

Abstract
Like azides, diazoacetamides undergo 1,3-dipolar cycloadditions with oxanorbornadienes (OND) in a reaction that is accelerated by the relief of strain in the transition state. The cycloaddition of a diazoacetamide with unstrained ethyl 4,4,4-trifluoro-2-butynoate is, however, 35-fold faster than with the analogous OND because of favorable interactions with the fluoro groups. Its rate constant (k = 0.53 M−1s−1 in methanol) is comparable to those of strain-promoted azide–cyclooctyne cycloadditions.
Graphical abstract

The Huisgen 1,3-dipolar cycloaddition dominates the landscape of reactions that are chemoselective in biological contexts.1 Although the copper-catalyzed azide–alkyne cycloaddition (CuAAC)2 and the strain-promoted azide–alkyne cycloaddition (SPAAC)1a fulfill many criteria of an ideal “click” reaction,2 the desire for increased reactivity and selectivity in non-catalyzed reactions remains strong.3
Recently, we demonstrated the use of a stabilized diazo group as a chemical reporter.1k,4,5 Despite significant conjugative stabilization, diazoacetamides are primed for rapid normal-electron-demand 1,3-dipolar cycloadditions, and show reaction rates with strained alkynes that surpass those of azides.6 Moreover, diazoacetamides endure cellular metabolism, and provide chemoselectivity in the presence of terminal alkynes, a feature unobtainable with an azido moiety.1k Still, overlapping reactivity with azido groups does not allow for selective reactivity of diazo groups in an SPAAC. We have found, however, that removing strain and tuning the LUMOs of the dipolarophile can provide diazo-selective reactivity.7 In addition, cycloaddition rates with α,β-unsaturated carbonyl compounds are increased by incorporating electron-withdrawing substituents.8
We sought to incorporate strain while retaining electronic tunability as a primary design feature. These attributes are enabled in the bicyclic oxanorbornadiene (OND) scaffold (Scheme 1). Electron-deficient ONDs are effective dipolarophiles in reactions with azides, displaying increased reactivity relative to their linear precursors.9 This increase is a consequence of the relief of ring-strain in the transition state (TS), along with a lower LUMO energy of the bicyclic alkene compared to that of an alkynyl π-bond.10 Their facile preparation from common starting materials and opportunity for alkenyl substitution make ONDs attractive in comparison to synthetically recalcitrant cyclooctynes. Moreover, the reactions of ONDs with nucleophilic species are rapid enough for numerous applications,11 and diazo groups are more nucleophilic than are azido groups.1j,6 Accordingly, we explored the reactivity of ONDs shown previously to undergo 1,3-dipolar cycloadditions with azides.9
Scheme 1.

Pyrazole product obtained from the 1,3-dipolar cycloaddition of diazoacetamide 1 with alkyne 3 and oxanorbornadiene 4. Azide 2 reacts analogously to form a triazole.
We found that, like azide 2, diazoacetamide 1 (R = Bn) undergoes 1,3-dipolar cycloadditions with OND 4 followed by a retro-Diels–Alder reaction to eliminate furan (Scheme 1). The resulting pyrazole product is identical to that formed upon reaction with analogous alkyne 3. Unlike with azides,9 however, we found that diazoacetamide 1 (R = Bn) does not react with the unsubstituted alkenyl group of ONDs 4a and 4b.
Cycloaddition reactions of an azido group with ONDs are known to be ~5-fold faster than are those with analogous alkynes.9a Accordingly, we determined rate constants for the cycloadditions of diazoacetamide 1 (R = Bn) with ONDs 4a (R1 = CO2Et, R2 = CF3) and 4b (R1 = R2 = CO2Me) and analogous alkynes 3a and 3b (Figure S1). We found the rate constants for the cycloaddition of diazoacetamide 1 (R = Bn) with ONDs 4a and 4b (Table 1) to be 102-fold faster than those of azides with ONDs.9a Moreover, the diazoacetamide–OND rate constants are comparable to those of known SPAACs.3 The incorporation of strain into OND 4b increases the rate constant by twofold relative to the reaction with alkyne 3b, a slightly smaller effect than observed in the analogous reactions of azides.9a
Table 1.
Rate Constants for the Cycloaddition of Diazoacetamide 1 (R = Bn) with Alkynes 3a and 3b and with ONDs 4a and 4ba
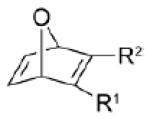
| dipolarophile | R1 | R2 | k (10−2 M−1s−1)a | |
|---|---|---|---|---|
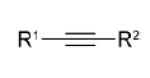
|
3a | CO2Et | CF3 | 53 |
| 3b | CO2Me | CO2Me | 0.46 | |
|
4a | CO2Et | CF3 | 1.5 |
| 4b | CO2Me | CO2Me | 1.1 | |
Values were determined in methanol with 1H NMR spectroscopy or ultraviolet spectroscopy.
The cycloadditions of diazoacetamide 1 with alkyne 3a and analogous OND 4a were dichotomous with those of azides.9a Rather than a modest increase in the reaction rate with an OND,9a we observed the reaction rate of diazoacetamide 1 with OND 4a to be 1/35th that with alkyne 3a. Moreover, the reaction rate of diazoacetamide 1 with alkyne 3a is comparable to that of the fastest of reported SPAACs,1f,12 and approaches that of the diazoacetamide–DIBAC cycloaddition.1k This result was unexpected, as the lower LUMO of alkenes relative to alkynes and the strain imposed by the OND framework led us to anticipate the opposite trend in reactivity. Accordingly, we pursued a computational analysis to better understand the surprising reactivity of alkyne 3a.
Calculated activation barriers for the cycloadditions of both alkynes and ONDs (Figure 1) correlate with their relative reactivity (Table 1). The 1,3-dipolar cycloaddition should be the rate-determining step in the cycloadditions with ONDs (Figure S2). Based on ΔE‡ values (Table 2), the origins of the increased reactivity of alkyne 3a are not immediately apparent. Enthalpic contributions suggest that OND 4a should react faster than alkyne 3a (Table S1). FMO energies and the increased strain of bicyclic systems13 relative to alkynes also predict a faster rate for the OND. To understand the increased reactivity of alkyne 3a, we turned to the distortion/interaction analysis developed by Ess and Houk (Table 2).14,15 In this approach, the overall reaction barrier can be understood as the sum of the free energy required to distort isolated reactants to transition state (TS) geometries and the free energy gained when these distorted reactants interact in the TS:15a
| (1) |
The origins of increased reactivity can be understood when free energies of distortion and interaction are taken into account (Table 2). The difference in total distortion free energy between the alkyne and OND TSs (ΔΔG‡dist < 1 kcal/mol for 1–3a TS and 1–4a TS), is much less than the difference in total distortion energy (ΔΔE‡dist = 1.7 kcal/mol for 1–3a TS and 1–4a TS; Figure S3), allowing interactions to dictate the observed reactivity (ΔΔG‡int = 1.4 kcal/mol versus ΔΔE‡int = 0.2 kcal/mol for 1–3a TS and 1–4a TS).
Figure 1.

Optimized transition-state geometries for the cycloaddition of diazoacetamide 1 (R = Me) with alkynes 3a and 3b and with ONDs 4a and 4b as calculated at the M06-2X/6-31+G(2d,p) level of theory. Free energies (kcal/mol) include solvation corrections (MeOH) on gas-phase geometries with the IEFPCM model (radii = UFF).
Table 2.
Distortion/Interaction Analysis for the Cycloaddition of Diazoacetamide 1 (R = Me) with Alkynes 3a and 3b and with ONDs 4a and 4ba
|
Δ
G
‡
dist
|
charge (e−) on the dipole (1) in the TSb |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
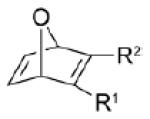
| dipolarophile | R1 | R2 | Δ E ‡ | Δ G ‡ | dipolarophile | dipole (1) | total | Δ G ‡ int | gas-phase | MeOH | Δ | |
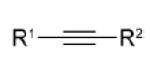
|
3a | CO2Me | CF3 | 9.6 | 21.5 | 10.4 | 14.8 | 25.2 | −3.6 | 0.25 | 0.28 | 0.3 |
| 3b | CO2Me | CO2Me | 9.8 | 22.8 | 12.7 | 13.1 | 25.8 | −3.0 | 0.26 | 0.28 | 0.2 | |
|
4a | CO2Me | CF3 | 8.2 | 22.1 | 8.1 | 16.2 | 24.3 | −2.2 | 0.24 | 0.25 | 0.1 |
| 4b | CO2Me | CO2Me | 7.8 | 22.5 | 6.4 | 17.7 | 24.1 | −1.6 | 0.22 | 0.23 | 0.1 | |
Calculations were performed at the M06-2X/6-31+G(2d,p) level of theory. Energies (kcal/mol) include solvation corrections (MeOH) on gas-phase geometries with the IEFPCM model (radii = UFF);
Calculated with natural bond orbital (NBO) analysis.18
In agreement with FMO energies, the distortion energies of ONDs 4a and 4b are significantly lower than are those of alkynes 3a and 3b.16 Interestingly, the distortion energy of fluorinated OND 4a is much larger than is that of bis-esterified OND 4b (8.1 versus 6.4 kcal/mol). This increase appears to be due to a slightly smaller dihedral angle of the alkene in the starting geometry of 4b along with increased steric crowding of electron-rich substituents in 4a (Figure 1). The closest O⋯O distance decreases by 0.07 Å from the starting material to the TS geometry in 4b (2.88 to 2.81 Å), whereas the closest O⋯F distance decreases by 0.12 Å in 4a (2.88 to 2.76 Å).
In alkyne 3a, which avoids deleterious steric interactions, fluorination facilitates bending, leading to decreased distortion energies.1h In addition, large interaction energies result from hyperconjugative assistance to the nascent bonds or hydrogen-bonding interactions (or both), depending on the regioisomer of the TS (Figure S4). Analogous effects of propargylic fluorination on alkyne bending and interaction energies have been reported previously during cycloadditions with azides1h,j and diazo compounds.8
Lowered distortion energies for diazoacetamide 1 in reactions with alkynes 3a and 3b (relative to reactions with ONDs 4a and 4b) and shorter incipient C–C bonds result in a highly asynchronous TS (Figure 1). The result is unidirectional charge-transfer amplified by propargylic fluoro groups in the reaction of alkyne 3a. The charge-transfer creates a more polar TS and larger solvent effects (Table 2).6,17 Only with alkyne 3a is the free energy of activation lower in solution than in the gas phase (Figure S5), suggesting that fluorine induces a more favorable ΔS‡ in polar media.
Conclusions
We have discovered an unexpectedly large effect of propargylic fluorination on 1,3-dipolar cycloadditions with diazoacetamides. These effects render the reaction with alkyne 3a to be much faster than that with an OND having analogous substitution and comparable in rate to the fastest known SPAACs. Others have noted that strain-relief is the most significant design principle for increasing the rate of azide–alkyne cycloadditions.19 In particular, alkyne bending decreases the HOMO–LUMO gap1j—a scenario that is optimal for azido dipoles, which are substantially more ambiphilic than are diazo dipoles.12b In essence, strain provides the opportunity to increase the donor and acceptor properties of the alkyne π-bond simultaneously, leading to cycloadditions that can be more synchronous than those of linear alkynes.
In comparison to their azido congeners, diazo dipoles are much more nucleophilic.6,7 Although strain-relief can increase cycloaddition rates,1k,4 a strong preference for electron-deficient dipolarophiles renders the diazo group much more sensitive to substituent effects, obviating strain-relief as a sine qua non for rapid reaction.20 This difference is advantageous in polar solvents (like water), where highly asynchronous TSs can lead to large rate enhancements. Accordingly, diazo groups are highly tunable moieties of broad potential utility in chemical biology.
Supplementary Material
Scheme 2.

ACKNOWLEDGMENT
This work was supported by Grant R01 GM044783 (NIH) and made use of the National Magnetic Resonance Facility at Madison, which is supported by Grant P41 GM103399 (NIH). Computational resources were supported in part by Grant CHE-0840494 (NSF).
Supporting Information. Synthetic methods and analytical data along with additional computational data, Cartesian coordinates, energies, and imaginary frequencies (for TSs). This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1) (a).Agard NJ, Prescher JA, Bertozzi CR. J. Am. Chem. Soc. 2004;126:15046–15047. doi: 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]; (b) Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Proc. Natl. Acad. Sci. USA. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ning X, Guo J, Wolfert MA, Boons G-J. Angew. Chem., Int. Ed. 2008;47:2253–2255. doi: 10.1002/anie.200705456. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Poloukhtine AA, Mbua NE, Wolfert MA, Boons G-J, Popik VV. J. Am. Chem. Soc. 2009;131:15769–15776. doi: 10.1021/ja9054096. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Debets MF, van Berkel SS, Schoffelen S, Rutjes FPJT, van Hest JCM, van Delft FL. Chem. Commun. 2010;46:97–99. doi: 10.1039/b917797c. [DOI] [PubMed] [Google Scholar]; (f) Jewett JC, Sletten EM, Bertozzi CR. J. Am. Chem. Soc. 2010;132:3688–3690. doi: 10.1021/ja100014q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Dommerholt J, Schmidt S, Temming R, Hendriks LJA, Rutjes FPJT, van Hest JCM, Lefeber DJ, Friedl P, van Delft FL. Angew. Chem., Int. Ed. 2010;49:9422–9425. doi: 10.1002/anie.201003761. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Gold B, Shevchenko NE, Bonus N, Dudley GB, Alabugin IV. J. Org. Chem. 2012;77:75–89. doi: 10.1021/jo201434w. [DOI] [PubMed] [Google Scholar]; (i) Varga BR, Kállay M, Hegyi K, Béni S, Kele P. Chem. Eur. J. 2012;18:822–828. doi: 10.1002/chem.201102329. [DOI] [PubMed] [Google Scholar]; (j) Gold B, Dudley GB, Alabugin IV. J. Am. Chem. Soc. 2013;135:1558–1569. doi: 10.1021/ja3114196. [DOI] [PubMed] [Google Scholar]; (k) Andersen KA, Aronoff MR, McGrath NA, Raines RT. J. Am. Chem. Soc. 2015;137:2412–2415. doi: 10.1021/ja5095815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kolb HC, Finn MG, Sharpless KB. Angew. Chem., Int. Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- (3) (a).Debets MF, van Berkel SS, Dommerholt J, Dirks AJ, Rutjes FPJT, van Delft FL. Acc. Chem. Res. 2011;44:805–815. doi: 10.1021/ar200059z. [DOI] [PubMed] [Google Scholar]; (b) Patterson DM, Nazarova LA, Prescher JA. ACS Chem. Biol. 2014;9:592–605. doi: 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- (4).McGrath NA, Raines RT. Chem. Sci. 2012;3:3237–3240. doi: 10.1039/C2SC20806G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5) (a).Caution! Unstabilized diazo compounds (e.g., diazomethane) are highly toxic and explosively reactive; their use should never be attempted in the context of chemical biology. See: Lewinn EB. Am. J. Med. Sci. 1949;218:556–562. de Boer TJ, Backer HJ. Org. Syn. 1956;36:16–18. Schoental R. Nature. 1960;188:420–421. doi: 10.1038/188420b0. Lewis CE. J. Occup. Environ. Med. 1964;6:91–92.
- (6).Gold B, Aronoff MR, Raines RT. J. Org. Chem. 2016;81:5998–6006. doi: 10.1021/acs.joc.6b00948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Aronoff MR, Gold B, Raines RT. Org. Lett. 2016;18:1538–1541. doi: 10.1021/acs.orglett.6b00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Aronoff MR, Gold B, Raines RT. Tetrahedron Lett. 2016;57:2347–2350. doi: 10.1016/j.tetlet.2016.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9) (a).van Berkel SS, Dirks AJ, Debets MF, van Delft FL, Cornelissen JJLM, Nolte RJM, Rutjes FPJT. ChemBioChem. 2007;8:1504–1508. doi: 10.1002/cbic.200700278. [DOI] [PubMed] [Google Scholar]; (b) van Berkel SS, Dirks AJ, Meeuwissen SA, Pingen DLL, Boerman OC, Laverman P, van Delft FL, Cornelissen JJLM, Rutjes FPJT. ChemBioChem. 2008;9:1805–1815. doi: 10.1002/cbic.200800074. [DOI] [PubMed] [Google Scholar]
- (10).Alabugin IV, Gold B. J. Org. Chem. 2013;78:7777–7784. doi: 10.1021/jo401091w. [DOI] [PubMed] [Google Scholar]
- (11) (a).Hong V, Kislukhin AA, Finn MG. J. Am. Chem. Soc. 2009;131:9986–9994. doi: 10.1021/ja809345d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kislukhin AA, Higginson CJ, Hong VP, Finn MG. J. Am. Chem. Soc. 2012;134:6491–6497. doi: 10.1021/ja301491h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12) (a).Gordon CG, Mackey JL, Jewett JC, Sletten EM, Houk KN, Bertozzi CR. J. Am. Chem. Soc. 2012;134:9199–9208. doi: 10.1021/ja3000936. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dommerholt J, van Rooijen O, Borrmann A, Guerra CF, Bickelhaupt FM, van Delft FL. Nat. Commun. 2014;5:5378. doi: 10.1038/ncomms6378. [DOI] [PubMed] [Google Scholar]
- (13).Lopez SA, Houk KN. J. Org. Chem. 2013;78:1778–1783. doi: 10.1021/jo301267b. [DOI] [PubMed] [Google Scholar]
- (14).Ess DH, Houk KN. J. Am. Chem. Soc. 2008;130:10187–10198. doi: 10.1021/ja800009z. [DOI] [PubMed] [Google Scholar]
- (15).For a tutorial review of the distortion/interaction model (which is also known as the activation strain model), see: Fernández I, Bickelhaupt FM. Chem. Soc. Rev. 2014;43:4953–4967. doi: 10.1039/c4cs00055b. Wolters LP, Bickelhaupt FM. WIREs Comput. Mol. Sci. 2015;5:324–343. doi: 10.1002/wcms.1221.
- (16).Liu F, Liang Y, Houk KN. J. Am. Chem. Soc. 2014;136:11483–11493. doi: 10.1021/ja505569a. [DOI] [PubMed] [Google Scholar]
- (17) (a).Breslow R. Acc. Chem. Res. 1991;24:159–164. [Google Scholar]; (b) Meijer A, Otto S, Engberts JBFN. J. Org. Chem. 1998;63:8989–8994. [Google Scholar]; (c) Chandrasekhar J, Shariffskul S, Jorgensen WL. J. Phys. Chem. B. 2002;106:8078–8085. [Google Scholar]; (d) Narayan S, Muldoon J, Finn MG, Fokin VV, Kolb HC, Sharpless KB. Angew. Chem., Int. Ed. 2005;44:3275–3279. doi: 10.1002/anie.200462883. [DOI] [PubMed] [Google Scholar]; (e) Ess DH, Jones GO, Houk KN. Adv. Synth. Catal. 2006;348:2337–2361. [Google Scholar]; (f) Jung Y, Marcus RA. J. Am. Chem. Soc. 2007;129:5492–5502. doi: 10.1021/ja068120f. [DOI] [PubMed] [Google Scholar]
- (18).Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Bohmann JA, Morales CM, Landis CR, Weinhold F. NBO 6.0. Theoretical Chemistry Institute, University of Wisconsin; Madison, WI: 2013. [Google Scholar]
- (19).Sletten EM, de Almeida G, Bertozzi CR. Org. Lett. 2014;16:1634–1637. doi: 10.1021/ol500260d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).The incorporation of highly electronegative substituents could, however, lead to undesirable side reactions, such as conjugate addition (ref. 11a).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.