

Abstract
In factory cells, the accumulation of misfolded protein provokes the unfolded protein response (UPR). For example, deletion of serine 63 (S63del) in myelin protein zero (P0) induces P0 accumulation in the endoplasmic reticulum (ER) of Schwann cells and a persistent UPR associated with Charcot-Marie-Tooth 1B (CMT1B) demyelinating peripheral neuropathy in human and mouse. PERK (protein kinase RNA-like ER kinase) is the ER stress sensor that attenuates global translation by phosphorylating eIF2α. Inhibition of the eIF2α holophosphatase GADD34:PP1, increases the phosphorylation of eIF2α in Schwann cells and largely rescues S63del neuropathy. Nonetheless, reducing phosphorylation of eIF2α, by Perk haploinsufficiency, also ameliorates the myelin defects of S63del nerves. This contradictory finding prompted us to investigate whether the beneficial effect of Perk deficiency on myelination could derive from neurons. To test this hypothesis, we generated and compared Schwann cell- and neuron-specific ablation of Perk in S63del nerves. Our data suggest that the detrimental effect of Perk in CMT1B derives primarily from Schwann cells. Furthermore, we show that Perk loss of function in Schwann cells restores myelination without diminishing accumulation of P0 or markers of ER stress, suggesting that Perk may modulate myelination through a pathway independent of the UPR.
SIGNIFICANCE STATEMENT In many endoplasmic reticulum (ER) stress-related disorders, activation of the unfolded protein sensor protein kinase RNA-like ER kinase (PERK) kinase is beneficial. Nonetheless, in Charcot-Marie-Tooth 1B neuropathy mice, we show that activation of PERK in Schwann cells, but not in neurons, is detrimental for myelination. PERK may interfere with myelination, independent of its role in ER stress.
Keywords: Charcot-Marie-Tooth, myelin, PERK, proteostasis, Schwann cells, unfolded protein response
Introduction
Myelin is a multilayered structure that protects axons and maintains the speed of electrical impulses along peripheral nerves (Sherman and Brophy, 2005). Schwann cells are the myelinating glia of the peripheral nervous system (PNS) and produce an enormous amount of proteins daily, among which myelin protein zero (P0) accounts for 20% (Patzig et al., 2011). Thus, myelinating Schwann cells require an efficient protein quality control to address translational burden. Deletion of serine 63 in P0 protein (P0S63del) causes Charcot-Marie-Tooth 1B (CMT1B) demyelinating neuropathy, which is characterized by demyelination, reduction in nerve conduction velocity, and motor impairment (Kulkens et al., 1993; Wrabetz et al., 2006; Miller et al., 2012). P0S63del protein is retained in the endoplasmic reticulum (ER) and triggers the unfolded protein response (UPR), which is pathogenetic in S63del neuropathy (Pennuto et al., 2008; D'Antonio et al., 2013).
The UPR restores cellular homeostasis by reducing the amount of aberrant proteins that accumulate in the ER (Schröder and Kaufman, 2005; Walter and Ron, 2011). The UPR is intermittently active and protective in physiological conditions. However, when it becomes persistent, the UPR can activate maladaptive effectors involved in cell death and dedifferentiation (Tabas and Ron, 2011; Hetz et al., 2013). Three ER stress sensors initiate the UPR pathways: the inositol-requiring protein-1 (IRE1), activating transcription factor-6 (ATF6), and protein kinase RNA-like ER kinase (PERK). Upon activation, IRE1 mediates the splicing of X-box binding protein 1 (Xbp1) mRNA (Bertolotti and Ron, 2001; Yoshida et al., 2001; Calfon et al., 2002). In mammals, XBP1s reprograms the transcription of genes involved in protein folding, degradation, and ER biogenesis (Friedlander et al., 2000; Travers et al., 2000; Lee et al., 2003; Yoshida et al., 2003; Sriburi et al., 2004). The ATF6 pathway promotes the expression of ER resident chaperones (Haze et al., 1999; Yoshida et al., 2000; Shen et al., 2002). Finally, PERK kinase phosphorylates the eukaryotic translation initiation factor 2 α (eIF2α) on Ser51 (Harding et al., 1999), which causes a global attenuation of protein translation, and thereby limits the accumulation of client proteins in the ER (Harding et al., 2000b).
Upon persistent stress, PERK-dependent eIF2α phosphorylation is beneficial for professional secretory cells; accordingly, Perk loss of function results in reduced numbers of differentiated pancreatic β cells or osteoblasts (Harding et al., 2000b, 2001b; Zhang et al., 2002, 2006; Wei et al., 2008). Downstream of PERK, growth arrest and DNA damage-inducible protein (GADD34):PP1 holophosphatase reduces eIF2α phosphorylation to limit translational arrest (Novoa et al., 2001). We have shown that loss of Gadd34 increases eIF2α phosphorylation and almost completely rescues S63del neuropathy (D'Antonio et al., 2013; Das et al., 2015). Nonetheless, Perk haploinsufficiency paradoxically also ameliorates the neuropathy of the CMT1B mouse model, despite that the levels of phosphorylated eIF2α (P-eIF2α) were significantly reduced (Musner et al., 2016).
These surprising results prompted us to investigate in S63del nerves whether the beneficial effect of Perk loss of function was Schwann cell or neuron autonomous. In this study, we demonstrate that Perk ablation in Schwann cells, but not in neurons, partially restores myelination in S63del nerves. The defects of S63del myelination are improved despite that P0 accumulates considerably in the ER, and the UPR markers remain upregulated. Our data suggest that the UPR may not be the only pathogenetic mechanism contributing to the S63del/CMT1B neuropathy. PERK may also perturb other pathways outside of the UPR.
Materials and Methods
Mice.
All experiments involving mice were performed in accord with the experimental protocols approved by the San Raffaele Scientific Institute, and Roswell Park Cancer Institute, and the University at Buffalo Institutional Animal Care and Use Committees. P0S63del transgenic mice include the S63del-L (129.4) transgenic line, overexpressing mutant MpzS63del mRNA by 60%, and the S63del-H (129.1) transgenic line, overexpressing mutant MpzS63del mRNA by 210%; Wrabetz et al., 2000, 2006). Mice containing Perk flanked by loxP conditional allele (Perkf/+ mice; Zhang et al., 2002) were crossed with mP0TOTA-Cre (P0Cre) mice (Feltri et al., 1999) and S63del mice, to obtain P0Cre/Perkf/+ and S63del/Perkf/+ breeders. Parents coming from these crosses were mated to generate S63del/P0Cre/Perkf/f (S63del/PerkSCKO) and S63del/P0Cre/Perkf/+ (S63del/PerkSCHet) and all other control genotypes within the same litter. To remove Perk from neurons, Perk floxed, NestinCre (Zimmerman et al., 1994; Tronche et al., 1999), and S63del mice were crossed, using the same mating strategy as for P0Cre to generate S63del/NestinCre/Perkf/f (S63del/PerkNRNKO) and control littermates. Chop-null mice (Zinszner et al., 1998) were used as controls and have been described previously. P0Cre, NestinCre, Perkf/+, and S63del-L mice were maintained on a C57BL/6NCrl background; S63del-H and Chop-null mice were maintained on an FVB/NCrl background. Male and female littermates were analyzed in all experiments. All genotypes were determined by PCR analysis of genomic DNA prepared from tail samples as described in the references above. Cre recombination PCR assays were performed on genomic DNA prepared from postnatal day 28 (P28) tissues with primer pairs generating 400 bp WT, 480 bp loxP, and 700 bp recombined loxP amplimers. PCR primers and conditions were as described previously (Zhang et al., 2002).
Morphology and electrophysiology.
For electrophysiology, mice were anesthetized with 20 mg/ml of tribromoethanol (Avertin), 0.02 ml g−1 of body weight. Nerve conduction velocity (NCV), F-wave latency, and distal motor action potential (CMAP) amplitude were analyzed at P28 as described previously (Wrabetz et al., 2006). Semithin section and electron microscopy were performed as described previously (Quattrini et al., 1996). Images for g ratio (axon diameter/fiber diameter), axonal distribution, and number of amyelinated fibers were acquired with a 100 × objective on a Leica DM 6000 microscope. Quantification was performed on four semithin images per sciatic nerve, 1200–1300 fibers in 12 fields for each genotype from WT, PerkSCKO, S63del, and S63del/PerkSCKO P28 sciatic nerves. The g ratio was measured using the Leica QWin software. Data were analyzed using GraphPad Prism, version 6.01. Ultrastructural images were acquired on an FEI Tecnai G2 Spirit BioTWIN electron microscope.
Western blot analysis.
Sciatic nerves from P28 mice were dissected and frozen in liquid nitrogen. Proteins were extracted in RIPA buffer (50 mm Tris HCl, pH 8.0, 150 mm NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm sodium orthovanadate, protease inhibitors; Krackeler). Lysates were sonicated and centrifuged at 13,200 rpm for 10 min at 4°C. Twenty micrograms of protein were resolved in SDS-polyacrylamide gel and blotted on PVDF membrane (PerkinElmer, GE Healthcare, Odyssey detection system). For the detection of phospho-IRE1 (P-IRE1), we used Phos-Tag (25 μm) acrylamide gel as described previously (Yang et al., 2010). Membranes were probed overnight with the following antibodies: rabbit poly-sera (Rb pAb) recognized α-tubulin (1:2000; Novus Biologicals), calnexin and GAPDH (Sigma, 1:2000), P-eIF2α (Cell Signaling Technology, 1:500), CCAAT/enhancer-binding homologous protein (CHOP) (Proteintech, 1:500), IRE1α (Cell Signaling Technology, 1:1000), activating transcription factor 4 (ATF4; Santa Cruz Biotechnology, 1:200), ATF6 (Enzo Life Sciences, 1:1000), and Rb mAb recognized PERK (Cell Signaling Technology, 1:1000). Membranes were incubated with HRP conjugated secondary antibodies against rabbit (DAKO, 1:5000). Rb pAb against calreticulin (Enzo Life Sciences, 1:1000), GRP78 (Novus Biologicals, 1:1000), GADD34 (Proteintech, 1:500), and rat mAb GRP94 (Abcam, 1:1000) were detected with the Odyssey CLx infrared imaging system (Li-Cor). IRDye 680 anti-mouse, 800 anti-rabbit, and 680 anti-rat were used as secondary antibodies (Li-Cor, 1:10,000). The densitometry was performed with ImageJ-64 software.
Immunohistochemistry.
Spinal cord from PerkNRNKO and sciatic nerves from P28 PerkSCKO were fixed overnight in 10% formalin buffer (Thermo Fisher Scientific). Tissues were infiltrated in sucrose (scale, 10–20-30%; 20 min each) and embedded in paraffin. Paraffin was removed from 1- to 2-μm-thick sections with three washings in xylene (Thermo Fisher Scientific; 10 min each) and ethanol (scale, 100-90-70%; 1 min each). Spinal cord sections were soaked in citrate buffer, pH 6.0 (95°C for 15 min), whereas sciatic nerve sections were immersed in Tris-EDTA, pH 9.0, buffer in an 87°C water bath for antigenic retrieval. Sections were washed in 10% H2O2 to block endogenous peroxidases, and were blocked with normal goat serum (Covance) for 20 min at room temperature. Sections were probed for 1 h with antibody against PERK (Lifespan, 1:500) in blocking buffer, followed by HRP-conjugated secondary antibody (DAKO Envision System-HRP) for 30 min at room temperature and diaminobenzadine (DAB) chromogen for 1 min at room temperature to reveal PERK. Finally, sections were counterstained with hematoxylin.
Immunofluorescence.
P28 sciatic nerves from WT, PerkSCKO, S63del, and S63del/PerkSCKO mice were fixed in 4% ice cold paraformaldehyde for 30 min before teasing on 3% 3-aminopropyl triethoxysilan-coated slides. Nerves were postfixed in 4% ice-cold paraformaldehyde for 15 min, permeabilized in cold methanol for 5 min, and blocked in 5% fish skin gelatin (Sigma), 0.5% Triton X-100 in 1 × PBS. Slides were incubated overnight with mouse antibody against KDEL (Enzo Life Sciences, 1:200) and chicken anti P0 (Aves, 1:300) in 0.2% Triton X-100/1 × PBS. After washing the primary antibodies in 1 × TBS, the slides were incubated for 1 h at room temperature with Jackson ImmunoResearch Alexa Fluor 594 secondary antibodies against mouse IgG sub2a and Alexa Fluor 488 secondary antibodies against chicken IgY (1:400). 4′,6-Diamidino-2-phenylindole (DAPI) was used to reveal nuclei. Adult dorsal root ganglia (DRGs) were dissected from P28 WT and PerkNRNKO mice and fixed in 4% ice cold paraformaldehyde for 30 min on ice. DRGs were then washed in 1 × PBS and left in 30% sucrose for at least 24 h at +4°C before embedding in tissue freezing media (OCT). After sectioning the DRGs (8 μm), 4% ice cold paraformaldehyde was applied for 15 min, and then acetone at −20°C for 5 min. DRGs were incubated overnight with the following primary antibodies: rabbit against phospho-eIF2α (Ser51; Cell Signaling Technology, 1:400) and mouse sub2a against Tuj-1 (BioLegend; 1:600) in blocking solution (5% filtered BSA, 1% donkey serum, and 0.2% Triton X-100 in 1 × TBS) at +4°C. Donkey secondary antibodies from Jackson ImmunoResearch against rabbit (Alexa Fluor 488) and against mouse IgG sub2a (Alexa Fluor 647) were used to finally reveal the staining. Pictures were acquired with the 100 × objective of the Leica confocal TCS SP5 microscope for sciatic nerve and the 20 × objective for adult DRGs. P0 pixel intensity within the KDEL area was quantified using ImageJ64 software as described previously (Fratta et al., 2011).
RNA isolation and TaqMan quantitative PCR.
RNA was isolated from P28 sciatic nerves using TRIzol reagent (Invitrogen), following the manufacturer's instructions. Briefly, pairs of nerves from single mice were pulverized in liquid nitrogen and added to 800 μl of TRIzol. Chloroform (160 μl) was added to the lysates and centrifuged at 12,000 × g for 15 min at 4°C. The aqueous phase was then placed overnight with 500 μl of isopropanol and 1 μl of glycogen (20 mg/ml). Pellets were collected by centrifugation at 12,000 × g for 10 min at 4°C and washed twice in 75% ethanol at 7500 × g for 15 min. One microgram of RNA was retrotranscribed to cDNA using the Superscript III kit (Invitrogen) following the manufacturer's instructions. TaqMan PCR analyses were performed on the Bio-Rad CFX96/384 real-time PCR machine. The PCRs for the target and reference genes were performed in separate test tubes with Assays-on-Demand (Applied Biosystems): Ddit3/Chop, Mm00492097_m1; Myd116/Gadd34, Mm00435119_m1; Xbp1s, Mm03464496_m1; Dnajc3/p58IPK, Mm00515299_m1; Dnajb9/ERdj4, Mm01622956_s1; Hspa5/BiP, Mm00517691_m1; Derl3/Derlin3, Mm00508292_m1; Car6/Doc1, Mm00486222_m1. Normalization was performed using rRNA 18s, Hs99999901_s1. Littermates were analyzed, and every genotype was compared to the WT of the same cohort.
Statistical analysis.
All statistical analysis was conducted using GraphPad Prism, version 6.01. In every experiment, at least three biological replicates were analyzed. To determine the significance between genotypes, Student's t test or one-way ANOVA was used. A value of p ≤ 0.05 was considered statistically significant. Graphical data are represented as mean ± SEM.
Results
Genetic ablation of Perk in S63del Schwann cells
Although Gadd34 impairment and increased levels of P-eIF2α are beneficial in S63del nerves (D'Antonio et al., 2013; Das et al., 2015), Perk haploinsuffiency and reduced P-eIF2α levels in nerves are paradoxically also beneficial in S63del mice (Musner et al., 2016). To investigate whether this contradiction results from effects of P-eIF2α in different cells in peripheral nerve in the two experiments, we exploited a conditional allele of Perk (Zhang et al., 2002) to produce mice lacking Perk in either Schwann cells or neurons.
To target ablation of Perk to Schwann cells, we crossed Perkf/+ mice (Zhang et al., 2002) with those with a P0Cre recombinase transgene to obtain P0Cre/Perkf/f mice (hereafter PerkSCKO). P0Cre activates recombination in all Schwann cells beginning at embryonic day 13.5 (Feltri et al., 1999). We then bred S63del onto the PerkSCKO background to obtain S63del/PerkSCKO mice.
To test the efficiency and specificity of the P0Cre, we analyzed the recombination of the PerkloxP allele by PCR assays in genomic DNA from WT, PerkSCHet, and PerkSCKO tissues (Fig. 1A). The Perk WT band appeared in all WT and PerkSCHet samples, whereas the nonrecombined Perk loxP band appeared in PerkSCHet as expected, but also in PerkSCKO samples, due to cells other than Schwann cells in nerve. The 700 bp recombination band (Zhang et al., 2002) appeared only in sciatic nerves from PerkSCHet and PerkSCKO mice, whereas it was not detected in brain, liver, kidney, muscle, or spinal cord samples (Fig 1A). Accordingly, PERK protein was almost absent from S63del/PerkSCKO sciatic nerve lysates (Fig 1B). Moreover, immunohistochemistry on cross-sections of sciatic nerves revealed that PERK cytoplasmic staining was notably reduced in Schwann cells from PerkSCKO mice (Fig. 1C, arrowheads). This evidence suggests that P0Cre efficiently removed PERK from myelinating Schwann cells.
Figure 1.
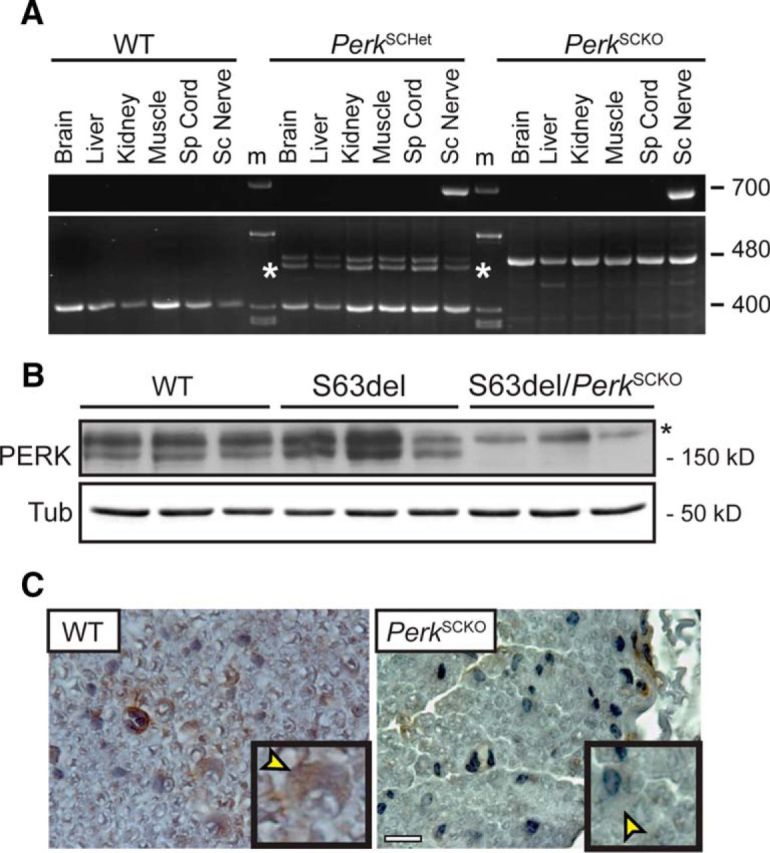
Genetic ablation of Perk in S63del Schwann cells. A, PCR analysis performed on genomic DNA from P28 WT, S63del/PerkSCHet, S63del/PerkSCKO nerves. The bands include a 400 bp WT allele, a 480 bp loxP allele, a probable 400 bp/480 bp heteroduplex marked by an asterisk, and a 700 bp recombined loxP allele. m, Markers. B, Western blot analysis for PERK was performed on P28 sciatic nerves from WT, S63del, and S63del/PerkSCKO mice. β-Tubulin (Tub) represents a loading control. An asterisk indicates a nonspecific band. C, Immunohistochemistry revealed PERK (brown) in P28 cross-sections of sciatic nerve. The insets show the presence of PERK in the cytoplasm (yellow arrowheads) of WT Schwann cells, but not in PerkSCKO Schwann cells. Nuclei were counterstained with hematoxylin (blue). Scale bar, 20 μm.
Perk ablation in Schwann cells improves S63del neuropathy
Schwann cells highly express P0 and P0S63del mRNA and protein at P28 (Wrabetz et al., 2006) and persistently activate PERK, which has detrimental effects in nerve (Musner et al., 2016). To confirm that PERK in Schwann cells contributes to the detrimental effect, we performed morphological analysis in P28 in PerkSCKO and S63del/PerkSCKO nerves. As shown previously, S63del nerves manifested hypomyelination compared to WT, as shown by semithin section (Fig. 2A) and g ratio analyses (Fig. 2B; 0.74 ± 0.02 in S63del vs 0.72 ± 0.01 in WT). Perk elimination in WT Schwann cells did not significantly alter myelination; however, its elimination in S63del Schwann cells returned myelin thickness to WT levels as measured by g-ratio analysis (Fig. 2B; 0.72 ± 0.02 in S63del/PerkSCKO vs 0.72 ± 0.01 in WT).
Figure 2.
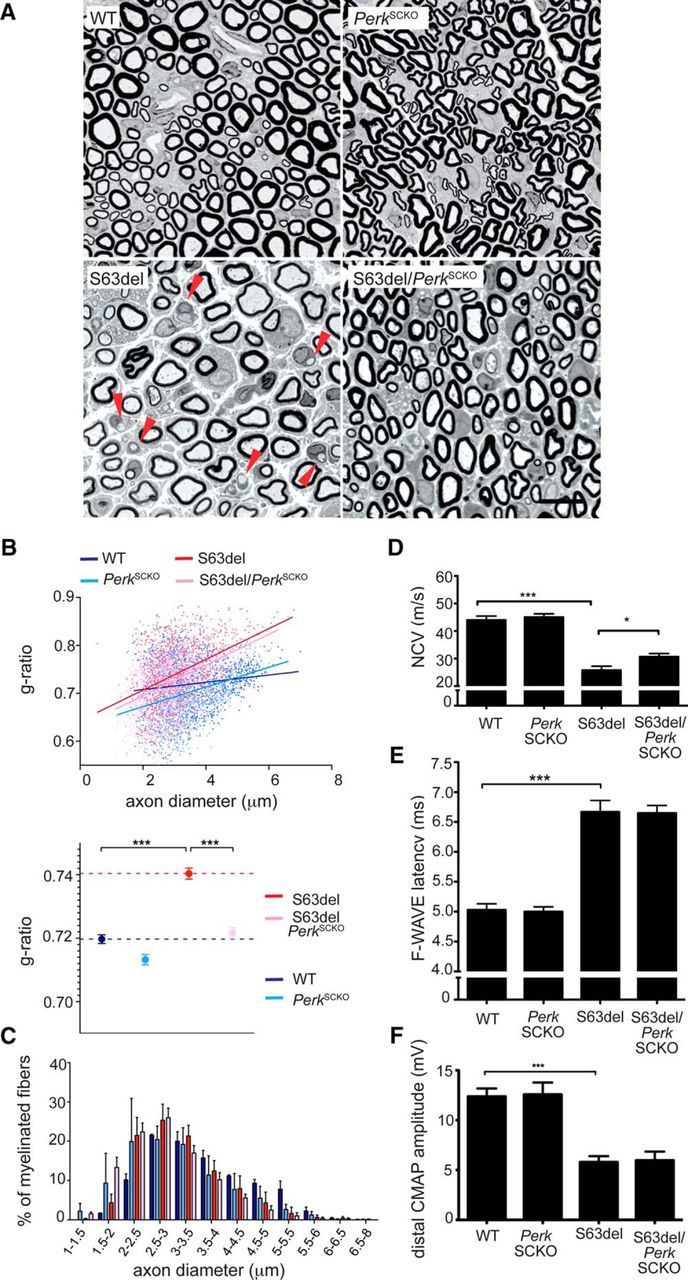
Perk ablation in Schwann cells ameliorates morphological and electrophysiological abnormalities in S63del mice. A, Semithin sections of P28 sciatic nerve stained with toluidine blue from WT, PerkSCKO, S63del, and S63del/PerkSCKO mice. S63del nerves show evidence of amyelinated fibers (red arrowheads). Scale bar, 10 μm. B, g ratio analysis (scatterplot above and graph below) confirmed hypomyelination in S63del (red) versus WT (blue) nerves, whereas in S63del/PerkSCKO(pink) nerves, myelin returns to WT thickness. ***p < 0.01 (one-way ANOVA with Bonferroni correction; 5 microscopic fields from 3 animals containing n = 1000–1200 fibers per genotype). C, Frequency distribution of myelinated axons as a function of axonal diameter shows that Perk ablation from S63del Schwann cells (pink) restores myelination, especially in small caliber axons (1–2 μm), compared to S63del (red). D–F, Electrophysiological analysis of P28 mouse sciatic nerve showing NCV (D), F-wave latency (E), and distal CMAP amplitude (F). *p < 0.05; ***p < 0.001 (Student's t test). n = 12–30 nerves. Error bars indicate SEM.
We noticed a slight shift toward smaller axon diameters in S63del compared to WT nerves, consistent with a secondary reduction of neurofilament phosphorylation due to mild hypomyelination (Fig. 1C; de Waegh et al., 1992). Interestingly, the number of the smallest myelinated fibers (1–2 μm in diameter) rose sharply in S63del/PerkSCKO compared to S63del nerves (Fig. 2C, compare pink, red bars). More generally, electron microscopic analysis confirmed the increment of myelin thickness in S63del/PerkSCKO compared to S63del nerves (Fig. 3A; axons pseudocolored blue), including in axons of equal diameter (Fig. 3B, a, a′). The nerves of C57BL6/N-S63del mice contain a considerable number of amyelinated axons. These axons are not myelinated despite their 1:1 relationship with Schwann cells and appropriate diameter (≥1 μm). In S63del/PerkSCKO nerves, the percentage of amyelinated fibers is reduced fourfold compared to S63del (Fig. 3C).
Figure 3.

Perk ablation decreases the number of amyelinated fibers in S63del nerves. A, Electron microscopy shows hypomyelination, especially in small caliber axons (pseudocolored blue), of S63del compared to WT sciatic nerves at P28. In S63del/PerkSCKO compared to S63del, myelin of small caliber axons appears thicker. Scale bar, 2 μm. B, Axons with similar diameter were magnified to show the difference between the hypomyelination in S63del (a) and its amelioration in S63del/PerkSCKO (a′). C, Quantification of amyelinated fibers (red arrowheads in Fig. 2A) on semithin sections of P28 sciatic nerves. Error bars indicate SEM. *p < 0.01; **p < 0.001 (one-way ANOVA with Bonferroni multiple comparison; 5 microscopic fields from 3 animals per genotype).
This improvement prompted us to investigate function. By electrophysiological analysis, we confirmed that NCV and distal CMAP amplitude were reduced (Fig. 2D,F), and F-wave latency was increased (E) in S63del compared to WT P28 mice. S63del/PerkSCKO nerves showed partially restored NCV compared to S63del (Fig. 2D), although the F-wave latency and distal CMAP amplitude did not differ between the two genotypes (Fig. 2E,F). Collectively, these data demonstrate that Perk ablation in S63del Schwann cells surprisingly prevents the developmental myelin abnormalities of the S63del neuropathy. Therefore, the detrimental effect of PERK on myelination does originate at least in part from Schwann cells.
Perk ablation in neurons does not alter S63del myelin defects
Perk haploinsufficiency surprisingly improved myelination and ameliorated the S63del phenotype (Musner et al., 2016). However, in the study by Musner et al. (2016), Perk was ablated in every cell type of the S63del mouse, including neurons. Moreover, axons are also damaged as a secondary consequence of demyelination in S63del nerves (Wrabetz et al., 2006). Therefore, we reasoned that Perk ablation in neurons might account for the paradoxical amelioration observed in S63del/Perk+/− mice. In particular, we asked whether Perk deletion in neurons could limit either the myelin defects or the secondary axonal defects as observed in S63del/PerkSCKO. We crossed Perkf/+ mice with mice expressing Cre recombinase in neural cells in the CNS, including motor neurons, and DRG neurons in the PNS (NestinCre; Tronche et al., 1999) to obtain NestinCre/Perkf/f mice (hereafter, PerkNRNKO). In this way, all neurons that contribute axons to sciatic nerves would lack PERK. We then bred S63del onto the PerkNRNKO background to obtain S63del/PerkNRNKO mice.
To test the efficiency of NestinCre-mediated ablation of Perk in neurons, PERK protein levels were detected by Western blot analysis of lumbar spinal cord (Fig. 4A) and by immunohistochemical analysis in ventral horn neurons (Fig. 4B, a, black arrowheads). In PerkNRNKO spinal cord, the level of PERK was significantly reduced in most motor neurons (Fig. 4A, B, a′). Importantly, we demonstrated that the eIF2α phosphorylation (PERK target; green) was also reduced in DRG sensory neurons (pseudocolor red) of PerkNRNKO, compared to WT (Fig. 4C). Together, these data indicate that PERK levels and kinase function have been effectively reduced in motor and DRG neurons in PerkNRNKO animals. In contrast, PERK was still easily detected in Schwann cell cytoplasm of PerkNRNKO sciatic nerves (Fig. 4B, a″, yellow arrowheads) confirming that NestinCre is not active in Schwann cells (Kao et al., 2009).
Figure 4.

Genetic ablation of Perk in neurons does not alter myelination in S63del nerves. A, PERK protein level was detected by Western blot analysis in whole lysates of spinal cord from P28 WT and PerkNRNKO mice. Neurofilament (NF) served as a loading control. The quantification of Western blot analysis confirms the decrease of PERK protein level in PerkNRNKO spinal cords. *p < 0.05 (Student's t test). n = 3. B, Immuohistochemistry on P28 spinal cord (ventral horns) and longitudinal sections of sciatic nerve revealed PERK in the cell body of neurons (a; black arrowheads indicate brown DAB precipitate) and Schwann cells (a″; yellow arrowheads), whereas PERK intensity strongly decreases in PerkNRNKO neurons (a′). Nuclei were counterstained with hematoxylin (blue). C, Immunofluorescence analysis on DRGs from P28 WT and PerkNRNKO mice shows P-eIF2α (green) in cell bodies of sensory DRG neurons (Tuj-1; pseudocolored red). D, Semithin sections of sciatic nerves stained with toluidine blue from P28 WT, PerkNRNKO, S63del, and S63del/PerkNRNKO mice. E, Quantification of amyelinated fibers was performed on semithin sections of P28 sciatic nerve. *p < 0.05 (one-way ANOVA; 5 microscopic fields from n = 3 animals per genotype). F, g ratio analysis (scatterplot) confirmed hypomyelination in S63del nerves (red) versus WT (blue) and PerkNRNKO (cyan). In S63del/PerkNRNKO (pink), myelin remains thin as in S63del. The graph represents the g ratio measured from over 700 myelinated axons (4–5 microscopic fields from n = 3 animals per genotype). G, H, Electrophysiological analysis on P28 sciatic nerves showing NCV (F) and distal CMAP amplitude (G). *p < 0.05; **p < 0.01; ***p < 0.001 (one-way ANOVA with Bonferroni's multiple comparison; n = 6–16 nerves; F–G). ns, Not significant. Scale bars: B, a, a′, 50 μm; B, a″, D, 10 μm; C, 20 μm. Error bars indicate SEM.
We analyzed myelin morphology and confirmed that S63del nerves show thinner myelin and an increased percentage of amyelinated fibers compared to WT nerves, as described previously (Fig. 4D–F). We did not find substantial differences between S63del and S63del/PerkNRNKO nerves for either the g ratio (Fig. 4E; 0.70 ± 0.005 vs 0.70 ± 0.010, respectively) or the amount of amyelinated fibers (Fig. 4E,F). Finally, Perk ablation in neurons did not alter NCV or distal CMAP amplitude of S63del (Fig. 4G,H). These data indicate that deletion of Perk in S63del neurons does not alter axons or myelination in S63del sciatic nerves.
Perk ablation reduces eIF2α phosphorylation but not CHOP activation in S63del nerves
Upon ER stress, PERK phosphorylates eIF2α to restore translational homeostasis (Harding et al., 1999, 2000a, 2001a). We showed previously that PERK is the main eIF2α kinase activated in S63del Schwann cells, and partial ablation of Perk reduced the level of eIF2α phosphorylation (Musner et al., 2016). Accordingly, we observed that eIF2α phosphorylation was remarkably reduced in S63del/PerkSCKO compared to S63del nerves (Fig. 5A). This reduction was noticeable also in PerkSCKO compared to WT control nerves. ATF4 mRNA escapes the translational block caused by P-eIF2α. In fact, ATF4 synthesis is ER stress dependent, and it decreases upon dephosphorylation of eIF2α (Harding et al., 2000a). The level of ATF4 was slightly reduced in S63del/PerkSCKO compared to S63del nerves, although the reduction was not statistically significant (Fig. 5B). Similarly, compared to S63del, Perk deficiency in S63del nerves did not cause a significant reduction of Chop mRNA or its target genes Gadd34, Doc1, and Derlin3 (Fig. 5C,E). Similarly, both CHOP and GADD34 protein levels remained high in S63del/PerkSCKO compared to S63del (Fig. 5D,F). Notably, we showed previously that only full Chop or Gadd34 ablation (Pennuto et al., 2008; D'Antonio et al., 2013), and not partial Chop ablation (data not shown), was sufficient to improve the S63del phenotype. Collectively, the data indicate that Perk ablation in Schwann cells remarkably diminishes eIF2α phosphorylation. However, removing Perk in S63del nerves does not alter Chop expression or activation of its target genes, suggesting that Chop does not account for the myelin improvement of S63del/PerkSCKO nerves.
Figure 5.

Perk ablation in S63del nerves reduced P-eIF2α but not CHOP activation. A, B, Western blot analysis on P28 sciatic nerves for P-eIF2α and ATF4. β-tubulin (Tub) served as a loading control. n = 4. C, Chop mRNA levels were measured on P28 sciatic nerves by RT-PCR TaqMan assay, normalized for 18S rRNA. S63delH (high), a different transgenic mouse that overexpresses the P0S63del transgene (210% expression), was used as positive control; S63del/Chop−/− nerves were used as a negative control. n = 6 RT from three independent sets of nerves of each genotype. D, CHOP protein levels were detected by Western blot analysis on P28 nerves. GAPDH served as a loading control. S63delH and S63del/Chop−/− sciatic nerves were used as positive and negative controls, respectively. One representative out of four independent experiments is shown. E, Gadd34, Doc1, and Derlin3 mRNA levels were measured on P28 sciatic nerves by RT-PCR TaqMan assay, normalized for 18S rRNA. n = 3–6 RT from three independent sets of nerves for each genotype. F, GADD34 protein levels were measured by Western blot analysis in P28 sciatic nerves and normalized to β-tubulin. n = 3. *p < 0.05; **p < 0.01 (Student's t test). ns, Not significant. Error bars indicate SEM.
Improved myelination in S63del/PerkSCKO nerves is not associated with compensation in the IRE1α and ATF6 pathways
IRE1α and ATF6 contribute to long-lasting adaptation to stress, and the amplification of these pathways is often beneficial upon ER stress. For example, persistent activation of IRE1α increases cell viability in HEK293 cells treated with tunicamycin or thapsigargin and in stressed neurons in the retina (Lin et al., 2007b). In addition, enhanced IRE1α expression and ATF6 activation upon stress have been reported in PERK-defective cells (Harding et al., 2000b; Yamaguchi et al., 2008). We therefore wondered whether Perk ablation in S63del Schwann cells might alter ATF6 or IRE1α signaling. ATF6 activation was estimated by the level of cleaved ATF6 (ATF6c) in nerve lysates from P28 WT, PerkSCKO, S63del, and S63del/PerkSCKO mice; ATF6c levels in S63del/PerkSCKO and S63del lysates were comparable (Fig. 6A). ATF6c promotes the expression of ER-resident chaperones, which are highly upregulated upon UPR (Okada et al., 2002; Wu et al., 2007; Yamamoto et al., 2007). Accordingly the expression of GRP94, BIP, and calreticulin were increased in S63del compared to WT mice. However, their levels in S63del/PerkSCKO and S63del lysates were again similar (Fig. 6B,C), suggesting that the ATF6 pathway is not affected by Perk ablation in S63del Schwann cells.
Figure 6.
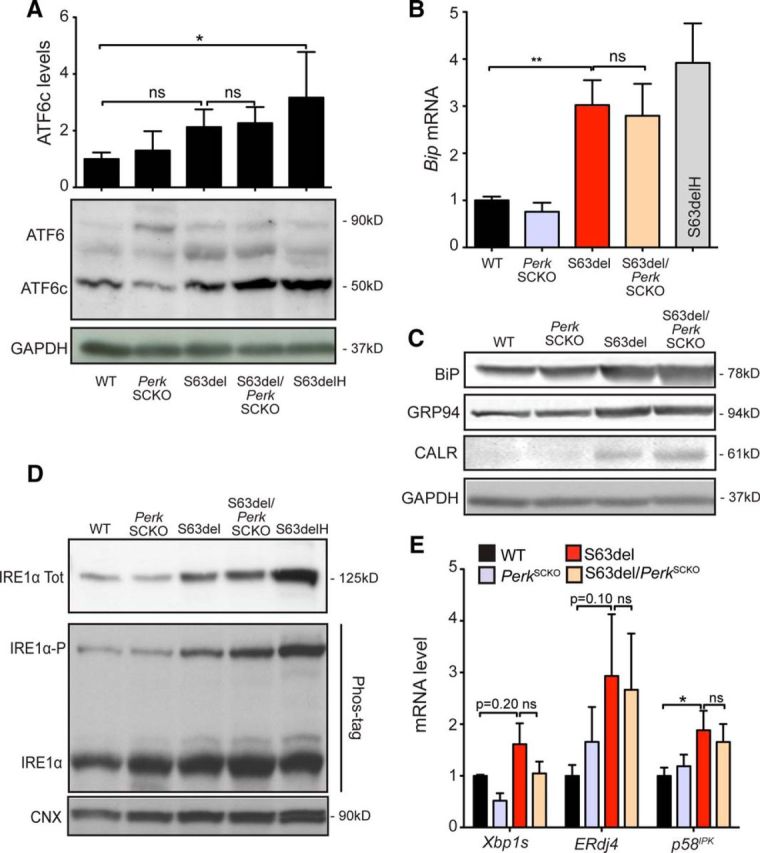
Perk loss of function in Schwann cells does not alter ATF6 or IRE1α pathways. A, ATF6 precursor form (90 kDa) and ATF6 cleaved form (50 kDa) and were quantified by Western blot analysis in P28 sciatic nerves. GAPDH was used as loading control. n = 3 lysates from independent pairs of nerves for each genotype. B, Bip mRNA was measured by RT-PCR TaqMan assay on P28 sciatic nerves, normalized for 18S rRNA. n = 6 RT from three independent sets of nerves for each genotype. C, Bip, Grp94, and calreticulin protein levels were identified by Western blot analysis, and GAPDH was used as loading control. One of five independent experiments is shown. D, IRE1α protein levels were detected by Western blot analysis. The phosphorylated active form of IRE1 (IRE1α-P) was separated from the unphosphorylated and inactive form (IRE1α) on a Phos-tag polyacrylamide gel (25 μm). One of three independent experiments is shown. Tot, total. E, Xbp1 splicing (Xbp1s), ERdj4, and p58IPK mRNA levels were measured by RT-PCR TaqMan assay on P28 sciatic nerves, normalized for 18S rRNA. n = 2–6 RT from three or four independent sets of nerves for each genotype. *p < 0.05; **p < 0.01 (Student's t test). Error bars indicate SEM.
To detect IRE1α activation, we measured the phosphorylation state of IRE1α and the mRNA levels of Xbp1s transcription factor (Yoshida et al., 2001; Calfon et al., 2002; Shen et al., 2002). We noticed an increase in expression of IRE1α total protein in S63del compared to WT, and the levels remained similarly high in S63del/PerkSCKO nerves (Fig. 6D). The separation of P-IRE1α from nonphosphorylated IRE1α was achieved by using the Phos-tag compound (Martinon et al., 2010; Yang et al., 2010). P-IRE1α levels increased in S63del compared to WT nerves, and the levels did not increase further in S63del/PerkSCKO nerves (Fig. 6D). Accordingly, Xbp1s mRNA did not increase in S63del/PerkSCKO compared to S63del nerves (Fig. 6E). Moreover, the expression of XBP1s-dependent ER enzymes, such as ERdj4 and p58IPK (Lee et al., 2003), was induced in S63del nerves and remained high in S63del/PerkSCKO nerves (Fig. 6E). Collectively, these data show that the UPR is activated in S63del/PerkSCKO nerves similarly to S63del nerves. In addition, IRE1α and ATF6 pathways are not significantly altered in S63del/PerkSCKO nerves, which suggests that compensation within the UPR does not account for the ameliorated myelination observed in S63del/PerkSCKO nerves.
P0 is retained at similar levels in the ER of S63del and S63del/PerkSCKO Schwann cells
The retention of P0S63del protein in the ER of Schwann cells triggers the UPR and causes the CMT1B neuropathy, and levels of P0 engulfment in the ER of Schwann cells is a direct measure of beneficial response (Wrabetz et al., 2006; Pennuto et al., 2008; D'Antonio et al., 2013). We showed that Perk elimination in S63del Schwann cells improves myelination without a substantial reduction of the UPR markers. Overall, our data indicate that these Schwann cells are subjected to persistent ER stress. Thus, we asked whether, despite the ongoing UPR, Perk ablation could reduce the amount of P0 in the ER and improve the proteostasis in S63del Schwann cells. We found that P0 colocalized with KDEL, marker of the ER, in both S63del and S63del/PerkSCKO teased fibers from P28 sciatic nerves (Fig. 7A, yellow arrowheads). The quantification of P0 pixel intensity within the KDEL area revealed lower levels of P0 colocalizing with KDEL in control nerves (Fig. 7B, blue and cyan dots). The amount of P0 localized in the ER was similarly greater in S63del and S63del/PerkSCKO Schwann cells (Fig. 7B, red and pink dots), clearly suggesting that P0 remains persistently retained in S63del/PerkSCKO Schwann cells. These data demonstrate that Perk ablation does not alleviate the ER overload in S63del Schwann cells and P0 retention likely contributes to the persistent UPR.
Figure 7.

P0 is retained at similar levels in the ER of S63del and S63del/PerkSCKO Schwann cells. A, Immunofluorescence for P0 (green) and KDEL (red) in teased fibers from P28 sciatic nerves revealed P0 localization in the endoplasmic reticulum of Schwann cells (yellow arrowheads). Nuclei were stained in blue (DAPI). Scale bar, 5 μm. B, The range of P0 pixel intensity was quantified within the KDEL area. Error bars indicate SD. n = 100–300 fibers from three or four different nerves for each genotype. A.U., Arbitrary unit.
Discussion
Here we characterized S63del/PerkSCKO and S63del/PerkNRNKO mice and showed that ablation of Perk in Schwann cells but not in neurons improves CMT1B neuropathy in mice. The improvement occurs despite a reduction in P-eIF2α. We also explored the signals downstream of PERK and the other UPR arms to identify any compensatory mechanism responsible for the surprising improvement, but found no evident changes. Comparing our data with the Gadd34-null amelioration of the S63del phenotype (D'Antonio et al., 2013), we conclude that UPR and translational homeostasis are not the only pathogenetic mechanisms involved in CMT1B-S63del Schwann cells. PERK, in fact, may interfere with other pathways outside of the UPR in S63del Schwann cells.
Improvement in S63del Schwann cells does not necessarily correlate with increased eIF2α phosphorylation
Here we show for the first time that a context with reduction of P-eIF2α specifically in Schwann cells can improve, rather than worsen, the myelin defect of S63del nerves. Our result seems to differ from other studies where perturbing the PERK P-eIF2α pathway has been shown to be extremely detrimental in different ER stress settings. For instance, Perk−/− and eIF2αA/A (not phosphorylatable) mice exhibit a significant reduction in the number of pancreatic cells, failure of hepatocytes to produce glycogen, and reduced osteoblast differentiation and collagen secretion (Harding et al., 2000a,b; Scheuner et al., 2001; Zhang et al., 2002; Wei et al., 2008; Saito et al., 2011). Mutations in human PERK result in Walcott-Rallison syndrome, associated with neonatal type I diabetes (Thornton et al., 1997; Rubio-Cabezas et al., 2009). Moreover, PERK-eIF2α function has been shown to promote neuroprotection in neurodegenerative disease models like Parkinson's disease and ALS (Wang et al., 2011; Colla et al., 2012).
Likewise, in myelin-related disorders, loss of function mutations in eIF2B, the target of P-eIF2α regulation, results in vanishing white matter disease, and Perk haploinsufficiency worsens the clinical outcome in the experimental allergic encephalomyelitis model of multiple sclerosis (Proud, 2001; Lin et al., 2007b). In agreement with these observations, reinforcing P-eIF2α in S63del peripheral demyelinating neuropathy, by genetically or pharmacologically impairing GADD34, rescues morphological and functional deficits in S63del nerves (D'Antonio et al., 2013; Das et al., 2015). Inhibiting GADD34 function increases the level of P-eIF2α, limits protein translation, reduces ER overload, and ultimately relieves S63del Schwann cells of chronic UPR. These data strongly suggest that reducing translation is sufficient to restore myelination in S63del neuropathy. Schwann cells rely on phosphorylation of eIF2α to achieve and maintain functional myelination in the face of persistent ER stress (D'Antonio et al., 2013).
The detrimental effect of Perk in CMT1B derives from Schwann cells
S63del nerves exhibit specific features that suggest a Schwann-cell-derived neuropathy (Wrabetz et al., 2006). For instance, at P28, myelin appears thinner and the number of amyelinating axons is increased compared to WT, suggesting a delay in Schwann cell development. Moreover, the NCV is reduced and the F-wave latency increased, which are hallmarks of demyelinating neuropathy. Interestingly, the distal CMAP is also reduced, indicating that the motor nerve action potential might be impaired due to the loss of axons or conduction block, a matter for further investigation.
We showed previously that reducing P-eIF2α by altering Perk dosage in S63del mice partially ameliorates the motor performance and demyelinating phenotype of S63del neuropathy (Musner et al., 2016). In that study, we eliminated one Perk allele in every cell of the S63del mouse, including brain, muscle, and pancreas, that could affect the motor performance (Musner et al., 2016). Nonetheless, there were no gross abnormalities in the cytoarchitecture of these tissues, suggesting that they did not contribute to the impaired motor performance. Moreover, Perk-null S63del neurons did not improve myelination in neuronal/Schwann cell cocultures, suggesting that activation of PERK in S63del neurons did not provoke aberrant S63del myelination (Musner et al., 2016). Here, we aimed to fully understand the role of Perk in S63del mice by asking whether Perk is detrimental in Schwann cells or neurons.
To directly address this question in S63del nerves, we eliminated Perk cell-specifically by taking advantage of a Cre-loxP strategy. Perk loss of function in normal Schwann cells does not impair physiological myelination. However, in pathological nerves, the absence of Perk specifically in Schwann cells results in the amelioration of S63del myelin defects. In S63del/PerkSCKO nerves, myelin thickness is increased, and the number of amyelinated axons is remarkably reduced compared to S63del. Furthermore, NCV is slightly improved, confirming that Perk ablation reversed hypomyelination. F-wave latency and distal CMAP are not restored to normal.
These data differ from those in our previous work, where Perk haploinsufficiency was not sufficient to improve myelin thickness and NCV (Musner et al., 2016). Beside Perk dosage and the extent of tissues in which Perk was ablated, other possible explanations for the difference include that the prior experiments were performed in 4-month-old animals in the FVB/N background, whereas the experiments here were performed in 1-month-old animals in the C57BL/6 background. In any case, the data here indicate that PERK, like CHOP and GADD34, contributes to the S63del neuropathic mechanism. Moreover, they show for the first time that the detrimental effect of the PERK pathway derives from Schwann cells.
Conversely, the ablation of Perk in neurons does not alter CMT1B neuropathy. In fact, neither myelination nor electrophysiological values were improved in S63del/PerkNRNKO compared to S63del nerves. Using NestinCre, PERK and eIF2α-P levels were reduced in motor and DRG neurons, respectively, indicating that neurons that contribute axons to sciatic nerves were appropriately targeted in S63del/PerkNRNKO animals. Other glia in the CNS are also targeted, but it is unlikely that this can explain the lack of phenotype in neurons. These data demonstrate that the detrimental role of Perk in CMT1B derives mainly from Schwann cells. Interestingly, the ablation of Perk in WT neurons slightly reduced the NCV and distal CMAP amplitude, suggesting a potentially beneficial role of Perk in normal neuronal function.
ER stress markers do not change upon Perk ablation in Schwann cells
To elucidate how Perk ablation ameliorates rather then worsens hypomyelination in S63del, we surveyed the UPR pathways. CHOP ablation in S63del mice strongly rescues neuropathy (Pennuto et al., 2008; D'Antonio et al., 2013). However, we found that CHOP protein level and target genes were still upregulated in S63del/PerkSCKO nerves similarly to S63del. This result indicates that CHOP is active in S63del/PerkSCKO nerves and cannot account for the S63del/PerkSCKO myelin phenotype. Our findings are also consistent with the idea that CHOP can be activated by other UPR pathways, like ATF6 (Yoshida et al., 2001). In Perk−/− ES cells, IRE1 activation is prolonged, and cells stably transfected with a dominant-negative form of PERK show enhanced ATF6 activation when challenged with tunicamycin (Harding et al., 2000b; Yamaguchi et al., 2008). Moreover, persistent activation of IRE1 promotes cell viability upon stress (Lin et al., 2007a). Hence, we hypothesized that perturbing Perk in S63del Schwann cells may trigger a beneficial compensatory response from the ATF6 or IRE1 arms. However, we found none of the main effectors of these two pathways were altered, indicating that IRE1 and ATF6 signals are not likely to compensate for Perk loss of function.
The effects of PERK and eIF2α are uncoupled
The scenarios described previously in S63del/GADD34ΔC/ΔC mice and here in S63del/PerkSCKO mice differ. GADD34 loss of function improves S63del myelination by reducing the accumulation of P0 in the ER of Schwann cells, which alleviates ER stress and UPR signaling (D'Antonio et al., 2013), whereas Perk ablation in Schwann cells ameliorates S63del myelination even if misfolded proteins in the ER and UPR remain. In other words, Perk ablation in Schwann cells alleviates the neuropathy without reverting the toxic pathological mechanism. Moreover, we find no evidence to suggest compensation from other parallel pathways of the UPR. This suggests that the UPR might not be the only pathological mechanism involved in CMT1B-S63del Schwann cells.
We propose that PERK might have P-eIF2α-independent effects in neuropathy, possibly through alternative pathways. For instance, there is evidence that PERK targets additional substrates to eIF2α, like the Nrf2 transcription factor and Mfn2, a mitochondrial protein that regulates ER morphology and Ca2+ flux between ER and mitochondria (Cullinan et al., 2003; Munoz et al., 2013). There may be other unrecognized PERK targets also in Schwann cells involved in metabolism and myelination.
Accordingly, other studies have suggested that PERK exerts a detrimental role in ER stress-related diseases, where the effect of Perk seems to be uncoupled from attenuation of translation. Perk haploinsufficiency slows the progression of diabetes in Akita mice probably by modulating ER associated degradation and degradation of proinsulin (Gupta et al., 2010). Pharmacological inhibition of PERK and reduction of P-eIF2α prevents the clinical outbreak of prion disease without altering the level of PrPSc in prion infected mice (Moreno et al., 2013). PrPSc mRNA carries several upstream open reading frames that allow it to escape the translational attenuation caused by eIF2α phosphorylation. Therefore, reducing P-eIF2α may actually reduce the synthesis of PrPSc in prion-infected mice (Moreno et al., 2012). PERK inhibition also reduces the neuronal toxicity of TDP-43 RNA-binding protein in a fly model of ALS (Kim et al., 2014). In this example, the increased level of P-eIF2α and resulting translational attenuation are thought to promote TDP-43 induced neuronal toxicity. Finally, transgenic mice with neurodegeneration due to overexpression of mutant tau protein may provide a similar example (Radford et al., 2015). These last three examples are similar to S63del mice and support a beneficial effect of Perk inhibition in neurodegenerative diseases. However, unlike the S63del mice where P-eIF2α is protective (D'Antonio et al., 2013), the proteotoxicity of PrP, TDP-43 and mutant tau is directly correlated to the increased level of P-eIF2α. This strongly supports that the mechanism responsible for S63del/PerkSCKO improvement is uncoupled from P-eIF2α. Experiments to specifically eliminate P-eIF2α in Schwann cells without altering PERK signaling (Back et al., 2009) and a search for other PERK targets in Schwann cells will address this hypothesis.
Footnotes
This work was supported by grants from the National Institutes of Health (R56NS096104 to L.W.; R01NS045630 to M.L.F.); Telethon, Italy (GGP071100 to L.W.; GGP08021 to M.L.F.); the European Community (FP7/2007-1013 under Grant Agreement HEALTH-F2-2008-201535 to L.W. and M.L.F.); and the Charcot Tooth Marie Association (L.W.). M.D. is the recipient of a Giovane Ricercatore Award from the Italian Ministry of Health (GR-2009-1548255). We thank GianGiacomo Conzalez for the NestinCre mice. We thank Stefania Saccucci, Desiree Zambroni, and Georgia Dina at San Raffaele Scientific Institute and Annette Featherstone of the South Campus Histology Core Facility, University at Buffalo, for excellent technical assistance.
The authors declare no competing financial interests.
References
- Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, Gildersleeve RD, Pennathur S, Kaufman RJ. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009;10:13–26. doi: 10.1016/j.cmet.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertolotti A, Ron D. Alterations in an IRE1-RNA complex in the mammalian unfolded protein response. J Cell Sci. 2001;114:3207–3212. doi: 10.1242/jcs.114.17.3207. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Colla E, Jensen PH, Pletnikova O, Troncoso JC, Glabe C, Lee MK. Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J Neurosci. 2012;32:3301–3305. doi: 10.1523/JNEUROSCI.5368-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi: 10.1128/MCB.23.20.7198-7209.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Antonio M, Musner N, Scapin C, Ungaro D, Del Carro U, Ron D, Feltri ML, Wrabetz L. Resetting translational homeostasis restores myelination in Charcot-Marie-Tooth disease type 1B mice. J Exp Med. 2013;210:821–838. doi: 10.1084/jem.20122005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das I, Krzyzosiak A, Schneider K, Wrabetz L, D'Antonio M, Barry N, Sigurdardottir A, Bertolotti A. Preventing proteostasis diseases by selective inhibition of a phosphatase regulatory subunit. Science. 2015;348:239–242. doi: 10.1126/science.aaa4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waegh SM, Lee VM, Brady ST. Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell. 1992;68:451–463. doi: 10.1016/0092-8674(92)90183-D. [DOI] [PubMed] [Google Scholar]
- Feltri ML, D'Antonio M, Previtali S, Fasolini M, Messing A, Wrabetz L. P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann N Y Acad Sci. 1999;883:116–123. doi: 10.1111/j.1749-6632.1999.tb08574.x. [DOI] [PubMed] [Google Scholar]
- Fratta P, Saveri P, Zambroni D, Ferri C, Tinelli E, Messing A, D'Antonio M, Feltri ML, Wrabetz L. P0S63del impedes the arrival of wild-type P0 glycoprotein to myelin in CMT1B mice. Hum Mol Genet. 2011;20:2081–2090. doi: 10.1093/hmg/ddr081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander R, Jarosch E, Urban J, Volkwein C, Sommer T. A regulatory link between ER-associated protein degradation and the unfolded-protein response. Nat Cell Biol. 2000;2:379–384. doi: 10.1038/35017001. [DOI] [PubMed] [Google Scholar]
- Gupta S, McGrath B, Cavener DR. PERK (EIF2AK3) regulates proinsulin trafficking and quality control in the secretory pathway. Diabetes. 2010;59:1937–1947. doi: 10.2337/db09-1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000a;6:1099–1108. doi: 10.1016/S1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000b;5:897–904. doi: 10.1016/S1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Harding HP, Novoa I, Bertolotti A, Zeng H, Zhang Y, Urano F, Jousse C, Ron D. Translational regulation in the cellular response to biosynthetic load on the endoplasmic reticulum. Cold Spring Harb Symp Quant Biol. 2001a;66:499–508. doi: 10.1101/sqb.2001.66.499. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D. Diabetes mellitus and exocrine pancreatic dysfunction in perk−/− mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001b;7:1153–1163. doi: 10.1016/S1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10:3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12:703–719. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- Kao SC, Wu H, Xie J, Chang CP, Ranish JA, Graef IA, Crabtree GR. Calcineurin/NFAT signaling is required for neuregulin-regulated Schwann cell differentiation. Science. 2009;323:651–654. doi: 10.1126/science.1166562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, Lee VM, Finkbeiner S, Gitler AD, Bonini NM. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46:152–160. doi: 10.1038/ng.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkens T, Bolhuis PA, Wolterman RA, Kemp S, te Nijenhuis S, Valentijn LJ, Hensels GW, Jennekens FG, de Visser M, Hoogendijk JE, Baas F. Deletion of the serine 34 codon from the major peripheral myelin protein P0 gene in Charcot-Marie-Tooth disease type 1B. Nat Genet. 1993;5:35–39. doi: 10.1038/ng0993-35. [DOI] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007a;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Bailey SL, Ho H, Harding HP, Ron D, Miller SD, Popko B. The integrated stress response prevents demyelination by protecting oligodendrocytes against immune-mediated damage. J Clin Invest. 2007b;117:448–456. doi: 10.1172/JCI29571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller LJ, Patzko A, Lewis RA, Shy ME. Phenotypic presentation of the Ser63Del MPZ mutation. J Peripher Nerv Syst. 2012;17:197–200. doi: 10.1111/j.1529-8027.2012.00398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, Barrett DA, Tsaytler P, Bertolotti A, Willis AE, Bushell M, Mallucci GR. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature. 2012;485:507–511. doi: 10.1038/nature11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Halliday M, Molloy C, Radford H, Verity N, Axten JM, Ortori CA, Willis AE, Fischer PM, Barrett DA, Mallucci GR. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med. 2013;5:206ra138. doi: 10.1126/scitranslmed.3006767. [DOI] [PubMed] [Google Scholar]
- Munoz JP, Ivanova S, Sánchez-Wandelmer J, Martínez-Cristóbal P, Noguera E, Sancho A, Díaz-Ramos A, Hernández-Alvarez MI, Sebastián D, Mauvezin C, Palacín M, Zorzano A. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013;32:2348–2361. doi: 10.1038/emboj.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musner N, Sidoli M, Zambroni D, Del Carro U, Ungaro D, D'Antonio M, Feltri ML, Wrabetz L. Perk ablation ameliorates myelination in S63del-Charcot-Marie-Tooth 1B neuropathy. ASN Neuro. 2016;8:1–18. doi: 10.1177/1759091416642351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Yoshida H, Akazawa R, Negishi M, Mori K. Distinct roles of activating transcription factor 6 (ATF6) and double-stranded RNA-activated protein kinase-like endoplasmic reticulum kinase (PERK) in transcription during the mammalian unfolded protein response. Biochem J. 2002;366:585–594. doi: 10.1042/bj20020391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patzig J, Jahn O, Tenzer S, Wichert SP, de Monasterio-Schrader P, Rosfa S, Kuharev J, Yan K, Bormuth I, Bremer J, Aguzzi A, Orfaniotou F, Hesse D, Schwab MH, Möbius W, Nave KA, Werner HB. Quantitative and integrative proteome analysis of peripheral nerve myelin identifies novel myelin proteins and candidate neuropathy loci. J Neurosci. 2011;31:16369–16386. doi: 10.1523/JNEUROSCI.4016-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennuto M, Tinelli E, Malaguti M, Del Carro U, D'Antonio M, Ron D, Quattrini A, Feltri ML, Wrabetz L. Ablation of the UPR-mediator CHOP restores motor function and reduces demyelination in Charcot-Marie-Tooth 1B mice. Neuron. 2008;57:393–405. doi: 10.1016/j.neuron.2007.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proud CG. Regulation of eukaryotic initiation factor eIF2B. Prog Mol Subcell Biol. 2001;26:95–114. doi: 10.1007/978-3-642-56688-2_4. [DOI] [PubMed] [Google Scholar]
- Quattrini A, Previtali S, Feltri ML, Canal N, Nemni R, Wrabetz L. Beta 4 integrin and other Schwann cell markers in axonal neuropathy. Glia. 1996;17:294–306. doi: 10.1002/(SICI)1098-1136(199608)17:4%3C294::AID-GLIA4%3E3.0.CO;2-%23. [DOI] [PubMed] [Google Scholar]
- Radford H, Moreno JA, Verity N, Halliday M, Mallucci GR. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015;130:633–642. doi: 10.1007/s00401-015-1487-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Cabezas O, Patch AM, Minton JA, Flanagan SE, Edghill EL, Hussain K, Balafrej A, Deeb A, Buchanan CR, Jefferson IG, Mutair A, Hattersley AT, Ellard S. Wolcott-Rallison syndrome is the most common genetic cause of permanent neonatal diabetes in consanguineous families. J Clin Endocrinol Metab. 2009;94:4162–4170. doi: 10.1210/jc.2009-1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito A, Ochiai K, Kondo S, Tsumagari K, Murakami T, Cavener DR, Imaizumi K. Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J Biol Chem. 2011;286:4809–4818. doi: 10.1074/jbc.M110.152900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/S1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Shen J, Chen X, Hendershot L, Prywes R. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev Cell. 2002;3:99–111. doi: 10.1016/S1534-5807(02)00203-4. [DOI] [PubMed] [Google Scholar]
- Sherman DL, Brophy PJ. Mechanisms of axon ensheathment and myelin growth. Nat Rev Neurosci. 2005;6:683–690. doi: 10.1038/nrn1743. [DOI] [PubMed] [Google Scholar]
- Sriburi R, Jackowski S, Mori K, Brewer JW. XBP1: a link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J Cell Biol. 2004;167:35–41. doi: 10.1083/jcb.200406136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton CM, Carson DJ, Stewart FJ. Autopsy findings in the Wolcott-Rallison syndrome. Pediatr Pathol Lab Med. 1997;17:487–496. doi: 10.1080/107710497174778,10.3109/15513819709168589,10.1080/15513819709168589,10.1080/713601302. [DOI] [PubMed] [Google Scholar]
- Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/S0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Wang L, Popko B, Roos RP. The unfolded protein response in familial amyotrophic lateral sclerosis. Hum Mol Genet. 2011;20:1008–1015. doi: 10.1093/hmg/ddq546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Sheng X, Feng D, McGrath B, Cavener DR. PERK is essential for neonatal skeletal development to regulate osteoblast proliferation and differentiation. J Cell Physiol. 2008;217:693–707. doi: 10.1002/jcp.21543. [DOI] [PubMed] [Google Scholar]
- Wrabetz L, Feltri ML, Quattrini A, Imperiale D, Previtali S, D'Antonio M, Martini R, Yin X, Trapp BD, Zhou L, Chiu SY, Messing A. P(0) glycoprotein overexpression causes congenital hypomyelination of peripheral nerves. J Cell Biol. 2000;148:1021–1034. doi: 10.1083/jcb.148.5.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrabetz L, D'Antonio M, Pennuto M, Dati G, Tinelli E, Fratta P, Previtali S, Imperiale D, Zielasek J, Toyka K, Avila RL, Kirschner DA, Messing A, Feltri ML, Quattrini A. Different intracellular pathomechanisms produce diverse Myelin Protein Zero neuropathies in transgenic mice. J Neurosci. 2006;26:2358–2368. doi: 10.1523/JNEUROSCI.3819-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GD, Kaufman RJ. ATF6alpha optimizes long-term endoplasmic reticulum function to protect cells from chronic stress. Dev Cell. 2007;13:351–364. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Larkin D, Lara-Lemus R, Ramos-Castañeda J, Liu M, Arvan P. Endoplasmic reticulum (ER) chaperone regulation and survival of cells compensating for deficiency in the ER stress response kinase, PERK. J Biol Chem. 2008;283:17020–17029. doi: 10.1074/jbc.M802466200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev Cell. 2007;13:365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Yang L, Xue Z, He Y, Sun S, Chen H, Qi L. A Phos-tag-based approach reveals the extent of physiological endoplasmic reticulum stress. PLoS One. 2010;5:e11621. doi: 10.1371/journal.pone.0011621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Okada T, Haze K, Yanagi H, Yura T, Negishi M, Mori K. ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol Cell Biol. 2000;20:6755–6767. doi: 10.1128/MCB.20.18.6755-6767.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/S0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Hosokawa N, Kaufman RJ, Nagata K, Mori K. A time-dependent phase shift in the mammalian unfolded protein response. Dev Cell. 2003;4:265–271. doi: 10.1016/S1534-5807(03)00022-4. [DOI] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab. 2006;4:491–497. doi: 10.1016/j.cmet.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Zimmerman L, Parr B, Lendahl U, Cunningham M, McKay R, Gavin B, Mann J, Vassileva G, McMahon A. Independent regulatory elements in the nestin gene direct transgene expression to neural stem cells or muscle precursors. Neuron. 1994;12:11–24. doi: 10.1016/0896-6273(94)90148-1. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]