

Abstract
Selective inhibitors of protein lysine methyltransferases, including SET domain-containing protein 8 (SETD8), are highly desired, as only a fraction of these enzymes are associated with high-quality inhibitors. From our previously discovered SETD8 inhibitor, we developed a more potent analog and solved a cocrystal structure, which is the first crystal structure of SETD8 in complex with a small-molecule inhibitor. This cocrystal structure allowed the design of a covalent inhibitor of SETD8 (MS453), which specifically modifies a cysteine residue near the inhibitor binding site, has an IC50 value of 795 nM, reacts with SETD8 with near-quantitative yield, and is selective for SETD8 against 28 other methyltransferases. We also solved the crystal structure of the covalent inhibitor in complex with SETD8. This work provides atomic-level perspective on inhibition of SETD8 by small molecules and will help identify high-quality chemical probes of SETD8.
Graphical Abstract
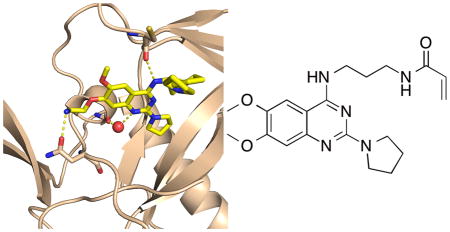
Introduction
Protein lysine methyltransferases (PKMT) control epigenetic gene regulation and carcinogenesis, and high quality small-molecule inhibitors of these enzymes are greatly desired as research tools and therapeutics.1–3 PKMTs utilize the cofactor S-adenosyl-L-methionine (SAM) to transfer methyl groups to lysine residues on target proteins, resulting in lysine mono-, di-, or trimethylation. Many different proteins are substrates of PKMTs, including histones. The differential methylation state of histone lysine residues provides a recognition platform for methyl-lysine binding proteins and is a fundamental part of the epigenetic system of gene regulation.4–6 Consequently, mutations or aberrant activity of PKMTs can dramatically change gene expression and cause cancer.7–10 High quality inhibitors of multiple PKMTs have been reported and some are now in clinical development.3, 11–14 However, selective inhibitors have not been identified for most PKMTs, despite significant effort.3, 11
Over 50 PKMTs are known, and all except DOT1L share the SET domain, named after Drosophila Su(var)3–9 (suppressor of variegation 3–9), E(z) (enhancer of zeste), and Trithorax.15 The SET domain binds the cofactor SAM and the peptide substrate, and catalyzes the methyl transfer reaction. The crystal structures of a number of PKMTs either in apo state or in complex with a substrate, the cofactor, and/or an inhibitor have been solved.16
SETD8 (also known as SET8, PR-SET7, or KMT5a) is the only methyltransferase known to catalyze monomethylation of histone H4 lysine 20 (H4K20). H4K20 monomethylation (H4K20me) has been shown to regulate the DNA damage response, the cell cycle, and chromatin condensation.4, 17, 18 The role of H4K20me in transcription is unclear; it has been associated with both activation and repression.17 SETD8 also catalyzes methylation of the cancer-related proteins proliferating cell nuclear antigen (PCNA) and p53.19 Methylation of p53 by SETD8 suppresses p53-mediated transcriptional activation.20 SETD8 knockdown enhances the proapoptotic function of p53. Methylation of PCNA by SETD8 stabilizes PCNA and likely contributes to its oncogenic functions.21 All of these observations strongly suggest that SETD8 inhibitors could be valuable anticancer agents.
Several SETD8 inhibitors have been reported. The natural product nauhoic acid A was reported as a SAM-competitive inhibitor of SETD8 with 6.5 μM potency in vitro.22 A set of quinone compounds were reported as cell-active inhibitors of SETD8.23 The quinones covalently modified one or more residues on SETD8. Given that the quinone moiety is redox-active and that quinones are known to be promiscuous assay-interference compounds,24 these compounds may have off-target activities. We previously reported UNC0379 (1), a substrate-competitive SETD8 inhibitor with an in vitro IC50 value of 7.3 μM.25 1 features the same quinazoline core found in high potency inhibitors of the lysine methyltransferases G9a and GLP.26–28 1 is selective for SETD8 over 15 other methyltransferases and its activity was confirmed in biophysical assays. Here we report the development of a more potent SETD8 inhibitor, the crystal structure of SETD8 in complex with this more potent inhibitor, and design, synthesis and biological evaluation of a covalent inhibitor, which specifically modifies a cysteine residue near the inhibitor binding site of SETD8.
Results and Discussion
The SETD8 inhibitor 1 is a quinazoline with amino substituents at the 2- and 4-positions and methoxy groups at the 6- and 7-positions, same as G9a/GLP inhibitors like BIX-01294.27, 29 Attempts to improve the potency of 1 by replacing either the 2- or 4-substituents were unsuccessful.25, 30 For G9a/GLP inhibitors, addition of an alkylamino group to the quinazoline 7-position greatly improved potency.26–28, 31, 32 In the G9a–inhibitor cocrystal structures, the alkylamino group occupies the lysine-binding channel of G9a.27, 28 We reasoned that since 1 and the G9a/GLP inhibitors share the quinazoline scaffold, it is possible that they have a similar binding mode to their cognate enzymes, and therefore, addition of an alkylamino group to the 7-position of 1 would improve potency for SETD8. This was indeed the case, and an ethyl group gave the greatest improvement in potency. The resulting compound, MS2177 (2) had an in vitro IC50 of 1.9 μM (σ = 1.05 μM, n = 4) in a scintillation proximity assay (Figure 1). Binding of 2 to SETD8 was confirmed by isothermal titration calorimetry (ITC), having a KD of 1.3 μM (Figure 2), close to its potency in the biochemical assay. The KD of 2 (1.3 μM) was much lower than that of 1 (18 μM).25 The ITC study also revealed that the binding of 2 is enthalpy driven. Competition assays were performed to determine the mechanism of action (MOA) of 2; 2 was competitive with the H4 peptide but not with SAM (Figure 3), the same MOA as 1. Synthesis of 2 was accomplished by alkylation of a substituted quinazoline at the 7-position (Scheme 1), following previously reported methods.30
Figure 1.

(Left) Competitive inhibitors of SETD8. (Right) Concentration–response curve of SETD8 inhibition by 2 in a scintillation proximity assay. Points from four separate experiments are shown, along with fitted curve and 95% confidence interval. Hillslope = 1.1
Figure 2.

ITC of 2 with SETD8.
Figure 3.

Mechanism of action of 2. IC50 values increased with the concentration of the H4 peptide but not with the cofactor SAM.
Scheme 1.

Synthesis of compounds 2–4.
Reagents and conditions: a) BocNHCH2CH2Br, K2CO3, acetone; b) HCl(aq), MeOH; c) 1,3-Diaminopropane, DIPEA, THF; d) Pyrrolidine, iPrOH, microwave; e) Acrylic acid, HOAt, EDCI, DIPEA, DMF; f) CH3NHCH2CH2CH2NHBoc, THF, DIPEA; g) Pyrrolidine, iPrOH, microwave; h) HCl (aq), MeOH; i) Acrylic acid, HOAt, EDCI, DIPEA, DMF.
We solved the crystal structure of 2 in complex with SETD8 (residues 234–380) (PDB code: 5T5G, Figure 4C). A C343S mutation was included in the SETD8 crystallization construct to increase stability and solubility of the protein. To our surprise, 2 binds to SETD8 in a different way than the quinazoline G9a inhibitors bind to G9a. The binding site for 2 overlaps that observed for the peptide substrate in the SETD8-SAH (S-adenosyl-L-homocysteine, the cofactor product)-peptide cocrystal structure (PDB code: 1ZKK) and the quinazoline rings fill the space that in the SETD8-SAH complex is occupied by the amino group of the substrate lysine side chain (Figure 4B).33 The pyrrolidinopentyl group is disordered in the crystal structure, and two possible conformations of this moiety are shown. The secondary amino group at the 4-position forms a hydrogen bond with the oxygen of C311 (Figure 4D). The crystal structure reveals why the addition of the alkylamino group increased potency: The amino group of the ethylamine tail forms a hydrogen bond with N339 on the surface of SETD8. In the crystal structure of SETD8 bound to SAM (PDB code: 4IJ8), this residue forms a hydrogen bond with the primary amino group of the amino acid portion of SAM. The quinazoline N1 nitrogen forms a solvent-mediated hydrogen bond with the oxygen of G335 and I338. Interestingly, binding of the inhibitor coincides with significant conformational changes of Y375 and Y314, resulting in the opening of an interior hydrophobic pocket. This expanded pocket accommodates the 2-pyrrolidine. The movement of these two tyrosine residues to form this hydrophobic space is illustrated in figure 4D. The volume of this hydrophobic space is filled near-optimally by the 2-pyrrolidine group, which explains why either smaller or larger rings at the quinazoline 2-position abrogated inhibition.25 From the crystal structure, it appears that 2 is a bisubstrate inhibitor, as it interacts with the substrate binding site and the ethylamino side chain extends into the cofactor binding site. However, MOA revealed that 2 is competitive with the peptide substrate and non-competitive with the cofactor SAM, suggesting that the ethylamino side chain, which somewhat overlaps with SAH in the crystal structure, might not prevent binding of SAM in vitro. The crystal structure also reveals a number of possible ligand-protein interactions that could be exploited for designing more potent analogs.
Figure 4.

The X-ray crystal structure of 2 in complex with SETD8. (A) Compound 2 bound to SETD8. (B) The crystal structure of SETD8 (surface shown) in complex with 2 aligned with the SETD8-SAH-peptide structure (PDB code: 1ZKK). SAH is shown in purple, peptide substrate is turquoise, and 2 is yellow. (C) Polar interactions between 2 and SETD8 are shown. (D) The crystal structure of 2 in complex with SETD8 (light brown) aligned with 4IJ8 (orange). Rotation of the two tyrosine residues (Y375 and Y314) is illustrated.
The disordered 4-(pentylpyrrolidine) group of 2 appears close to the C311 residue on the protein’s surface. Surface cysteine residues can be exploited for targeted development of covalent inhibitors.34 The addition of an electrophilic group to the quinazoline 4-position produced a covalent inhibitor, MS453 (3) (Figure 5). We selected acrylamide as the electrophile because of its convenient synthesis and inertness towards glutathione.35 We examined the linker length and found that a propyl chain was ideal for linking the acrylamide to the 4-amino group. In the scintillation proximity assay, with a one-hour preincubation time, 3 has an IC50 value of 6.90 μM. With a five-hour preincubation time, the IC50 value of 3 was improved to 804 nM (σ = 285 nM, n = 2) (Figure 5). The lower IC50 value with increased preincubation time is consistent with covalent inhibition. To confirm that 3 specifically reacts with C311, 3 was incubated with purified SETD8 containing the full SET domain for 3 hours and then analyzed by mass spectrometry (Figure 6 and Supplemental Figure 1). We observed an adduct with mass equal to SETD8 plus 3, with no detectable unmodified SETD8, demonstrating that 3 efficiently forms a single-modified covalent adduct (Figure 6). Importantly, 3 did not form an adduct with the C311S mutant of SETD8 (Figure 6). On the other hand, 3 formed a single-modified adduct with the C343S mutant of SETD8 (Supplemental Figure 1), confirming that it does not react with the distal C343 residue, which is not reachable by the acrylamide group of 3. Taken together, these results have conclusively demonstrated that 3 covalently modifies C311 specifically, thus validating our structure-based design of this covalent inhibitor. Furthermore, the covalent modification of SETD8 by 3 was not affected by the presence of 5 mM glutathione (GSH), which mimics cellular conditions (Supplemental Figure 1). In addition, 3 was incubated with other protein lysine methyltransferases including PRC2, SMYD2, and SMYD3, and no labeling of these proteins was observed by MS analysis (Supplemental Figures 2,3, and 4), suggesting that the covalent modification by 3 is specific to SETD8. Synthesis of 3 was accomplished by disubstitution of 2,4-dichloroquinazoline with 1,3-diaminopropane then pyrrolidine, followed by coupling acrylic acid to the free primary amine. (Scheme 1)
Figure 5.

(Left) Structure and IC50 values for 3 and 4. IC50 values are the result from a scintillation proximity assay following preincubation with SETD8 for the specified duration. (Right) Concentration response curves for 3 following incubation with SETD8 for 1 or 5 hours. Hillslope = 1.2 for both incubation times.
Figure 6.

Mass Spectrometry results of covalent modification of SETD8 by 3. (A) WT SETD8. (B) WT SETD8 + 3. (C) SETD8 C311S mutant. (D) SETD8 C311S + 3.
We also obtained a crystal structure of the SETD8-3 adduct (PDB code: 5TH7), which reveals a homodimer of adducts where each covalently modified C311 residue presents the inhibitor to the quinazoline binding site of the complementary adduct (Figure 7). The inhibitor forms some of the same hydrogen bonds that were observed in the crystal structure of SETD8 in complex with 2, including the hydrogen bond between the 4-position secondary amino group and the carbonyl oxygen of C311 on the peptide backbone. We synthesized a negative control compound, 4 (Scheme 1), in which the secondary amino group at the 4-position is methylated, and cannot form the hydrogen bond with C311. This compound was indeed inactive in the SETD8 biochemical assay (Figure 5) and did not covalently modify SETD8 (Supplemental Figure 1). 3 also forms a solvent-mediated hydrogen bonds between the quinazoline N1 nitrogen and carbonyl groups on the protein backbone. In addition, the 2-pyrrolidine also occupies the induced-fit interior hydrophobic pocket seen with 2. As 3 is excluded from the active site of the SETD8 molecule to which it is attached, it is possible that the enzyme-inhibitor adduct is unstable in solution. This possibility is consistent with the relatively low in vitro potency of 3 after prolonged preincubation.
Figure 7.

The crystal structure of 3 covalently bound to SETD8.
The C311A mutation significantly reduces SETD8 activity, but does not completely deactivate SETD8.36 We measured residual enzymatic activity of the 3-SETD8 adduct. SETD8 was incubated with 3 for sufficient time to allow complete formation of the adduct, as observed by MS analysis. The resulting adduct was purified by gel filtration, and both a monomer and a dimer were observed. The purified monomer and dimer, which interconvert after a short time, were tested in enzymatic assays, where they retained about 10% of the enzymatic activity of wild-type SETD8 (Supplemental Figure 5). The Kcat/Km of C311A is about 8% of wildtype SETD8,36 which is similar to the SETD8-3 adduct. This residual potency explains why the IC50 of 3 is still not very low even though it quantitatively reacts with SETD8. The crystal structure of SETD8-3 may represent the SETD8 dimer in solution induced by covalent modification.
We also assessed selectivity of 3 for SETD8 over a broad range of methyltransferases. As illustrated in Figure 8, 3 is selective for SETD8 against 28 other methyltransferases. However, we did not observe significant reduction in the H4K20me mark when HeLa cells were treated with 3 at up to 10μM. The lack of cellular activity is likely resulted from its poor membrane permeability (4.4 nm/s) and high efflux ratio (137). Future work will attempt to establish cellular activity by improving membrane permeability and reducing efflux.
Figure 8.

Selectivity of 3 against 29 methyltransferase enzymes. % Activity is shown.
Conclusion
By adding an alkylamino group to our original SETD8 screening hit, an addition that improved potency for our G9a/GLP inhibitors, we identified an improved SETD8 inhibitor, 2. We solved the cocrystal structure of 2 in complex with SETD8, the first crystal structure of SETD8 in complex with a small-molecule inhibitor. Interestingly, the structure revealed that the binding mode of 2 is dissimilar from other PKMT inhibitors, and that inhibitor binding is accompanied by conformational adjustments of two tyrosine residues to form a hydrophobic pocket, which is occupied by the 2-pyrrolidine. Based on the cocrystal structure, we designed and synthesized 3, an analog of 1 containing an electrophilic acrylamide group, to specifically react with a surface exposed cysteine residue. Indeed, 3 specifically modifies the cysteine residue in the active site nearly quantitatively. It has an IC50 value of 795 nM following extended preincubation and is selective for SETD8 over 30 other methyltransferases. The crystal structure of 3 in complex with SETD8 revealed that the inhibitor was flipped out from the active site and occupied the active site of the other subunit of a SETD8 homodimer. The cocrystal structures provide valuable new insight into the inhibition of PKMTs by multiple inhibitory modes and may guide the development of improved SETD8 inhibitors.
Experimental
Chemistry General Procedures
HPLC spectra for all compounds were acquired using an Agilent 1200 Series system with DAD detector. Analytical HPLC chromatography was performed on a 2.1×150 mm Zorbax 300SB-C18 5 μm column with water containing 0.1% formic acid as solvent A and acetonitrile containing 0.1% formic acid as solvent B at a flow rate of 0.4 mL/min. The gradient program was as follows: 1% B (0–1 min), 1–99% B (1–4 min), and 99% B (4–8 min). High resolution mass spectra (HRMS) data were acquired in positive ion mode using an Agilent G1969A API-TOF with an electrospray ionization (ESI) source. Flash column chromatography was performed on a Teledyne ISCO CombiFlash Rf system equipped with a variable wavelength UV detector and a fraction collector using RediSep Rf normal phase silica columns. Microwave reactions were performed using a Discover SP CEM. Nuclear Magnetic Resonance (NMR) spectra were acquired on a Bruker DRX-600 spectrometer with 600 MHz for proton (1H NMR) and 150 MHz for carbon (13C NMR); chemical shifts are reported in ppm (δ). Preparative HPLC was performed on Agilent Prep 1200 series with UV detector set to 254 nm. Samples were injected onto a Phenomenex Luna 75 × 30 mm, 5 μm, C18 column at room temperature. The flow rate was 30 mL/min. A linear gradient was used with 10% (or 50%) of MeOH (A) in H2O (with 0.1 % TFA) (B) to 100% of MeOH (A). HPLC was used to establish the purity of target compounds. All final compounds had > 95% purity using the HPLC methods described above.
Generic Procedure 1
The dichloroquinazoline (1.0 eq), diisopropylethylamine (2.0 eq) and R1-amine freebase (1.5 eq) were stirred in THF (10 mL per mmol) for 24 hours. The reaction mixture was taken up in EtOAc, washed with sat. aq. NaHCO3 then brine, and then concentrated. The intermediate was purified by column chromatography. The main UV-active peak was concentrated and taken up in pyrrolidine (5.0 eq) and iPrOH (3 mL per mmol). This mixture was heated to 160 °C in a microwave reactor for 30 minutes. The resulting solution was concentrated under vacuum and the product was purified by column chromatography.
7-(2-Aminoethoxy)-6-methoxy-2-(pyrrolidin-1-yl)-N-(5-(pyrrolidin-1-yl)pentyl)quinazolin-4-amine. (2)
6-Methoxy-2-(pyrrolidin-1-yl)-4-((5-(pyrrolidin-1-yl)pentyl)amino)quinazolin-7-ol was synthesized according to the procedures reported previously.30 To a suspension of 6-methoxy-2-(pyrrolidin-1-yl)-4-((5-(pyrrolidin-1-yl)pentyl)amino)quinazolin-7-ol (189 mg, 0.47 mmol), K2CO3 (173 mg, 1.3 mmol) and acetone (10 mL) was added 2-(Boc-amino)ethyl bromide (commercially available, 75 mg, 0.33 mmol). The resulting suspension was stirred for 3 days at 70 °C until LCMS showed that most of the starting material had disappeared. After purification by reverse phase ISCO, tert-butyl (2-((6-methoxy-2-(pyrrolidin-1-yl)-4-((5-(pyrrolidin-1-yl)pentyl)amino)quinazolin-7-yl)oxy)ethyl)carbamate was obtained as a TFA salt. To the tert-butyl (2-((6-methoxy-2-(pyrrolidin-1-yl)-4-((5-(pyrrolidin-1-yl)pentyl)amino)quinazolin-7-yl)oxy)ethyl)carbamate and methanol (2.0 mL) was added hydrochloric acid (37%, 0.2 mL). The resulting solution was stirred for 6 hours at 60 °C until LCMS showed that most of the starting material had disappeared. After removal of the solvent in vacuo, the residue was purified by HPLC to give the title compound 2 as a TFA salt. The product was further mixed with excess solid NaHCO3 and purified by ISOC using alumina basic column to give the title compound 2 as free amine, yellow semi-solid (104 mg, yield 40% for two steps). 1H NMR (600 MHz, CD3OD): δ 7.32 (s, 1H), 6.92 (s, 1H), 4.07 (t, J = 5.0 Hz, 2H), 3.89 (s, 3H), 3.61 – 3.55 (m, 6H), 3.05 (s, 2H), 2.50 (br.s, 4H), 2.45 – 2.40 (m, 2H), 1.96 (br.s, 4H), 1.83 – 1.67 (m, 6H), 1.62 – 1.54 (m, 2H), 1.44 – 1.37 (m, 2H). 13C NMR (151 MHz, CD3OD) δ 160.6, 158.6, 154.8, 148.6, 146.6, 105.9, 104.8, 104.2, 71.2, 68.5, 57.5, 56.8, 55.0 (two carbons), 47.7, 41.9, 41.7, 30.2, 29.4, 26.5, 26.3, 24.1 (two carbons), 22.4. HRMS calcd for [C24H38N6O2 + H]+, 443.3129; found, 443.3136 [M + H]+.
N-(3-((6,7-Dimethoxy-2-(pyrrolidin-1-yl)quinazolin-4-yl)amino) propyl)acrylamide. (3)
N1-(6,7-Dimethoxy-2-(pyrrolidin-1-yl)quinazolin-4-yl)propane-1,3-diamine was synthesized according to generic procedure 1 from 2,4-dichloro-6,7-dimethoxylquinazoline, 1,3-diaminopropane, and pyrrolidine.25 (HRMS calcd for C17H25N5O2 + H, 332.2081; found, 332.2071 [M+H]+) To the solution of N1-(6,7-dimethoxy-2-(pyrrolidin-1-yl)quinazolin-4-yl)propane-1,3-diamine (268 mg, 0.81 mmol) in DMF (4 mL) was added acrylic acid (67 μL, 0.97 mmol). To this solution were added HOAt (110 mg, 0.81 mmol), EDCI (186 mg, 0.97 mmol) and N,N-diisopropylethylamine (494 μL, 2.8 mmol) respectively at 0 °C. The resulting solution was stirred at room temperature overnight. TLC indicated the completion of the reaction. After removal of the solvent in vacuo, the residue was redissolved in dichloromethane, washed with brine. The organic layer was dried with sodium sulfate, concentrated and purified by ISCO to give the title compound 3, white solid (234 mg, yield 51% for three steps). 1H NMR (600 MHz, CD3OD): δ 7.40 (s, 1H), 6.95 (s, 1H), 6.24 – 6.14 (m, 2H), 5.63 (dd, J = 9.4, 2.6 Hz, 1H), 3.88 (s, 3H), 3.84 (s, 3H), 3.63 (t, J = 6.9 Hz, 2H), 3.52 (s, 4H), 3.34 (t, J = 6.8 Hz, 2H), 2.18 – 1.95 (m, 4H), 1.92 (p, J = 6.9 Hz, 2H). 13C NMR (151 MHz, CD3OD) δ 168.2, 160.2, 156.9, 151.3, 148.6, 136.8, 132.0, 126.8, 104.7, 103.4, 99.5, 56.9, 56.8, 47.4 (two carbons), 40.4, 38.1, 29.7, 26.7, 25.6. HRMS calcd for [C20H27N5O3 + H]+, 386.2187; found, 386.2175 [M + H]+.
N-(3-((6,7-Dimethoxy-2-(pyrrolidin-1-yl)quinazolin-4-yl)(methyl)amino)propyl)acrylamide (4)
To the solution of tert-butyl (3-((6,7-dimethoxy-2-(pyrrolidin-1-yl)quinazolin-4-yl)(methyl)amino)propyl)carbamate (5) (267 mg, 0.6 mmol) and methanol (5 mL) was added hydrochloric acid (37%, 0.5 mL). The resulting solution was stirred for 6 hours at 60 °C until LCMS showed that most of the starting material had disappeared. After removal of the solvent in vacuo, the residue obtained (251 mg, 0.60 mmol) and acrylic acid (85 μL, 1.2 mmol) were dissolved in DMF (5 mL). To this solution were added HATU (456 mg, 1.2 mmol) and N,N-diisopropylethylamine (523 μL, 3.0 mmol) respectively at 0 °C. The resulting solution was stirred at room temperature overnight. TLC indicated the completion of the reaction. After removal of the solvent in vacuo, the residue was redissolved in dichloromethane, washed with brine. The organic layer was dried, concentrated and purified by ISCO to give the title compound 5, yellow solid (144 mg, yield 60% over two steps). 1H NMR (600 MHz, CD3OD): δ 7.41 (s, 1H), 7.11 (s, 1H), 6.26 – 6.16 (m, 2H), 5.66 (dd, J = 8.5, 3.4 Hz, 1H), 3.95 (s, 3H), 3.90 (s, 3H), 3.89 – 3.86 (m, 2H), 3.63 (br.s, 4H), 3.54 (s, 3H), 3.37 (t, J = 6.7 Hz, 2H), 2.14 – 1.99 (m, 6H). 13C NMR (151 MHz, CD3OD) δ 168.1, 162.2, 156.7, 150.4, 147.1, 140.3, 132.0, 126.8, 109.1, 104.0, 100.1, 56.9, 56.8, 52.5, 47.7 (two carbons), 41.0, 38.0, 27.7, 26.1. HRMS calcd for [C21H29N5O3 + H]+, 400.2343; found, 400.2352 [M + H]+.
tert-Butyl (3-((6,7-dimethoxy-2-(pyrrolidin-1-yl)quinazolin-4-yl)(methyl)amino)propyl)carbamate (5)
tert-Butyl (3-((6,7-dimethoxy-2-(pyrrolidin-1-yl)quinazolin-4-yl)(methyl)amino)propyl)carbamate was prepared according generic procedure 1 from 2,4-dichloro-6,7-dimethoxylquinazoline, tert-butyl (3-(methylamino)propyl)carbamate (commercially available) and pyrrolidine. Yield: 48% over two steps. 1H NMR (600 MHz, CDCl3): δ 7.13 (s, 1H), 6.96 (s, 1H), 4.01 – 3.94 (s, 3H), 3.94 – 3.83 (s, 3H), 3.68 (m, 6H), 3.27 (m, 2H), 3.20 (m, 2H), 2.02 – 1.87 (m, 6H), 1.44 (s, 9H). HRMS calcd for [C23H35N5O4 + H]+, 446.2762; found, 446.3092 [M + H]+
SETD8 purification and radioactivity assays
SETD8 catalytic domain (232–393) for enzyme assay was expressed in E. Coli and purified as reported.25 SETD8 mutations of C311S and C324S were purified with same method. SETD8 enzyme assay was developed as reported.25 To prepare the proteins for cocrystallization, a construct of human SETD8 covering residues 234–380 with a C343S mutation was subcloned into a modified pET28-MHL vector with an N-terminal His tag. The protein was overexpressed in E.coli BL21 (DE3) V2R-pRARE in Terrific Broth medium in the presence of 50 μg/mL of kanamycin and chloramphenicol. Cells were grown at 37°C to an OD600 of 1.5, induced by isopropyl-1-thio-D-galactopyranoside (IPTG, final concentration 1 mM) and incubated overnight at 15°C. The cell pellets were frozen in liquid nitrogen and stored at −80°C. For purification, the cell paste was thawed and resuspended in lysis buffer with 1mM phenylmethyl sulfonyl fluoride (PMSF). SETD8 (234–380) was purified by Ni-NTA column (Qiagen) and processed by TEV protease to remove the His tag. The protein was further purified by a Superdex-75 gel filtraton column pre-equilibrated with gel filtration buffer, 50 mM Tris-HCl, 100 mM NaCl, 5 mM DTT, pH 8.0. MOA experiments were performed as previously described.25
Methyltransferase selectivity assays
Selectivity assays were performed as previously described.37
Crystallization
Purified protein SETD8 (234–380) with a C343S mutation was concentrated to 16 mg/mL.
Complex of SETD8 and compound 2. A 25 mM stock solution of 2 was prepared from the TFA salt of 2 and the pH adjusted to 7 using 10 M NaOH. The protein was mixed with a 5 fold excess of compound 2 stock solution and incubated on ice for 1 hour. The reservoir solution for crystallization contained 30% w/v PEG 2000 monomethyl ether, 0.1 M potassium thiocyanate, 0.1 M BIS-TRIS, pH 6.5.
Complex of SETD8 and compound 3: Protein was mixed with a 5 fold excess of a 25 mM inhibitor stock and incubated on ice for more than 5 hours until protein was completely covalently modified as confirmed by Mass spectrometry. The reservoir solution for crystallization contained 20% w/v PEG 3350, 0.2 M NH4F, 0.1 M sodium cacodylate, pH 5.2. The samples were spun at 13,000 rpm (Eppendorf 5415R) for 15 minutes to remove insoluble components. Crystals were grown at 18 °C using the sitting drop vapor diffusion method and briefly soaked in a 4:1 mixture of reservoir solution and glycerol before freezing by immersion in liquid nitrogen
Structure determination
Diffraction data were collected under cooling at beam line 19ID of the Advanced Photon Source, processed with XDS or HKL3000,38, 39 and merged with POINTLESS/AIMLESS.40 Structure solution was by molecular replacement with PHASER software.41 Geometry restraints were calculated with PRODRG,42 for later stages of refinement, with GRADE,43 which uses MOGUL.44 The structure of the complex with 2 was solved using data from an isomorphous crystal and coordinates from PDB entry 4IJ8. The protein model was automatically rebuilt with ARP/wARP. 45, 46 Restrained coordinate and temperature factor refinement was performed with REFMAC.47 The structure of the complex with 3 was solved using preliminary coordinates from the compound 2 complex model. REFMAC and AUTOBUSTER were used for restrained refinement.48 COOT and MOLPROBITY were used for interactive rebuilding and geometry validation, respectively.49–51 Model deposition in the PDB and summaries were prepared with PDB_EXTRACT, PHENIX and CCP4 programs and the IOTBX library.52–54
Mass spectrometry
To investigate the covalent modification of SETD8 by compounds, 7 μM SETD8 catalytic domain (or mutations C311S, C343S) was incubated with 20–200 μM compounds in water for over 3 hours. The samples were analysed by an Agilent LC/MSD Time-Of-Flight (TOF) mass spectrometer (Agilent Technologies, Santa Clara, CA, USA) equipped with an electrospray ion source. The molecular weights of the proteins without compounds were also tested as control (SETD8 wildtype: 18759 Da; C311S with His tag: 21580 Da; C343S: 18743 Da).
Isothermal titration calorimetry
The SETD8 catalytic domain was diluted into a buffer containing 50 mM Tris–HCl buffer (pH 7.5), 100 mM NaCl at 20 mM. Compound 2 was prepared in water at 25 mM first with adjusted pH ~ 7.0. The compound was also diluted into same buffer at 0.5 mM. ITC measurements were carried out in a VP-ITC instrument at 25 °C. Binding constants were calculated by fitting the data using the ITC data analysis module of Origin 7.0 (OriginLab Corp.).
Associated Content
Supplemental Figures, 1H and 13C spectra of 2 and 3, and data table for crystallography.
Supplementary Material
Acknowledgments
The research described here was supported by the grant R01GM103893 (to J.J.) from the U.S. National Institutes of Health. The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck & Co., Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, São Paulo Research Foundation-FAPESP, Takeda, and the Wellcome Trust. K.V.B. was supported by a postdoctoral fellowship from the American Cancer Society (PF-14-021-01-CDD). We thank Dr. Raymond C. Trievel of the University of Michigan for providing us with the SETD8 vector. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Abbreviations
- PKMT
Protein lysine methyltransferase
- SAM
S-Adenosyl-methionine
- DIPEA
Diisopropylethylamine
- THF
Tetrahydrofuran
- HOAt
1-Hydroxy-7-azabenzotriazole
- EDCI
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
- DMF
Dimethylformamide
- ITC
Isothermal titration calorimetry
- MOA
Mechanism of action
Footnotes
PDB ID Codes: The structure of SETD8 in complex with 2 has been deposited under ID: 5T5G. The structure of SETD8 in complex with 3 has been deposited under ID: 5TH7. Authors will release the atomic coordinates and experimental data upon article publication.
References
- 1.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discovery. 2009;8:724–732. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- 2.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: A new frontier for drug discovery. Nat Rev Drug Discovery. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 3.Kaniskan HU, Konze KD, Jin J. Selective inhibitors of protein methyltransferases. J Med Chem. 2015;58:1596–1629. doi: 10.1021/jm501234a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callen E, Celeste A, Pagani M, Opravil S, De La Rosa-Velazquez IA, Espejo A, Bedford MT, Nussenzweig A, Busslinger M, Jenuwein T. A chromatin-wide transition to h4k20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008;22:2048–2061. doi: 10.1101/gad.476008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greer EL, Shi Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012;13:343–357. doi: 10.1038/nrg3173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yap KL, Zhou MM. Structure and mechanisms of lysine methylation recognition by the chromodomain in gene transcription. Biochemistry. 2011;50:1966–1980. doi: 10.1021/bi101885m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krivtsov AV, Armstrong SA. Mll translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 9.Simon JA, Lange CA. Roles of the ezh2 histone methyltransferase in cancer epigenetics. Mutat Res. 2008;647:21–29. doi: 10.1016/j.mrfmmm.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 10.Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, Shah S, Zhu H, Kimbara M, Shashkin P, Charlot JF, Tcherpakov M, Corbett R, Tam A, Varhol R, Smailus D, Moksa M, Zhao Y, Delaney A, Qian H, Birol I, Schein J, Moore R, Holt R, Horsman DE, Connors JM, Jones S, Aparicio S, Hirst M, Gascoyne RD, Marra MA. Somatic mutations altering ezh2 (tyr641) in follicular and diffuse large b-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaniskan HU, Jin J. Chemical probes of histone lysine methyltransferases. ACS Chem Biol. 2015;10:40–50. doi: 10.1021/cb500785t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitchell LH, Boriack-Sjodin PA, Smith S, Thomenius M, Rioux N, Munchhof M, Mills JE, Klaus C, Totman J, Riera TV, Raimondi A, Jacques SL, West K, Foley M, Waters NJ, Kuntz KW, Wigle TJ, Scott MP, Copeland RA, Smith JJ, Chesworth R. Novel oxindole sulfonamides and sulfamides: Epz031686, the first orally bioavailable small molecule smyd3 inhibitor. ACS Med Chem Lett. 2016;7:134–138. doi: 10.1021/acsmedchemlett.5b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sweis RF, Wang Z, Algire M, Arrowsmith CH, Brown PJ, Chiang GG, Guo J, Jakob CG, Kennedy S, Li FL, Maag D, Shaw B, Soni NB, Vedadi M, Pappano WN. Discovery of a-893, a new cell-active benzoxazinone inhibitor of lysine methyltransferase smyd2. ACS Med Chem Lett. 2015;6:695–700. doi: 10.1021/acsmedchemlett.5b00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campbell JE, Kuntz KW, Knutson SK, Warholic NM, Keilhack H, Wigle TJ, Raimondi A, Klaus CR, Rioux N, Yokoi A, Kawano S, Minoshima Y, Choi HW, Scott MP, Waters NJ, Smith JJ, Chesworth R, Moyer MP, Copeland RA. Epz011989, a potent, orally-available ezh2 inhibitor with robust in vivo activity. ACS Med Chem Lett. 2015;6:491–495. doi: 10.1021/acsmedchemlett.5b00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schapira M. Structural chemistry of human set domain protein methyltransferases. Curr Chem Genomics. 2011;5:85–94. doi: 10.2174/1875397301005010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campagna-Slater V, Mok MW, Nguyen KT, Feher M, Najmanovich R, Schapira M. Structural chemistry of the histone methyltransferases cofactor binding site. J Chem Inf Model. 2011;51:612–623. doi: 10.1021/ci100479z. [DOI] [PubMed] [Google Scholar]
- 17.Beck DB, Oda H, Shen SS, Reinberg D. Pr-set7 and h4k20me1: At the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 2012;26:325–337. doi: 10.1101/gad.177444.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu S, Wang W, Kong X, Congdon LM, Yokomori K, Kirschner MW, Rice JC. Dynamic regulation of the pr-set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev. 2010;24:2531–2542. doi: 10.1101/gad.1984210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamamoto R, Saloura V, Nakamura Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat Rev Cancer. 2015;15:110–124. doi: 10.1038/nrc3884. [DOI] [PubMed] [Google Scholar]
- 20.Shi X, Kachirskaia I, Yamaguchi H, West LE, Wen H, Wang EW, Dutta S, Appella E, Gozani O. Modulation of p53 function by set8-mediated methylation at lysine 382. Mol Cell. 2007;27:636–646. doi: 10.1016/j.molcel.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takawa M, Cho HS, Hayami S, Toyokawa G, Kogure M, Yamane Y, Iwai Y, Maejima K, Ueda K, Masuda A, Dohmae N, Field HI, Tsunoda T, Kobayashi T, Akasu T, Sugiyama M, Ohnuma S, Atomi Y, Ponder BA, Nakamura Y, Hamamoto R. Histone lysine methyltransferase setd8 promotes carcinogenesis by deregulating pcna expression. Cancer Res. 2012;72:3217–3227. doi: 10.1158/0008-5472.CAN-11-3701. [DOI] [PubMed] [Google Scholar]
- 22.Williams DE, Dalisay DS, Li F, Amphlett J, Maneerat W, Chavez MA, Wang YA, Matainaho T, Yu W, Brown PJ, Arrowsmith CH, Vedadi M, Andersen RJ. Nahuoic acid a produced by a streptomyces sp. Isolated from a marine sediment is a selective sam-competitive inhibitor of the histone methyltransferase setd8. Org Lett. 2013;15:414–417. doi: 10.1021/ol303416k. [DOI] [PubMed] [Google Scholar]
- 23.Blum G, Ibanez G, Rao X, Shum D, Radu C, Djaballah H, Rice JC, Luo M. Small-molecule inhibitors of setd8 with cellular activity. ACS Chem Biol. 2014;9:2471–2478. doi: 10.1021/cb500515r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (pains) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 25.Ma A, Yu W, Li F, Bleich RM, Herold JM, Butler KV, Norris JL, Korboukh V, Tripathy A, Janzen WP, Arrowsmith CH, Frye SV, Vedadi M, Brown PJ, Jin J. Discovery of a selective, substrate-competitive inhibitor of the lysine methyltransferase setd8. J Med Chem. 2014;57:6822–6833. doi: 10.1021/jm500871s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu F, Barsyte-Lovejoy D, Li FL, Xiong Y, Korboukh V, Huang XP, Allali-Hassani A, Janzen WP, Roth BL, Frye SV, Arrowsmith CH, Brown PJ, Vedadi M, Jin J. Discovery of an in vivo chemical probe of the lysine methyltransferases g9a and glp. J Med Chem. 2013;56:8931–8942. doi: 10.1021/jm401480r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu F, Chen X, Allali-Hassani A, Quinn AM, Wasney GA, Dong A, Barsyte D, Kozieradzki I, Senisterra G, Chau I, Siarheyeva A, Kireev DB, Jadhav A, Herold JM, Frye SV, Arrowsmith CH, Brown PJ, Simeonov A, Vedadi M, Jin J. Discovery of a 2,4-diamino-7-aminoalkoxyquinazoline as a potent and selective inhibitor of histone lysine methyltransferase g9a. J Med Chem. 2009;52:7950–7953. doi: 10.1021/jm901543m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, Dimaggio PA, Wasney GA, Siarheyeva A, Dong A, Tempel W, Wang SC, Chen X, Chau I, Mangano TJ, Huang XP, Simpson CD, Pattenden SG, Norris JL, Kireev DB, Tripathy A, Edwards A, Roth BL, Janzen WP, Garcia BA, Petronis A, Ellis J, Brown PJ, Frye SV, Arrowsmith CH, Jin J. A chemical probe selectively inhibits g9a and glp methyltransferase activity in cells. Nat Chem Biol. 2011;7:566–574. doi: 10.1038/nchembio.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kubicek S, O’Sullivan RJ, August EM, Hickey ER, Zhang Q, Teodoro ML, Rea S, Mechtler K, Kowalski JA, Homon CA, Kelly TA, Jenuwein T. Reversal of h3k9me2 by a small-molecule inhibitor for the g9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 30.Ma A, Yu W, Xiong Y, Butler KV, Brown PJ, Jin J. Structure-activity relationship studies of setd8 inhibitors. MedChemComm. 2014;5:1892–1898. doi: 10.1039/C4MD00317A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu F, Chen X, Allali-Hassani A, Quinn AM, Wigle TJ, Wasney GA, Dong AP, Senisterra G, Chau I, Siarheyeva A, Norris JL, Kireev DB, Jadhav A, Herold JM, Janzen WP, Arrowsmith CH, Frye SV, Brown PJ, Simeonov A, Vedadi M, Jin JA. Protein lysine methyltransferase g9a inhibitors: Design, synthesis, and structure activity relationships of 2,4-diamino-7-aminoalkoxy-quinazolines. J Med Chem. 2010;53:5844–5857. doi: 10.1021/jm100478y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu F, Barsyte-Lovejoy D, Allali-Hassani A, He YL, Herold JM, Chen X, Yates CM, Frye SV, Brown PJ, Huang J, Vedadi M, Arrowsmith CH, Jin J. Optimization of cellular activity of g9a inhibitors 7-aminoalkoxy-quinazolines. J Med Chem. 2011;54:6139–6150. doi: 10.1021/jm200903z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Couture JF, Dirk LMA, Brunzelle JS, Houtz RL, Trievel RC. Structural origins for the product specificity of set domain protein methyltransferases. P Natl Acad Sci U S A. 2008;105:20659–20664. doi: 10.1073/pnas.0806712105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu QS, Sabnis Y, Zhao Z, Zhang TH, Buhrlage SJ, Jones LH, Gray NS. Developing irreversible inhibitors of the protein kinase cysteinome. Chem Biol. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flanagan ME, Abramite JA, Anderson DP, Aulabaugh A, Dahal UP, Gilbert AM, Li C, Montgomery J, Oppenheimer SR, Ryder T, Schuff BP, Uccello DP, Walker GS, Wu Y, Brown MF, Chen JM, Hayward MM, Noe MC, Obach RS, Philippe L, Shanmugasundaram V, Shapiro MJ, Starr J, Stroh J, Che Y. Chemical and computational methods for the characterization of covalent reactive groups for the prospective design of irreversible inhibitors. J Med Chem. 2014;57:10072–10079. doi: 10.1021/jm501412a. [DOI] [PubMed] [Google Scholar]
- 36.Couture JF, Collazo E, Brunzelle JS, Trievel RC. Structural and functional analysis of set8, a histone h4 lys-20 methyltransferase. Genes Dev. 2005;19:1455–1465. doi: 10.1101/gad.1318405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eram MS, Shen YD, Szewczyk MM, Wu H, Senisterra G, Li FL, Butler KV, Kaniskan HU, Speed BA, dela Sena C, Dong AP, Zeng H, Schapira M, Brown PJ, Arrowsmith CH, Barsyte-Lovejoy D, Liu J, Vedadi M, Jin J. A potent, selective, and cell-active inhibitor of human type i protein arginine methyltransferases. ACS Chem Biol. 2016;11:772–781. doi: 10.1021/acschembio.5b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. Hkl-3000: The integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr. 2006;62:859–866. doi: 10.1107/S0907444906019949. [DOI] [PubMed] [Google Scholar]
- 40.Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr. 2013;69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schuttelkopf AW, van Aalten DM. Prodrg: A tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 43.Smart OS, Womack TO, Sharff A, Flensburg C, Keller P, Paciorek W, Vonrhein C, Bricogne G. Grade version 1.2.9. Cambridge, united kingdom: global phasing ltd; 2014. [Google Scholar]
- 44.Bruno IJ, Cole JC, Kessler M, Luo J, Motherwell WD, Purkis LH, Smith BR, Taylor R, Cooper RI, Harris SE, Orpen AG. Retrieval of crystallographically-derived molecular geometry information. J Chem Inf Comput Sci. 2004;44:2133–2144. doi: 10.1021/ci049780b. [DOI] [PubMed] [Google Scholar]
- 45.Langer G, Cohen SX, Lamzin VS, Perrakis A. Automated macromolecular model building for x-ray crystallography using arp/warp version 7. Nat Protoc. 2008;3:1171–1179. doi: 10.1038/nprot.2008.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Perrakis A, Sixma TK, Wilson KS, Lamzin VS. Warp: Improvement and extension of crystallographic phases by weighted averaging of multiple-refined dummy atomic models. Acta Crystallogr D Biol Crystallogr. 1997;53:448–455. doi: 10.1107/S0907444997005696. [DOI] [PubMed] [Google Scholar]
- 47.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. Refmac5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bricogne G, BE, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, Womack TO. Buster version 2.10.2. Cambridge, united kingdom: Global phasing ltd; 2016. [Google Scholar]
- 49.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. Molprobity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. Phenix: A comprehensive python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang H, Guranovic V, Dutta S, Feng Z, Berman HM, Westbrook JD. Automated and accurate deposition of structures solved by x-ray diffraction to the protein data bank. Acta Crystallogr D Biol Crystallogr. 2004;60:1833–1839. doi: 10.1107/S0907444904019419. [DOI] [PubMed] [Google Scholar]
- 53.Gildea RJ, Bourhis LJ, Dolomanov OV, Grosse-Kunstleve RW, Puschmann H, Adams PD, Howard JA. Iotbx.Cif: A comprehensive cif toolbox. J Appl Crystallogr. 2011;44:1259–1263. doi: 10.1107/S0021889811041161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the ccp4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.