decreased modestly beyond this time point: the mean fEPSP slope for minutes 85 and 90 was −16.3 ± 2.6% smaller than baseline (p < 0.01, paired t test). The comparable value for CORT-treated slices was +2.0 ± 2.2%, significantly different from the decrease produced by CRH (p < 0.01).
Figure 4.

Coordinate actions of CORT and CRH on synaptic physiology recapitulate the effects of multiple concurrent acute stresses. CRH infusion (100 nm) into hippocampal slices depressed the size (slope) of EPSPs but only after a delay of 1 h (green). CORT (100 nm) had no detectable effect on synaptic responses (blue). Infusion of CORT+CRH resulted in rapid, profound depression of synaptic responses (orange). The difference between the three groups at 50–55 min of the infusion was highly significant (p < 0.01, one-way ANOVA), with the combination treatment distinct from either of the CORT or CRH alone groups. The response size versus time of infusion interaction for the three groups from minutes 10 through 40 was significantly different (p < 0.01, two-way ANOVA; n = 4 animals per group, 2–3 slices per animal).
Because CORT and CRH bathe hippocampal synapses concurrently during stress (Joëls and Baram, 2009; Popoli et al., 2011), we investigated the possibility that they act in concert. Combined application of the two hormones led to rapid and profound attenuation of fEPSP slope that was evident already 35 min after the start of infusion and reached a stable level by 75 min (Fig. 4). The percentage decrease at 50–55 min after introduction of the combination treatment was −22.2 ± 3.8% from baseline (p = 0.01). The comparable values for CORT or CRH alone were +4.4 ± 1.1% and −3.2 ± 1.4%, respectively, and the difference between the three groups was highly significant (p < 0.01, one-way ANOVA), with the combination treatment group distinct from either the CORT-alone or CRH-alone group (Fig. 4). As expected, the response size versus time of infusion interaction for the three groups from minutes 10 through 40 was highly significant (p < 0.01, two-way ANOVA). These findings indicate that CORT and CRH exert synergistic, negative effects on synaptic function.
We then examined the individual and combined actions of CORT and CRH on dendritic spines and synapses in the same pathway that was adversely influenced by stress and where combined CORT and CRH provoked electrophysiological impairments. We used transgenic mice expressing YFP in principal hippocampal neurons to visualize dendritic spines directly. Compared with control preparations, application of CORT (100 nm) had little effect on the density of spines (14.47 ± 0.67 and 14.14 ± 0.57 spines/10 μm in CORT and control, respectively; p > 0.05) or the density of the subset of thin spines (8.75 ± 0.54 and 8.07 ± 0.76/10 μm in CORT and control, respectively; p > 0.05) (Fig. 5A,B). Application of CRH led to a reduction in the density of thin spines (5.31 ± 0.38 in CRH vs 8.07 ± 0.76 in vehicle; repeated measures one-way ANOVA, F(3,39) = 24.87, p < 0.0001; post hoc, p < 0.05), as described previously (Chen et al., 2008, 2013). In contrast, combined application of CORT+CRH recapitulated the full effect of the acute concurrent stresses: the combined treatment reduced total spine density (4.47 ± 0.55 vs 8.07 ± 0.76; repeated measures one-way ANOVA, F(3,39) = 20.45, p < 0.001; post hoc, p < 0.05), and significantly increased the size of mushroom-type spine heads (repeated measures one-way ANOVA, F(3,33) = 25.28, p < 0.0001; post hoc, p < 0.01; Fig. 5A–C) relative to controls or to values from hippocampal slices treated with CRH or CORT individually (post hoc, p < 0.01; Fig. 5A, arrows and arrowheads). Accordingly, the shift in the size distribution of dendritic spine heads was dramatic (Fig. 5C), driven by both loss of thin spines and increased head size of mushroom-type spines. The use of wide-field FDT of PSD-95-immunoreactive synapses in hippocampal slices exposed to CORT, CRH, or both confirmed these results (Fig. 5D–F). Application of CORT+CRH reduced total synapse number compared with controls and compared with each hormone alone (one-way ANOVA, F(3,116) = 54.03, p < 0.0001; post hoc, p < 0.01). The combined hormone infusion reduced the number of small (0.15 μm3) synapses (one-way ANOVA, F(3,60) = 14.66; p < 0.0001; post hoc, p < 0.001), increased the proportion of larger (>0.30 μm3) synapses (one-way ANOVA, F(3,196) = 30.32, p < 0.0001; post hoc, p < 0.01), and thus shifted the size-distribution of PSD-95-positive synapses toward larger diameters (Fig. 5F).
Figure 5.
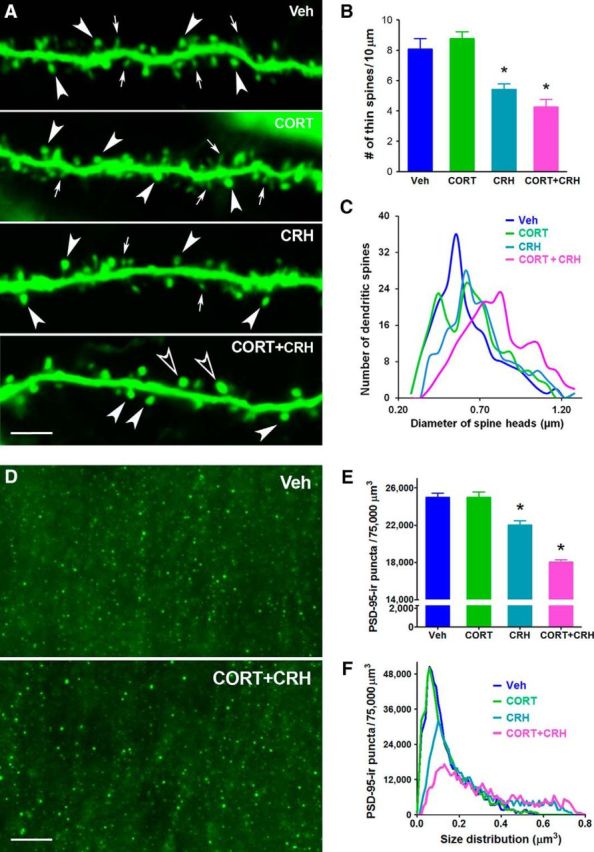
Concerted actions of CORT and CRH on dendritic spines recapitulate the effects of multiple acute stresses. A, Combined CORT+CRH at levels presumed to obtain in hippocampus during stress (Chen et al., 2012; Maras and Baram, 2012) recapitulate the effects of concurrent short stresses on dendritic spine integrity in hippocampal CA1. CORT has little effect on thin spines (arrows) and tends to increase the size of mushroom-type spines (arrowheads). CRH reduces the numbers of thin spines, whereas the combined application of both hormones diminishes thin spine density and promotes aberrantly large mushroom-type spine heads (empty arrowheads). Scale bar, 6 μm. B, Reduced density of thin spines on apical dendrites of CA1 pyramidal neurons exposed to both hormones (repeated-measures one-way ANOVA, F(3, 39) = 24.87, p < 0.0001; post hoc, p < 0.05). C, Exposure to CORT+CRH promotes a shift in the size–frequency distributions of YFP-labeled mushroom-type spines in CA1 (repeated-measures one-way ANOVA, F(3, 33) = 25.28, p < 0.0001; post hoc, p < 0.01). D, An independent method (wide-field microscopy and FDT) demonstrates reduction of PSD-95-positive synapses by combined application of CORT+CRH. Scale bar, 8 μm. E, Combined application of CORT+CRH reduced total synapse number compared with vehicle and with each hormone applied alone (one-way ANOVA, F(3, 116) = 54.03, p < 0.0001; Bonferroni's post hoc test, *p < 0.01). The number of PSD-95 synapses was reduced by CRH alone compared with vehicle (*p < 0.05). F, CORT+CRH exposure reduced the number of small (0.15 μm3) synapses (one-way ANOVA, F(3, 60) = 14.66; p < 0.0001; post hoc, p < 0.001), increased the proportion of larger (>0.30 μm3) synapses (one-way ANOVA, F(3, 196) = 30.32, p < 0.0001; post hoc, p < 0.01), and shifted the size–frequency distribution of PSD-95-positive synapses toward larger diameters.
Mechanisms of CORT and CRH actions converge on the spine actin-regulating RhoGTPase, RhoA
Proteins that sever filamentous actin control its polymerization state and remodeling, contributing to changes in spine shape and size. The same molecules though, if unopposed or highly active, can provoke spine destruction. Actin-modulating proteins are largely regulated by the small Rho GTPases, including Rac1, RhoA, and cdc42, which constitute molecular switches controlling spine structure (Newey et al., 2005; van Galen and Ramakers, 2005; Rex et al., 2009; Penzes et al., 2011). Specifically, activation of RhoA enables actin remodeling and is required for spine plasticity, growth, and changes in synaptic strength (Kramár et al., 2009). We found previously that CORT alone reduces downstream signaling in the RhoA pathway, including cofilin phosphorylation (Jafari et al., 2012). We also reported that CRH alone activates the calcium-regulated enzyme calpain via NMDA-receptor and calcium-dependent mechanisms (Andres et al., 2013). Calpain1, in turn, has been shown to degrade RhoA (Briz et al., 2015). Together, these prior observations suggested mechanisms for the convergent and mutually potentiating actions of CORT and CRH on spine integrity and plasticity (Fig. 6A). CRH might induce calpain-mediated degradation of RhoA, leading to reduced spine levels of the total and active GTPase, whereas CORT might prevent activation without influencing total RhoA levels. Reduced levels of active RhoA as a result of the putative convergent actions of the two hormones would promote activation (dephosphorylation) of the actin-severing enzyme cofilin and consequent degradation of F-actin polymers. This hypothetical mechanism predicts that, individually, CRH but not CORT will reduce total RhoA at excitatory synapses, whereas a decrease in active RhoA will require both hormones. Increased spine-head size might arise as well because, unopposed by RhoA, Rac1 should promote actin branching and spine growth (Jafari et al., 2012).
Figure 6.

CORT and CRH effects converge on mechanisms regulating spine stability and specifically on the actin-regulating RhoA. A, Putative convergent mechanisms by which CORT and CRH influence spine integrity. CORT, via GR, prevents activation of RhoA in dendritic spine heads (Jafari et al., 2012). Activation of CRFR1 by CRH engages calpain in the spine through an NMDA-mediated mechanism (Andres et al., 2013), which destroys the Rho-like GTPase RhoA. This leads to pCofilin dephosphorylation, generating the active cofilin, which can cleave F-actin. Together, these hormone actions promote depolymerization of F-actin and spine collapse. B, The ratio of RhoA-positive+PSD-95-positive puncta to those immunolabeled for PSD-95 alone was reduced by coadministration of CORT and CRH (one-way ANOVA, F(3, 13) = 7.45, p < 0.005; Newman–Keuls multiple-comparisons test, p < 0.01), as well as by CRH alone, but not by CORT alone (post hoc, p < 0.01), as predicted by the model (A). C, In contrast, active RhoA at synapses, measured as a ratio of dually labeled active RhoA+PSD-95/single label PSD-95 puncta was reduced significantly only when both hormones acted in concert (one-way ANOVA, F(3, 31) = 5.27, p < 0.005; post hoc, p < 0.05). D, Representative raw images for colocalizion of total RhoA/PSD-95 (left) and active RhoA/PSD-95(right). PSD-95 is labeled in green and RhoA, active or total, is labeled in red. Groups are arrayed vertically: control, CORT, CRH, or both. An effect of treatment group on the proportion of RhoA-labeled PSD-95 puncta (appearing yellow) within the whole PSD-95-positive population is apparent and is quantified in B and C. CRH at 100 nm, CORT at 100 nm, and CORT+CRH at 100 nm each were applied for 15–30 min. Scale bars, 5 μm.
To test this hypothetical scenario, we quantified numbers of spine synapses associated with total and activated RhoA in hippocampal CA1 stratum radiatum. We used wide-field imaging and FDT, which supports measures of >30,000 synapses per image z-stack and hundreds of thousands of synapses per mouse (Lauterborn et al., 2009; van Gemert et al., 2009; Seese et al., 2013). We then quantified the colocalization of active RhoA+PSD-95 for all four groups (n = 8–12 per group), as well as of total RhoA+PSD-95 (n = 3–5 per group). To address whether, in any given slice, the treatment led to a reduced amount of activated RhoA, which should promote shifts between double and single-labeled PSD-95 puncta, we assessed the ratio of double-labeled to single-labeled PSD-95 positive puncta. This measure also obviates potential variance in total numbers of PSD-95-positive puncta in any given slice. In vehicle-infused hippocampal sections, colocalization of total RhoA with PSD-95 was prominent (Fig. 6B,D). As predicted, application of CORT (10 nm, 15 min) did not reduce the incidence of RhoA-positive synapses significantly, whereas CRH application for a similar duration did (one-way ANOVA, F(3,13) = 7.45, p < 0.005; post hoc, p < 0.01; Fig. 6A,B,D). The combined application of these hormones yielded the same effect as CRH applied alone (post hoc, p < 0.01). In contrast, the proportion of synapses associated with active RhoA (active RhoA-positive PSDs/total PSD-95 contacts) was reduced significantly only in the presence of both hormones (one-way ANOVA, F(3,31) = 5.27, p < 0.005; post hoc, p < 0.05; Fig. 6C,D). These findings support the presence of synergistic effects of CORT and CRH on the availability of active, actin-stabilizing RhoA within dendritic spines.
Blocking the actions of both CORT and CRH abrogates stress-induced memory impairments
We then reasoned that, if the coordinated actions of CORT and CRH mediate the profound effects of acute concurrent stresses on hippocampus-dependent memory, then interference with the actions of both hormones, but not each alone, should abrogate these deficits. To avoid the potential effects on spine and synapse structure and function that might arise from genetic manipulation of these crucial structures, we used acute pharmacological tools. Mice were exposed to acute simultaneous stresses immediately after one of four treatments applied directly into the brain. The control group received vehicle; a second group received RU38486, a blocker of the CORT/GR that mediates the hormone's effect on RhoA (Jafari et al., 2012); a third group received NBI 30775, a selective blocker of the CRH receptor CRFR1 that resides within the PSD (Chen et al., 2012), and the fourth group received a mixture of RU38486+NBI 30775. Plasma CORT levels were measured before and during the concurrent stresses to ascertain that this central treatment did not impair stress-induced release of CRH from the hypothalamus and of CORT from the adrenal gland.
Control mice explored the object in the novel location significantly more than expected by chance (novel/familiar location ratio = 1.72 ± 0.14; one-sample t test, t(19) = 4.98, p < 0.0001) and significantly more than stressed mice (p = 0.001). The latter had little preference for the relocated object (ratio: 1.08 ± 0.097; one-sample t test, t(17) = 0.819; p = 0.42). Blocking hippocampal GRs did not ameliorate impairments of object location memory caused by acute stresses (main effect of stress, F(1,21) = 8.94, p < 0.01; Fig. 7A). The GR blocker had little effect in stressed mice (no main effect of antagonist, F(1,21) = 0.17, p = 0.69) and object exploration still did not differ from chance (t(3) = 1.25, p = 0.303). Similarly, after selective blocking of CRFR1 receptors using NBI 30775, stressed mice explored the novel location object less than the controls (main effect of stress, F(1,16) = 6.86, p < 0.05), not more than predicted by chance (t(4) = 1.26, p = 0.28), and the antagonist had little effect (no main effect of antagonist, F(1,16) = 0.48, p = 0.50; one-sample t test, t(3) = 0.53, p = 0.63; Fig. 7B). In contrast, blocking both GR and CRFR1 via RU38486+NBI 30775 abrogated the stress-induced deficits in spatial memory (trend for stress × drug interaction, F(1,16) = 2.90, p = 0.1; post hoc, p < 0.05; Fig. 7C). Stressed mice given the mixture spent significantly more time exploring objects that had been moved (one-sample t test, t(4) = 12.61, p < 0.01), whereas untreated stressed mice did not. In all experiments, control, stressed, and blocker-treated mice had comparable periods of exploration during both training and testing sessions (p > 0.05; Fig. 8). Importantly, in these experiments, the central administration of individual or combined receptor blockers did not influence the magnitude of hypothalamic–pituitary axis activation as measured by plasma CORT levels (one-way ANOVA, F(4,19) = 38.02, p < 0.001; post hoc, p < 0.0001, for control vehicle being lower than all stress groups. Post hoc tests did not distinguish the stress vehicle from the stress antagonist groups; Fig. 7D).
Figure 7.

Blocking the actions of both CORT and CRH, but not each individually, abrogates memory impairments induced by acute concurrent stresses (MAS). A, Stressed mice explored the object in the novel location not more than expected by chance (one-sample t test, t(7) = 1.161, p = 0.238) and less than controls (F(1, 21) = 8.94; p < 0.01). The GR blocker RU38486 had little effect (F(1, 21) = 0.17, p = 0.69) and object exploration did not differ from chance (t(3) = 1.25, p = 0.303). B, After selective blocking of CRFR1 receptors using NBI 30775, stressed mice explored the object in the novel location less than controls (F(1, 16) = 6.86, p < 0.05) and the antagonist had little effect (F(1, 16) = 0.48, p = 0.50). C, Blocking both GR and CRFR1 by a mixture of RU38486+NBI 30775 prevented stress-induced deficits in spatial memory (stress × drug interaction, F(1, 15) = 6.22, p < 0.05; post hoc, p < 0.05, for MAS + vehicle < MAS + both antagonists). Stressed mice explored the object in the novel location less than controls, yet mixture-treated stressed mice spent significantly more time exploring the novel-located object than chance (one-sample t test, t(4) = 12.61, p < 0.01). In all cases, mice were trained after they recovered from the stress and receptor blockers were administered centrally before the stresses. Asterisks in A–C denote values that are different from those expected by chance. D, Blocking brain GR, CRFR1, or both did not interfere with activation of the hypothalamic–pituitary adrenal axis, as apparent from robust increase of plasma levels of CORT in all MAS groups (one-way ANOVA, F(4, 19) = 38.02, p < 0.001; post hoc, p < 0.0001, for control vehicle being lower than all stress groups; post hoc tests did not distinguish the stress vehicle from the stress antagonist groups). CORT levels were measured at 1 h from the onset of the ongoing stresses (Maras et al., 2014; n = 3–6 per group; *p < 0.05 vs stress groups).
Figure 8.
The total duration of exploring the novel and familiar objects are comparable in stress-free and stressed mice with/without antagonist-treatment. Cannulas were implanted 5–6 d before the experiments. Blockers including RU38486 (750 ng in 1 μl), a specific blocker of glucocorticoid receptors (A); NBI 30775 (5 μg in 1 μl), a specific antagonist of CRH receptor type 1, CRFR1 (B); and a RU38486+NBI 30775 mixture (750 ng + 5 μg in 1 μl, respectively) (C) were delivered via the cannulas 30 min before a 2 h MAS. After a recovery period of 1.5–2 h, animals were trained (10 min) for exploring 2 objects (training session), followed by a 5 min testing (testing session) 6 h later the training (n = 8 mice per group, p > 0.05).
Discussion
The present results demonstrate the potent coordinate actions of CORT and CRH on hippocampal synapses. They suggest that both hormones may function during acute concurrent stresses and interact to disrupt dendritic spine integrity and synaptic potentiation, with significant consequences on memory functions. In addition, these findings may shed light on the origin of spatial memory problems identified in posttraumatic stress disorder (PTSD).
Excitatory postsynaptic elements of principal neurons reside on dendritic spines and the integrity of these, as well as changes in their dimensions and receptor type and numbers, have been critically implicated in memory processes (Yuste and Bonhoeffer, 2001; Segal, 2005; Bourne and Harris, 2008; Holtmaat and Svoboda, 2009; Makino and Malinow, 2009; Kasai et al., 2010). Not surprisingly, stress hormones that influence memory have striking effects on dendritic spines (Joëls and Baram, 2009; Lupien et al., 2009; Krugers et al., 2010; Popoli et al., 2011; Maras and Baram, 2012; Conboy and Sandi, 2010). CORT, the canonical adrenal stress hormone, increases spine formation and turnover in mature cortex within hours (Liston and Gan, 2011) and induces spine and dendrite atrophy chronically (Lupien et al., 2009; Popoli et al., 2011). Here, we show that a short CORT exposure tends to shift the distribution of spine size, which is consistent with the established effects of the hormone on trafficking of AMPA receptors into dendritic spine heads (Krugers et al., 2010; Conboy and Sandi, 2010). CRH also contributes to mechanisms of synapse and dendritic spine stability (Pawlak et al., 2005; Reyes et al., 2008; Chen et al., 2010) and the actions of the peptide, which is released during stress from hippocampal interneurons (Chen et al., 2012), take place within minutes to hours (Chen et al., 2013). We now show for the first time that, together, CORT and CRH provoke a collapse of synaptic function and plasticity accompanied by rapid loss of spines. Particularly affected are smaller PSD-95-expressing synapses, where CORT seems to potentiate the effects of CRH. These are thought to undergo insertion of AMPA receptors and growth during processes of memory formation (Bourne and Harris, 2008; Holtmaat and Svoboda, 2009; Makino and Malinow, 2009; Kasai et al., 2010). Therefore, the synergistic actions of nanomolar levels of CORT and CRH, estimated to be present in hippocampus during stress (Joëls and Baram, 2009; Maras and Baram, 2012), lead to accelerated deterioration of spine and synapse integrity.
Spine structure and synaptic plasticity are both dependent upon dynamics of the actin spine skeleton (Newey et al., 2005; van Galen and Ramakers, 2005; Rex et al., 2009; Penzes et al., 2011). Remodeling of filamentous, polymeric actin (F-actin) via actin-interacting proteins is required for actin branching and spine growth and stabilization. In contrast, runaway cleavage of F-actin promotes spine collapse. The small Rho-GTPases, including RhoA, Rac1, and cdc42, interact to enable actin dynamics and stability (Newey et al., 2005; van Galen and Ramakers, 2005; Kramár et al., 2009). Therefore, LTP induction leads to actin polymerization via RhoA-cofilin signaling and to F-actin stabilization via a Rac1 cascade (Kramár et al., 2009; Rex et al., 2009). We have found previously that CORT prevents the RhoA activation that is required for spine growth and synapse potentiation (Jafari et al., 2012) and that stress levels of CRH promote spine loss via activation of calpain (Andres et al., 2013), an enzyme that degrades RhoA (Briz et al., 2015). Together, these two hormones should deplete active RhoA rapidly from dendritic spine synapses, promoting their collapse (Fig. 5A). Indeed, the current data indicate that CRH alone reduces levels of total RhoA in the spine. However, reduction of the active RhoA requires concerted actions of both stress hormones and leads to rapid and catastrophic spine destabilization.
The complexity of the cellular and molecular machinery engaged during simultaneous acute stresses to disrupt memory processes is remarkable, although, in the current study, we have not tested in parallel the possibility that a single severe stress might activate similar processes. This complex, integrated set of mechanisms suggests that the phenomenon may have an adaptive role, yet this role remains unclear. Memory of a manageable acute stressful event may enable its future avoidance, whereas memory of concurrent stresses may be overwhelming (Hines et al., 2014; Baglietto-Vargas et al., 2015; Blore et al., 2015) and amnesia of the event may be adaptive. Here, we found that acute concurrent stresses disrupted encoding of hippocampus-dependent episodic memory (the “what” of narrative memory), with unclear biological advantage. Notably, selective deficits in spatial memory have been identified in stress-related memory disorders including PTSD (Beers and De Bellis, 2002; Gilbertson et al., 2001; Miller and McEwen et al., 2006; Luethi et al., 2011; Tempesta et al., 2012; St Jacques et al., 2013; Ramaswamy et al., 2015; Smith et al., 2015). Therefore, the profound and enduring memory deficits engendered by acute concurrent stresses may shed light into potential mechanisms underlying these memory defects.
Footnotes
This work was supported by the National Institutes of Health (Grants P50 MH096889, NS28912, and NS45260) and the Hewitt Foundation for Biomedical Research. We thank Ben Gunn for excellent discussions.
The authors declare no competing financial interests.
References
- Adler AB, Keane TM, Bliese PD. Measurement of posttraumatic stress disorder in an occupational health context. In: Sinclair RR, Wang M, Tetrick LE, editors. Research methods in occupational health psychology: measurement, design, and data analysis. New York: Routledge/Taylor and Francis Group; 2013. pp. 122–137. [Google Scholar]
- Andres AL, Regev L, Phi L, Seese RR, Chen Y, Gall CM, Baram TZ. NMDA receptor activation and calpain contribute to disruption of dendritic spines by the stress neuropeptide CRH. J Neurosci. 2013;33:16945–16960. doi: 10.1523/JNEUROSCI.1445-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baglietto-Vargas D, Chen Y, Suh D, Ager RR, Rodriguez-Ortiz CJ, Medeiros R, Myczek K, Green KN, Baram TZ, LaFerla FM. Short-term modern life-like stress exacerbates Aβ-pathology and synapse loss in 3xTg-AD mice. J Neurochem. 2015;134:915–926. doi: 10.1111/jnc.13195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beers SR, De Bellis MD. Neuropsychological function in children with maltreatment-related posttraumatic stress disorder. Am J Psychiatry. 2002;159:483–486. doi: 10.1176/appi.ajp.159.3.483. [DOI] [PubMed] [Google Scholar]
- Blore JD, Sim MR, Forbes AB, Creamer MC, Kelsall HL. Depression in Gulf War veterans: a systematic review and meta-analysis. Psychol Med. 2015;45:1565–1580. doi: 10.1017/S0033291714001913. [DOI] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briz V, Zhu G, Wang Y, Liu Y, Avetisyan M, Bi X, Baudry M. Activity-dependent rapid local RhoA synthesis is required for hippocampal synaptic plasticity. J Neurosci. 2015;35:2269–2282. doi: 10.1523/JNEUROSCI.2302-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadbent NJ, Gaskin S, Squire LR, Clark RE. Object recognition memory and the rodent hippocampus. Learn Mem. 2009;17:5–11. doi: 10.1101/lm.1650110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Moser EI. Memory, navigation and theta rhythm in the hippocampal-entorhinal system. Nat Neurosci. 2013;16:130–138. doi: 10.1038/nn.3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Bender RA, Frotscher M, Baram TZ. Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: a quantitative spatiotemporal analysis. J Neurosci. 2001;21:7171–7181. doi: 10.1523/JNEUROSCI.21-18-07171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Fenoglio KA, Dubé CM, Grigoriadis DE, Baram TZ. Cellular and molecular mechanisms of hippocampal activation by acute stress are age-dependent. Mol Psychiatry. 2006;11:992–1002. doi: 10.1038/sj.mp.4001863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Dubé CM, Rice CJ, Baram TZ. Rapid loss of dendritic spines after stress involves derangement of spine dynamics by corticotropin-releasing hormone. J Neurosci. 2008;28:2903–2911. doi: 10.1523/JNEUROSCI.0225-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Rex CS, Rice CJ, Dubé CM, Gall CM, Lynch G, Baram TZ. Correlated memory defects and hippocampal dendritic spine loss after acute stress involve corticotropin-releasing hormone signaling. Proc Natl Acad Sci U S A. 2010;107:13123–13128. doi: 10.1073/pnas.1003825107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Andres AL, Frotscher M, Baram TZ. Tuning synaptic transmission in the hippocampus by stress: the CRH system. Front Cell Neurosci. 2012;6:13. doi: 10.3389/fncel.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Kramár EA, Chen LY, Babayan AH, Andres AL, Gall CM, Lynch G, Baram TZ. Impairment of synaptic plasticity by the stress mediator CRH involves selective destruction of thin dendritic spines via RhoA signaling. Mol Psychiatry. 2013;18:485–496. doi: 10.1038/mp.2012.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conboy L, Sandi C. Stress at learning facilitates memory formation by regulating AMPA receptor trafficking through a glucocorticoid action. Neuropsychopharmacology. 2010;35:674–685. doi: 10.1038/npp.2009.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corneil W, Beaton R, Murphy S, Johnson C, Pike K. Exposure to traumatic incidents and prevalence of posttraumatic stress symptomatology in urban firefighters in two countries. J Occup Health Psychol. 1999;4:131–141. doi: 10.1037/1076-8998.4.2.131. [DOI] [PubMed] [Google Scholar]
- Gilbertson MW, Gurvits TV, Lasko NB, Orr SP, Pitman RK. Multivariate assessment of explicit memory function in combat veterans with posttraumatic stress disorder. J Trauma Stress. 2001;14:413–432. doi: 10.1023/A:1011181305501. [DOI] [PubMed] [Google Scholar]
- Hines LA, Sundin J, Rona RJ, Wessely S, Fear NT. Posttraumatic stress disorder post Iraq and Afghanistan: prevalence among military subgroups. Can J Psychiatry. 2014;59:468–479. doi: 10.1177/070674371405900903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Svoboda K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci. 2009;10:647–658. doi: 10.1038/nrn2699. [DOI] [PubMed] [Google Scholar]
- Ivy AS, Rex CS, Chen Y, Dubé C, Maras PM, Grigoriadis DE, Gall CM, Lynch G, Baram TZ. Hippocampal dysfunction and cognitive impairments provoked by chronic early-life stress involve excessive activation of CRH receptors. J Neurosci. 2010;30:13005–13015. doi: 10.1523/JNEUROSCI.1784-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafari M, Seese RR, Babayan AH, Gall CM, Lauterborn JC. Glucocorticoid receptors are localized to dendritic spines and influence local actin signaling. Mol Neurobiol. 2012;46:304–315. doi: 10.1007/s12035-012-8288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M, Baram TZ. The neuro-symphony of stress. Nat Rev Neurosci. 2009;10:459–466. doi: 10.1038/nrn2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joëls M, Fernandez G, Roozendaal B. Stress and emotional memory: a matter of timing. Trends Cogn Sci. 2011;15:280–288. doi: 10.1016/j.tics.2011.04.004. [DOI] [PubMed] [Google Scholar]
- Kasai H, Hayama T, Ishikawa M, Watanabe S, Yagishita S, Noguchi J. Learning rules and persistence of dendritic spines. Eur J Neurosci. 2010;32:241–249. doi: 10.1111/j.1460-9568.2010.07344.x. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Diamond DM. The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci. 2002;3:453–462. doi: 10.1038/nrn849. [DOI] [PubMed] [Google Scholar]
- Kramár EA, Chen LY, Brandon NJ, Rex CS, Liu F, Gall CM, Lynch G. Cytoskeletal changes underlie estrogen's acute effects on synaptic transmission and plasticity. J Neurosci. 2009;29:12982–12993. doi: 10.1523/JNEUROSCI.3059-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krugers HJ, Hoogenraad CC, Groc L. Stress hormones and AMPA receptor trafficking in synaptic plasticity and memory. Nat Rev Neurosci. 2010;11:675–681. doi: 10.1038/nrn2913. [DOI] [PubMed] [Google Scholar]
- Lauterborn JC, Pineda E, Chen LY, Ramirez EA, Lynch G, Gall CM. Ampakines cause sustained increases in brain-derived neurotrophic factor signaling at excitatory synapses without changes in AMPA receptor subunit expression. Neuroscience. 2009;159:283–295. doi: 10.1016/j.neuroscience.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liston C, Gan WB. Glucocorticoids are critical regulators of dendritic spine development and plasticity in vivo. Proc Natl Acad Sci U S A. 2011;108:16074–16079. doi: 10.1073/pnas.1110444108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luethi M, Meier B, Sandi C. Stress effects on working memory, explicit memory, and implicit memory for neutral and emotional stimuli in healthy men. Front Behav Neurosci. 2009;2:5. doi: 10.3389/neuro.08.005.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luksys G, Sandi C. Neural mechanisms and computations underlying stress effects on learning and memory. Curr Opin Neurobiol. 2011;21:502–508. doi: 10.1016/j.conb.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 2009;10:434–445. doi: 10.1038/nrn2639. [DOI] [PubMed] [Google Scholar]
- Maggio N, Segal M. Differential modulation of long-term depression by acute stress in the rat dorsal and ventral hippocampus. J Neurosci. 2009;29:8633–8638. doi: 10.1523/JNEUROSCI.1901-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino H, Malinow R. AMPA receptor incorporation into synapses during LTP: the role of lateral movement and exocytosis. Neuron. 2009;64:381–390. doi: 10.1016/j.neuron.2009.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maras PM, Baram TZ. Sculpting the hippocampus from within: stress, spines, and CRH. Trends Neurosci. 2012;35:315–324. doi: 10.1016/j.tins.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maras PM, Molet J, Chen Y, Rice C, Ji SG, Solodkin A, Baram TZ. Preferential loss of dorsal-hippocampus synapses underlies memory impairments provoked by short, multimodal stress. Mol Psychiatry. 2014;19:811–822. doi: 10.1038/mp.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MM, McEwen BS. Establishing an agenda for translational research on PTSD. Ann N Y Acad Sci. 2006;1071:294–312. doi: 10.1196/annals.1364.023. [DOI] [PubMed] [Google Scholar]
- Newey SE, Velamoor V, Govek EE, Van Aelst L. Rho GTPases, dendritic structure, and mental retardation. J Neurobiol. 2005;64:58–74. doi: 10.1002/neu.20153. [DOI] [PubMed] [Google Scholar]
- Pawlak R, Rao BS, Melchor JP, Chattarji S, McEwen B, Strickland S. Tissue plasminogen activator and plasminogen mediate stress-induced decline of neuronal and cognitive functions in the mouse hippocampus. Proc Natl Acad Sci U S A. 2005;102:18201–18206. doi: 10.1073/pnas.0509232102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Cahill ME, Jones KA, VanLeeuwen JE, Woolfrey KM. Dendritic spine pathology in neuropsychiatric disorders. Nat Neurosci. 2011;14:285–293. doi: 10.1038/nn.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2011;13:22–37. doi: 10.1038/ncb2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy S, Madabushi J, Hunziker J, Bhatia SC, Petty F. An open-label trial of memantine for cognitive impairment in patients with posttraumatic stress disorder. J Aging Res. 2015 doi: 10.1155/2015/934162. 934162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refojo D, et al. Glutamatergic and dopaminergic neurons mediate anxiogenic and anxiolytic effects of CRHR1. Science. 2011;333:1903–1907. doi: 10.1126/science.1202107. [DOI] [PubMed] [Google Scholar]
- Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, Lynch G. Different Rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J Cell Biol. 2009;186:85–97. doi: 10.1083/jcb.200901084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes BA, Valentino RJ, Van Bockstaele EJ. Stress-induced intracellular trafficking of corticotropin-releasing factor receptors in rat locus coeruleus neurons. Endocrinology. 2008;149:122–130. doi: 10.1210/en.2007-0705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi B, Cordero MI, Sandi C. Learning under stress: the inverted-U-shape function revisited. Learn Mem. 2010;17:522–530. doi: 10.1101/lm.1914110. [DOI] [PubMed] [Google Scholar]
- Seese RR, Chen LY, Cox CD, Schulz D, Babayan AH, Bunney WE, Henn FA, Gall CM, Lynch G. Synaptic abnormalities in the infralimbic cortex of a model of congenital depression. J Neurosci. 2013;33:13441–13448. doi: 10.1523/JNEUROSCI.2434-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M. Dendritic spines and long-term plasticity. Nat Rev Neurosci. 2005;6:277–284. doi: 10.1038/nrn1649. [DOI] [PubMed] [Google Scholar]
- Sheng H, Zhang Y, Sun J, Gao L, Ma B, Lu J, Ni X. Corticotropin-releasing hormone depresses n-methyl-D-aspartate receptor-mediated current in cultured rat hippocampal neurons via CRH receptor type 1. Endocrinology. 2008;149:1389–1398. doi: 10.1210/en.2007-1378. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Chua C, Falduto J. Sex differences and opposite effects of stress on dendritic spine density in the male versus female hippocampus. J Neurosci. 2001;21:6292–6297. doi: 10.1523/JNEUROSCI.21-16-06292.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KV, Burgess N, Brewin CR, King JA. Impaired allocentric spatial processing in posttraumatic stress disorder. Neurobiol Learn Mem. 2015;119:69–76. doi: 10.1016/j.nlm.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Jacques PL, Kragel PA, Rubin DC. Neural networks supporting autobiographical memory retrieval in posttraumatic stress disorder. Cogn Affect Behav Neurosci. 2013;13:554–566. doi: 10.3758/s13415-013-0157-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempesta D, Mazza M, Iaria G, De Gennaro L, Ferrara M. A specific deficit in spatial memory acquisition in post-traumatic stress disorder and the role of sleep in its consolidation. Hippocampus. 2012;22:1154–1163. doi: 10.1002/hipo.20961. [DOI] [PubMed] [Google Scholar]
- Thoeringer CK, Henes K, Eder M, Dahlhoff M, Wurst W, Holsboer F, Deussing JM, Moosmang S, Wotjak CT. Consolidation of remote fear memories involves Corticotropin-releasing hormone receptor type 1-mediated enhancement of AMPA receptor GluR1 signaling in the dentate gyrus. Neuropsychopharmacology. 2012;37:787–796. doi: 10.1038/npp.2011.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trieu BH, Kramár EA, Cox CD, Jia Y, Wang W, Gall CM, Lynch G. Pronounced differences in signal processing and synaptic plasticity between piriform-hippocampal network stages: a prominent role for adenosine. J Physiol. 2015;593:2889–2907. doi: 10.1113/JP270398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Galen EJ, Ramakers GJ. Rho proteins, mental retardation and the neurobiological basis of intelligence. Prog Brain Res. 2005;147:295–317. doi: 10.1016/S0079-6123(04)47022-8. [DOI] [PubMed] [Google Scholar]
- van Gemert NG, Carvalho DM, Karst H, van der Laan S, Zhang M, Meijer OC, Hell JW, Joëls M. Dissociation between rat hippocampal CA1 and dentate gyrus cells in their response to corticosterone: effects on calcium channel protein and current. Endocrinology. 2009;150:4615–4624. doi: 10.1210/en.2009-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel-Ciernia A, et al. The neuron-specific chromatin regulatory subunit BAF53b is necessary for synaptic plasticity and memory. Nat Neurosci. 2013;16:552–561. doi: 10.1038/nn.3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KV, Hartmann J, Mangold K, Wang XD, Labermaier C, Liebl C, Wolf M, Gassen NC, Holsboer F, Rein T, Müller MB, Schmidt MV. Homer1 mediates acute stress-induced cognitive deficits in the dorsal hippocampus. J Neurosci. 2013;33:3857–3864. doi: 10.1523/JNEUROSCI.4333-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XD, Chen Y, Wolf M, Wagner KV, Liebl C, Scharf SH, Harbich D, Mayer B, Wurst W, Holsboer F, Deussing JM, Baram TZ, Müller MB, Schmidt MV. Forebrain CRHR1 deficiency attenuates chronic stress-induced cognitive deficits and dendritic remodeling. Neurobiol Dis. 2011;42:300–310. doi: 10.1016/j.nbd.2011.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XD, Su YA, Wagner KV, Avrabos C, Scharf SH, Hartmann J, Wolf M, Liebl C, Kühne C, Wurst W, Holsboer F, Eder M, Deussing JM, Müller MB, Schmidt MV. Nectin-3 links CRHR1 signaling to stress-induced memory deficits and spine loss. Nat Neurosci. 2013;16:706–713. doi: 10.1038/nn.3395. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T. Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci. 2001;24:1071–1089. doi: 10.1146/annurev.neuro.24.1.1071. [DOI] [PubMed] [Google Scholar]