

Abstract
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are two apparently distinct neurodegenerative diseases, the former characterized by selective loss of motor neurons in the brain and spinal cord and the latter characterized by selective atrophy of frontal and temporal lobes. Over the years, however, growing evidence from clinical, pathological and genetic findings has suggested that ALS and FTD belong to the same clinic-pathological spectrum disorder. This concept has been further supported by the identification of the most common genetic cause for both diseases, an aberrantly expanded hexanucleotide repeat GGGGCC sequence located in a non-coding region of the gene C9orf72. Three hypotheses have been proposed to explain how this repeats expansion causes diseases: 1) C9orf72 haploinsufficiency-expanded repeats interfere with transcription or translation of the gene, leading to decreased expression of C9orf72 protein; 2) RNA gain of function-RNA foci formed by sense and antisense transcripts of expanded repeats interact and sequester essential RNA binding proteins, causing neurotoxicity; 3) Repeat associated non-ATG initiated (RAN) translation of GGGGCC repeat expansion-RAN translation of expanded sense and antisense repeats produces potential toxic dipeptide repeat protein (DPR). In this review, we assess current evidence supporting or arguing against each proposed mechanism in C9 ALS/FTD disease pathogenesis. Additionally, controversial findings are also discussed. Lastly, we discuss the possibility that the three pathogenic mechanisms are not mutually exclusive and all three might be involved in disease.
Keywords: C9orf72, ALS, FTD, repeat expansion disease, RNA foci, dipeptide repeat protein
1. ALS and FTD: One Clinicopathological Spectrum Disorder
Amyotrophic lateral sclerosis (ALS), a fatal neurodegenerative disease, causes the degeneration of upper motor neurons in the cortex and lower motor neurons in the medulla and spinal cord. Although few juvenile cases have been reported, ALS has generally an adult onset and progresses rather rapidly in most patients (few years from onset to end-stage). There are two forms of ALS: sporadic (sALS) and familiar (fALS). About 5% of ALS cases are inherited, in which there is a clear family history of disease [1]. In familial ALS cases, an autosomal dominant hereditary pattern is usually observed. Since the identification of the first causative mutation in SOD1 in 1993, more than 30 genes have been identified to cause familial ALS worldwide [2]. These genetic mutations have been found to induce disturbances in protein homeostasis, RNA metabolism and/or cause abnormalities in axonal structures and functions [2, 3]. ALS involves also non-cell autonomous components, meaning cell types other than motor neurons are also affected by the disease pathogenic mechanisms and contribute to the demise of the motor neurons.
Frontal temporal dementia (FTD) is second most common form of dementia after Alzheimer’s disease in the under 65-age group. As its name implies, FTD is characterized by atrophy of the frontal and temporal lobes. Symptoms include changes in personality and behaviors, disturbances in language, and cognitive deficits, which worsen progressively [4]. Like ALS, there are two types of FTD: familial (fFTD) and sporadic (sFTD). FTD has a strong genetic component with up to 50% of the cases being hereditary. Clinical subtypes include behavioral variant FTD (bvFTD), progressive non-fluent aphasia (PNFA) and progressive aphasia semantic dementia (SD) [5]. The average survival is 8–10 years after disease onset [6, 7]. Selective serotonin reuptake inhibitors and serotonin norepinephrine reuptake inhibitors have been shown to have positive effects only in some bvFTD patients [5]. Over the past twenty years, our understanding of the genetic causes and molecular pathogenic mechanisms has been growing at a surprisingly fast pace. Neuropathological classification of FTD has also been transformed. Mutations in MAPT (microtubule-associated protein tau), GRN (progranulin), VCP (AAA-type ATPase valosin-containing protein), CHMP2B (charged multiversicular protein 2B), the FET family (DNA/RNA binding proteins), including FUS, EWS, and TAF15, have been identified to cause FTD [8]. A variety of mechanisms have been implicated in FTD including impairments in neurotrophic support, autophagy and proteasome pathways, RNA metabolisms and neurofilament structures [7].
Although ALS and FTD present with distinct clinical features, strong associations have been found between them. ALS and FTD often occur in the same family and same patients sometimes. Indeed, there are overlaps in clinical presentations, genetic causes, pathological findings and pathogenic mechanisms. Both diseases are age dependent, and symptoms usually start in fifth decade of life. Both diseases relentlessly progress over time, and eventually lead to death. More than 50% of sALS patients and 60% of fALS patients have cognitive deficits; and around 15% of sALS patients are actually diagnosed with concomitant FTD [9, 10]. Similarly, 12.5–14% of FTD patients have concomitant motor neuron disease (MND), and another 27–36% of FTD patients have clinical evidence of motor system dysfunction [11, 12]. Disease progression seems to be more rapid in FTD patients with concomitant motor neuron disease, as these patients were reported to have an earlier onset age and a shorter mean survival [13]. ALS and FTD are not only heterogeneous in clinical presentations; they are highly heterogeneous in genetic causes. A number of genetic mutations have been found to cause both ALS and FTD, namely mutations in FUS, TDP-43, SQSTM1, UBQLN2, CHCHD10, VCP and C9orf72 [14–18]
2. Discovery of GGGGCC/CCCCGG Hexanucleotide Repeat Expansions in C9orf72
Multiple groups conducted extensive sequencing in families with hereditary ALS/FTD with unknown mutations, and linked the diseases to a 3.7 Mb region in the short arm of chromosome 9. Later the region was narrowed down to an 80kb area by genome wide association studies (GWAS) [4]. Deep sequencing of exons and exon-intron boundaries failed to identify the mutation. However, two independent groups pinpointed the nature and location of the mutation by carefully examining non-coding regions and manually aligning individual sequences [14, 16].
The mutation turned out to be a vastly expanded GGGGCC repeat (CCCCGG in the antisense direction) located in a non-coding region of the C9orf72 gene. Since C9orf72 has three different transcript variants, the mutation resides in different regions in different variants: Promoter region for variant 2 and first intron for variant 1 and 3.
C9orf72 is located on chromosome 9, in the 72nd open reading frame and consists of 12 exons. C9orf72 (also referred here as C9) is a highly conserved gene with three transcript variants, encoding two protein isoforms. Variants 2 and 3 encode C9orf72 isoform a, the longer version with 481 amino acids, and variant 1 encodes C9orf72 isoform b, the shorter version containing 222 amino acids. Both isoforms are still of unknown biological function (Figure 1).
Figure 1. The three C9orf72 transcripts and GGGGCC hexanuleotide repeat expansion.
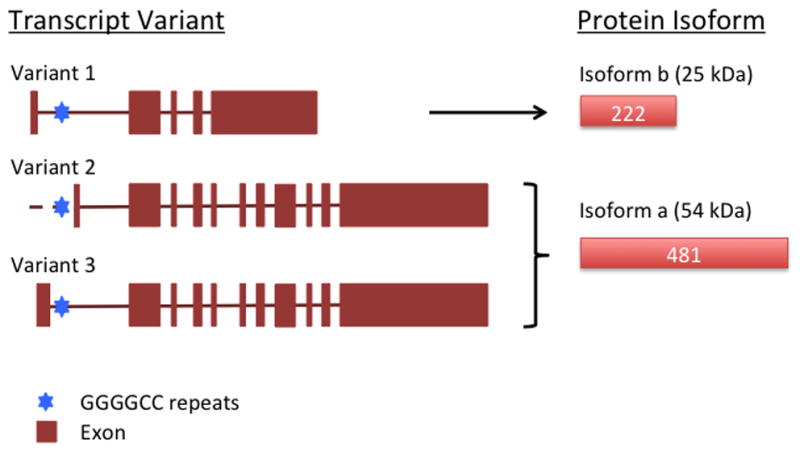
Three C9 transcript variants and two C9 protein isoforms are listed and named as in PubMed. GGGGCC hexanucleotide repeat (blue hexagon) and exons (brown block) are indicated. Predicted molecular weight for each protein isoform is listed.
The most common GGGGCC repeat size in C9orf72 was reported to be 2 in normal, non-diseased individuals [19]. However, variable ranges of repeat number have been reported for controls and patients: fewer than 14 repeats in controls as 20–22 repeats were associated with FTD [20]; fewer than 23 repeats in controls [15]; 800–4400 repeats in patients [19]; 0–30 repeats in controls and more than 500 repeats in patients [21]. And finally a more recent study employing a southern blot based semi-automated quantification showed that most control carry fewer than 10 repeats, while patients carry 300–3800 repeats [22].
Similar to what was observed in other repeat expansion diseases, the repeat size was also found to be associated with sample collection time [22, 23]. C9orf72 repeat expansion sizes have been widely reported to vary in different tissue types and cell types [19, 22–24], indicating the expansion of repeats might be caused by some cell division-independent mechanism in addition to somatic instability. The repeat size variation among different tissues within a same carrier was larger than that between different carriers, suggesting that intrinsic properties of specific tissue might affect the stability of the expanded repeats. In some cases, the difference in size between peripheral blood and CNS tissues was several thousand [21, 25]. It was also shown that repeat sizes smaller than 16 were somatically stable, implying higher repeats were prone to expansion [25]. In addition to the repeat size variation within the same patient, there are also technical challenges and limited DNA available, thus it remains to be answered what is the minimal size for pathogenesis and what is the normal range.
3. Disease Mechanisms
Discovery of the expanded GGGGCC repeat in a non-coding region of the C9orf72 gene, the most common genetic cause for both ALS and FTD, brought researchers’ attention to a group of diseases called repeat expansion disease. Repeat expansions were known to cause a number of hereditary diseases, such as fragile X syndrome (FXS), fragile X tremor ataxia syndrome (FXTAS), myotonic dystrophy (DM), Huntington’s disease (HD), and spinocerebellar ataxia 1 (SCA1) [26]. Three common pathogenic mechanisms underly repeat expansion diseases:
Loss-of-function, as seen in FXS, where expanded CGG repeats in FMR1 causes hypermethylation of the promoter region and subsequent loss of FMRP protein, which is an essential RNA binding protein that regulates local protein synthesis leading to abnormal synaptic function [27];
RNA mediated gain-of-function, as seen in DM1, where expanded CTG repeats in the gene DMPK form nuclear RNA foci in muscle cells, sequester essential RNA binding proteins and causes misregulated RNA splicing [28];
Protein mediated gain-of-function, as seen in HD, where expanded CAG repeats in the coding region of the huntingtin gene are translated into poly-glutamine (poly-Q) tract, which leads to proteins aggregation and disruption of the normal functions of huntingtin [29, 30]. Protein mediated gain-of-function can also arise from repeats located in a non-coding region, (e.g. intron, 3′UTR, 5′ UTR) where expanded repeats are translated into toxic protein through a novel translational mechanism called Repeat-Associated Non-ATG initiated (RAN) translation, which circumvents the requirement for a canonical start codon [31]. RAN translation was first reported in myotonic dystrophy type 1 (DM1) and in spinocerebellar ataxia type 8 (SCA8), where expanded CAG•CTG repeats in non-coding regions were translated into poly-Gln and poly-Ala [31].
The characteristics of C9orf72 mutation suggest all three potential mechanisms. Here, findings in favor of or against each potential mechanism are discussed.
3.1 C9orf72 Haploinsufficiency
C9orf72 expression levels were thoroughly assessed in repeat expansion carriers and control population. C9orf72 transcripts were found to be significantly reduced in lymphobast cells, frontal cortex, spinal cord, cerebellum and induced pluripotent stem cells (iPSCs) derived neurons from patients carrying the repeat expansion, and variant 2 was found to be mostly affected [16, 17, 32–36]. In a separate study, variant 2 was identified to be the primary transcript for C9orf72 [37], indicating loss of variant 2 would contribute greatly to loss of overall C9orf72 protein levels. Furthermore, higher variant 1 expression levels in frontal cortex and cerebellum were found to be survival favorable [32].
Using a customized antibody recognizing both C9orf72 protein isoforms, decreased levels of the long isoform were found in patients frontal cortex samples but not in cerebellum samples compared to control samples [38]. In another publication, two customized antibodies against C9orf72 short and long isoforms respectively were developed, and researchers found that C9orf72 long isoform was down regulated in temporal and frontal lobes and no changes were detected in cerebellum, motor cortex and lumber spinal cord (low signal) while the short isoform was up regulated in C9-ALS patients [39].
However, a case report on a homozygous repeats expansion patient argued against the C9 loss-of-function (LOF) hypothesis. If C9 LOF were the major disease mechanism, more severe clinical course and neuropathological abnormality would be expected in a homozygous mutation carrier. In this case, homozygosity of C9 repeat expansion did cause a further reduction of C9orf72 transcript levels, but clinical phenotype and neuropathological features were not unusual when compared to heterozygous carrier [40].
Do decreased C9orf72 expression levels contribute to disease pathogenesis? To answer this question, C9 knockdown or C9 knockout models have been developed. Deletion of alf-1, the c. elegans ortholog of C9orf72, led to age-dependent progressive motor deficits similar to those seen in TDP-43 and FUS models and increased sensitivity to stress-induced paralysis, and GABAergic motor neuron was primarily affected [41]. Similarly, knockdown of C9orf72 in zebrafish caused motor deficits and impaired axonal projection [35]. Although c. elegans and zebrafish C9 knockdown models suggested loss of C9orf72 was sufficient to cause disease phenotype, this hypothesis remained to be further tested since neither C9orf72 homologs of c. elegans nor zebrafish shared high homology with human C9orf72. Later, a series of C9 knockdown and knockout mouse models were generated, where mouse ortholog of C9orf72 is highly similar to human C9orf72. Mouse C9orf72 was either conditionally removed in neuronal and glial cells using Nestin-Cre system or constitutively removed in all cell types using gene targeting or Crispr/Cas9 systems. Heterozygous and homozygous mice were analyzed to understand the functional consequences of decreased C9 protein level (resembling most of the patients) vs. full knockout of C9 protein. All together, mice with decreased or abrogated C9orf72 level failed to show ALS/FTD-like features including motor deficits, behavioral abnormality, shortened life span, pathological hallmarks of ALS/FTD (TDP-43-positive inclusions, and p62-positive inclusions) [42–47]. To be noted, two C9 knockout mouse models showed mild motor deficits after 40 weeks [45] and decreased survival [47], but these phenotypes were suggested to be caused by other underlying conditions rather than ALS/FTD. Using a different approach, C9 level was also successfully reduced when antisense oligonucleotides (ASOs) against C9orf72 were injected into adult (8 week old) mouse intracerebral ventricles, yet no ALS/FTD phenotypes were observed [43]. Taken together, absence of ALS/FTD-like phenotypes in C9orf72 knockdown or knockout mouse models indicates C9orf72 haploinsufficiency is not the primary cause of ALS/FTD.
The above C9 knockout mouse models also provide insights into physiological functions of C9orf72, albeit they failed to display ALS/FTD related disease phenotypes. Upon close examination, these mice showed enlarged spleens and lymph nodes and they also showed microglia activation with abnormal lysosomal activity and altered cytokine production, suggesting that C9orf72 protein might be involved in regulating immune homeostasis, and autophagy/lysosome pathway [44–48]. Surprisingly, neoplastic events were also reported seen in one C9 knockout mouse model, which might be triggered by defects in immune systems [47].
Understanding the physiological function of C9orf72 is not only important for defining the role of C9 haploinsufficiency but also crucial for developing therapeutic strategies to mitigate the repeat expansion toxcity. In the hope that knowing where C9orf72 protein is located would provide some insight into its functionality, several groups made attempts to characterize C9orf72 protein localization in cells, with variable localization profiles reported: nuclear [14], primarily cytosolic [49], nuclear and cytoplasmic vesicles [50], nuclear, cytosolic and synaptic vesicles [51], and nuclear, P-bodies and stress granules [52]. Different cells and tissues were evaluated in these studies. Additionally, various commercial antibodies were used, which are still poorly characterized. These differences most likely led to different observations. Thus, a more specific antibody is crucial for an accurate characterization. Recent studies using customized antibodies provided more information on this matter. Using two newly developed antibodies against C9 short or long isoforms, researchers demonstrated that C9 long isoform showed diffuse cytoplasmic staining pattern and C9 short isoform was found to localize to nuclear membrane [39]. The long isoform of the C9orf72 protein also showed distinct speckle-pattern staining only in cytoplasm of cerebellar Purkinje neurons-the less affected neuronal cell type in diseases, not in spinal motor neurons-the primarily affected cell type [39]. The implications of these interesting findings remain to be explored.
Computational algorithms based on sequence and structural similarity predicts that C9orf72 is a guanine exchange factor with a DENN (Differentially Expressed in Normal and Neoplastic) like domain [53]. Consistent with the computational prediction of its function, C9orf72 protein was found to colocalize and interact with Rab proteins and was involved in endosomal trafficking in cells [50]. The role of C9orf72 protein in autophagy regulation was further established in a series of follow up studies: C9orf72 protein was implied in regulation of autophagy initiation by functioning as Rab1a effector and interacting with ULK1 initiation complex [54]; C9orf72 protein interacted with SMCR8 and WDR41 to participate in autophagy initiation [46, 55, 56].
Taken together, C9orf72 haploinsufficiency might not be driving disease pathogenesis, but it is highly likely involved in it. It would be of great interest to further characterize the function of C9orf72 protein and investigate whether C9orf72 is beneficial in stabilizing expanded repeats.
3.2 RNA Gain of Function
Nuclear and cytoplasmic RNA foci formed by sense or anti-sense transcript of the GGGGCC repeat expansion were found in C9 patient lymphoblast cells and fibroblast cells. In the CNS, RNA foci were found in glia cells and neurons (more frequently than glia cells) of frontal cortex, cerebellum, hippocampus and spinal cord; the RNA foci most likely consisted of expanded GGGGCC sequence without flanking regions upstream or downstream of the repeat [14, 16, 33, 43, 57–59]. Besides forming RNA foci, the GGGGCC repeat expansion was also revealed to form highly stable G-quadruplex structure and R-loops in vitro [34, 60–62]. G-quadruplex consists of planar stacks formed by four guanine residues that are stabilized by Hoogsteen hydrogen bonding (Figure 2A). R-loops are hybrids of DNA and RNA formed by nascent RNA strand and template DNA strand during transcription [62] (Figure 2B).
Figure 2. GGGGCC/CCCCGG repeat expansions form G quadruplex and R-loops and interact with RBPs.

(A) Four guanine residues (pink ball) form a G-quartet through eight pairs of hoogsteen hydrogen bonding. G-quartets stack on top of each other, forming helical G-quadruplex structure. (B) R-loops are formed newly transcribed RNA transcript and unwound DNA template during transcription. (C) A summary of RBPs reported to be sequestered in RNA foci or to interact with GGGGCC or CCCCGG RNA repeats.
Characterization of functional consequences of expanded repeats in C9 patients and their iPSC-derived neurons (iPSNs) supported the toxic role of RNA foci. Sequestration of ADARB2 protein by RNA foci and altered gene expression profiles were detected in C9 patient iPSNs and motor cortex; C9-iPSNs’ susceptibility to glutamate toxicity can be alleviated by ASOs against C9orf72 accompanied by decreased RNA foci number and continuing presence of RAN-DPR (RAN translation product poly-GlyPro level was evaluated), suggesting that RNA foci were the primary toxic species [33]. So far, a list of RNA binding proteins (RBPs) was identified to co-localize with sense and anti-sense RNA foci in patient tissues, including ALYREF, ADARB2, hnRNP-A1, hnRNP-H/F, hnRNP-K, hnRNP-U, nucleolin, Pur-α, SC35, SRSF2 [33, 34, 63–66] (summarized in Figure 2C). Another group examined the role of G-quadruplex formed by GGGGCC repeats in disease pathogenesis. They found that GGGGCC repeat expansion formed G-quadruplex and R-loops in vitro, which led to aberrant interaction with nucleolar proteins, resulting in nucleolar stress, reminiscent of traits seen in C9 patients and their cells, as well as loss of full length transcripts and the accumulation of abortive transcripts due to disrupted RNA polymerase processivity [34]. To be noted, a different group reported contrary findings that the intron containing expanded repeats were transcribed and spliced out correctly in C9 patients [36]. Overt cellular toxicity, altered RNA processing and nucleolar stress caused by expressing GGGGCC repeats in vitro and in vivo lend support to the notion that C9 repeat expansion causes disease phenotype through a RNA gain-of-function pathway. A higher number of RNA foci in the frontal cortex positively correlated with a younger disease onset age in C9 FTD patients, adding further support to RNA toxicity in disease pathogenesis [59].
To explore whether these abundant RNA foci seen in patients would cause neurodegeneration, transgenic Drosophila and cell models were developed. When 30 GGGGCC repeats were cloned into a 5′ UTR, toxicity was observed in vitro in N2a cells, as well as decreased locomotor activity and disrupted eye morphology when expressed in motor neurons and retinal cells, respectively, in aged flies [67]. In the same study, the purine-rich element binding protein A, Pur-α was found to interact with GGGGCC repeats and overexpression of Pur-α rescued the degenerative phenotypes. Later, using the same fly model, a different group showed expanded GGGGCC transcripts and RNA foci interacted with and sequestered the Ran GTPase-activating protein RanGAP, leading to reduced nuclear import in vitro and in vivo [68]. Moreover, expression of 38 or longer GGGGCC repeats caused apoptosis in neuronal cell lines and fish embryos via sequestration of hnRNP-H by RNA foci [64]. Besides hnRNP-H, several other translation regulators like eIF2a, FUS and ILF3 were also shown to be sequestered into RNA foci leading to translation arrest in cells expressing 31 repeats of GGGGCC [69]. Most of the RBPs sequestered in RNA foci would fall into two categories: RNA processing regulators and translation regulators. Gene profiling of C9 patient cells and tissues revealed that splicing error rate was associated with disease severity, providing support for RBP sequestration by RNA foci as a potential disease mechanism [70]. Nevertheless, direct evidence showing functional consequences resulted from sequestration of these critical proteins are somewhat lacking. Interestingly, GGGGCC repeat RNA particles were also found in neurites of iPSNs from C9 patients, and these repeat RNA particles correlated with neuritic arborization defect seen in primary neurons expressing expanded repeats and Drosophila carrying 48 GGGGCC repeats (no RAN translation product was detected) [71].
And it was suggested that the toxicity might be due to dysfunctional granule transport caused by GGGGCC repeat expansion [71]. Contradictory results, however, were also reported in several studies where in vitro and in vivo models were used, suggesting RNA foci alone were not sufficient to cause neurodegeneration. In a transgenic fly model carrying up to 288 pure GGGGCC repeats (RNA only without production of RAN translated proteins), neither neurodegeneration nor decreased viability was observed [72]. More recently, in another fly model expressing up to 160 GGGGCC repeats with intronic and exonic flanking sequences from human C9orf72, which closely resembled the mutation in C9 patients, displayed no toxicity when expressed in a variety of tissues. More importantly, the average number of sense RNA foci per cell was a lot higher than that in C9 patient iPSCs derived neurons [36], suggesting that it was unlikely for RNA foci to be toxic. Similar claims have been made from several mouse models. In a mouse model expressing 80 repeats of GGGGCC in a 5′-UTR region, the repeat size was proven to remain stable through multiple generations and no RAN translation product was detected, however, researchers did not find motor/behavior phenotypes, or neuronal cell loss [73]. Moreover, two independently generated BAC transgenic mouse models carrying either the full human C9orf72 gene with expanded GGGGCC repeats or first six exons of the human C9orf72 gene with expanded GGGGCC repeats. These two mouse models both showed formation of abundant sense and anti-sense RNA foci as well as Gly-Pro dipeptides similar to pathological findings seen in C9 patients, but the mice lived a normal life span without developing any cognitive, behavioral or motor defects [74, 75]. In contrast, another two BAC transgenic mouse models carrying human C9orf72 gene together with the expanded GGGGCC repeats showed ALS/FTD like phenotypes, including cognitive deficits, motor deficits and decreased survival [48, 76]. Similarly, these mice also showed pathological and molecular signatures of C9 ALS/FTD, including sense and anti-sense RNA foci, and RNA translation products (poly-GP, poly-GA and poly-GR inclusions). Moreover, RAN translation was dependent on length of repeats, expression levels, age, and disease course [48, 76]. However, it remains possible that the observed toxicity could come from RAN translation product.
3.3 RAN Translated Dipeptide Repeat Protein Toxicity
RAN translation of expanded repeats has become an emerging mechanism underlying nucleotide repeat disorders since the initial discovery in DM1 and SCA8. Efforts were also made to determine whether these unexpected protein products resulted from RAN translation would contribute to disease. In fragile X-associated tremor ataxia syndrome (FXTAS), CGG repeats in the 5′ UTR of the fragile X mental retardation gene (FMR1) were shown to undergo RAN translation and produce poly-Ala protein and poly-Gly protein from different reading frames, the latter of which caused neural toxicity in Drosophila model [77] and was shown to trigger impairment in ubiquitin proteasome system (UPS) [78], supporting the role of novel protein products from RAN translation in disease pathogenesis. Recent progress on how RAN translation occurred in FXTAS provided mechanistic support. It was shown that RAN translation of CGG repeats was 30–40% efficient compared to canonical translation, and it utilized the canonical translation machinery and was dependent on repeat length and surrounding sequence [79].
If both sense and anti-sense transcripts of expanded GGGGCC repeats underwent RAN translation, five different dipeptide proteins (DPRs), namely poly-Gly-Ala(GA), poly-Gly-Pro(GP), poly-Gly-Arg(GR), poly-Pro-Ala(PA), and poly-Pro-Arg(PR), would have been produced from different reading frames (Figure 3). In C9 ALS/FTD patient tissues, DPRs were initially observed in p62 positive neuronal cytoplasmic inclusions (NCI), which stained negatively for the nuclear transcription factor TDP-43 [14, 16, 80–82]. Since their discovery, DPR pathology has been a highly consistent and definitive feature for C9orf72 cases. Later, the presence of each potential DPR has been described in C9 ALS/FTD patient brain tissues and less frequently in spinal cord tissues, using a spectrum of antibodies, targeting either specific C-terminal regions or dipeptide-repeat regions of the individual DPRs [57, 58, 83–87]. DPR inclusions were not limited to neurons, and they were also shown in glial cells [83, 85, 88, 89]. Most studies reported that individual DPRs shared similar localization patterns but displayed different abundances: from highest to lowest are poly-GA, poly-GP, poly-GR, and poly-PA/poly-PR [89, 90]. Similar findings were also reported in C9 patient iPSCs derived neurons and C9 patient CSF (poly-GP was detected) [33, 49, 91–94]. Fundamental questions regarding the role of these C9-DPRs remain: Is the presence of DPRs a mere pathological hallmark? Are these DPRs actually causing neurodegeneration?
Figure 3. RAN translated DPRs impair cellular functions.

RAN translation of sense and anti-sense transcripts of expanded GGGGCC repeats in all possible reading frames gives rise to five DPRs. Sense strand: poly-GA, poly-GP and poly-GR. Anti-sense strand: poly-PA, poly-PR and poly-GP (same as sense strand). Impairments in multiple cellular functions are found caused by C9-DPRs.
To clearly dissect the pathogenic mechanisms, researchers used a “random-alternative codon” strategy to design DNA constructs encoding each specific DPR or synthetic dipeptides, so they could study DPRs independent of one another and from potential interference from pure GC sequences at transcript level. Using this approach, GR and PR, the two arginine rich DPRs, have been shown to be strongly toxic by multiple groups when overexpressed in yeast, cultured neurons and Drosophila, while other DPRs were not or were only mildly toxic [49, 72, 95–99]. Interestingly, GR and PR showed distinct localization pattern: predominantly nucleolar accompanied by occasional cytoplasmic localization. Further analysis revealed that these arginine rich DPRs interact with a number of nucleolar/nuclear proteins and cause global translational dysregulations, including induction of nucleolar stress, suppression of rRNA synthesis, abnormal stress granule formation, splicing aberration, and inhibition of protein translation [49, 95, 96, 100]. Several cellular pathways have also been reported as potential disease mechanisms: Dysfunction of UPS [101]; Disturbance in nucleocytoplasmic transport [99, 102]; Suppression of Notch signaling pathway [98]. Surprisingly, co-expression of GA was found to partially mitigate toxicity caused by GR in Drosophila cells and human iPSNs [98]. In addition, Drosophila carrying 160 intronic GGGGCC repeats showed abundant RNA foci but no neurodegeneration when grown at 25°C, while Drosophila expressing 36 GGGGCC repeats with a poly(A) tail, which readily produced GR and GP, displayed neurodegeneration phenotype [36]. More interestingly, when grown at a higher temperature, decreased survival was observed in Drosophila carrying 160 intronic GGGGCC repeats, which correlated with the production of GR, lending further support to the theory that R-rich DPRs induce neurotoxicity [36].
Tandem repeats of Gly-Ala is characteristic of EBNA1 protein of Ebstein-Barr virus and has been shown to impair proteasome functions [103, 104]. Likewise, studies showed that overexpression of poly-GA in cultured cells induced sequestration of Unc119, inhibition of proteasome activity, endoplasmic reticulum (ER) stress, reduced dendritic branching complexity, and apoptosis; Overexpression of Unc119 or inhibitors of ER stress ameliorated toxicity caused by GA expression [105, 106]. Another study showed that overexpression of GA or GP caused cellular toxicity via disrupting the UPS [101]. More intriguingly, 15 repeats of GA dipeptides were shown to form toxic amyloid fibrils when incubated in test tubes and could be transmitted from cell to cell [107]. However, it remains to be tested whether GA would spontaneously form amyloid fibrils when expressed in cells. Recently, mice expressing GA dipeptides were generated using a viral delivery system by injecting AAV1 encoding GFP tagged 50 repeats of GA into intracerebral ventricles [108]. These mice showed brain atrophy, motor deficits, and behavioral abnormality indicative of ALS/FTD like symptoms, confirming that expression of GA causes neurotoxicity in vivo. Consistent with the previous study, 50 repeats of GA dipeptides were found to form toxic fibrils, and the toxicity was conformation dependent since disruption of fibril formation would abolish toxicity. Furthermore, sequestration of HR23 proteins and nuclear pore proteins by GA aggregates were implied as potential mechanisms. It is of notice that each research group employed a unique sequence with different combination of alternative codons, different G/C content and different levels of repetitive sequences to encode the same DPR. These differences might lead to varied expression levels of the same DPR species and other unknown effects, which might explain the inconsistency in DPR toxicity seen by different groups.
In summary, overexpression of arginine rich DPRs cause neurotoxicity in vitro and in vivo by disturbing global translational regulation, nucleocytoplasmic transport, UPS, and by suppressing Notch signaling pathway; whether other DPRs cause direct neurotoxicity remains conflicting. Nevertheless, expression of C9-RAN DPR (GA) alone is sufficient to cause neurodegenerative phenotypes in vivo, strengthening the role of DPR toxicity in disease pathogenesis (summarized in Figure 3).
How relevant is DPR toxicity to human diseases remains debatable. If DPR toxicity causes direct neurodegeneration in disease, one would intuitively expect to see an overall heavier DPR burden or more toxic DPR species in regions or cell types that are affected most in the disease, or correlation between DPR pathology load and disease severity. The main argument is that there is low abundance of DPRs in key regions, and DPR distribution patterns and abundances do not differ between disease types (ALS vs. FTLD), nor do they correlate with disease onset, neurodegeneration, or phenotype [85, 89, 90, 109]. Exceptions were also reported in a few studies. In one study, the poly-GA positive dystrophic neurites were found associated with frontal cortex degeneration in FTLD and total poly-GA loads were associated with disease onset [90]. Cerebellar poly-GP levels were significantly lower in ALS patient samples compared to FTLD and FTLD-MND patient samples and was also associated with cognitive deficits [110]. Although poly-PR inclusions were rare, the total burden was shown to be significantly higher in CA3/4 regions in FTLD cases than MND cases [89]. Moreover, the lack of abundant DPRs pathology, especially toxic arginine rich DPRs, in patient post-mortem tissues could also be explained by the possibility that toxic DPRs caused neuronal death, thus neurons “survived” and showed up in pathology tissue sections were those who did not have abundant DPRs. So, non-invasive or minimally invasive assays that could detect R-rich DPRs in patients throughout disease course would provide invaluable information. Interestingly, pathological findings from three C9orf72-FTLD patients who prematurely died shortly after disease onset suggested that DPRs were abundant throughout the cerebral cortical regions, hippocampus, and cerebellum, while TDP-43 pathological changes were sparse [111].
Additionally, some studies state that TDP-43 cytoplasmic inclusions were more abundant than DPRs, and that the occurrence of TDP-43 correlates to degeneration in specific areas in C9ORF72-positive patients [85, 109]. However, a recent systematic review of a large cohort of pathological studies looking at TDP-43, p62, and DPR inclusions in the brains and spinal cords of C9ORF72-positive patients reported different findings [112]. A total of 261 C9orf72-positive patients were analyzed after excluding studies that only looked at one type of inclusion or one specific CNS region, and it was shown that there was greatly lower prevalence of TDP-43 inclusions in the cerebellum and hippocampus than DPRs and p62. Additionally, there was little to no difference between the prevalence of TDP-43, DPRs, and p62 inclusions in the frontal lobe, temporal lobe, spinal cord, and brainstem [112].
4. Concluding Remarks
Despite the excitement of identifying C9orf72 mutation as the most common genetic cause for ALS and FTD, we need to acknowledge that it is critical and challenging to decipher how the enormously expanded GGGGCC repeats in a non-coding region of C9orf72 gene cause neurodegeneration. Three pathogenic mechanisms were proposed. The research field of C9orf72 has been moving at a fast pace to determine which mechanism induce neurotoxicity and benefited greatly from lessons learned from other repeat expansion diseases. Abundant pathological and experimental findings have emerged. It remains to be investigated which pathogenic mechanism strikes neurons first and if there is a sequential cascade of neurotoxic events. Successful development of targeted therapeutics also relies on defining the contribution of each pathogenic mechanism.
Recent development of several lines of BAC transgenic mice carrying partial or full human C9orf72 gene and expanded GGGGCC repeats has greatly advanced modeling of the diseases in rodents. What is more intriguing is the discrepancy of phenotypes of different lines-some of the lines showed ALS/FTD like phenotypes [48, 76] while others did not show any phenotypes [74, 75]. For the line that showed more robust phenotypes, one obvious difference is the different strain background-FVB/NJ was used in this line as opposed to C57BL/6 or SJL/B6 in other lines. The mutation specific for one genetic strain has led to the discovery of novel functions of GTPBP2 and potentials of mutations in tRNAs in causing neurodegeneration [113]. Although the reasons for the discordant results are complicated and still unknown, there is a possibility that the different genetic background could be one underlying cause like in the case. A thorough comparison of different lines would provide more mechanistic insights.
Keeping in mind that these pathogenic mechanisms are not mutually exclusive, one should also explore the consequences of two or more mechanisms co-existing in the same model, e.g. crossing the BAC mouse with the C9 knockout mouse. It is possible that one mechanism might protect neurons against the toxicity caused by other mechanism(s). It is also possible that one mechanism might enhance the toxicity induced by other mechanism(s). There might also be convergence of mechanisms. Indeed, loss of C9orf72 protein sensitized cells to toxicity induced by expression of 30 repeats and 60 repeats of GGGGCC [52]. Moreover, decreased level of C9orf72 short isoform nuclear membrane staining has been found in C9-ALS patients, and the short isoform of the C9orf72 protein has also been found to physically interact with importin-b and RAN-GTPase [39]. Disruption of nucleocytoplasmic transport was associated with RNA toxicity as well as protein toxicity from R-rich DPRs [68, 97, 99]. Although it is premature to say C9 loss-of-function induce neurodegeneration together with toxic RNA and DPRs via disruption of nuclear transport, it is definitely worth further exploration.
Acknowledgments
We thank Lauren Rosenblum for her critical reading. This work was supported by the Farber Family Foundation (to D.T. and P.P.), the National Institutes of Health (NIH) grants R21-NS090912 (to D.T.), R56-NS092572 (to P.P.), and the Muscular Dystrophy Association (MDA) research grant (to D.T.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Kiernan MC, Vucic S, Cheah BC, Turner MR, Eisen A, Hardiman O, Burrell JR, Zoing MC. Amyotrophic lateral sclerosis. The Lancet. 2011;377:942–955. doi: 10.1016/S0140-6736(10)61156-7. [DOI] [PubMed] [Google Scholar]
- 2.Peters OM, Ghasemi M, Brown RH., Jr Emerging mechanisms of molecular pathology in ALS. J Clin Invest. 2015;125:1767–1779. doi: 10.1172/JCI71601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nature reviews. Neuroscience. 2013;14:248–264. doi: 10.1038/nrn3430. [DOI] [PubMed] [Google Scholar]
- 4.Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nature reviews. Neurology. 2012;8:423–434. doi: 10.1038/nrneurol.2012.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Waldo ML. The frontotemporal dementias. Psychiatr Clin North Am. 2015;38:193–209. doi: 10.1016/j.psc.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 6.van Blitterswijk M, DeJesus-Hernandez M, Rademakers R. How do C9ORF72 repeat expansions cause amyotrophic lateral sclerosis and frontotemporal dementia: can we learn from other noncoding repeat expansion disorders? Current opinion in neurology. 2012;25:689–700. doi: 10.1097/WCO.0b013e32835a3efb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bang J, Spina S, Miller BL. Frontotemporal dementia. The Lancet. 2015;386:1672–1682. doi: 10.1016/S0140-6736(15)00461-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lashley T, Rohrer JD, Mead S, Revesz T. Review: An update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathology and applied neurobiology. 2015;41:858–881. doi: 10.1111/nan.12250. [DOI] [PubMed] [Google Scholar]
- 9.Ringholz GM, Appel SH, Bradshaw M, Cooke NA, Mosnik DM, Schulz PE. Prevalence and patterns of cognitive impairment in sporadic ALS. Neurology. 2005;65:586–590. doi: 10.1212/01.wnl.0000172911.39167.b6. [DOI] [PubMed] [Google Scholar]
- 10.Wheaton MW, Salamone AR, Mosnik DM, McDonald RO, Appel SH, Schmolck HI, Ringholz GM, Schulz PE. Cognitive impairment in familial ALS. Neurology. 2007;69:1411–1417. doi: 10.1212/01.wnl.0000277422.11236.2c. [DOI] [PubMed] [Google Scholar]
- 11.Burrell JR, Kiernan MC, Vucic S, Hodges JR. Motor neuron dysfunction in frontotemporal dementia. Brain : a journal of neurology. 2011;134:2582–2594. doi: 10.1093/brain/awr195. [DOI] [PubMed] [Google Scholar]
- 12.Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59:1077–1079. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- 13.Josephs KA, Knopman DS, Whitwell JL, Boeve BF, Parisi JE, Petersen RC, Dickson DW. Survival in two variants of tau-negative frontotemporal lobar degeneration: FTLD-U vs FTLD-MND. Neurology. 2005;65:645–647. doi: 10.1212/01.wnl.0000173178.67986.7f. [DOI] [PubMed] [Google Scholar]
- 14.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ Consortium I. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Byrne S, Elamin M, Bede P, Shatunov A, Walsh C, Corr B, Heverin M, Jordan N, Kenna K, Lynch C, McLaughlin RL, Iyer PM, O’Brien C, Phukan J, Wynne B, Bokde AL, Bradley DG, Pender N, Al-Chalabi A, Hardiman O. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. The Lancet Neurology. 2012;11:232–240. doi: 10.1016/S1474-4422(12)70014-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR, Rademakers R. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S, Engelborghs S, Sieben A, De Jonghe P, Vandenberghe R, Santens P, De Bleecker J, Maes G, Bäumer V, Dillen L, Joris G, Cuijt I, Corsmit E, Elinck E, Van Dongen J, Vermeulen S, Van den Broeck M, Vaerenberg C, Mattheijssens M, Peeters K, Robberecht W, Cras P, Martin J-J, De Deyn PP, Cruts M, Van Broeckhoven C. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. The Lancet Neurology. 2012;11:54–65. doi: 10.1016/S1474-4422(11)70261-7. [DOI] [PubMed] [Google Scholar]
- 18.Lattante S, Ciura S, Rouleau GA, Kabashi E. Defining the genetic connection linking amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD) Trends Genet. 2015;31:263–273. doi: 10.1016/j.tig.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 19.Beck J, Poulter M, Hensman D, Rohrer JD, Mahoney CJ, Adamson G, Campbell T, Uphill J, Borg A, Fratta P, Orrell RW, Malaspina A, Rowe J, Brown J, Hodges J, Sidle K, Polke JM, Houlden H, Schott JM, Fox NC, Rossor MN, Tabrizi SJ, Isaacs AM, Hardy J, Warren JD, Collinge J, Mead S. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. American journal of human genetics. 2013;92:345–353. doi: 10.1016/j.ajhg.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gomez-Tortosa E, Gallego J, Guerrero-Lopez R, Marcos A, Gil-Neciga E, Sainz MJ, Diaz A, Franco-Macias E, Trujillo-Tiebas MJ, Ayuso C, Perez-Perez J. C9ORF72 hexanucleotide expansions of 20–22 repeats are associated with frontotemporal deterioration. Neurology. 2013;80:366–370. doi: 10.1212/WNL.0b013e31827f08ea. [DOI] [PubMed] [Google Scholar]
- 21.Fratta P, Polke JM, Newcombe J, Mizielinska S, Lashley T, Poulter M, Beck J, Preza E, Devoy A, Sidle K, Howard R, Malaspina A, Orrell RW, Clarke J, Lu CH, Mok K, Collins T, Shoaii M, Nanji T, Wray S, Adamson G, Pittman A, Renton AE, Traynor BJ, Sweeney MG, Revesz T, Houlden H, Mead S, Isaacs AM, Fisher EM. Screening a UK amyotrophic lateral sclerosis cohort provides evidence of multiple origins of the C9orf72 expansion. Neurobiology of aging. 2015;36:546 e541–547. doi: 10.1016/j.neurobiolaging.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suh E, Lee EB, Neal D, Wood EM, Toledo JB, Rennert L, Irwin DJ, McMillan CT, Krock B, Elman LB, McCluskey LF, Grossman M, Xie SX, Trojanowski JQ, Van Deerlin VM. Semi-automated quantification of C9orf72 expansion size reveals inverse correlation between hexanucleotide repeat number and disease duration in frontotemporal degeneration. Acta neuropathologica. 2015;130:363–372. doi: 10.1007/s00401-015-1445-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, Murray ME, Heckman MG, Diehl NN, Brown PH, Baker MC, Finch NA, Bauer PO, Serrano G, Beach TG, Josephs KA, Knopman DS, Petersen RC, Boeve BF, Graff-Radford NR, Boylan KB, Petrucelli L, Dickson DW, Rademakers R. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. The Lancet Neurology. 2013;12:978–988. doi: 10.1016/S1474-4422(13)70210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xi Z, van Blitterswijk M, Zhang M, McGoldrick P, McLean JR, Yunusova Y, Knock E, Moreno D, Sato C, McKeever PM, Schneider R, Keith J, Petrescu N, Fraser P, Tartaglia MC, Baker MC, Graff-Radford NR, Boylan KB, Dickson DW, Mackenzie IR, Rademakers R, Robertson J, Zinman L, Rogaeva E. Jump from pre-mutation to pathologic expansion in C9orf72. American journal of human genetics. 2015;96:962–970. doi: 10.1016/j.ajhg.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nordin A, Akimoto C, Wuolikainen A, Alstermark H, Jonsson P, Birve A, Marklund SL, Graffmo KS, Forsberg K, Brannstrom T, Andersen PM. Extensive size variability of the GGGGCC expansion in C9orf72 in both neuronal and non-neuronal tissues in 18 patients with ALS or FTD. Human molecular genetics. 2015;24:3133–3142. doi: 10.1093/hmg/ddv064. [DOI] [PubMed] [Google Scholar]
- 26.La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nature reviews. Genetics. 2010;11:247–258. doi: 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bagni C, Tassone F, Neri G, Hagerman R. Fragile X syndrome: causes, diagnosis, mechanisms, and therapeutics. J Clin Invest. 2012;122:4314–4322. doi: 10.1172/JCI63141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahimov F, Kunkel LM. The cell biology of disease: cellular and molecular mechanisms underlying muscular dystrophy. J Cell Biol. 2013;201:499–510. doi: 10.1083/jcb.201212142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nelson DL, Orr HT, Warren ST. The unstable repeats--three evolving faces of neurological disease. Neuron. 2013;77:825–843. doi: 10.1016/j.neuron.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orr HT, Zoghbi HY. Trinucleotide repeat disorders. Annual review of neuroscience. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 31.Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M, Ranum LP. Non-ATG-initiated translation directed by microsatellite expansions. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:260–265. doi: 10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Blitterswijk M, Gendron TF, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, Daughrity LM, Murray ME, Heckman MG, Jiang J, Lagier-Tourenne C, Edbauer D, Cleveland DW, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Petrucelli L, Boeve BF, Graff-Radford NR, Boylan KB, Dickson DW, Rademakers R. Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta neuropathologica. 2015 doi: 10.1007/s00401-015-1480-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Donnelly CJ, Zhang PW, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM, Maragakis N, Tienari PJ, Petrucelli L, Traynor BJ, Wang J, Rigo F, Bennett CF, Blackshaw S, Sattler R, Rothstein JD. RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron. 2013;80:415–428. doi: 10.1016/j.neuron.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, Wang J. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014;507:195–200. doi: 10.1038/nature13124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ciura S, Lattante S, Le Ber I, Latouche M, Tostivint H, Brice A, Kabashi E. Loss of function of C9orf72 causes motor deficits in a zebrafish model of amyotrophic lateral sclerosis. Annals of neurology. 2013;74:180–187. doi: 10.1002/ana.23946. [DOI] [PubMed] [Google Scholar]
- 36.Tran H, Almeida S, Moore J, Gendron TF, Chalasani U, Lu Y, Du X, Nickerson JA, Petrucelli L, Weng Z, Gao FB. Differential Toxicity of Nuclear RNA Foci versus Dipeptide Repeat Proteins in a Drosophila Model of C9ORF72 FTD/ALS. Neuron. 2015;87:1207–1214. doi: 10.1016/j.neuron.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu EY, Russ J, Wu K, Neal D, Suh E, McNally AG, Irwin DJ, Van Deerlin VM, Lee EB. C9orf72 hypermethylation protects against repeat expansion-associated pathology in ALS/FTD. Acta neuropathologica. 2014;128:525–541. doi: 10.1007/s00401-014-1286-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waite AJ, Baumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiology of aging. 2014;35:1779 e1775–1779 e1713. doi: 10.1016/j.neurobiolaging.2014.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao S, MacNair L, McGoldrick P, McKeever PM, McLean JR, Zhang M, Keith J, Zinman L, Rogaeva E, Robertson J. Isoform-specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Annals of neurology. 2015;78:568–583. doi: 10.1002/ana.24469. [DOI] [PubMed] [Google Scholar]
- 40.Fratta P, Poulter M, Lashley T, Rohrer JD, Polke JM, Beck J, Ryan N, Hensman D, Mizielinska S, Waite AJ, Lai MC, Gendron TF, Petrucelli L, Fisher EM, Revesz T, Warren JD, Collinge J, Isaacs AM, Mead S. Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta neuropathologica. 2013;126:401–409. doi: 10.1007/s00401-013-1147-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Therrien M, Rouleau GA, Dion PA, Parker JA. Deletion of C9ORF72 results in motor neuron degeneration and stress sensitivity in C. elegans. PloS one. 2013;8:e83450. doi: 10.1371/journal.pone.0083450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sa R, Schellevis RD, Waite AJ, Blake DJ, Veldink JH, van den Berg LH, Pasterkamp RJ. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Annals of neurology. 2015;78:426–438. doi: 10.1002/ana.24453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lagier-Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M, Qiu J, Sun Y, Ling SC, Zhu Q, Polymenidou M, Drenner K, Artates JW, McAlonis-Downes M, Markmiller S, Hutt KR, Pizzo DP, Cady J, Harms MB, Baloh RH, Vandenberg SR, Yeo GW, Fu XD, Bennett CF, Cleveland DW, Ravits J. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E4530–4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Rourke JG, Bogdanik L, Yanez A, Lall D, Wolf AJ, Muhammad AK, Ho R, Carmona S, Vit JP, Zarrow J, Kim KJ, Bell S, Harms MB, Miller TM, Dangler CA, Underhill DM, Goodridge HS, Lutz CM, Baloh RH. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016;351:1324–1329. doi: 10.1126/science.aaf1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y, Fury W, Burfeind P, Zamfirova R, Warshaw G, Orengo J, Oyejide A, Fralish M, Auerbach W, Poueymirou W, Freudenberg J, Gong G, Zambrowicz B, Valenzuela D, Yancopoulos G, Murphy A, Thurston G, Lai KM. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Scientific reports. 2016;6:23204. doi: 10.1038/srep23204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sullivan PM, Zhou X, Robins AM, Paushter DH, Kim D, Smolka MB, Hu F. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta neuropathologica communications. 2016;4:51. doi: 10.1186/s40478-016-0324-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sudria-Lopez E, Koppers M, de Wit M, van der Meer C, Westeneng HJ, Zundel CA, Youssef SA, Harkema L, de Bruin A, Veldink JH, van den Berg LH, Pasterkamp RJ. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta neuropathologica. 2016;132:145–147. doi: 10.1007/s00401-016-1581-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, Stauffer JE, Jafar-Nejad P, Drenner K, Schulte D, Chun S, Sun S, Ling SC, Myers B, Engelhardt J, Katz M, Baughn M, Platoshyn O, Marsala M, Watt A, Heyser CJ, Ard MC, De Muynck L, Daughrity LM, Swing DA, Tessarollo L, Jung CJ, Delpoux A, Utzschneider DT, Hedrick SM, de Jong PJ, Edbauer D, Van Damme P, Petrucelli L, Shaw CE, Bennett CF, Da Cruz S, Ravits J, Rigo F, Cleveland DW, Lagier-Tourenne C. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron. 2016;90:535–550. doi: 10.1016/j.neuron.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB, Pasinelli P, Ichida JK, Trotti D. Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron. 2014;84:1213–1225. doi: 10.1016/j.neuron.2014.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Farg MA, Sundaramoorthy V, Sultana JM, Yang S, Atkinson RA, Levina V, Halloran MA, Gleeson PA, Blair IP, Soo KY, King AE, Atkin JD. C9ORF72, implicated in amytrophic lateral sclerosis and frontotemporal dementia, regulates endosomal trafficking. Human molecular genetics. 2014;23:3579–3595. doi: 10.1093/hmg/ddu068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Atkinson RA, Fernandez-Martos CM, Atkin JD, Vickers JC, King AE. C9ORF72 expression and cellular localization over mouse development. Acta neuropathologica communications. 2015;3:59. doi: 10.1186/s40478-015-0238-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Maharjan N, Kunzli C, Buthey K, Saxena S. C9ORF72 Regulates Stress Granule Formation and Its Deficiency Impairs Stress Granule Assembly, Hypersensitizing Cells to Stress. Molecular neurobiology. 2016 doi: 10.1007/s12035-016-9850-1. [DOI] [PubMed] [Google Scholar]
- 53.Levine TP, Daniels RD, Gatta AT, Wong LH, Hayes MJ. The product of C9orf72, a gene strongly implicated in neurodegeneration, is structurally related to DENN Rab-GEFs. Bioinformatics. 2013;29:499–503. doi: 10.1093/bioinformatics/bts725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, Myszczynska MA, Higginbottom A, Walsh MJ, Whitworth AJ, Kaspar BK, Meyer K, Shaw PJ, Grierson AJ, De Vos KJ. The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J. 2016 doi: 10.15252/embj.201694401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb-Cheynel I, Oulad-Abdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E, Charlet-Berguerand N. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016;35:1276–1297. doi: 10.15252/embj.201593350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiao S, MacNair L, McLean J, McGoldrick P, McKeever P, Soleimani S, Keith J, Zinman L, Rogaeva E, Robertson J. C9orf72 isoforms in Amyotrophic Lateral Sclerosis and Frontotemporal Lobar Degeneration. Brain research. 2016 doi: 10.1016/j.brainres.2016.04.062. [DOI] [PubMed] [Google Scholar]
- 57.Gendron TF, Bieniek KF, Zhang YJ, Jansen-West K, Ash PE, Caulfield T, Daughrity L, Dunmore JH, Castanedes-Casey M, Chew J, Cosio DM, van Blitterswijk M, Lee WC, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat-associated non-ATG translation in c9FTD/ALS. Acta neuropathologica. 2013;126:829–844. doi: 10.1007/s00401-013-1192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, Ostrow LW, Rothstein JD, Troncoso JC, Ranum LP. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E4968–4977. doi: 10.1073/pnas.1315438110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, Isaacs AM. C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta neuropathologica. 2013;126:845–857. doi: 10.1007/s00401-013-1200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fratta P, Mizielinska S, Nicoll AJ, Zloh M, Fisher EM, Parkinson G, Isaacs AM. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Scientific reports. 2012;2:1016. doi: 10.1038/srep01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reddy K, Zamiri B, Stanley SY, Macgregor RB, Jr, Pearson CE. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. The Journal of biological chemistry. 2013;288:9860–9866. doi: 10.1074/jbc.C113.452532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reddy K, Schmidt MH, Geist JM, Thakkar NP, Panigrahi GB, Wang YH, Pearson CE. Processing of double-R-loops in (CAG).(CTG) and C9orf72 (GGGGCC).(GGCCCC) repeats causes instability. Nucleic acids research. 2014;42:10473–10487. doi: 10.1093/nar/gku658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cooper-Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D, Ince PG, Wharton SB, Wilson SA, Kirby J, Hautbergue GM, Shaw PJ. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain : a journal of neurology. 2014;137:2040–2051. doi: 10.1093/brain/awu120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C, Adachi Y, Sardone V, Miller JW, Smith BN, Gallo JM, Ule J, Hirth F, Rogelj B, Houart C, Shaw CE. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell reports. 2013;5:1178–1186. doi: 10.1016/j.celrep.2013.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sareen D, O’Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A, Gendron T, Petrucelli L, Baughn M, Ravits J, Harms MB, Rigo F, Bennett CF, Otis TS, Svendsen CN, Baloh RH. Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Science translational medicine. 2013;5:208ra149. doi: 10.1126/scitranslmed.3007529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cooper-Knock J, Higginbottom A, Stopford MJ, Highley JR, Ince PG, Wharton SB, Pickering-Brown S, Kirby J, Hautbergue GM, Shaw PJ. Antisense RNA foci in the motor neurons of C9ORF72-ALS patients are associated with TDP-43 proteinopathy. Acta neuropathologica. 2015;130:63–75. doi: 10.1007/s00401-015-1429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS, Jin P. Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:7778–7783. doi: 10.1073/pnas.1219643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE, Rothstein JD. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61. doi: 10.1038/nature14973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rossi S, Serrano A, Gerbino V, Giorgi A, Di Francesco L, Nencini M, Bozzo F, Schinina ME, Bagni C, Cestra G, Carri MT, Achsel T, Cozzolino M. Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. Journal of cell science. 2015;128:1787–1799. doi: 10.1242/jcs.165332. [DOI] [PubMed] [Google Scholar]
- 70.Cooper-Knock J, Bury JJ, Heath PR, Wyles M, Higginbottom A, Gelsthorpe C, Highley JR, Hautbergue G, Rattray M, Kirby J, Shaw PJ. C9ORF72 GGGGCC Expanded Repeats Produce Splicing Dysregulation which Correlates with Disease Severity in Amyotrophic Lateral Sclerosis. PloS one. 2015;10:e0127376. doi: 10.1371/journal.pone.0127376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schweizer Burguete A, Almeida S, Gao FB, Kalb R, Akins MR, Bonini NM. GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. Elife. 2015;4 doi: 10.7554/eLife.08881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IO, Pietrzyk J, Cleverley K, Nicoll AJ, Pickering-Brown S, Dols J, Cabecinha M, Hendrich O, Fratta P, Fisher EM, Partridge L, Isaacs AM. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345:1192–1194. doi: 10.1126/science.1256800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hukema RK, Riemslagh FW, Melhem S, van der Linde HC, Severijnen LA, Edbauer D, Maas A, Charlet-Berguerand N, Willemsen R, van Swieten JC. A new inducible transgenic mouse model for C9orf72-associated GGGGCC repeat expansion supports a gain-of-function mechanism in C9orf72-associated ALS and FTD. Acta neuropathologica communications. 2014;2:166. doi: 10.1186/s40478-014-0166-y. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 74.O’Rourke Jacqueline G, Bogdanik L, Muhammad AKMG, Gendron Tania F, Kim Kevin J, Austin A, Cady J, Liu Elaine Y, Zarrow J, Grant S, Ho R, Bell S, Carmona S, Simpkinson M, Lall D, Wu K, Daughrity L, Dickson Dennis W, Harms Matthew B, Petrucelli L, Lee Edward B, Lutz Cathleen M, Baloh Robert H. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron. 2015;88:892–901. doi: 10.1016/j.neuron.2015.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Peters Owen M, Cabrera Gabriela T, Tran H, Gendron Tania F, McKeon Jeanne E, Metterville J, Weiss A, Wightman N, Salameh J, Kim J, Sun H, Boylan Kevin B, Dickson D, Kennedy Z, Lin Z, Zhang Y-J, Daughrity L, Jung C, Gao F-B, Sapp Peter C, Horvitz HR, Bosco Daryl A, Brown Solange P, de Jong P, Petrucelli L, Mueller C, Brown Robert H. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron. 2015;88:902–909. doi: 10.1016/j.neuron.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, Yachnis AT, Ranum LP. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron. 2016;90:521–534. doi: 10.1016/j.neuron.2016.04.005. [DOI] [PubMed] [Google Scholar]
- 77.Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V, Elenitoba-Johnson K, Vonsattel JP, Louis ED, Sutton MA, Taylor JP, Mills RE, Charlet-Berguerand N, Paulson HL. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron. 2013;78:440–455. doi: 10.1016/j.neuron.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oh SY, He F, Krans A, Frazer M, Taylor JP, Paulson HL, Todd PK. RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X-associated tremor ataxia syndrome. Human molecular genetics. 2015;24:4317–4326. doi: 10.1093/hmg/ddv165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kearse MG, Green KM, Krans A, Rodriguez CM, Linsalata AE, Goldstrohm AC, Todd PK. CGG Repeat-Associated Non-AUG Translation Utilizes a Cap-Dependent Scanning Mechanism of Initiation to Produce Toxic Proteins. Mol Cell. 2016;62:314–322. doi: 10.1016/j.molcel.2016.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Al-Sarraj S, King A, Troakes C, Smith B, Maekawa S, Bodi I, Rogelj B, Al-Chalabi A, Hortobagyi T, Shaw CE. p62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta neuropathologica. 2011;122:691–702. doi: 10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- 81.Murray ME, DeJesus-Hernandez M, Rutherford NJ, Baker M, Duara R, Graff-Radford NR, Wszolek ZK, Ferman TJ, Josephs KA, Boylan KB, Rademakers R, Dickson DW. Clinical and neuropathologic heterogeneity of c9FTD/ALS associated with hexanucleotide repeat expansion in C9ORF72. Acta neuropathologica. 2011;122:673–690. doi: 10.1007/s00401-011-0907-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mann DM, Rollinson S, Robinson A, Bennion Callister J, Thompson JC, Snowden JS, Gendron T, Petrucelli L, Masuda-Suzukake M, Hasegawa M, Davidson Y, Pickering-Brown S. Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta neuropathologica communications. 2013;1:68. doi: 10.1186/2051-5960-1-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW, 3rd, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gendron TF, Belzil VV, Zhang YJ, Petrucelli L. Mechanisms of toxicity in C9FTLD/ALS. Acta neuropathologica. 2014;127:359–376. doi: 10.1007/s00401-013-1237-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K, Weng SM, Haass C, Kretzschmar HA, Edbauer D, Neumann M. Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations. Acta neuropathologica. 2013;126:859–879. doi: 10.1007/s00401-013-1181-y. [DOI] [PubMed] [Google Scholar]
- 86.Mori K, Arzberger T, Grasser FA, Gijselinck I, May S, Rentzsch K, Weng SM, Schludi MH, van der Zee J, Cruts M, Van Broeckhoven C, Kremmer E, Kretzschmar HA, Haass C, Edbauer D. Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta neuropathologica. 2013;126:881–893. doi: 10.1007/s00401-013-1189-3. [DOI] [PubMed] [Google Scholar]
- 87.Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C, Edbauer D. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 88.Mackenzie IR, Frick P, Neumann M. The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta neuropathologica. 2014;127:347–357. doi: 10.1007/s00401-013-1232-4. [DOI] [PubMed] [Google Scholar]
- 89.Schludi MH, May S, Grasser FA, Rentzsch K, Kremmer E, Kupper C, Klopstock T, Arzberger T, Edbauer D. Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing. Acta neuropathologica. 2015;130:537–555. doi: 10.1007/s00401-015-1450-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mackenzie IR, Frick P, Grasser FA, Gendron TF, Petrucelli L, Cashman NR, Edbauer D, Kremmer E, Prudlo J, Troost D, Neumann M. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta neuropathologica. 2015 doi: 10.1007/s00401-015-1476-2. [DOI] [PubMed] [Google Scholar]
- 91.Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, Tapper AR, Sellier C, Charlet-Berguerand N, Karydas A, Seeley WW, Boxer AL, Petrucelli L, Miller BL, Gao FB. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta neuropathologica. 2013;126:385–399. doi: 10.1007/s00401-013-1149-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang WY, Fostvedt E, Jansen-West K, Belzil VV, Desaro P, Johnston A, Overstreet K, Oh SY, Todd PK, Berry JD, Cudkowicz ME, Boeve BF, Dickson D, Floeter MK, Traynor BJ, Morelli C, Ratti A, Silani V, Rademakers R, Brown RH, Rothstein JD, Boylan KB, Petrucelli L, Disney MD. Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron. 2014;83:1043–1050. doi: 10.1016/j.neuron.2014.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME, Bieniek KF, Bauer PO, Whitelaw EC, Rousseau L, Stankowski JN, Stetler C, Daughrity LM, Perkerson EA, Desaro P, Johnston A, Overstreet K, Edbauer D, Rademakers R, Boylan KB, Dickson DW, Fryer JD, Petrucelli L. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015;348:1151–1154. doi: 10.1126/science.aaa9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dafinca R, Scaber J, Ababneh N, Lalic T, Weir G, Christian H, Vowles J, Douglas AG, Fletcher-Jones A, Browne C, Nakanishi M, Turner MR, Wade-Martins R, Cowley SA, Talbot K. C9orf72 Hexanucleotide Expansions are Associated with Altered ER Calcium Homeostasis and Stress Granule Formation in iPSC-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells. 2016 doi: 10.1002/stem.2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y, McKnight SL. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science. 2014;345:1139–1145. doi: 10.1126/science.1254917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tao Z, Wang H, Xia Q, Li K, Li K, Jiang X, Xu G, Wang G, Ying Z. Nucleolar stress and impaired stress granule formation contribute to C9orf72 RAN translation-induced cytotoxicity. Human molecular genetics. 2015;24:2426–2441. doi: 10.1093/hmg/ddv005. [DOI] [PubMed] [Google Scholar]
- 97.Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB, Taylor JP. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature. 2015;525:129–133. doi: 10.1038/nature14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang D, Abdallah A, Li Z, Lu Y, Almeida S, Gao FB. FTD/ALS-associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions. Acta neuropathologica. 2015;130:525–535. doi: 10.1007/s00401-015-1448-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jovicic A, Mertens J, Boeynaems S, Bogaert E, Chai N, Yamada SB, Paul JW, 3rd, Sun S, Herdy JR, Bieri G, Kramer NJ, Gage FH, Van Den Bosch L, Robberecht W, Gitler AD. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nature neuroscience. 2015;18:1226–1229. doi: 10.1038/nn.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kanekura K, Yagi T, Cammack AJ, Mahadevan J, Kuroda M, Harms MB, Miller TM, Urano F. Poly-dipeptides encoded by the C9ORF72 repeats block global protein translation. Human molecular genetics. 2016 doi: 10.1093/hmg/ddw052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yamakawa M, Ito D, Honda T, Kubo K, Noda M, Nakajima K, Suzuki N. Characterization of the dipeptide repeat protein in the molecular pathogenesis of c9FTD/ALS. Human molecular genetics. 2015;24:1630–1645. doi: 10.1093/hmg/ddu576. [DOI] [PubMed] [Google Scholar]
- 102.Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovicic A, De Baets G, Scheveneels W, Steyaert J, Cuijt I, Verstrepen KJ, Callaerts P, Rousseau F, Schymkowitz J, Cruts M, Van Broeckhoven C, Van Damme P, Gitler AD, Robberecht W, Van Den Bosch L. Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Scientific reports. 2016;6:20877. doi: 10.1038/srep20877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang M, Coffino P. Repeat sequence of Epstein-Barr virus-encoded nuclear antigen 1 protein interrupts proteasome substrate processing. The Journal of biological chemistry. 2004;279:8635–8641. doi: 10.1074/jbc.M310449200. [DOI] [PubMed] [Google Scholar]
- 104.Hoyt MA, Zich J, Takeuchi J, Zhang M, Govaerts C, Coffino P. Glycine-alanine repeats impair proper substrate unfolding by the proteasome. EMBO J. 2006;25:1720–1729. doi: 10.1038/sj.emboj.7601058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, Grasser FA, Mori K, Kremmer E, Banzhaf-Strathmann J, Mann M, Meissner F, Edbauer D. C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration. Acta neuropathologica. 2014;128:485–503. doi: 10.1007/s00401-014-1329-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang YJ, Jansen-West K, Xu YF, Gendron TF, Bieniek KF, Lin WL, Sasaguri H, Caulfield T, Hubbard J, Daughrity L, Chew J, Belzil VV, Prudencio M, Stankowski JN, Castanedes-Casey M, Whitelaw E, Ash PE, DeTure M, Rademakers R, Boylan KB, Dickson DW, Petrucelli L. Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress. Acta neuropathologica. 2014;128:505–524. doi: 10.1007/s00401-014-1336-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chang YJ, Jeng US, Chiang YL, Hwang IS, Chen YR. Glycine-Alanine Dipeptide Repeat from C9orf72 Hexanucleotide Expansions Forms Toxic Amyloids Possessing Cell-to-cell Transmission Property. The Journal of biological chemistry. 2016 doi: 10.1074/jbc.M115.694273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu YF, Katzman RB, Gass J, Murray ME, Shinohara M, Lin WL, Garrett A, Stankowski JN, Daughrity L, Tong J, Perkerson EA, Yue M, Chew J, Castanedes-Casey M, Kurti A, Wang ZS, Liesinger AM, Baker JD, Jiang J, Lagier-Tourenne C, Edbauer D, Cleveland DW, Rademakers R, Boylan KB, Bu G, Link CD, Dickey CA, Rothstein JD, Dickson DW, Fryer JD, Petrucelli L. C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins. Nature neuroscience. 2016 doi: 10.1038/nn.4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gomez-Deza J, Lee YB, Troakes C, Nolan M, Al-Sarraj S, Gallo JM, Shaw CE. Dipeptide repeat protein inclusions are rare in the spinal cord and almost absent from motor neurons in C9ORF72 mutant amyotrophic lateral sclerosis and are unlikely to cause their degeneration. Acta neuropathologica communications. 2015;3:38. doi: 10.1186/s40478-015-0218-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gendron TF, van Blitterswijk M, Bieniek KF, Daughrity LM, Jiang J, Rush BK, Pedraza O, Lucas JA, Murray ME, Desaro P, Robertson A, Overstreet K, Thomas CS, Crook JE, Castanedes-Casey M, Rousseau L, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Boeve BF, Graff-Radford NR, Rademakers R, Lagier-Tourenne C, Edbauer D, Cleveland DW, Dickson DW, Petrucelli L, Boylan KB. Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers. Acta neuropathologica. 2015;130:559–573. doi: 10.1007/s00401-015-1474-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Baborie A, Griffiths TD, Jaros E, Perry R, McKeith IG, Burn DJ, Masuda-Suzukake M, Hasegawa M, Rollinson S, Pickering-Brown S, Robinson AC, Davidson YS, Mann DM. Accumulation of dipeptide repeat proteins predates that of TDP-43 in frontotemporal lobar degeneration associated with hexanucleotide repeat expansions in C9ORF72 gene. Neuropathology and applied neurobiology. 2015;41:601–612. doi: 10.1111/nan.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schipper LJ, Raaphorst J, Aronica E, Baas F, de Haan R, de Visser M, Troost D. Prevalence of brain and spinal cord inclusions, including dipeptide repeat proteins, in patients with the C9ORF72 hexanucleotide repeat expansion: A systematic neuropathological review. Neuropathology and applied neurobiology. 2015 doi: 10.1111/nan.12284. [DOI] [PubMed] [Google Scholar]
- 113.Ishimura R, Nagy G, Dotu I, Zhou H, Yang XL, Schimmel P, Senju S, Nishimura Y, Chuang JH, Ackerman SL. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014;345:455–459. doi: 10.1126/science.1249749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mori K, Lammich S, Mackenzie IR, Forne I, Zilow S, Kretzschmar H, Edbauer D, Janssens J, Kleinberger G, Cruts M, Herms J, Neumann M, Van Broeckhoven C, Arzberger T, Haass C German Consortium for Frontotemporal Lobar D, Bavarian Brain Banking A. hnRNP A3 binds to GGGGCC repeats and is a constituent of p62-positive/TDP43-negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta neuropathologica. 2013;125:413–423. doi: 10.1007/s00401-013-1088-7. [DOI] [PubMed] [Google Scholar]