

Abstract
The emergence of drug resistant malaria parasites continues to hamper efforts to control this lethal disease. Dihydroorotate dehydrogenase has recently been validated as a new target for the treatment of malaria and a selective inhibitor (DSM265) of the Plasmodium enzyme is currently in clinical development. With the goal of identifying a backup compound to DSM265, we explored replacement of the SF5-aniline moiety of DSM265 with a series of CF3-pyridinyls, while maintaining the core triazolopyrimidine scaffold. This effort led to the identification of DSM421, which has improved solubility, lower intrinsic clearance and increased plasma exposure after oral dosing compared to DSM265, while maintaining a long predicted human half-life. Its improved physical and chemical properties will allow it to be formulated more readily than DSM265. DSM421 showed excellent efficacy in the SCID mouse model of P. falciparum malaria that supports the prediction of a low human dose (<200 mg). Importantly DSM421 showed equal activity against both P. falciparum and P. vivax field isolates, while DSM265 was more active on P. falciparum. DSM421 has the potential to be developed as a single dose cure or once-weekly chemopreventative for both P. falciparum and P. vivax malaria leading to its advancement as a preclinical development candidate.
Keywords: malaria, Plasmodium, pyrimidine biosynthesis, dihydrorotate dehydrogenase
Graphical Abstract
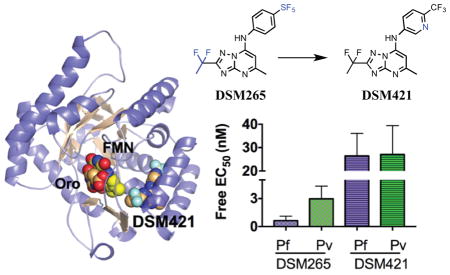
Introduction
Malaria is a mosquito-transmitted disease caused by protozoan parasites of the Plasmodium species.1–2 Despite modern medicine and insect control programs it remains endemic in 97 countries, with an exposed population of 3.2 billion.3 While five species of Plasmodium infect humans, P. falciparum predominates in Africa and is responsible for most of the severe disease and deaths. P. vivax is more widely distributed across other regions of the tropics and while it causes significantly fewer deaths, this strain is associated with relapse through a dormant stage that resides in hepatocytes. The WHO estimates that 214 million cases of malaria occurred worldwide in 2015 resulting in almost half a million recorded deaths, the vast majority in sub-Saharan African children.3 The case numbers and deaths from malaria have been declining due mainly to the effectiveness of current artemisinin combination therapies (ACTs). However resistance to the artemisinin derivatives is now prevalent in parts of Southeast Asia and poses a serious threat to malaria control programs globally.4–7 The ability of the parasite to evade drug therapies through the development of resistance has compromised most previously used antimalarials (e.g. chloroquine and pyrimethamine)2, underscoring the importance of new drug discovery to combat the disease.
A robust portfolio of new antimalarial agents is being developed spanning from early discovery work to clinical development.8 Several important strategies have been outlined to promote the goals of malaria control programs by developing combination therapies that will be effective against all Plasmodium species and life cycle stages and that will reduce the potential for resistance to develop.8–10 The goal of either single dose treatment or once weekly prophylaxis to improve patient compliance has also been promoted.8, 11 The first of the new chemical entities to reach clinical development are currently in Phase II and include a long half-life synthetic ozonide (OZ43912–13), a spiroindolone targeting the P-type cation-transporter ATPase4 (KAE609 14–16), an imidazolopiperazine (KAF15617) and DSM26518 (Fig. 1), a triazolopyrimidine-based dihydroorotate dehydrogenase (DHODH) inhibitor.
Figure 1.

A. Chemical structures of DSM265 and DSM421. B. X-ray structure of PfDHODH bound to DSM421. The inhibitor binding-site showing limited residues within the 4Å shell of DSM421 is displayed. DSM421 is displayed with a dot surface representative of the van der Waals surface. Colors are as follows: DSM421, FMN and orotate carbons tan, protein carbons purple, oxygens red, nitrogens blue, sulfurs yellow, phosphates orange and fluorines turquoise.
DHODH catalyzes the fourth step in the de novo pyrimidine biosynthetic pathway and it is essential to Plasmodium species since they lack pyrimidine salvage pathways that are found in other cells including the human host.19 Using a target-based high throughput screen our group identified the triazolopyrimidines as potent and selective P. falciparum DHODH inhibitors that showed good antimalarial activity.20–21 The series was subsequently optimized to improve both potency and in vivo pharmacokinetic and pharmacologic properties leading to the identification of DSM265, which subsequently was advanced to clinical development.22–23 DSM265 is a potent and selective PfDHODH inhibitor, with good in vivo activity against P. falciparum blood stage infection and pharmacokinetic properties that support efficacy after a single dose treatment or once-weekly for prophylaxis.18 DSM265 was also shown to have liver stage activity, supporting the use of DHODH inhibitors in chemoprevention.18
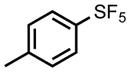
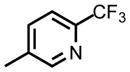
Given the high attrition rate of compounds in clinical development we sought to identify backup compounds targeting DHODH that could be advanced if DSM265 fails to progress during clinical development. Specifically, we aimed to identify a triazolopyrimidine with improved solubility and better species selectivity compared to DSM265. While DSM265 does not have activity on human DHODH, inhibition of mouse and rat DHODH has complicated its development18 since these species are heavily utilized for toxicological studies. With these goals in mind, the SF5-aniline ring was replaced with a series of CF3-pyridinyls leading to the identification of DSM421, which has improved drug-like properties and better species selectivity relative to DSM265, whilst maintaining efficacy and pharmacokinetic properties to support a similar product profile (single dose treatment or once-weekly chemopreventative). Taken together, the data described herein supported the advancement of DSM421 as a preclinical development candidate for the treatment of malaria.
Results
Medicinal chemistry to identify novel triazolopyrimidines with antimalarial activity
We hypothesized that many of the DSM265 liabilities could be addressed by identifying less lipophilic analogs. In addition to the obvious benefits this strategy would have for improving solubility and decreasing hERG activity, we also found that loss of species selectivity (e.g. activity on animal DHODHs) was influenced by lipophilicity, with the more lipophilic analogs showing the most mammalian DHODH activity.24 We therefore replaced the SF5-aniline with a series of CF3-pyridinyls and initially assessed them for activity against P. falciparum and P. vivax DHODH, human DHODH and against P. falciparum 3D7 in whole cell blood stage assays (Table 1). We also conducted a preliminary assessment of ADME characteristics and determined the LogDpH7.4, the kinetic solubility in aqueous buffer, and the in vitro metabolic stability in human and mouse liver microsomes. In comparison to DSM265, substitution of the pyridinyl ring reduced potency on both PfDHODH and P. falciparum parasites by 3–50-fold. DSM421 (para CF3-pyridin-3-yl) was identified as the most potent of the pyridinyl analogs and it was only 3-fold less potent than DSM265 against P. falciparum 3D7 parasites. Within the series activity on P. vivax DHODH was 2–5-fold lower than for PfDHODH, with DSM421 showing a 2-fold difference, which is similar to the difference observed for DSM265. Typically, compounds in this series including DSM421 did not inhibit the human enzyme up to the maximum concentration tested (100 μM), with the exception that DSM456 and DSM457 showed weak inhibition (IC50 = 50 – 60 μM) (Table 1). Analogs with nitrogen in the pyridin-3-yl position (DSM421 and DSM464) had lower LogD values, better solubility and higher potency against P. falciparum 3D7 parasites than those with nitrogen in the pyridin-2-yl position (DSM456 and DSM471). Compounds with meta-CF3 (DSM451, DSM457) were less potent than para-CF3 analogs (e.g. DSM421). Analogs with 2 nitrogens in the ring (DSM458, DSM463, DSM472) were less potent overall, with substitutions at positions 3 and 5 (DSM458 and DSM472) reducing potency less than substitution at positions 2 and 6 (DSM463). Compounds with CF3 at position C2 on the triazolopyrimidine ring were slightly less potent against the parasite than those with CF2CH3, and in comparing the two most potent examples from these groups, DSM421 had better solubility than DSM464. Metabolic stability was assessed in human and mouse liver microsomes where there was minimal degradation (<15%) for all compounds (DSM451 and DSM463 were not tested) resulting in in vitro intrinsic clearance values of < 7 μL/min/mg protein suggesting that these compounds are likely to be metabolically stable and exhibit good plasma exposure in vivo. Based on the combination of the best potency and solubility of the series, DSM421 (Fig. 1) was selected for more extensive profiling.
Table 1.
SAR of select compounds from the triazolopyrimidine series
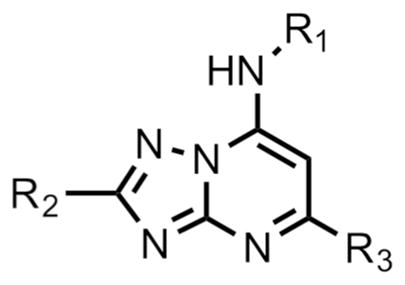
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | R3 | 1Pf 3D7 EC50 (μM) | Pfalcip. DHODH IC50 (μM) | Pvivax DHODH IC50 (μM) | Human DHODH IC50 (μM) | LogD pH7.4 | Kinetic solubility pH 6.5 (μg/ml) |
| DSM265 |
|
CF2CH3 | CH3 | 0.00432 | 0.0242 | 0.0482 | ~1003 | 3.6 | 12.5 |
| DSM421 |
|
CF2CH3 | CH3 | 0.014 (0.013 – 0.015) | 0.053 (0.048 – 0.059) | 0.094 (0.085 – 0.10) | >100 | 2.5 | >100 |
| DSM456 |
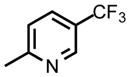
|
CF2CH3 | CH3 | 0.039 (0.037 – 0.041 | 0.051 (0.046 – 0.060) | 0.35 (0.30 – 0.41) | 58 (53 – 64) | 3.4 | 3.1 |
| DSM451 |

|
CF2CH3 | CH3 | 0.9 (0.74 – 1.1) | 0.12 (0.12 – 0.13) | nd | nd | nd | nd |
| DSM457 |
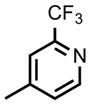
|
CF2CH3 | CH3 | 0.076 (0.067 – 0.085) | 0.079 (0.070 – 0.090) | 0.34 (0.31 – 0.36) | 53 (46 – 59) | 2.3 | >100 |
| DSM458 |
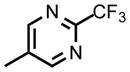
|
CF2CH3 | CH3 | 0.070 (0.061 – 0.082) | 0.070 (0.060 – 0.090) | 0.33 (0.027 – 0.033) | >100 | 2.0 | >100 |
| DSM463 |
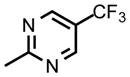
|
CF2CH3 | CH3 | 0.21 (0.020 – 0.024) | 0.13 (.12 – 0.14) | nd | nd | nd | nd |
| DSM464 |
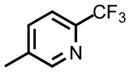
|
CF3 | CH3 | 0.032 (0.011 – 0.097) | 0.041 (0.033 – 0.050) | 0.10 (0.092 – 0.11) | >100 | 2.7 | 25 |
| DSM471 |
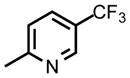
|
CF3 | CH3 | 0.070 (0.061 – 0.08) | 0.037 (0.030 – 0.04) | 0.15 (0.12 – 0.17) | >50 | 3.6 | 0.78 |
| DSM472 |
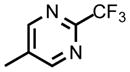
|
CF3 | CH3 | 0.082 (0.069 – 0.096) | 0.040 (0.030 – 0.04) | 0.23 (0.21 – 0.25) | >100 | 1.9 | >100 |
| DSM565 |
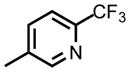
|
CF2CH3 | CH2OH | 0.62 (0.58– 0.66) | 0.088 (0.077 – 0.10) | nd | nd | nd | nd |
Activity of DSM421 on a panel of Plasmodium and animal DHODHs
As noted above, DSM421 is a potent and selective inhibitor of PfDHODH and like DSM265 shows a 2-fold higher IC50 against P. vivax DHODH (Fig. 2 and Tables 1 and 2). DSM421 is 100-fold less active against P. berghei (PbDHODH) than PfDHODH due to amino acid differences in the inhibitor binding-site. For this reason, studies using P. berghei models (both in blood and liver stages) are unsuitable for testing the efficacy of DSM421. DSM421 does not significantly inhibit human, rabbit, monkey or minipig DHODH (IC50>100 μM) while it has modest activity on mouse, rat and dog DHODH (Fig. 2 and Tables 1 and 2). The inhibitory activity of DSM421 is reduced 10-fold towards the rodent enzymes and 2.2-fold versus the dog enzyme in comparison to DSM265. Thus, DSM421 shows improved species selectivity versus animal DHODHs that should provide an easier path to development with respect to toxicology studies than has been the case for DSM265.
Figure 2.

Inhibitory activity of DSM421 on a panel of Plasmodium and mammalian DHODHs. A. Plasmodium enzymes. B. Mammalian enzymes. Triplicate data were collected per concentration point and data were fitted in GraphPad Prism to log(inhibitor) vs. response (three parameter equation) equation for the Plasmodium enzymes, and to the standard IC50 equation (v=vo/(1+(X/IC50)), where v is the enzyme velocity) for the mammalian enzymes. The fitted IC50 values with error estimates are shown in Tables 1 and 2. Error bars represent the standard deviation of the mean.
Table 2.
Comparative kinetic analysis of DSM265 and DSM421 on a panel of P. falciparum and mammalian DHODHs.
| Compd | DSM265 | DSM421 |
|---|---|---|
|
| ||
| DHODH | IC50 (μM) | |
| P. falciparum | 0.00891 | 0.0531 |
| P. vivax | 0.0271 | 0.0941 |
| P. cynomolgi | 0.0122 | 0.012 (0.011 – 0.014) |
| P. berghei | 2.52 | 5.0 (3.5 – 7.1) |
| Human | ~ 1002 | > 100 |
| Mouse | 2.33 | 18 (15 – 20) |
| Rat | 2.73 | 22 (18 – 26) |
| Dog | 163 | 35 (31 – 39) |
| Rabbit | > 1002 | > 100 |
| Minipig | ~ 1002 | > 100 |
| Monkey | ~ 1002 | > 100 |
X-ray structure of PfDHODH bound to DSM421
The X-ray structure of PfDHODH bound to DSM421 was solved to 2.15 Å resolution (Fig. 1B, and Table S1). Good density for the entirety of the DSM421 ligand was evident prior to refinement (Fig. S1). DSM421 occupies the binding site adjacent to the flavin mononucleotide phosphate (FMN) cofactor in the same orientation and binding mode as previously solved X-ray structures of triazolopyrimidine analogs bound to DHODH.18, 24–25 The binding mode is nearly identical to that observed for DSM265 and the two structures align with an RMSD of 0.19 Å (compared to PDB 4RX0). The DSM421 and DSM265 ligands in these structures are nearly superimposable, with only a slight tilt of the aniline rings relative to each other (Fig. S2). In the DSM421 structure the pyridin-3-yl ring was positioned so that the ring nitrogen was placed into the stronger observed electron density (on the F227 side of the ring), however we cannot rule out the possibility that the ring is bound in the flipped orientation with the nitrogen pointing towards L531 or that potentially both conformations are present within the crystal. However if the inhibitor was bound with the pyridinyl nitrogen facing L531, the nitrogen would be 3.4Å from the backbone oxygen of L531, which would be a less favorable interaction than if it is oriented towards F227. Neither orientation provides the opportunity of an H-bond interaction between the pyridinyl nitrogen and the protein. Indeed the hydrophobic nature of the pocket and the lack of interacting residues for the pyridinyl nitrogen likely contribute to the reduced potency of DSM421 relative to DSM265. In comparing the amino acid sequence between P. falciparum and P. vivax, all residues within the 4Å shell of DSM421 are conserved, with the exception of C175 (L175 in P. vivax). The side chain of C175 points away from the inhibitor, suggesting it is not a major determinant of species differences in binding.
DSM421 Plasmodium blood and liver stage activity
Evaluation of DSM421 efficacy against P. falciparum parasites in whole cell blood stage in vitro assays was extended to multiple strains, including strains that are known to be drug resistant to clinically used compounds (e.g. chloroquine and pyrimethamine) and to several compounds in preclinical or clinical development. DSM421 inhibited the panel of P. falciparum laboratory strains with an EC50 ranging from 0.010 – 0.080 μM (average 0.028 ± 0.020 μM) in the standard 72 h in vitro albumax-based assays, similar to the results for 3D7 parasites above (Fig. 3A and Tables 1 and S2). DSM421 was effective against isolates that are resistant to standard antimalarial agents and no significant cross-resistance was observed. The transgenic D10w/yDHODH P. falciparum cell line, which expresses yeast DHODH making it resistant to DHODH inhibitors 26, was also tested (Fig. 3A). DSM421 did not inhibit this line demonstrating that its parasite killing mechanism is on target and due to DHODH inhibition. Finally, DSM421 was also tested for activity against P. falciparum Dd2 isolates that were selected for resistance to DSM265, including a line that harbors a G181C mutation in PfDHODH and a second line that has 3-fold gene amplification of the DHODH gene (Table S2). As expected from the similarity of their structures and similar binding-modes (below), DSM421 shares cross resistance with DSM265, providing further validation of the target.
Figure 3.

P. falciparum and mammalian cell growth inhibition by DSM421. A. Dose response of DSM421 versus P. falciparum 3D7 cells EC50 = 0.014 (0.013 – 0.015) μM and on D10 cells transfected with yeast DHODH (EC50 > 10 μM). B. Dose response of DSM421 (EC50 > 50 μM) and 5-fluoroorotate (EC50 = 0.77 (0.71 – 0.83) μM versus Human HepG2 cells. Data in A and B were fitted in GraphPad Prism to log(inhibitor) vs. response – Variable slope (four parameter) fit equation to determine the EC50 (data in parenthesis show the 95% confidence interval for the fit). Triplicate data were collected per concentration point and error represents the standard deviation of the mean.
DSM421 liver stage activity was evaluated against P. cynomolgi large and small forms in both prophylactic and radical cure models for parasites grown in primary rhesus monkey hepatocytes in human serum-based media. DSM421 did not show radical cure activity at concentrations at or below 10 μM, but it did show prophylactic activity that was particularly potent on large forms (Large form EC50 = 0.19 μM; small form EC50 = 0.59 μM). When data were corrected for protein binding, the unbound EC50 (0.028 and 0.089 μM on the large and small forms, respectively) for the large form liver stage was similar to the unbound EC50 on the blood stages (Table 3), suggesting that DSM421 will block formation of the liver stage schizont and have good prophylactic activity.
Table 3.
Comparative DSM421 and DSM265 activity on P. falciparum and P. vivax.
| Assay | EC50(μM) | Fraction unbound in assay media | unbound EC50 (μM) | EC50 (μM) | Fraction unbound in assay media | unbound EC50 (μM) |
|---|---|---|---|---|---|---|
| DSM265 | DSM421 | |||||
| Blood-stage: P. falciparum 3D7 in vitro 72 h growth assay | 0.0043 | 0.17a | 0.00073 | .014 | 0.5a | 0.0070 |
| Blood-stage: P. falciparum 3D7 in vitro 48 h growth assay (PRR) | 0.011 | 0.17a | 0.0019 | .043 | 0.5a | 0.022 |
| Blood-stage: P. falciparum field isolates, 72 h assay | ND | ND | ND | 0.023 | 0.5a | 0.012 |
| Blood-stage: schizont maturation assay P. falciparum Field isolates | 0.19 | 0.003b | 0.00057 | 0.15 | 0.15b | 0.023 |
| Blood-stage: schizont maturation assay P. vivax Field isolates | 0.92 | 0.003b | 0.0028 | 0.18 | 0.15b | 0.027 |
DSM421 kill rates in whole cell parasite assays
In order to determine how quickly DSM421 killed blood stage parasites, kill rates were determined using a standard drug wash out assay.27 P. falciparum 3D7A parasites were incubated with 10xEC50 of DSM421 for 24–120 h before washing and replating in fresh drug-free media to determine the fraction of viable cells. The EC50 in the 48 h growth assay in this study was 0.043 ± 0.006 μM. A 48 h lag time was observed for DSM421 before killing began and the kill rate was similar to that of atovaquone (Table S3) and to DSM26518, providing supporting evidence that the kill rate is a property of the target. While DSM421 is only about 3-fold less potent than DSM265 in these in vitro assays (48 and 72 h assays), the intrinsic potency is 10-fold lower than DSM265 based on unbound concentrations (Table 3).
Assessment of cytotoxicity against a human cell line
DSM421 was tested to determine if it showed cytotoxicity against human HepG2 cells (Fig. 3B). DSM421 did not inhibit the growth of this cell line (EC50 > 50 μM), thus providing a selectivity window of >600–5000 fold over its activity against P. falciparum.
Efficacy of DSM421 against P. falciparum field isolates
DSM421 was tested against fresh P. falciparum isolates obtained from patients visiting health care facilities in the Districts of Yopougon and Attecoube, Abidjan, Côte d’Ivoire using the standard 72 h SYBR Green assay (Fig. 3C). DSM421 showed equal efficacy against these isolates (EC50 = 0.023 ± 0.0026 μM) compared to the laboratory strains tested above, and there was no evidence for a resistant population. Chloroquine (average EC50 = 0.035 μM), lumefantrine (average EC50 = 0.029 μM) and artesunate (average EC50 =0.0038 μM) were assayed as controls in the study (Fig. 3C).
Activity of DSM421 and DSM265 on P. falciparum and P. vivax blood stages – ex vivo schizont maturation assay
Unlike P. falciparum, which can be sustained in culture, continuous culture of P. vivax is not yet feasible. Thus to test the activity of DSM421 and DSM265 on P. vivax blood stages, parasite isolates were collected from patients and evaluated for drug susceptibility using a short-term 48 h schizont maturation assay. P. falciparum isolates were collected in parallel to serve as a control and additionally two lab strains (F27 and K1) of P. falciparum were evaluated to control for methodology differences compared to the more commonly used methods (above). Chloroquine, piperaquine, mefloquine, artesunate and amodiaquine were also tested in parallel. Field isolates were collected from patients attending malaria clinics in Timika, Papua Province, Indonesia, an area with known high grade multidrug resistance in P. falciparum and P. vivax 28. Ring stage parasites were exposed to drug for 48 h and matured using a candle jar until 40% of parasites had reached the schizont stage (35–56 h). Notably, P. vivax field isolates were 5-fold less sensitive to DSM265 than the P. falciparum field isolates (Fig. 4 and Table S4). In contrast, DSM421 showed equivalent activity towards both species (Fig. 4 and Table S4). The magnitude of the difference for DSM265 was surprising given the finding that the difference between the IC50 on the two enzymes (PfDHODH and PvDHODH) was only 2-fold, and was similar for both DSM421 and DSM265. When comparing the schizont maturation data obtained from the P. falciparum field isolates with that from the lab strains, the IC50’s were similar for both DSM265 and DSM421, however, they were significantly higher for both compounds compared to those obtained in the standard 72 h blood-stage albumax assay (Fig. 4 and Tables 1 and S4). The schizont maturation assays were conducted in human serum-based media and when unbound concentrations were compared, the IC50 values between the various P. falciparum assays were comparable, especially when comparing assays that were conducted for 48 h of drug exposure (Table 3).
Figure 4.

Activity of tirazolopyrimidines on P. falciparum and P. vivax field isolates. A. Activity of DSM421 on P. falciparum blood stage field isolates collected from patients in Côte d’Ivoire. EC50’s were measured in the standard 72 h growth assay in RPMI media supplemented with 0.5% Albumax. Data are shown as a scatterplot of compound EC50s for individual patient isolates. AS, artesunate, CQ, chloroquine, and LUM, lumefantrine were tested as controls. Bars represent the mean. The mean ± standard deviation, number of isolates (n) and the range for each drug were as follows: DSM421 (0.023 ± 0.012, n=20, 6.9 – 48); AS (0.0038 ± 0.0039, n=18, 0.2 – 15.7); CQ (0.035 ± 0.030, n=18, 3.9–116); LUM (0.029 ± 0.031, n=7, 0.9 – 94). B. Activity of DSM421 and DSM265 on P. falciparum and P. vivax blood stage field isolates collected from patients in Papua, Indonesia. Drug efficacy was tested in a 48 h schizont maturation assay in 10% human serum based media. Bars represent the mean. Median values and ranges are reported in Table S4.
In vivo activity of DSM421 in the P. falciparum SCID mouse model
DSM421 was tested for in vivo efficacy using the P. falciparum SCID mouse model.29 DSM421 was dosed orally once daily (QD) for 4 days at a range of doses. DSM421 was well tolerated and no adverse reactions were noted. Blood samples for the measurement of DSM421 concentrations were collected over the first 24 h after dosing to establish the pharmacokinetic/pharmacodynamic (PK/PD) relationship. DSM421 displayed potent in vivo antimalarial activity; the ED90 24 h after the last dose was 2.6 mg/kg/day (AUC0–23h 12.9 μg.h/mL, Cav 0.53 μg/mL, Cmin 0.1μg/mL) (Fig. 5A and Table S5A) and compared very favorably with the activity of chloroquine and mefloquine. Across the full dose range, the maximum blood concentrations (Cmax) and area under the blood concentration vs. time profiles (Day 1 AUC0–23 h) were approximately dose proportional (Fig. 5B and Table S5B). Maximum parasite killing occurred at or slightly above a dose of 5 mg/kg/day (AUC0–23h 30.8 μg.h/mL; Cav 1.3μg/mL; Cmin 0.33μg/mL) (Fig. 5 and Table S5B). DSM421 showed similar efficacy to DSM265 based on comparison of the total blood AUC required to achieve the ED90 in the SCID model (Table S5B). As noted above, DSM265 is intrinsically ~10-fold more potent than DSM421, however DSM421 has significantly lower intrinsic clearance leading to higher unbound plasma concentrations for DSM421 at a similar total blood exposure and dose.
Figure 5.

P. falciparum SCID mouse in vivo efficacy data. DSM421 was dosed QD for 4 days starting 3 days post infection. A. Blood parasitemia levels versus days post infection, with the indicated dose levels administered in mg/kg. The lower limit of detection was 0.01% parasitemia. B. Blood concentrations (μg/mL) of DSM421 from 0.25 – 24 h after the first dose.
Absorption, distribution, excretion and metabolism (ADME) studies
The physicochemical properties of DSM421 were compared to DSM265 (Table S6). The measured Log D7.4 (2.36) indicates moderate lipophilicity that is significantly less than that for DSM265 (Log D7.4 4.0). The aqueous solubility of DSM421 was reasonably independent of media conditions with generally good solubility (80 – 160 μg/ml) evident in all physiologically relevant media tested. DSM421 is considerably more soluble (6–80-fold) than DSM265, providing a significant advantage from a formulation perspective. Permeability values for DSM421 across Caco-2 cell monolayers were high in both directions with no evidence for significant efflux (Table S7). Collectively, these results suggest that neither solubility nor permeability would be expected to limit the oral absorption of DSM421.
The in vitro metabolism of DSM421 was studied using human, dog, rat and mouse liver microsomes and cryopreserved hepatocytes. The intrinsic clearance (CLint) was very low in both microsomes and hepatocytes across all species (Table S8). The in vitro intrinsic clearance was <7μL/min/mg protein across all species, and corresponded to a low predicted in vivo unbound CLint of approximately 2.3 mL/min/kg in humans.
Metabolite identification studies were conducted in human cryopreserved hepatocytes. DSM421 showed minimal degradation in human hepatocyte incubations conducted at a substrate concentration of 10μM over 120 minutes. Several putative metabolites consistent with mono-oxygenation (two M+16 metabolites), glucuronidation (M+176) and mono-oxygenation and glucuronidation (M+192) were identified based on accurate mass measurement. However, due to the low extent of metabolite formation and subsequent weak MS signals, the sites of metabolism could not be conclusively determined. As hydroxylation of the methyl on the triazolopyrimidine ring was the major site of metabolism for DSM26518, we synthesized the analogous compound (DSM565; Table 1) in the context of the DSM421 aniline. Comparison of the MS/MS fragmentation patterns of the metabolites with the authentic standard for DSM565 confirmed that DSM565 is the most abundant M+16 metabolite formed in hepatocytes based on peak area. DSM565 showed similar activity as an inhibitor of PfDHODH to DSM421 (1.6-fold higher IC50), but similar to the hydroxyl metabolite of DSM265, DSM565 showed much poorer activity versus P. falciparum in whole parasite assays suggesting that this metabolite is unlikely to contribute to parasite killing in vivo (Table 1).
Plasma protein binding was assessed in human, dog, rat and mouse plasma. DSM421 was stable in plasma at 37°C over the 5 h period used for the binding experiments and in blood over a 4 h incubation period. Plasma protein binding was higher in human plasma (~98%) than in the remaining species (all ~90%) (Table S9). Blood to plasma partitioning ratios indicated that partitioning into red blood cells was lower in human blood than in rat, mouse and dog blood, most likely due to the higher plasma protein binding in human plasma.
Cytochrome P450 inhibition studies showed mild inhibition of CYP2C9 and CYP2D6 (22% and 45% inhibition respectively) and no measurable inhibition of the other isoforms at the maximum DSM421 concentration tested (20μM) (Table S11). Time-dependent inhibition studies were conducted to assess the potential for mechanism-based inhibition. Similar to above, there was no evidence of mechanism-based inhibition of any of the CYP isoforms tested and IC50 values were all greater than 20μM (Table S13).
Pharmacokinetic analysis in mice, rats and dogs
In order to support the advancement of DSM421, pharmacokinetic analysis in multiple species was conducted to aid in human dose predictions and the design of animal toxicological studies. Studies were conducted in mice, rats and dogs following IV and oral dosing using the dosing conditions described in Table S14. DSM421 was well tolerated in all species and no adverse reactions were noted. In mice, DSM421 exhibited low plasma clearance (5.1 mL/min/kg, or about 4% of mouse hepatic bloodflow), moderate Vss (0.9 L/kg) and a half-life of approximately6 2 h (Fig. 6A and Table S15). Following oral dosing, Tmax ranged from 1–2 h following doses of 4.5 to 41.5 mg/kg, with Cmax ranging from 3.11 to 33.3 μg/mL over the same dose range. Plasma AUC values following oral dosing increased 13-foldfor a 9-fold increase in dose. Oral bioavailability exceeded 100% at all dose levels indicating dose-dependency in clearance processes. In rats, DSM421 exhibited low plasma clearance (2.4 mL/min/kg, orabout 4% of rat hepatic blood flow), moderate Vss (0.8 L/kg) and a moderate half-life of approximately 5 h (Fig. 6B and Table S16). Tmax ranged from 1.8 to 7.5 h following oral doses of 4.3 to 53.7 mg/kg suggesting prolonged absorption with increasing dose. Over the oral dose range of 4.3 to 53.7 mg/kg, plasma AUC values increased 31-fold for a 12-fold increase in dose indicating dose-dependent kinetics. At the lowest dose, oral bioavailability was approximately 80% and increased with increasing dose. Only minor amounts of DSM421 were detected in urine as intact drug suggesting minimal contribution of renal elimination to the overall clearance. In dogs, DSM421 exhibited low plasma clearance (0.5mL/min/kg, or about 2% of dog hepatic blood flow), moderate Vss (0.8 L/kg) and a long half-life of 15–18 h (Fig. 6C and Table S17). There was evidence of double peaking in dogs following IV dosingsuggesting enterohepatic recirculation, which has also been observed for DSM265 in dogs.18 Following oral administration of 1 mg/kg, Tmax was 4 h and bioavailability was ~100%.
Figure 6.

Oral (PO) and intravenous (IV) pharmacokinetic profiles after a single dose of DSM421 in A) mice, B) rats and C) dogs. Dose levels are indicated on the graph. Data represent the mean ± range of n=2 samples per time point for mice and n= 2 rats, or mean ± SD for n=3 dogs. Dosing conditions are described in Table S14.
Human pharmacokinetic and dose predictions
Human PK parameters were estimated using a physiologically based pharmacokinetic (PBPK)approach. The predicted unbound human clearance based on studies in human liver microsomes was 2.3 mL/min/kg and the total plasma clearance was predicted to be 0.045 mL/min/kg. For comparison, human unbound clearance was also estimated using allometric scaling of unbound clearance values measured after IV administration to mice, rats and dogs, giving a predicted unbound human clearance of 2.2 mL/min/kg and a predicted total plasma clearance of 0.042 mL/min/kg. The volume of distribution (Vss) was estimated to be 0.3 L/h giving an estimated human half-life of 78 h.
The target minimum plasma concentration needed to achieve a near maximal rate of kill (minimum parasiticidal concentration or MPC) was estimated using the in vitro PRR data and in vivo efficacy data from SCID mice accounting for differences in binding in each of the different test systems. The estimated target plasma concentration was 1.2 μg/mL based on the in vitro PRR data compared to approximately 1 μg/mL based on the SCID data. Simulated plasma concentration versus time profiles (Fig. 7) suggested that a single dose of 200 mg would maintain a target plasma concentration of 1–2 μg/mL for more than 8 days.
Figure 7.

Simulated human plasma concentration versus time profiles following a single oral dose of 200 mg administered in the fasted state (GastroPlus).
Safety Pharmacology
DSM421 was evaluated in a series of safety pharmacology studies (Table S18). Profiling in a CEREP diversity panel against a set of 100 human channels and enzymes indicated no significant inhibition at a concentration of 10 μM (3.6 μg/mL). DSM421 was evaluated for genotoxicity in a GLP Ames 5 strain test with and without metabolic activation on a batch of highly purified compound (99.6% pure by HPLC) where it exhibited no obvious cytotoxicity in any tester strain at any tested dose level, either in the presence or absence of metabolic activation. In a standard hERG channel patch clamp study and Ca_1.2 and Na_V1.5 channel assays, DSM421 exhibited no activity on either the Ca_1.2 orNa_V1.5 channels. For the hERG channel, the IC50 was 26.2 μM (9.4 μg/mL) and 20% inhibition occurred at ~10 μM (3.6 μg/mL). Since the maximum concentration tested in the assay was 33 μM, these values were calculated based only on a partial dose response curve where 50% inhibition wasobserved at the highest tested concentration. Assuming a plasma Cmax of approximately 9 μg/mL(unbound Cmax = 0.17 μg/mL) from the human dose prediction, the ratios of hERG IC50/Cmax unbound andIC20/Cmax unbound in humans are predicted to be ~55 and ~21, respectively, suggesting a low risk for interaction with the channel in humans.
Discussion
DHODH is a novel target for the treatment of malaria and the triazolopyrimidines represent a new class of antimalarial agents that do not share cross-resistance with current therapies. A key feature of the DHODH target is that its inhibition leads to both blood and liver stage activity opening up the potential for DHODH inhibitors to be used for either treatment or chemoprevention.18 While DSM265 has progressed to clinical development, backup candidates were sought to improve on its potential liabilities. We identified DSM421, which like DSM265, is a potent and selective inhibitor ofPlasmodium DHODH with excellent in vivo activity. However, DSM421 has the following advantages over DSM265: 1) it has equivalent activity on P. vivax and P. falciparum in the ex vivo model of field isolates whereas DSM265 was 5 times less potent on P. vivax in a similar study; 2) it is considerably more soluble than DSM265 resulting in higher in vivo plasma exposure, which is likely to simplify development of an oral formulation; and 3) it has better selectivity than DSM265 vs. non-humanmammalian DHODH enzymes.
The PK properties of DSM421 were profiled in mice, rats and dogs and results were compared to previous data for DSM265.18 Systemic plasma clearance was similarly low across species for both compounds, however the unbound intrinsic clearance for DSM421 was considerably lower than that for DSM265. For example, systemic plasma clearance values for DSM421 and DSM265 in dogs were 0.5 and 1.2 mL/min/kg, respectively, whereas the unbound intrinsic clearances were 4.5 and 183 mL/min/kg. The lower unbound intrinsic clearance most likely reflects the lower lipophilicity forDSM421 (Log D7.4 of 2.4 compared to 4.0 for DSM265). Importantly, the lower intrinsic clearance leads to higher unbound concentrations for DSM421 compared to DSM265 at a similar dose. As discussed below, this difference in exposure in spite of lower intrinsic potency for DSM421 means that the required dose is expected to be similar to possibly lower than for DSM265. Another important comparison for DSM421 and DSM265 is that the oral bioavailability was consistently >80% for DSM421 across species tested compared to bioavailability values of 20–70% for DSM265 across species. These results likely reflect the improved physicochemical properties of DSM421, most notably its improved aqueous solubility, and the potential impact this has on oral absorption. These data support the conclusion that, unlike DSM265, formulation of DSM421 will be readily achieved through relatively standard approaches. Indeed, preliminary studies suggested that dissolution was rapid using simple formulation excipients in a capsule preparation providing a viable option for dosing in clinical studies.
Collectively, the preclinical PK data for DSM421 suggest that it is likely to show similar prolonged plasma exposure in humans compared to DSM265 (predicted human half-life ~ 78 h for DSM421 versus 100 h for DSM265). A single oral dose of approximately 200 mg is predicted to provide plasma concentrations above the estimated MPC for a period of more than 8 days. Thus we concluded that DSM421 has the potential to be developed in combination with a partner drug for either a single dose treatment or for once-weekly chemoprevention, and that it will have a similar product profile compared to DSM265, with the notable exception that DSM421 is expected to have equal activity on both P. falciparum and P. vivax.
During the medicinal chemistry program to identify DSM421 we evaluated a series of CF3-pyridinyl replacements to the SF5-aniline of DSM265 and found that the position and number of nitrogens added to the ring had significant effects on the potency and the physicochemical properties. The para CF3-pyridin-3-yl of DSM421 yielded both the most potent and most soluble derivatives in the series. There was good correlation between the IC50 on PfDHODH and its potency on the parasite in whole cell assays for the compounds in the series. The inhibitor binding-site in Plasmodium DHODH contains very few polar residues, and the reduced potency of DSM421 relative to DSM265 is likely caused by the fact that the nitrogen in the CF3-pyridin-3-yl ring is unable to make any polar contacts within the binding site. However despite the lower intrinsic potency of DSM421, its lower unbound intrinsic clearance led to higher unbound plasma concentrations, and as a consequence, the dose required for efficacy in the SCID mouse model was similar to that for DSM265. Thus the reduced potency is more than compensated for by the improved in vivo pharmacokinetic properties of DSM421.
Prior reported studies for DSM265 had not evaluated its activity on P. vivax parasites, but the data presented herein show that DSM265 has 5-fold lower activity on P. vivax field isolates compared to P. falciparum. In contrast, DSM421 is equally active on both parasites. This difference represents a major point of superiority for DSM421 over DSM265, and suggests that DSM421 will be equally efficacious against both species while DSM265 may show greater efficacy towards P. falciparum in the clinic. The underlying mechanistic factors contributing to the observed differences between DSM421 and DSM265 on P. vivax are not immediately clear. Both compounds show similar activity on PfDHODH versus PvDHODH (both showing only a 2-fold difference in IC50), and the two compounds bind in the inhibitor binding-site with a near perfect overlap. There are no significant amino acid differences between PfDHODH and PvDHODH within 4Å of the bound inhibitor, so if amino acid differences between the enzymes were a contributing factor, second shell interactions would have to be playing a role. While we cannot rule out the possibility of enzyme potency differences within the context of the full-length mitochondrial enzyme, the data suggest that the DSM265 activity difference on P. vivax vs. P. falciparum does not result from DHODH species differences.
A notable difference between P. falciparum and P. vivax is that while P. falciparum can be cultured in vitro in human erythrocytes at all stages of development, P. vivax and P. berghei (mouse malaria) infect mostly reticulocytes, which are an early cell type along the erythrocyte developmental pathway. Mature erythrocytes lack or have reduced pools of many metabolic precursors that are found in reticulocytes and these differences have been shown to be a contributing factor in differential sensitivity to gene knockout in P. berghei.30 In these studies the first four enzymes in pyrimidine biosynthesis, including DHODH were refractory to gene knockout, implying that they are essential even for parasites such as P. berghei and P. vivax that infect young erythrocytes. Our findings that DSM421 has equal activity on P. vivax and P. falciparum are consistent with the reported knockout studies, demonstrating that DHODH is indeed essential for growth of malaria parasites in both reticulocytes and erythrocytes. Furthermore, DHODH inhibitors from the thiophene-2-carboxamide class that have good activity on both P. falciparum and P. berghei DHODH have been shown to have good efficacy against P. berghei in a mouse model.31 Thus the reduced activity of DSM265 on P. vivax does not appear to be a consequence of difference in P. vivax and P. falciparum biology due to metabolic rescue by more complex metabolite pools in the immature reticulocytes. While it seems unlikely that cell permeability would be a limiting factor for either DSM265 or DSM421, it remains possible that the different physicochemical properties of the two molecules lead to better uptake (or higher concentrations) of DSM421 into reticulocytes than for DSM265, leading to the observed differences in drug sensitivities between the species.
Outside of the physicochemical differences between DSM421 and DSM265, many aspects of their biological activity are similar and are a property of the target. Both compounds show similar parasite kill rates and show a 24–48 h lag time before killing begins. While DSM421 P. falciparum liver stage activity has not been evaluated, it showed good activity on P. cynomolgi large form liver stages and therefore would be expected to have a similar product profile to DSM26518 with the potential to be useful for both treatment of acute malaria and for casual prophylaxis.
Mammalian DHODH species selectivity was evaluated with the goal of identifying compounds with less activity on the rodent and dog enzymes than DSM265. While DSM265 is not an inhibitor of human DHODH, it has significant activity on rodent DHODH, which has complicated the choice of suitable species for toxicity testing. The rat in particular has not been a useful species for DSM265 toxicity testing because of the combination of enzyme inhibition and lower plasma protein binding than for other species.18 DSM421 in contrast has maintained the good selectivity against human DHODH while also showing 10-fold reduced activity on rat and mouse DHODH, and 2-fold reduced activity on dog DHODH, suggesting that all three species will be suitable for toxicological testing of DSM421.
In conclusion, DSM421 appears to have all the beneficial properties of DSM265, but importantly, overcomes the key liabilities in relation to P. vivax activity, solubility, and rodent DHODH species selectivity. What makes compounds from this class particularly promising is that they have both blood and liver stage activity and the predicted long half-life and excellent oral exposure suggest that DSM421 will be able to meet the target product profile of either a single dose cure or once-weekly prophylaxis with a suitable partner drug. The compound has cleared the list of safety pharmacology assessments and it was well tolerated in a rat 7-day exploratory toxicity study (data not shown), clearing the path for its advancement to preclinical development. On the strength of these data, DSM421 has been advanced as a preclinical development candidate for malaria.
Materials and Methods
DHODH expression, purification and inhibitor IC50 determination
P. falciparum, P. vivax, human, and animal DHODHs were expressed as recombinant proteins in E. coli and purified by Ni+ and gel filtration column chromatography as previously described.18, 21–22, 24–25 Steady-state kinetic analysis was performed using the 2,5-dichloroindophenol (DCIP) based spectrophotometric method as previously described 21–22 employing the following conditions: DHODH(E = 5–10 nM), substrates (0.2 mM L-dihydroorotate and 0.02 mM CoQd). 100 x compound stock solutions were prepared in DMSO (range 0.001 – 100 mM) and then diluted 1 to 100 into assay buffer to the final concentration. DMSO solutions were protected from light. Data were fitted to the log [I] vs. response (three parameters) equation (Y=Bottom + (Top−Bottom)/(1+10^((X−LogIC50)))) to determine the IC50 or for compounds where the IC50 >10 μM, data were fit to the standard IC50 equation (Y=1/(1+X/(IC50))) using Graph Pad Prism.
X-ray structure determination of PfDHODH in complex with DSM421
Purified loop truncated PfDHODHΔ384–413 was used for crystallization as previously described.31 Preliminary crystallization conditions were found using the random crystallization screen Cryos suite (Nextal) and conditions were then refined by variation of pH and precipitant concentration by hanging drop vapor diffusion at 20°C. Crystallization drops were mixtures of equal volume of reservoir solution and PfDHODΔ384–413 (27 mg/ml) pre-equilibrated with 1 mM DSM421 (0.1 M stock solution in DMSO) and 2 mM dihydroorotate (DHO, 0.1 M stock solution in DMSO). Crystals of thePfDHODΔ384–413-DSM421 grew overnight from 0.16 M ammonium sulfate, 0.1 M sodium acetate, pH 4.4, 10% PEG4000 (w/v), 24% glycerol (v/v), and 10 mM DTT.
Diffraction data were collected at 100K on beamline 19ID at Advanced Photon Source (APS)using an ADSC Q315 detector. The crystal of PfDHODΔ384–413-DSM421 diffracted to 2.15 Å and has a space group of P64 with the cell dimension of a=b=86.4, c=138.6. The structure contains only one molecule of PfDHODH in the asymmetric unit. Diffraction data were integrated and intensities were scaled with HKL2000 package.32
Crystallographic phases for PfDHODH inhibitors were solved by molecular replacement with Phaser 33 using the previously reported structure of PfDHODΔ384–413 bound to DSM1 (PDB ID 3I65 25) as a search model (Table S1). Structures were rebuilt with COOT 34 and refined in PHENIX 35 to Rwork and Rfree of 0.18 and 0.22, respectively. Electron density for the 344–355 loop was missing. The structure was displayed with PyMOL Molecular Graphics System (Version 1.7, Schrödinger). The coordinates have been submitted to the Protein Data Bank (PDB 5TBO).
In vitro parasite blood stage activity assays
Blood stage P. falciparum parasites were grown in RPMI 1640 containing 0.5% Albumax II and human red cells using a 72 h assay as previously described.20, 22 Determination of compound EC50 was performed using the SYBR Green method 24 except for assays performed by Swiss TPH which were conducted using the [3H]hypoxanthine incorporation method as described previously.36 In vitro killing rates to assess the Parasite Reduction Ratio (PRR) were performed on P. falciparum 3D7 cells at 10xEC50 of DSM421 using the [3H] hypoxanthine based assay as described.27 Parasites were plated with drug, washed free of drug at the indicated time and then replated to assess viability. The reported EC50 from this study was determined from a 48 h growth assay.
In vitro P. cynomolgi liver stage activity assays
P. cynomolgi M strain liver-stage parasite assays to test for DSM265 activity were performed as described 37–38, except that 10% human serum was used in place of fetal calf serum in the William’s E growth medium.
P. falciparum in vitro activity studies using clinical isolates
Studies were approved by the National Committee for Ethics and Research (CNER) of Côte d’Ivoire under the number 15/MSLS/CNER-dkn dated April 10th 2013. Participants were covered by risk insurance (GNAN°30105811210003) during the full duration of the study.
P. falciparum isolates were collected form patients attending local medical centers namely (i) Formation Sanitaire Urbaine Communautaire of Wassakara (FSU-Com) in the district of Yopougon and (ii) Centre de Santé Urbain (CSU) of Abobodoume-Locodjro in the district of Attecoube, Abidjan, Southern Côte d’Ivoire. Patients were over 18 years old, were not yet treated, and were singly infected with P. falciparum as determined by a Rapid Diagnostic Test (Type CareStart Malaria HRP2/PLDH (Pf/VOM) COMBO). Only samples with >0.1% parasitemia were used in the study. Parasitized RBCs were washed in complete RPMI medium (RPMI 1640 supplemented with 0.5% Albumax II, 25 mM Hepes, 25 mM NaHCO3 (pH 7.3), 0.36 mM hypoxanthine and 100 g/ml neomycin) and incubated in 96-well plates with freshly prepared serial 2-fold dilutions of the compounds to be tested (from 10 mg/ml DMSO stock solutions, except for chloroquine which was prepared in sterile water) for 72 h at 37°C under hypoxic conditions (5% CO2). Parasite growth in the presence of antimalarial compounds was determined by incorporation of SYBR Green into the parasite’s DNA followed by measurement of fluorescence of the resulting DNA-dye complex as described above.
Ex vivo schizont maturation drug susceptibility assay against P. vivax and P. falciparum field isolates
Ethical approval for this study was obtained from the Eijkman Institute Research Ethics Commission, Eijkman Institute for Molecular Biology, Jakarta, Indonesia (EIREC-47), and the Human Research Ethics Committee of the Northern Territory (NT), Department of Health & Families and Menzies School of Health Research, Darwin, Australia (HREC 2010-1396).
Plasmodium isolates were collected from patients attending malaria clinics in Timika (Papua, Indonesia) and drug susceptibility was measured using a protocol modified from the WHO microtest as previously described.28, 39–40 Assays were performed in RPMI 1640 medium plus 10% AB+ human serum (for P. falciparum) or McCoy’s 5A medium plus 20% AB+ human serum (for P. vivax) using 2% hematocrit. Details can be found in supplemental methods.
P. falciparum SCID mouse efficacy studies
The human biological samples were sourced ethically and their research use was in accord with the terms of the informed consents. All animal studies were ethically reviewed and carried out in accordance with European Directive 86/609/EEC and the GSK Policy on the Care, Welfare and Treatment of Animals.
P. falciparum Pf3D70087/N9 (20 × 106) parasites were used to infect NOD-scid IL-2Rγnull (NSG) mice (Jackson Laboratory, USA) (23 – 26 g) engrafted with human erythrocytes as previously described.29 DSM421 was administered orally QD in vehicle (0.5% w/v sodium carboxymethylcellulose, 0.5% v/v benzyl alcohol, 0.4% v/v Tween 80 in water) starting from day 3 post infection. Flow cytometry was used to monitor parasitemia levels and the effective dose to obtain 90 and 99% reduction in parasitemia (ED90 and ED99, respectively) was calculated as previously described. 41 Concentrations of DSM421 were assessed by LCMSMS as described in the supplemental methods.
Physicochemical measurements
Estimation of the partition coefficient at pH 7.4 was conducted using either a gradient chromatography method (gLog D7.4)22 or a shake flask method42 as described previously. Solubility measurements were determined using either a kinetic method in pH 6.5 buffer22 or by equilibration in physiologically-relevant media 18 as described previously. Methods for measurement of Caco-2 cell permeability are described in supplemental materials.
Binding studies
Binding to plasma proteins (human, dog, rat and mouse), microsomes (human), and media (0.5% Albumax II culture medium, 10% FCS in DMEM and 10% human serum in RPMI) was assessed by ultracentrifugation at 37°C with a DSM421 concentration of 1000 ng/mL using previously described methods.22 Samples were assayed for DSM421 using the methods described for plasma in the supplemental methods.
In vitro metabolism in liver microsomes and cryopreserved hepatocytes
In vitro studies using hepatic microsomes (BD Gentest, BD Biosciences, Bedford, MA or Sekisui XenoTech, LLC, Kansas City, KS) and cryopreserved hepatocytes (Sekisui XenoTech) from humans, dogs, rats and mice were conducted at an initial substrate of 1 μM and either 0.4 mg/mL microsomal protein or 1 × 106 hepatocytes/mL. Metabolite identification studies were conducted in human cryopreserved hepatocytes (1 × 106 cells/mL) and a substrate concentration of 10 μM. Methodologies for microsomes22 and hepatocytes 18 have been previously described. Additional analytical details and data analysis methods can be found in the supplemental methods.
CYP Inhibition
Reversible cytochrome P450 inhibition studies were conducted using a substrate specific approach as previously described.18 Further details and methods used for time-dependent inhibition are provided in the supplemental methods.
Pharmacokinetics
Pharmacokinetic studies in mice and rats were conducted at Monash University, Parkville, Australia and were performed in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, and study protocols were approved by the Monash Institute of Pharmaceutical Sciences Animal Ethics Committee. DSM421 was administered intravenously and orally at doses of approximately 2 mg/kg and 5–50 mg/kg, respectively. Animal, formulation and dosing details are described in Table S14. Blood samples were collected over 30–48 h post-dosing, and plasma separated by centrifugation and assayed by LC/MS as described in the supplemental methods. Pharmacokinetic parameters were calculated using non-compartmental methods as described previously.22 Pharmacokinetic studies in dogs were conducted at Abbvie Inc., North Chicago, IL under approval of the institutional ethics committee. DSM421 was administered intravenously and orally at doses of 1 mg/kg (see Table S14 for formulation and dosing details). Blood samples were collected over 144 h, plasma separated by centrifugation and assayed by LC/MS as described in the supplemental methods. Pharmacokinetic parameters were calculated using non-compartmental methods as described for mice and rats.
Human half-life and dose predictions
Human PK parameters were estimated using a physiologically-based pharmacokinetic (PBPK) approach as described in the supplemental methods.
Safety Pharmacology
CEREP assays (Express S profile) were performed by CEREP (Bois l’Evêque, France), hERG, Ca_1.2 and NaV1.5 channel assays were performed using IonWorks patch clamp electrophysiology at Essen Labs (Hertfordshire, UK) and the GLP Ames study was performed by WuXi AppTec Cp. (Suzhou, China) as described in the supplemental methods.
Chemical Synthesis of DSM421 and analogs
DSM421(2-(1,1-Difluoroethyl)-5-methyl-N-(6-(trifluoromethyl)pyridin-3-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine) and analogs were prepared using previously reported methods 22–23 to generate the common intermediates 7-Chloro-2-(1,1-difluoroethyl)-5-methyl[1,2,4]triazolo[1,5-a] pyrimidine (5a) or 7-Chloro-2-(trifluoromethyl)-5-methyl[1,2,4]triazolo[1,5-a] pyrimidine (5b), which were then reacted with individual amines to yield the final products (Scheme S1). All final compounds were >95% pure based on LC/MS. Detailed chemistry methods and compound experimental details are described in supplemental methods.
Supplementary Material
Acknowledgments
The authors thank Dr. Sergio Wittlin, Swiss Tropical and Public Health Institute, for contributing P. falciparum blood stage assays on multiple strains. This work was supported by funds from the United States National Institutes of Health grant, R01AI103947 (to MAP and PKR) and from Medicines for Malaria Venture (MMV). MAP acknowledges the support of the Welch Foundation (I-1257). MAP holds the Beatrice and Miguel Elias Distinguished Chair in Biomedical Science and the Carolyn R. Bacon Professorship in Medical Science and Education. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Footnotes
Author Contributions.
MAP and SAC wrote the paper and contributed to overall study design and data interpretation; MAP and FEM designed, performed and or analyzed studies on enzyme inhibition; XD, DM, MAP and DRT performed the protein X-ray crystallography and or interpreted the data; SAC, KLW, DMS, FCKC, MC, KM designed, supervised and or analyzed ADME and PK studies; PKR, SK, MJP, DW, JNB, KM, KRR performed or supervised chemical synthesis; JW, PKR, GW, RN, RNP, JM, MBJ, SFB, IAB, MSM, MLM, KS, AMZ, CK, DL, BB designed, performed and or analyzed in vitro and in vivo parasite efficacy studies; ER and TR advised on study design related to late lead candidate assessment.
Competing interests: MAP, SAC, PKR, DW and DM hold a pending patent covering DSM421 and related compounds. DW, DM and MJP are paid consultants to MMV.
References
- 1.Miller LH, Ackerman HC, Su XZ, Wellems TE. Malaria biology and disease pathogenesis: insights for new treatments. Nat Med. 2013;19(2):156–67. doi: 10.1038/nm.3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.White NJ, Pukrittayakamee S, Hien TT, Faiz MA, Mokuolu OA, Dondorp AM. Malaria. Lancet. 2014;383(9918):723–35. doi: 10.1016/S0140-6736(13)60024-0. [DOI] [PubMed] [Google Scholar]
- 3.WHO. Fact Sheet No 94. WHO; 2015. Malaria. 10/2015 ed. [Google Scholar]
- 4.Straimer J, Gnadig NF, Witkowski B, Amaratunga C, Duru V, Ramadani AP, Dacheux M, Khim N, Zhang L, Lam S, Gregory PD, Urnov FD, Mercereau-Puijalon O, Benoit-Vical F, Fairhurst RM, Menard D, Fidock DA. Drug resistance. K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science. 2015;347(6220):428–31. doi: 10.1126/science.1260867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mok S, Ashley EA, Ferreira PE, Zhu L, Lin Z, Yeo T, Chotivanich K, Imwong M, Pukrittayakamee S, Dhorda M, Nguon C, Lim P, Amaratunga C, Suon S, Hien TT, Htut Y, Faiz MA, Onyamboko MA, Mayxay M, Newton PN, Tripura R, Woodrow CJ, Miotto O, Kwiatkowski DP, Nosten F, Day NP, Preiser PR, White NJ, Dondorp AM, Fairhurst RM, Bozdech Z. Drug resistance. Population transcriptomics of human malaria parasites reveals the mechanism of artemisinin resistance. Science. 2015;347(6220):431–5. doi: 10.1126/science.1260403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miotto O, Amato R, Ashley EA, MacInnis B, Almagro-Garcia J, Amaratunga C, Lim P, Mead D, Oyola SO, Dhorda M, Imwong M, Woodrow C, Manske M, Stalker J, Drury E, Campino S, Amenga-Etego L, Thanh TN, Tran HT, Ringwald P, Bethell D, Nosten F, Phyo AP, Pukrittayakamee S, Chotivanich K, Chuor CM, Nguon C, Suon S, Sreng S, Newton PN, Mayxay M, Khanthavong M, Hongvanthong B, Htut Y, Han KT, Kyaw MP, Faiz MA, Fanello CI, Onyamboko M, Mokuolu OA, Jacob CG, Takala-Harrison S, Plowe CV, Day NP, Dondorp AM, Spencer CC, McVean G, Fairhurst RM, White NJ, Kwiatkowski DP. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet. 2015 doi: 10.1038/ng.3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Menard D, Ariey F. Towards real-time monitoring of artemisinin resistance. Lancet Infect Dis. 2015;15(4):367–8. doi: 10.1016/S1473-3099(15)70046-0. [DOI] [PubMed] [Google Scholar]
- 8.Wells TN, Hooft van Huijsduijnen R, Van Voorhis WC. Malaria medicines: a glass half full? Nat Rev Drug Discov. 2015;14(6):424–42. doi: 10.1038/nrd4573. [DOI] [PubMed] [Google Scholar]
- 9.Burrows JN, Burlot E, Campo B, Cherbuin S, Jeanneret S, Leroy D, Spangenberg T, Waterson D, Wells TN, Willis P. Antimalarial drug discovery - the path towards eradication. Parasitology. 2014;141(1):128–39. doi: 10.1017/S0031182013000826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith PW, Diagana TT, Yeung BK. Progressing the global antimalarial portfolio: finding drugs which target multiple Plasmodium life stages. Parasitology. 2014;141(1):66–76. doi: 10.1017/S0031182013000747. [DOI] [PubMed] [Google Scholar]
- 11.Duparc S, Lanza C, Ubben D, Borghini-Fuhrer I, Kellam L. Optimal dose finding for novel antimalarial combination therapy. Trop Med Int Health. 2012;17(4):409–13. doi: 10.1111/j.1365-3156.2012.02963.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Charman SA, Arbe-Barnes S, Bathurst IC, Brun R, Campbell M, Charman WN, Chiu FC, Chollet J, Craft JC, Creek DJ, Dong Y, Matile H, Maurer M, Morizzi J, Nguyen T, Papastogiannidis P, Scheurer C, Shackleford DM, Sriraghavan K, Stingelin L, Tang Y, Urwyler H, Wang X, White KL, Wittlin S, Zhou L, Vennerstrom JL. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc Natl Acad Sci U S A. 2011;108(11):4400–5. doi: 10.1073/pnas.1015762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moehrle JJ, Duparc S, Siethoff C, van Giersbergen PL, Craft JC, Arbe-Barnes S, Charman SA, Gutierrez M, Wittlin S, Vennerstrom JL. First-in-man safety and pharmacokinetics of synthetic ozonide OZ439 demonstrates an improved exposure profile relative to other peroxide antimalarials. Br J Clin Pharmacol. 2013;75(2):524–37. doi: 10.1111/j.1365-2125.2012.04368.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leong FJ, Li R, Jain JP, Lefevre G, Magnusson B, Diagana TT, Pertel P. A first-inhuman randomized, double-blind, placebo-controlled, single- and multiple-ascending oral dose study of novel antimalarial Spiroindolone KAE609 (Cipargamin) to assess its safety, tolerability, and pharmacokinetics in healthy adult volunteers. Antimicrob Agents Chemother. 2014;58(10):6209–14. doi: 10.1128/AAC.03393-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rottmann M, McNamara C, Yeung BK, Lee MC, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, Gonzalez-Paez GE, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck HP, Brun R, Nosten F, Renia L, Dartois V, Keller TH, Fidock DA, Winzeler EA, Diagana TT. Spiroindolones, a potent compound class for the treatment of malaria. Science. 2010;329(5996):1175–80. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spillman NJ, Allen RJ, McNamara CW, Yeung BK, Winzeler EA, Diagana TT, Kirk K. Na(+) regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe. 2013;13(2):227–37. doi: 10.1016/j.chom.2012.12.006S1931-3128(13)00035-8. [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuhen KL, Chatterjee AK, Rottmann M, Gagaring K, Borboa R, Buenviaje J, Chen Z, Francek C, Wu T, Nagle A, Barnes SW, Plouffe D, Lee MC, Fidock DA, Graumans W, van de Vegte-Bolmer M, van Gemert GJ, Wirjanata G, Sebayang B, Marfurt J, Russell B, Suwanarusk R, Price RN, Nosten F, Tungtaeng A, Gettayacamin M, Sattabongkot J, Taylor J, Walker JR, Tully D, Patra KP, Flannery EL, Vinetz JM, Renia L, Sauerwein RW, Winzeler EA, Glynne RJ, Diagana TT. KAF156 is an antimalarial clinical candidate with potential for use in prophylaxis, treatment, and prevention of disease transmission. Antimicrob Agents Chemother. 2014;58(9):5060–7. doi: 10.1128/AAC.02727-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phillips MA, Lotharius J, Marsh K, White J, Dayan A, White KL, Njoroge JW, El Mazouni F, Lao Y, Kokkonda S, Tomchick DR, Deng X, Laird T, Bhatia SN, March S, Ng CL, Fidock DA, Wittlin S, Lafuente-Monasterio M, Benito FJ, Alonso LM, Martinez MS, Jimenez-Diaz MB, Bazaga SF, Angulo-Barturen I, Haselden JN, Louttit J, Cui Y, Sridhar A, Zeeman AM, Kocken C, Sauerwein R, Dechering K, Avery VM, Duffy S, Delves M, Sinden R, Ruecker A, Wickham KS, Rochford R, Gahagen J, Iyer L, Riccio E, Mirsalis J, Bathhurst I, Rueckle T, Ding X, Campo B, Leroy D, Rogers MJ, Rathod PK, Burrows JN, Charman SA. A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Sci Transl Med. 2015;7(296):296ra111. doi: 10.1126/scitranslmed.aaa6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phillips MA, Rathod PK. Plasmodium dihydroorotate dehydrogenase: a promising target for novel anti-malarial chemotherapy. Infect Disord Drug Targets. 2010;10(3):226–39. doi: 10.2174/187152610791163336. BSP/ID DT/E-Pub/-0025-10-3 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gujjar R, Marwaha A, El Mazouni F, White J, White KL, Creason S, Shackleford DM, Baldwin J, Charman WN, Buckner FS, Charman S, Rathod PK, Phillips MA. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J Med Chem. 2009;52(7):1864–72. doi: 10.1021/jm801343r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Phillips MA, Gujjar R, Malmquist NA, White J, El Mazouni F, Baldwin J, Rathod PK. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite, Plasmodium falciparum. J Med Chem. 2008;51:3649–3653. doi: 10.1021/jm8001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Coteron JM, Marco M, Esquivias J, Deng X, White KL, White J, Koltun M, El Mazouni F, Kokkonda S, Katneni K, Bhamidipati R, Shackleford DM, Angulo-Barturen I, Ferrer SB, Jimenez-Diaz MB, Gamo FJ, Goldsmith EJ, Charman WN, Bathurst I, Floyd D, Matthews D, Burrows JN, Rathod PK, Charman SA, Phillips MA. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J Med Chem. 2011;54(15):5540–5561. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gujjar R, El Mazouni F, White KL, White J, Creason S, Shackleford DM, Deng X, Charman WN, Bathurst I, Burrows J, Floyd DM, Matthews D, Buckner FS, Charman SA, Phillips MA, Rathod PK. Lead-optimization of aryl and aralkyl amine based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in mice. J Med Chem. 2011;54(11):3935–49. doi: 10.1021/jm200265b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deng X, Kokkonda S, El Mazouni F, White J, Burrows JN, Kaminsky W, Charman SA, Matthews D, Rathod PK, Phillips MA. Fluorine modulates species selectivity in the triazolopyrimidine class of Plasmodium falciparum dihydroorotate dehydrogenase inhibitors. Journal of medicinal chemistry. 2014;57(12):5381–94. doi: 10.1021/jm500481t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deng X, Gujjar R, El Mazouni F, Kaminsky W, Malmquist NA, Goldsmith EJ, Rathod PK, Phillips MA. Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J Biol Chem. 2009;284:26999–27009. doi: 10.1074/jbc.M109.028589. M109.028589 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ganesan SM, Morrisey JM, Ke H, Painter HJ, Laroiya K, Phillips MA, Rathod PK, Mather MW, Vaidya AB. Yeast dihydroorotate dehydrogenase as a new selectable marker for Plasmodium falciparum transfection. Mol Biochem Parasitol. 2011;177(1):29–34. doi: 10.1016/j.molbiopara.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanz LM, Crespo B, De-Cozar C, Ding XC, Llergo JL, Burrows JN, Garcia-Bustos JF, Gamo FJP. falciparum in vitro killing rates allow to discriminate between different antimalarial mode-of-action. PLoS One. 2012;7(2):e30949. doi: 10.1371/journal.pone.0030949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ratcliff A, Siswantoro H, Kenangalem E, Wuwung M, Brockman A, Edstein MD, Laihad F, Ebsworth EP, Anstey NM, Tjitra E, Price RN. Therapeutic response of multidrug-resistant Plasmodium falciparum and P. vivax to chloroquine and sulfadoxine-pyrimethamine in southern Papua, Indonesia. Trans R Soc Trop Med Hyg. 2007;101(4):351–9. doi: 10.1016/j.trstmh.2006.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jimenez-Diaz MB, Mulet T, Viera S, Gomez V, Garuti H, Ibanez J, Alvarez-Doval A, Shultz LD, Martinez A, Gargallo-Viola D, Angulo-Barturen I. Improved murine model of malaria using Plasmodium falciparum competent strains and non-myelodepleted NOD-scid IL2Rgammanull mice engrafted with human erythrocytes. Antimicrob Agents Chemother. 2009;53(10):4533–6. doi: 10.1128/AAC.00519-09. AAC.00519-09 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Srivastava A, Creek DJ, Evans KJ, De Souza D, Schofield L, Muller S, Barrett MP, McConville MJ, Waters AP. Host reticulocytes provide metabolic reservoirs that can be exploited by malaria parasites. PLoS Pathog. 2015;11(6):e1004882. doi: 10.1371/journal.ppat.1004882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Booker ML, Bastos CM, Kramer ML, Barker RH, jr, Skerlj R, Bir Sdhu A, Deng X, Celatka C, Cortese JF, Guerrero Bravo JE, Krespo Llado KN, Serrano AE, Angulo-Barturen I, Belén Jiménez-Díaz M, Viera S, Garuti H, Wittlin S, Papastogiannidis P, Lin J, Janse CJ, Khan SM, Duraisingh M, Coleman B, Goldsmith EJ, Phillips MA, Munoz B, Wirth DF, Klinger JD, Wiegand R, Sybertz E. Novel inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in the mouse model. J Biol Chem. 2010;285(43):33054–33064. doi: 10.1074/jbc.M110.162081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 33.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D Biol Crystallogr. 2007;63(Pt 1):32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 35.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vennerstrom JL, Arbe-Barnes S, Brun R, Charman SA, Chiu FC, Chollet J, Dong Y, Dorn A, Hunziker D, Matile H, McIntosh K, Padmanilayam M, Santo Tomas J, Scheurer C, Scorneaux B, Tang Y, Urwyler H, Wittlin S, Charman WN. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature. 2004;430(7002):900–4. doi: 10.1038/nature02779. [DOI] [PubMed] [Google Scholar]
- 37.Dembele L, Gego A, Zeeman AM, Franetich JF, Silvie O, Rametti A, Le Grand R, Dereuddre-Bosquet N, Sauerwein R, van Gemert GJ, Vaillant JC, Thomas AW, Snounou G, Kocken CH, Mazier D. Towards an in vitro model of Plasmodium hypnozoites suitable for drug discovery. PloS one. 2011;6(3):e18162. doi: 10.1371/journal.pone.0018162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zeeman AM, van Amsterdam SM, McNamara CW, Voorberg-van der Wel A, Klooster EJ, van den Berg A, Remarque EJ, Plouffe DM, van Gemert GJ, Luty A, Sauerwein R, Gagaring K, Borboa R, Chen Z, Kuhen K, Glynne RJ, Chatterjee AK, Nagle A, Roland J, Winzeler EA, Leroy D, Campo B, Diagana TT, Yeung BK, Thomas AW, Kocken CH. KAI407, a potent non-8-aminoquinoline compound that kills Plasmodium cynomolgi early dormant liver stage parasites in vitro. Antimicrob Agents Chemother. 2014;58(3):1586–95. doi: 10.1128/AAC.01927-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marfurt J, Chalfein F, Prayoga P, Wabiser F, Wirjanata G, Sebayang B, Piera KA, Wittlin S, Haynes RK, Mohrle JJ, Anstey NM, Kenangalem E, Price RN. Comparative ex vivo activity of novel endoperoxides in multidrug-resistant plasmodium falciparum and P. vivax. Antimicrob Agents Chemother. 2012;56(10):5258–63. doi: 10.1128/AAC.00283-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wirjanata G, Sebayang BF, Chalfein F, Prayoga, Handayuni I, Trianty L, Kenangalem E, Noviyanti R, Campo B, Poespoprodjo JR, Mohrle JJ, Price RN, Marfurt J. Potent Ex Vivo Activity of Naphthoquine and Methylene Blue against Drug-Resistant Clinical Isolates of Plasmodium falciparum and Plasmodium vivax. Antimicrob Agents Chemother. 2015;59(10):6117–24. doi: 10.1128/AAC.00874-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jimenez-Diaz MB, Mulet T, Gomez V, Viera S, Alvarez A, Garuti H, Vazquez Y, Fernandez A, Ibanez J, Jimenez M, Gargallo-Viola D, Angulo-Barturen I. Quantitative measurement of Plasmodium-infected erythrocytes in murine models of malaria by flow cytometry using bidimensional assessment of SYTO-16 fluorescence. Cytometry A. 2009;75(3):225–35. doi: 10.1002/cyto.a.20647. [DOI] [PubMed] [Google Scholar]
- 42.Baragana B, Hallyburton I, Lee MC, Norcross NR, Grimaldi R, Otto TD, Proto WR, Blagborough AM, Meister S, Wirjanata G, Ruecker A, Upton LM, Abraham TS, Almeida MJ, Pradhan A, Porzelle A, Martinez MS, Bolscher JM, Woodland A, Norval S, Zuccotto F, Thomas J, Simeons F, Stojanovski L, Osuna-Cabello M, Brock PM, Churcher TS, Sala KA, Zakutansky SE, Jimenez-Diaz MB, Sanz LM, Riley J, Basak R, Campbell M, Avery VM, Sauerwein RW, Dechering KJ, Noviyanti R, Campo B, Frearson JA, Angulo-Barturen I, Ferrer-Bazaga S, Gamo FJ, Wyatt PG, Leroy D, Siegl P, Delves MJ, Kyle DE, Wittlin S, Marfurt J, Price RN, Sinden RE, Winzeler EA, Charman SA, Bebrevska L, Gray DW, Campbell S, Fairlamb AH, Willis PA, Rayner JC, Fidock DA, Read KD, Gilbert IH. A novel multiple-stage antimalarial agent that inhibits protein synthesis. Nature. 2015;522(7556):315–20. doi: 10.1038/nature14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kokkonda S, Deng X, White KL, Coteron JM, Marco M, de Las Heras L, White J, El Mazouni F, Tomchick DR, Manjalanagara K, Rudra KR, Chen G, Morizzi J, Ryan E, Kaminsky W, Leroy D, Martinez-Martinez MS, Jimenez Diaz MB, Bazaga SF, Angulo-Barturen I, Waterson D, Burrows JN, Matthews D, Charman SA, Phillips MA, Rathod PK. Tetrahydro-2-naphthyl and 2-indanyl triazolopyrimidines targeting Plasmodium falciparum dihydroorotate dehydrogenase display potent and selective antimalarial activity. J Med Chem. 2016;59(11):5416–31. doi: 10.1021/acs.jmedchem.6b00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.