

Abstract
In the context of tumor suppression, p53 is an undisputedly critical protein. Functioning primarily as a transcription factor, p53 helps fend off the initiation and progression of tumors by inducing cell cycle arrest, senescence or programmed cell death (apoptosis) in cells at the earliest stages of precancerous development. Compelling evidence, however, suggests that p53 is involved in other aspects of human physiology, including metabolism. Indeed, recent studies suggest that p53 plays a significant role in the development of metabolic diseases, including diabetes, and further that p53’s role in metabolism may also be consequential to tumor suppression. Here we present a review of the literature on the role of p53 in metabolism, diabetes, pancreatic function, glucose homeostasis, and insulin resistance. Additionally, we discuss the emerging role of genetic variation in the p53 pathway (single nucleotide polymorphisms) on the impact of p53 in metabolic disease and diabetes. A better understanding of the relationship between p53, metabolism and diabetes may one day better inform the existing and prospective therapeutic strategies to combat this rapidly growing epidemic.
Keywords: p53, metabolism, diabetes, insulin resistance
Introduction
Long regarded as the “guardian of the genome”, the functions of TP53 (hereafter p53) as a tumor suppressor have been extensively studied (Levine and Oren 2009). More than half of human cancers possess mutations in either p53 or in genes whose protein products control p53 activity (MDM2 or CDKN2A). p53 exerts its tumor suppressive activities mainly by acting as a transcription factor to transactivate gene expression following genotoxic or oncogenic stress. Under mild stress, activated p53 induces expression of growth-arrest genes, such as CDKN1A/p21, to facilitate DNA damage repair. When encountering prolonged or catastrophic DNA damage, p53 triggers programmed cell death (apoptosis), primarily by transactivating genes that encode pro-apoptotic proteins, such as BBC3 (PUMA) and PMAIP1 (NOXA). Additionally, it has recently become appreciated that the significance of p53 reaches beyond the limits of cancer biology. An impact of p53 has been seen in a variety of physiological functions, including aging, innate and adaptive immunity, development, reproduction, and neuronal degeneration (Chang, et al. 2012; Danilova, et al. 2008; Levine, et al. 2011; Menendez, et al. 2013; Poyurovsky and Prives 2010). Additionally, the relationship between p53, metabolism, and metabolic diseases has become a new focal point for p53 researchers (Berkers, et al. 2013; Sano, et al. 2007; Vousden and Ryan 2009). This review will cover the role of p53 in metabolism, with focus on its role in diabetes.
Structure and function of the p53 tumor suppressor protein
p53 is a 393 amino acid protein that uses a zinc-coordinated DNA binding domain to bind in a sequence-specific manner to DNA consensus elements that contain two copies of the sequence PPPCWWGYYY (where P=purine, W=A or T, Y=pyrimidine) separated by a spacer of 0–13 nucleotides. Though it is intrinsically disordered, the amino terminus of p53 is the business end of the molecule; it is responsible for interaction with the negative regulator MDM2, which targets p53 for degradation. This domain becomes phosphorylated following stress, thereby reducing the affinity for MDM2, and increasing the affinity for transcriptional co-activators like p300/CBP. The sequence-specific DNA binding domain of p53 encompasses approximately 2/3 of this protein, and this is where the majority of mutations occur in tumors. These mutations are often missense mutations, and six commonly mutated residues account for up to 30% of the mutations in p53 found in human tumors; these are called “hotspot” mutations (Figure 1A). p53 binds to its consensus element as a dimer of dimers, and this is mediated by a C-terminal oligomerization domain. Finally, at the extreme C terminus is a highly basic disordered domain that binds to DNA in non-specific manner with high affinity; this domain is believed to ‘scan’ the DNA for double strand breaks or gaps, whereupon p53 stalls and becomes phosphorylated by kinases that are activated by DNA damage, such as ATM (for review see (Vousden and Prives 2009)).
Figure 1. p53 structure and function.
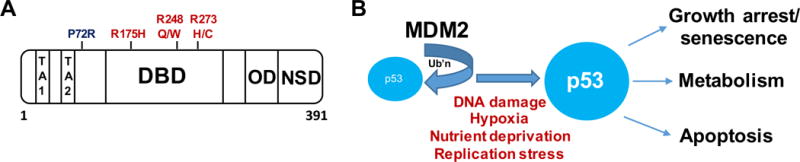
A. Functional domains of p53. Outlined are the domains responsible for transactivation (TA1 and TA2), the codon 72 polymorphism (P72R), the DNA binding domain (DBD, with some of the most common p53 ‘hotspot’ mutations shown), the oligomerization domain (OD), and the non-specific DNA binding domain (NSD).
B. Overview of the p53 pathway. Ub’n: ubiquitin
There are multiple stresses that stabilize p53 and activate it as a transcription factor: these include DNA damage, hypoxia, nutrient deprivation, and DNA replication stress (Figure 1B). Following these stresses p53 becomes stabilized and activated as a transcription factor, leading to the sequence specific transactivation of genes such as the cyclin dependent kinase inhibitor CDKN1A (p21/waf1) which leads to growth arrest and senescence, or to pro-apoptotic genes like PUMA, PMAIP1 (NOXA) or BAX, which lead to cell death. In addition, p53 has a transcription-independent role in cell death, mediated by direct binding and oligomerization of BAX and BAK at the mitochondria (for review see (Pietsch, et al. 2008)). In addition to target genes involved in growth arrest and cell death, p53 is also well-known to regulate target genes with roles in metabolism, such as the genes GLS2, SCO2, RRAD, and TIGAR (for review see (Berkers et al. 2013)). Whereas the role of p53 in metabolism is only now becoming appreciated, recent data from genetically-engineered mouse models of p53 function implicate this set of p53 target genes in tumor suppression (Brady, et al. 2011; Li, et al. 2012).
Type 1 and type 2 diabetes
Diabetes refers to a series of metabolic conditions characterized by high blood glucose, caused either by inadequate insulin production in the body or by impaired response to insulin. Type 1 diabetes accounts for approximately 10% of cases, and is caused by death and dysfunction of pancreatic beta islet cells, leading to an absence of insulin production. Type 2 diabetes, or adult onset diabetes, is characterized by impaired or insufficient insulin response; this disease accounts for 90% of cases of diabetes, and it is estimated that there are close to over 340 million individuals in the world with type 2 diabetes. Risk factors for both forms of diabetes include family history, genetics, and age. Risk factors for type 2 diabetes also include obesity and poor diet. Complications of diabetes include glaucoma, poor wound healing, hypertension, neuropathy, nephropathy (kidney disease) and higher risk of stroke (for review see (Polonsky 2012)).
Both type 1 and type 2 diabetes are severe diseases that are better managed with healthy diet and exercise. Whereas type I diabetes requires treatment with insulin, in some cases type II diabetes may not require insulin, and can be managed with non-insulin medications that increase the response to insulin or reduce blood glucose. These non-insulin medications include the agents metformin and thiazolidinediones, which increase the body’s sensitivity to insulin, sulfonylureas and meglitinides, which stimulate the pancreas to secrete more insulin, DPP-4 inhibitors and GLP-1 receptor agonists, which help lower blood glucose levels, and SGLT2 inhibitors, which prevent the kidneys from reabsorbing glucose into the blood (for review see (Polonsky 2012)).
The tumor suppressor p53 influences diabetes
The first evidence linking p53 to the development of type 2 diabetes came in 2009, when Minamino and colleagues demonstrated that diet-induced insulin resistance in Ay transgenic mice, which are susceptible to diabetes, is mediated by p53 (Minamino, et al. 2009). This group showed that inhibition of p53 activity, either by siRNA knock-down in cells, or by TP53 gene knock-out in mice, alleviated senescence and caused decreased inflammatory cytokine expression in the adipose tissue of mice, ultimately preventing them from developing insulin resistance. A year later, in a study focusing on the connection between non-homologous end-joining (NHEJ) DNA repair mechanisms and p53, Tavana and colleagues discovered another unexpected connection between p53 and diabetes (Tavana, et al. 2010). Specifically, knockout of Lig4 in mice resulted in NHEJ deficiency and embryonic lethality; not surprisingly, this embryonic lethality was rescued by p53 deficiency. The authors found that Lig4−/−; p53−/− mice developed B-cell lymphoma, but that introduction of a hypomorphic p53 mutant that fails to induce apoptosis but retains the ability to induce growth arrest and senescence (p53R172P) prevented the lymphomagenesis. However, the result was severe diabetes and early fatality in these mice; these were attributed to senescence of the pancreatic beta cells in these Lig4−/−; p53R172P mice. In sum, these studies implicated p53-mediated senescence of adipocytes and pancreatic beta cells, respectively, in the development of insulin resistance and diabetes.
Diabetes phenotypes regulated by p53
In general, there are three major causes of diabetes: 1) functional deficiencies in pancreatic insulin production; 2) aberration of glucose homeostasis; and 3) development of insulin resistance. The available in vitro and in vivo evidence suggests that p53 is capable of having a significant impact on all three of these pathways.
p53 regulates pancreatic function and survival
One of the most common features of both type 1 and type 2 diabetes is dysfunction of pancreatic beta cells. Pancreatic beta cells are responsible for secreting insulin into the bloodstream; this lowers the circulating glucose level by stimulating glucose uptake (Cnop, et al. 2005). Loss of pancreatic beta cell function reduces insulin secretion and results in hyperglycemia and diabetes. Multiple signaling pathways converge upon p53 to regulate pancreatic beta cell function (Figure 2). Wrede et al. demonstrated that free fatty acids (FFA) increase apoptosis of pancreatic beta cells through reduced phosphorylation/activation of AKT (PKB) (Wrede, et al. 2002); AKT is well known to normally protect cells from p53-mediated apoptosis by phosphorylating and activating the main negative regulator of p53, MDM2 (Mayo and Donner 2001; Zhou, et al. 2001). In a study that used FFA-treated beta cells to mimic visceral obesity-induced pancreatic dysfunction and apoptosis, p53 activation led to induction of the microRNA miR34a, which sensitized beta cells to apoptosis and inhibited the insulin exocytosis pathway, resulting in impaired insulin secretion (Lovis, et al. 2008). In a pancreatic beta cell line and in insulin-producing islet cells, the pro-inflammatory cytokines IFN-γ and TNF-α were shown to synergistically function to increase ROS production, p53 induction, and apoptosis (Kim, et al. 2005). Finally, reactive oxygen species (ROS) were also found to be involved in FFA-mediated apoptosis of beta cells, and both ROS generation and p53 activation were shown to be downstream effects of NAPDH oxidase 2 (NOX2) (Koshkin, et al. 2003; Yuan, et al. 2010). In sum, these researchers found that free fatty acids, inflammatory cytokines and ROS can all act upstream of p53, to induce p53-mediated apoptosis of pancreatic beta cells.
Figure 2. The regulation of pancreatic function by p53.
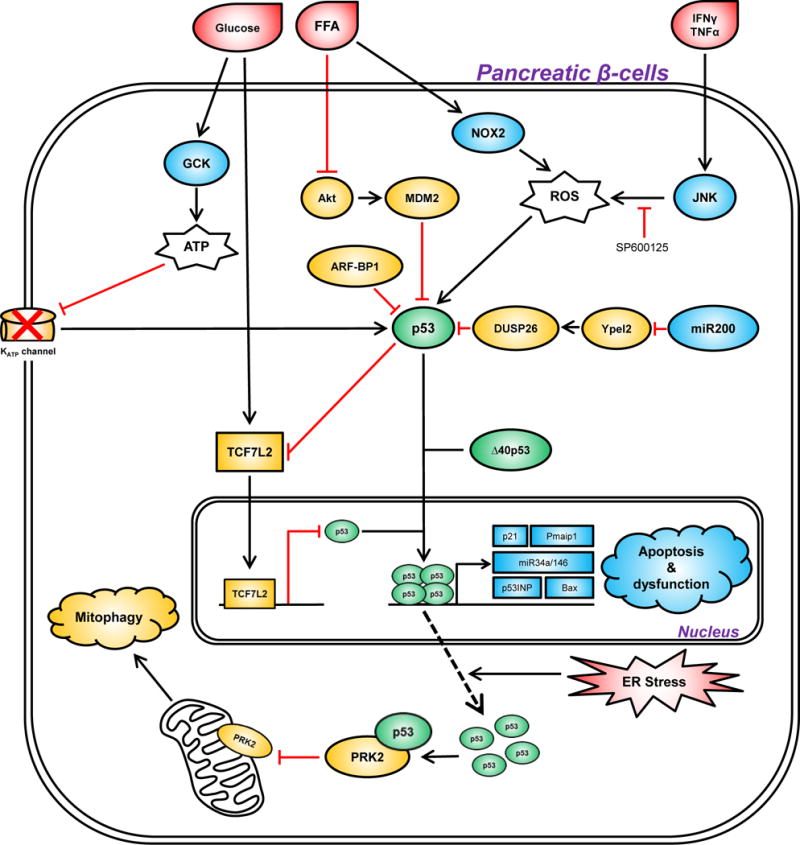
In response to extrinsic (hyperglycemia, free fatty acid, inflammation) and intrinsic (ER stress) stresses (colored red), p53 is activated to trigger pancreatic dysfunction and impair insulin production through several mechanisms. The genes depicted in squares represent transcriptional targets of p53. The genes colored in blue positively contribute to p53 activation, while genes colored in orange inhibit p53-mediated pancreatic dysfunction. Note that the KATP channel only activates p53 signaling in response to excessive ATP. FFA, free fatty acid; ROS, reactive oxygen species.
Several proteins that regulate p53 are also influential in the death and survival of pancreatic beta cells. These include N-terminal truncated isoforms of p53, the transcription factor TCF7L2, and the microRNA miR-200. Hinault et al. showed that ectopically overexpressing the delta40p53 isoform of p53 in mice led to hypoinsulinemia and glucose intolerance en route to premature death (Hinault, et al. 2011). delta40p53 is typically known to negatively regulate p53 transcriptional activity, but in some cell types it can positively regulate p53 function (Bourdon, et al. 2005). In the case of pancreatic islet cells, delta40p53 led to increased expression of the cell cycle inhibitor p21/CDKN1A, a p53 target gene (Hinault et al. 2011). p53 function in the pancreas is also regulated by the transcription factor T-cell factor 7-like 2 (TCF7L2, also known as TCF-4). TCF7L2 maintains pancreatic beta cell survival by physically binding to the p53 promoter and down-regulating p53 expression (Zhou, et al. 2012). Silencing of TCF7L2 leads to increased apoptosis by inducing expression of p53 and its apoptotic targets, including the pro-apoptotic target gene p53INP1. Notably, a single nucleotide polymorphism (SNP) of TCF7L2 (rs7903146; C>T) is associated with impaired insulin secretion and increased risk of type 2 diabetes in Caucasians (Grant, et al. 2006). Interestingly, TCF7L2 is also a p53-repressed target gene, suggesting there is feedback regulation between these two proteins (Rother, et al. 2004). Finally, a recent study connected p53 with a microRNA, miR-200, along with pancreatic beta cell apoptosis and lethal type 2 diabetes in vivo (Belgardt, et al. 2015). Overexpression of miR-200 in beta cells activates p53 and creates a pro-apoptotic gene-expression signature in diabetic mice. miR-200 inhibits the expression of its direct target YPEL2, which is responsible for maintaining the expression of DUSP26. Dusp26 (dual specificity phosphatase 26) is a negative regulator of p53, and miR-200-mediated reduction of DUSP26 induces p53 activation and apoptosis (Shang, et al. 2010). There are thus several proteins that regulate the p53 pathway, which can also regulate its ability to mediate pancreatic beta cell death.
p53 regulates dysfunctional beta cells during pre-diabetes
By studying a rare monogenic disease, congenital hyperinsulinism (CHI), p53 activity was found to be elevated during dysregulated beta cell replication, a phenotype shared by CHI and pre-diabetes (Tornovsky-Babeay, et al. 2014). In patients and mice with CHI, an activating mutation of glucokinase GCK and dysfunctional ATP-sensitive potassium channels (KATP channel) result in beta cell membrane depolarization, hypersecretion of insulin and overactive glycolysis. Glucotoxicity triggers DNA damage and p53 activation, which ultimately results in beta cell death and late-onset diabetes (Tornovsky-Babeay et al. 2014). Two other processes regulated by p53, endoplasmic reticulum (ER) stress and dysfunctional autophagy, have also been implicated in beta cell failure and hyperglycemia (Karunakaran, et al. 2012; Kung, et al. 2011; Las and Shirihai 2010; Stavridi and Halazonetis 2004). A recent study clarified this complex network by connecting diabetes-associated glucolipotoxicity with p53-mediated disruption of mitochondrial function (Hoshino, et al. 2014). Specifically, in mouse models of both type 1 and type 2 diabetes, increased ER stress stimulates nuclear exportation and cytosolic accumulation of p53. This cytosolic p53 interacts with Parkin (PARK2) and inhibits Parkin-mediated mitophagy, resulting in mitochondrial dysfunction and insulin deficiency. Deletion or inhibition of p53 restores mitochondrial function and increases glucose tolerance (Hoshino et al. 2014).
p53 regulates glucose homeostasis: glucose transport, glycolysis and gluconeogenesis
Frank (or mature) diabetes is defined by an elevated level of circulating glucose. It often occurs concomitant with impaired glucose uptake in peripheral tissues such as muscle, adipose tissue, liver and the brain (Cherrington 1999). These phenotypes are in direct contrast to the enhanced cellular glucose uptake and glycolysis, better known as the “Warburg effect”, that is frequently observed in human tumors that contain mutations in p53 (Vander Heiden, et al. 2009). Recent studies suggest that p53 exerts its tumor suppressive activity, at least in part, through its ability to regulate glucose homeostasis (Madan, et al. 2011). In the sections below we summarize the data indicating that p53 is an important regulator of glucose transport, glycolysis and gluconeogenesis, as well as insulin resistance.
p53 regulates glucose transporters
Maintaining proper function of glucose transporters is critical in glucose homeostasis and suppression of diabetes (Shepherd and Kahn 1999). p53 regulates the function of glucose transporters by affecting both their transcription and their translocation (Figure 3A). For example, p53 activated by genotoxic stress can directly bind to the promoters of Glut1 and Glut4 and repress their transcription (Schwartzenberg-Bar-Yoseph, et al. 2004). p53 also represses the expression of GLUT3, but this occurs through an indirect mechanism by inhibition of IkB kinase, or IKK (Kawauchi, et al. 2008). Recent studies have also identified a role for p53 in the control of the localization of glucose transporters. Tumor-associated mutant forms of p53, such as the common ‘hotspot’ mutants R175H and R273H, can induce the Warburg effect by promoting translocation of GLUT1 to the plasma membrane (Zhang, et al. 2013). This pathway is mediated by activation of the GTPases RhoA and ROCK. Conversely, wild-type p53 negatively regulates GLUT1 translocation to the plasma membrane by virtue of its ability to transcriptionally induce the p53 target gene RRAD (Ras-related associated with diabetes) (Zhang, et al. 2014). Interestingly, RRAD was first identified as a gene up-regulated in tissues of type 2 diabetics, and overexpression of RRAD impairs glucose uptake and results in insulin resistance in muscle and fat tissues (Moyers, et al. 1996; Reynet and Kahn 1993). In sum, wild type p53 negatively regulates, and mutant p53 positively regulates, glucose transporters and glucose import.
Figure 3. Three pathways for the p53-mediated regulation of glucose homeostasis.
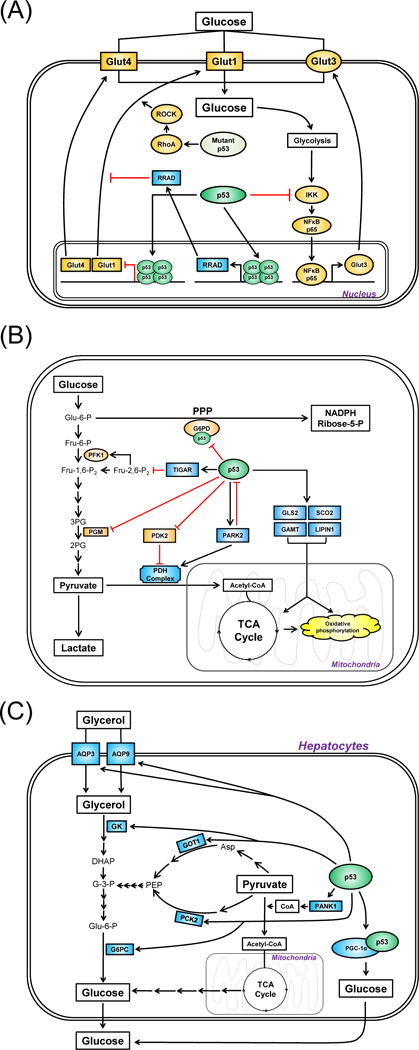
Wild-type p53 generally functions to induce circulating glucose level by (A) suppressing glucose uptake; (B) inhibiting aerobic glycolysis; and (C) promoting gluconeogenesis. The genes in squares represent transcriptional targets of p53. In (A), the genes colored in orange and blue represent positive and negative regulators of glucose uptake, respectively. In (B), the genes colored in orange and blue represent positive and negative regulators of glycolysis. In (C), the genes colored in blue represents positive regulators of gluconeogenesis. PPP, pentose phosphatase pathway; TCA cycle, the citric acid cycle; PDH, pyruvate dehydrogenase; PG, phosphoglycerate.
p53 negatively regulates glycolysis
p53 influences glucose levels by directly regulating both its degradation (glycolysis) and synthesis (gluconeogenesis). Following DNA damage, p53 can reprogram the cell’s energy-producing strategies from glycolysis to mitochondrial respiration (or oxidative phosphorylation) in order to suppress tumor progression (Figure 3B). The first p53 target gene identified to inhibit glycolysis is TIGAR (TP53-induced glycolysis and apoptosis regulator) (Bensaad, et al. 2006). Overexpression of TIGAR lowers the level of fructose-2,6-bisphosphate (Fru-2,6-P2), which activates the glycolysis stimulator PFK1 (6-phosphofructo-1-kinase). p53 also negatively regulates the stability of phosphoglycerate mutase (PGM), another enzyme critical for the completion of glycolysis by converting 3-phosphoglycerate (3-PG) to 2-phosphoglycerate (2-PG) (Kondoh, et al. 2005). Analysis of the pentose phosphate pathway (PPP), an alternative mechanism to consume glucose for energy production, showed that it is blocked by p53, in part through direct interaction between p53 and the rate-limiting enzyme, glucose 6-phosphate dehydrogenase (G6PD) (Jiang, et al. 2011). p53 also negatively regulates pyruvate dehydrogenase kinase-2 (PDK2) through both transcriptional and post-translational pathways to activate the PDH complex, which converts pyruvate to acetyl-CoA to tip the balance from glycolysis to mitochondrial respiration (Contractor and Harris 2012; Kolobova, et al. 2001). Finally, p53 transactivates Parkin (PARK2), a gene associated with Parkinsons disease, in order to inhibit glycolysis (Zhang, et al. 2011). Parkin inhibits glycolysis by positively regulating the expression level of PDHA1, which is the catalytic subunit of the PDH complex (Palacino, et al. 2004).
There are other ways that p53 negatively regulates glycolysis. For example, p53 transcriptionally induces target genes that favor mitochondrial respiration: these include SCO2, GLS2, GAMT and LIPIN1 (Assaily, et al. 2011; Hu, et al. 2010; Ide, et al. 2009; Matoba, et al. 2006). It has also been suggested that p53 can indirectly control glycolysis by regulating the mTOR and PI3K/Akt pathways. Specifically, these pathways are negatively regulated by the p53 target genes AMPK-B, Sestrin-1/2, TSC-2, REDD1 (which negatively regulate the mTOR pathway), and IGF-BP3 and PTEN (which negatively regulate the PI3K/AKT pathway) (see review (Liang, et al. 2013)). Finally, p53 regulates the expression of enzymes important for glucose uptake or glycolysis, including hexokinase-2 and phosphoglycerate mutase, PGM (Mathupala, et al. 1997; Ruiz-Lozano, et al. 1999). In the case of PGM, p53-mediated induction of PGM is muscle-specific, suggesting that tissue-specific functions of p53 exist within the context of glucose homeostasis regulation (Kondoh et al. 2005; Ruiz-Lozano et al. 1999).
p53 positively regulates gluconeogenesis
Recent studies have uncovered a direct way for p53 to increase the level of glucose, by promoting gluconeogenesis (Figure 3C). In an attempt to define p53-regulated metabolic pathways in liver-derived HepG2 cells using microarray analysis, Goldstein et al. identified multiple p53 target genes involved in the synthesis of glucose (Goldstein, et al. 2013). They showed that activation of p53 by nutlin-3a, a compound that stabilizes p53 by virtue of its ability to inhibit MDM2, causes the increased expression of genes involved in gluconeogenesis (G6PC, PCK2) and in the supply of glucogenic precursors (GK, AQP3, AQP9 and GOT1). Moreover, this group found that active p53 augments hepatic glucose production (HGP) in both HepG2 cells and primary mouse hepatocytes in glucogenic medium. p53-mediated induction of gluconeogenic enzymes in the liver was also shown in the Ay transgenic mouse model, which develops dietary obesity and diabetes, providing physiological relevance for p53-regulated gluconeogenesis in diabetes development (Minamino et al. 2009).
To identify the mechanisms underlying p53’s ability to induce gluconeogenesis, two separate groups used p53-deficient mice as tools, and found that these mice displayed reduced glucogenic capacity compared with wild-type mice in response to metabolic stress, such as starvation. Sen and colleagues found that p53 forms a complex with the transcriptional cofactor Peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1α) to facilitate the gluconeogenesis-related transcriptional machinery (Sen, et al. 2011). Another group showed that p53 directly transactivates pantothenate kinase-1 (PANK1) to promote gluconeogenesis in the liver (Wang, et al. 2013).
p53 regulates insulin resistance
Insulin resistance is the most reliable indicator of pre-diabetes and type 2 diabetes (Samuel and Shulman 2012), and p53 regulates insulin resistance in a number of ways (Figure 4). Many years ago it was discovered that p53 negatively regulates the transcription of the insulin receptor (Webster, et al. 1996). This was the first clue that p53 could influence insulin signaling directly. Impaired insulin sensitivity is accompanied by reduction of insulin signaling markers, including AKT phosphorylation, in peripheral tissues that are responsible for glucose uptake, such as adipose tissue, muscle, liver and brain. In rats fed with a high-fat diet, p53 was found to be activated in the peripheral tissues of obese rats that developed insulin resistance. As in the Ay mouse model of diabetes, an inhibitor of p53 was shown to ameliorate insulin resistance in this rat model (Homayounfar, et al. 2015).
Figure 4. The regulation of insulin resistance by p53.
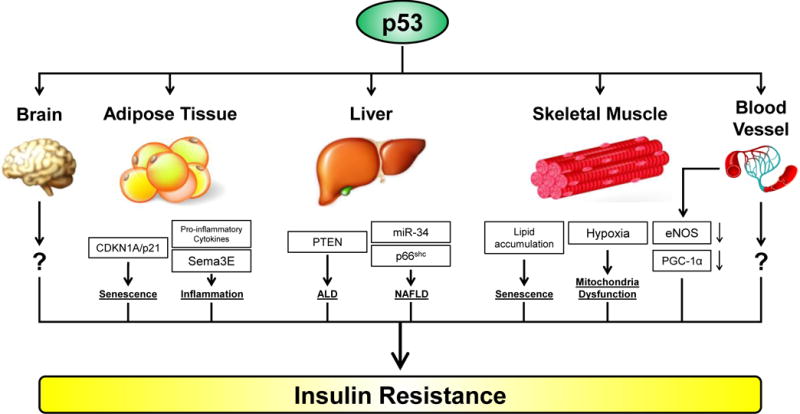
The activation of p53 induces insulin resistance through multiple tissues/organs, including adipose tissue, liver, skeletal muscle, endothelial cells, and the brain. Cross-talk between these tissues also occurs; for example endothelial cell-mediated insulin resistance in the skeletal muscle occurs via reduction in the former of eNOS and PGC-1α. ALD, alcoholic liver disease; NAFLD, non-alcoholic fatty liver disease.
Increased expression and activity of p53 is well known to occur in the adipocytes of obese mice (Yahagi, et al. 2003). As mentioned above, in Ay transgenic mice fed with a high-fat/high-sucrose diet, p53-mediated senescence in adipose tissue is critical for the development of diabetes-like phenotypes and insulin resistance (Minamino et al. 2009). p53-mediated senescence in adipose tissue is correlated with increased expression of p21/CDKN1A and pro-inflammatory cytokines, as well as decreased expression of anti-inflammatory cytokines. This group showed that adipose tissue-specific overexpression of p53 influences liver functions by regulating the expression of gluconeogenic enzymes in a paracrine fashion. The same group later found that p53-mediated chronic inflammation in adipose tissue leads to cardiac failure, signifying a connection between insulin resistance/diabetes and cardiovascular disease (Shimizu, et al. 2012). Using a diet-induced obesity (DIO) mouse model, this group went on to show that p53 regulates adipose tissue inflammation and insulin resistance by virtue, in part, of transactivation of class 3 semaphorin E (Sema3E), which acts as a chemo-attractant to promote infiltration of macrophages (Shimizu, et al. 2013).
A recent study suggests the convergence of p53 and the growth hormone (GH) pathway in adipose tissue during the development of insulin resistance. In response to a high-fat diet, p53 expression in adipose tissue is increased through activation of p38MAPK; the latter can be inhibited by GH antagonists (Bogazzi, et al. 2013). This finding provides a potential link between diet-induced obesity and p53-mediated adipose tissue inflammation and insulin resistance. As mentioned previously, the role of p21/CDKN1A in insulin resistance is thought to be due to its role in p53-mediated senescence. In an interesting study however, Inoue et al. demonstrated that p21/CDKN1A is critical for maintaining obesity-induced insulin resistance by promoting adipocyte differentiation and hypertrophy (Inoue, et al. 2008). This group showed that knockdown of p21/CDKN1A in the adipose tissue of mice with diet-induced obesity rendered these tissues susceptible to p53-induced apoptosis. These data suggest that inhibition of apoptosis, in addition to induction of p53-mediated senescence, may underlie the role of p21/CDKN1A in the development of insulin resistance.
p53 in the liver and skeletal muscle
Hepatic steatosis, or fatty liver, is one of the most common manifestations of obesity and type 2 diabetes (Williams, et al. 2013). Despite current uncertainty about whether hepatic steatosis alone is sufficient to drive insulin resistance, there is little doubt that steatosis-mediated liver dysfunction significantly contributes to diabetic phenotypes (Gruben, et al. 2014). Taking advantage of transgenic mouse models, Yahagi et al. showed that activation of p53 plays an important role in the pathogenesis of fatty liver disease, regardless of the status of obesity (Yahagi, et al. 2004). Similarly, Derdak et al. demonstrated that activation of p53 enhances hepatic steatosis and insulin resistance in both alcoholic liver disease (ALD) and non-alcoholic fatty liver disease (NAFLD); notably, this group found that both ALD and NAFLD can be ameliorated by inhibiting p53 activity (Derdak, et al. 2011; Derdak, et al. 2013). These studies also offered possible mechanistic underpinnings, and suggested that p53-mediated induction of PTEN and miRNA34a are responsible for the development of ALD and NAFLD, respectively. Using p53-knockout mice, another group confirmed a role for p53 in the progression of NAFLD, potentially through its downstream target p66shc (Tomita, et al. 2012). It is important to note that these phenotypes observed in mouse models are physiologically relevant, as the level of steatosis and p53 expression are positively correlated in human liver samples (Panasiuk, et al. 2006).
The roles of p53 in regulating mitochondrial function and oxidative stress are well documented (Puzio-Kuter 2011). In Goto-Kakizaki rats that develop early insulin resistance and type 2 diabetes, exercise training decreased p53 protein levels and restored mitochondrial function in skeletal muscle (Qi, et al. 2011). These findings suggested that, in the context of diabetes, p53 might contribute positively to the development of insulin resistance through inducing oxidative stress in skeletal muscle. Consistent with this hypothesis, p53 was found to be an important target to control peripheral artery disease (PAD), which is an ischemic limb syndrome strongly associated with type 2 diabetes (Morimoto, et al. 2011). Deferoxamine (DFX), a hypoxia mimetic and iron chelator, was used to mimic PAD in vitro and found to promote p53 accumulation in myoblast cells by suppressing MDM2 expression (Morimoto et al. 2011).
Other tissues and pathways involved in p53-mediated insulin resistance
The contribution of p53 to insulin resistance in other tissues was rarely studied until recently. Although not known as one of the main tissues to uptake glucose, dysfunctional microvascular tissue has been linked to the development of insulin resistance in obese and diabetic patients (Muris, et al. 2012). Impaired insulin signaling in endothelial cells is believed to promote insulin resistance by reducing blood flow and glucose-uptake capability of skeletal muscle (Clark, et al. 2003; Kubota, et al. 2011). Moreover, vascular p53 is activated as a transcription factor in diabetic patients (Orimo, et al. 2009). A direct connection between vascular p53 and insulin resistance was not uncovered until 2014, when Yokoyama et al. showed that in mice fed with a high-calorie diet, endothelial p53 promotes insulin resistance by suppressing the expression of endothelial nitric oxide synthase (eNOS) and peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) in skeletal muscle (Yokoyama, et al. 2014).
It is possible that p53 also impacts insulin sensitivity through other mechanisms, such as obesity. Obesity is a strong risk factor for type 2 diabetes and insulin resistance (Wang, et al. 2005). p53 transcriptionally regulates many genes associated with lipid metabolism (Goldstein, et al. 2012; Goldstein and Rotter 2012). Whether p53-mediated regulation of lipid-controlling genes plays a direct role in obesity remains to be determined. Recent evidence suggests that p53 might influence obesity by controlling food intake. Velasquez et al. showed that p53-deficient mice failed to respond to the ‘hunger hormone’ ghrelin to induce food intake (Velasquez, et al. 2011). This group posited that hypothalamic p53 is essential to mediate the orexigenic action of ghrelin. A later study showed that p53, by altering the expression levels of lipogenic and adipogenic genes, is necessary for ghrelin-mediated lipid storage in fat and liver (Porteiro, et al. 2013). These combined studies implicate p53 as a potentially important player in the regulation of feeding behavior and obesity. Interestingly, as if it serves to counteract the “obesity-driving” functions of p53, it was shown that p53 in the brown adipose tissue protects against diet-induced obesity (Molchadsky, et al. 2013). Therefore, the role of p53 in obesity may be tissue-specific.
There is some information available concerning the role of post-translational modifications of p53 on insulin sensitivity. For example, Ser15 phosphorylation of p53 (equivalent to Ser18 in mouse) is the most commonly used indicator for p53 activation (Banin, et al. 1998). Mutation at this site was predicted to inhibit p53’s ability to induce apoptosis or cell cycle arrest. Interestingly, however, mice possessing the Ser18 to alanine mutation did not display significant changes in growth arrest or apoptosis, but rather displayed phenotypes of impaired glucose homeostasis and increased insulin resistance (Armata, et al. 2010). Notably, this phenotype mimics the metabolic syndrome seen in Ataxia telangiectasia patients, who possess mutations in the p53 Ser15 kinase, ATM (ataxia telangiectasia mutated).
p53 lies both upstream and downstream of a key regulator of metabolic stress, AMPK
A master regulator of metabolism is the mammalian Target of Rapamycin, or mTOR. mTOR is a member of the phosphatidylinositol 3-kinase-related kinase protein family. It is a serine/threonine protein kinase that positively regulates protein synthesis, cell growth, and proliferation, and negatively regulates autophagy, which is a key catabolic pathway that is activated under conditions of nutrient starvation (Shaw and Cantley 2006). The p53 tumor suppressor negatively regulates mTOR in part by transactivating its negative regulator, AMPK (5′ AMP-activated protein kinase). In response to DNA damage p53 directly transactivates the beta1 and beta2 subunits of AMPK, as well as other negative regulators of mTOR, including the tuberous sclerosis complex protein TSC2, and the phosphatase PTEN (Feng, et al. 2007). All of these can inhibit mTOR activity, and therefore lead to growth cessation and induction of autophagy. Additionally, p53 transcriptionally regulates the genes Sestrin1 and Sestrin2; the protein product of these genes binds to and activates AMPK, which in turn phoshophorylates TSC2 and consequently inhibits mTOR. The loss of Sestrin2 during nutrient deprivation (Budanov and Karin 2008) or pharmacological inhibition of AMPK (Feng, et al. 2005) both significantly reduce p53-mediated inhibition of mTOR.
In normal healthy cells, p53 is kept at low levels by the E3 ubiquitin ligase MDM2 (HDM2 in mice), which ubiquitylates p53 and targets it for proteasomal degradation. In response to various forms of stress, phosphorylation of the amino terminus of p53 prevents interaction with MDM2, leading to p53 stabilization. Metabolic stress (nutrient deprivation) is known to induce p53 through phosphorylation on serine 15 mediated by the kinase AMPK, which responds to low AMP levels following ATP depletion. Once activated, p53 transactivates gene involved in cell cycle arrest and apoptosis, thereby eliminating cells with prolonged activation of AMPK (Feng et al. 2007; Jones, et al. 2005).
Polymorphisms in the p53 pathway: the codon 72 polymorphism of p53
Genetic polymorphisms arise over time and can undergo natural selection. Among these, some single nucleotide polymorphisms (SNPs) can significantly affect biological functions. In the context of p53, a common SNP occurs at amino acid 72, where nucleotide sequence CCC or CGC encodes for Proline (P72) or Arginine (R72), respectively (rs1042522; P72R). This polymorphism can have significant impact on p53 function. In response to DNA damage, the P72 variant has been shown to promote a stronger cell cycle arrest phenotype, while the R72 variant is a superior inducer of apoptosis (Dumont, et al. 2003; Kung, et al. 2015), although tissue-specific differences to these phenotypes have been observed (Azzam, et al. 2011). The impact of the codon 72 polymorphism on metabolic disease was not well studied, until recent reports linked this p53 SNP to susceptibility to diabetes. It was first reported that the codon 72 polymorphism was associated with type 1 diabetes in a Russian population (Spitsina, et al. 2007). Similar results were later found in an Italian cohort (Bitti, et al. 2011). In 2008, the R72 variant was identified as one of the strongest risk factors in more than 2,000 Finnish patients for type 2 diabetes (Gaulton, et al. 2008). Using clinical data from over 55,000 Europeans, a subsequent meta-analysis study confirmed this finding, that the R72 variant was linked to type 2 diabetes (Burgdorf, et al. 2011). Similar findings were made in the Chinese Han population, indicating that R72-increased susceptibility to type 2 diabetes is not a race-restricted phenomenon (Qu, et al. 2011). Finally, the R72 variant is associated with increased insulin resistance, even among diabetes patients (Bonfigli, et al. 2013).
A cohort study of over 2,500 Dutch and Finnish subjects found that the R72 variant is associated with increased waist circumference (Reiling, et al. 2012). These data suggested that the R72 variant might predispose individuals to adiposity/obesity, and that this might then lead to increased risk for the development of type 2 diabetes. A recent GWAS (genome-wide association study) found that the R72 variant of p53 is associated with increased body mass index (BMI) (Speliotes, et al. 2010). Consistent with these reports, a separate study showed that the association between BMI and diabetes incidence is much stronger in individuals of the R72 genotype (Gloria-Bottini, et al. 2011). Recently, our group used a mouse model for the codon 72 polymorphism of p53 in order to investigate the role of this SNP in obesity and diabetes. To understand this role, we monitored these mice following challenge with a high-fat diet (HFD). Mice with the R72 variant of p53 developed more severe obesity and glucose intolerance on a HFD, compared to mice with the proline 72 variant (P72). R72 mice on a HFD developed insulin resistance, islet hypertrophy, increased infiltration of immune cells, and fatty liver disease, but the earliest difference between R72 and P72 mice on a HFD was fat accumulation (obesity). Gene expression analyses indicated that two p53 target genes, with roles in inflammation (Tnf) and cholesterol metabolism (Npc1l1), were in part responsible for this phenotype (Kung, et al. 2016).
Other polymorphisms in p53 pathway genes
Several genetic polymorphisms in genes involved in p53 signaling pathways have been shown to affect p53 function (Whibley, et al. 2009), and some of these may likewise affect diabetes risk or severity. The A17708T polymorphism of p53 intron 10 is associated with the development of early uremic complications in diabetic patients (Szoke, et al. 2009). A recent genome wide association study in type 2 diabetic patients identified a SNP in ATM (rs11212617) that controls treatment response to metformin, the most commonly used drug for type 2 diabetes (GoDarts, et al. 2011). The authors suggested that this ATM variant might affect metformin efficacy by regulating downstream target of metformin, AMPK (AMP-activated protein kinase). ATM is an important regulator of p53, and ATM SNPs have been shown to affect p53 function (Takagi, et al. 2004). As noted above, p53 activity is regulated by AMPK (Jones et al. 2005). Therefore, it is possible that p53 plays an active role in the impact of rs11212617 on the glycemic response to metformin.
As mentioned previously, the T allele of TCF7L2 (rs7903146) confers the strongest risk for type 2 diabetes known in Caucasians (Florez 2007). TCF7L2 serves a unique dual-role in the p53 pathway, as it serves both as a regulatory transcription factor for p53, and a transcriptional target of p53 (Rother et al. 2004; Zhou et al. 2012). TCF7L2 regulates the expression of p53 and the p53 target gene p53INP1 to control the survival of pancreatic beta cells. Interestingly, a SNP in p53INP1 (rs896854) has also recently been identified as a type 2 diabetes susceptibility locus through a large scale GWAS project (Voight, et al. 2010) (Table 1). The overall data suggest that multiple SNPs in p53 and in genes involved in the p53 pathway can be predicted to play roles in metabolism and diabetes.
Table 1.
p53 pathway SNPs and diabetes
| Gene | SNP (Allele) | MAF* | Relationship with p53 | Impact | References |
|---|---|---|---|---|---|
| TP53 | rs1042522 (C>G) | 0.46(C) | G allele (Arg72) is a risk factor for obesity and diabetes | (Bitti et al. 2011; Bonfigli et al. 2013; Burgdorf et al. 2011; Gaulton et al. 2008; Gloria-Bottini et al. 2011; Qu et al. 2011; Reiling et al. 2012; Spitsina et al. 2007) | |
| TP53 | rs17880847 (A>T) | 0.01(T) | T allele predisposes to uremic complications in diabetic patients | (Szoke et al. 2009) | |
| TCF7L2 | rs7903146 (C>T) | 0.23(T) | Transcription factor and transcriptional target | T allele is a risk factor for diabetes in Caucasians | (Florez 2007) |
| P53INP1 | rs896854 (T>C) | 0.48(T) | Transcriptional target | C allele is a risk factor for diabetes | (Voight et al. 2010) |
| ATM | rs11212617 (C>A) | 0.47(A) | Activator | Regulates treatment response to metformin | (GoDarts et al. 2011) |
Source: Genome 1000 project database (1000genomes.org)
SNP, Single Nucleotide Polymorphism; MAF, Minor Allele Frequency.
p53 and the efficacy of diabetes treatment
The magnitude of the impact of diabetes on human health is often characterized by its associated complications, such as cardiovascular diseases, impaired wound healing, nephropathy, blindness and infertility (Brownlee 2005). In addition to its roles in diabetes risk and severity, p53 has also been shown to play an active role in these conditions by regulating apoptosis, cell cycle arrest, senescence and inflammation (Gurel, et al. 2014; Jazayeri, et al. 2008; Morimoto et al. 2011; Nguyen, et al. 2010; Samarakoon, et al. 2012; Zhao, et al. 2011). Targeting p53 to treat diabetes potentially offers extra benefits as an inclusive approach to alleviate both diabetes and downstream symptoms. The majority of studies outlined in this review have shown that inhibiting p53 using gene silencing, knock-out or pharmacological approaches can alleviate diabetes and its phenotypes; however, this trend has not been consistent. For example, nutlin-3a, a non-genotoxic activator of p53 that works via inhibition of the MDM2-p53 interaction, was able to improve pancreatic function and glucose homeostasis in a streptozotocin-induced mouse model of diabetes (Secchiero, et al. 2013). Similarly, targeting the peroxisome proliferator activated receptor-γ (PPARγ) has been a proven strategy for the drug development against obesity and diabetes (Blaschke, et al. 2006). PPARγ forms a heterodimer with retinoid X receptor (RXR), and PPARγ/RXR complexes can be targeted by RXR antagonists to confer anti-obesity and anti-diabetes activities (Yamauchi, et al. 2001). Nakatsuka et al. showed that the RXR antagonist HX531 up-regulated the p53-p21/CDKN1A pathway in adipocytes to induce cell cycle arrest and inhibit cellular hypertrophy that could otherwise lead to obesity (Nakatsuka, et al. 2012). Therefore, the therapeutic benefit of inhibiting or inducing p53 is likely to be influenced by cell type and stage of disease.
Conclusions
Given the magnitude of evidence presented in this review, it is clear that p53 is a key player in diabetes and the severity of diabetic phenotypes. For the most part, it is activation of p53 that was shown to exacerbate diabetic phenotypes. For example this protein is induced and activated in many cell types in animals and humans with diabetes, and in many cases pharmacological inhibition of p53 led to an amelioration of diabetic phenotypes. However, clearly the role of p53 in diabetes is complex, and in some settings, activation of p53 has been shown to be beneficial to disease outcome (Secchiero et al. 2013). Therefore, despite the magnitude of evidence suggesting that inhibitors of p53 might ameliorate diabetes, it is clearly premature to propose this avenue. Rather, identifying the key pathways and target genes controlled by p53, and targeting these, might be more advisable. Along these lines, in a diet-induced obesity model of diabetes, we identified the p53 target genes Tnf (TNF-alpha) and NPC1L1 (controls cholesterol metabolism) as markedly induced by p53 after as little of seven days on a high fat diet. Notably, we found that the use of antibody or small molecule inhibitors to either of these proteins caused reduced fat accumulation and ameliorated diabetic phenotypes (Kung et al. 2016). Therefore, there may be more merit in the complete identification of p53 target genes that are specifically induced in diabetic tissues, with the goal of targeting these genes for therapy.
Acknowledgments
Funding
This work was supported by the NIH (R01 CA102184 and CA201430 to M.M.). The authors wish to thank Subhasree Basu and Thibaut Barnoud for critical reading of this manuscript.
Footnotes
Declaration of Interest
The authors declare there are no conflicts of interest.
References
- Armata HL, Golebiowski D, Jung DY, Ko HJ, Kim JK, Sluss HK. Requirement of the ATM/p53 tumor suppressor pathway for glucose homeostasis. Mol Cell Biol. 2010;30:5787–5794. doi: 10.1128/MCB.00347-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assaily W, Rubinger DA, Wheaton K, Lin Y, Ma W, Xuan W, Brown-Endres L, Tsuchihara K, Mak TW, Benchimol S. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol Cell. 2011;44:491–501. doi: 10.1016/j.molcel.2011.08.038. [DOI] [PubMed] [Google Scholar]
- Azzam GA, Frank AK, Hollstein M, Murphy ME. Tissue-specific apoptotic effects of the p53 codon 72 polymorphism in a mouse model. Cell Cycle. 2011;10:1352–1355. doi: 10.4161/cc.10.9.15344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–1677. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- Belgardt BF, Ahmed K, Spranger M, Latreille M, Denzler R, Kondratiuk N, von Meyenn F, Villena FN, Herrmanns K, Bosco D, et al. The microRNA-200 family regulates pancreatic beta cell survival in type 2 diabetes. Nat Med. 2015;21:619–627. doi: 10.1038/nm.3862. [DOI] [PubMed] [Google Scholar]
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, Gottlieb E, Vousden KH. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–120. doi: 10.1016/j.cell.2006.05.036. [DOI] [PubMed] [Google Scholar]
- Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. Metabolic regulation by p53 family members. Cell Metab. 2013;18:617–633. doi: 10.1016/j.cmet.2013.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitti ML, Saccucci P, Capasso F, Piccinini S, Angelini F, Rapini N, Porcari M, Arcano S, Petrelli A, Del Duca E, et al. Genotypes of p53 codon 72 correlate with age at onset of type 1 diabetes in a sex-specific manner. J Pediatr Endocrinol Metab. 2011;24:437–439. doi: 10.1515/jpem.2011.058. [DOI] [PubMed] [Google Scholar]
- Blaschke F, Takata Y, Caglayan E, Law RE, Hsueh WA. Obesity, peroxisome proliferator-activated receptor, and atherosclerosis in type 2 diabetes. Arterioscler Thromb Vasc Biol. 2006;26:28–40. doi: 10.1161/01.ATV.0000191663.12164.77. [DOI] [PubMed] [Google Scholar]
- Bogazzi F, Raggi F, Russo D, Bohlooly YM, Sardella C, Urbani C, Lombardi M, Manetti L, Lupi I, Tornell J, et al. Growth hormone is necessary for the p53-mediated, obesity-induced insulin resistance in male C57BL/6J × CBA mice. Endocrinology. 2013;154:4226–4236. doi: 10.1210/en.2013-1220. [DOI] [PubMed] [Google Scholar]
- Bonfigli AR, Sirolla C, Testa R, Cucchi M, Spazzafumo L, Salvioli S, Ceriello A, Olivieri F, Festa R, Procopio AD, et al. The p53 codon 72 (Arg72Pro) polymorphism is associated with the degree of insulin resistance in type 2 diabetic subjects: a cross-sectional study. Acta Diabetol. 2013;50:429–436. doi: 10.1007/s00592-012-0450-x. [DOI] [PubMed] [Google Scholar]
- Bourdon JC, Fernandes K, Murray-Zmijewski F, Liu G, Diot A, Xirodimas DP, Saville MK, Lane DP. p53 isoforms can regulate p53 transcriptional activity. Genes Dev. 2005;19:2122–2137. doi: 10.1101/gad.1339905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 2008;134:451–460. doi: 10.1016/j.cell.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgdorf KS, Grarup N, Justesen JM, Harder MN, Witte DR, Jorgensen T, Sandbaek A, Lauritzen T, Madsbad S, Hansen T, et al. Studies of the association of Arg72Pro of tumor suppressor protein p53 with type 2 diabetes in a combined analysis of 55,521 Europeans. PLoS One. 2011;6:e15813. doi: 10.1371/journal.pone.0015813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JR, Ghafouri M, Mukerjee R, Bagashev A, Chabrashvili T, Sawaya BE. Role of p53 in neurodegenerative diseases. Neurodegener Dis. 2012;9:68–80. doi: 10.1159/000329999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherrington AD. Banting Lecture 1997. Control of glucose uptake and release by the liver in vivo. Diabetes. 1999;48:1198–1214. doi: 10.2337/diabetes.48.5.1198. [DOI] [PubMed] [Google Scholar]
- Clark MG, Wallis MG, Barrett EJ, Vincent MA, Richards SM, Clerk LH, Rattigan S. Blood flow and muscle metabolism: a focus on insulin action. Am J Physiol Endocrinol Metab. 2003;284:E241–258. doi: 10.1152/ajpendo.00408.2002. [DOI] [PubMed] [Google Scholar]
- Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(Suppl 2):S97–107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012;72:560–567. doi: 10.1158/0008-5472.CAN-11-1215. [DOI] [PubMed] [Google Scholar]
- Danilova N, Sakamoto KM, Lin S. p53 family in development. Mech Dev. 2008;125:919–931. doi: 10.1016/j.mod.2008.09.003. [DOI] [PubMed] [Google Scholar]
- Derdak Z, Lang CH, Villegas KA, Tong M, Mark NM, de la Monte SM, Wands JR. Activation of p53 enhances apoptosis and insulin resistance in a rat model of alcoholic liver disease. J Hepatol. 2011;54:164–172. doi: 10.1016/j.jhep.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdak Z, Villegas KA, Harb R, Wu AM, Sousa A, Wands JR. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J Hepatol. 2013;58:785–791. doi: 10.1016/j.jhep.2012.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont P, Leu JI, Della Pietra AC, 3rd, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet. 2003;33:357–365. doi: 10.1038/ng1093. [DOI] [PubMed] [Google Scholar]
- Feng Z, Hu W, de Stanchina E, Teresky AK, Jin S, Lowe S, Levine AJ. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007;67:3043–3053. doi: 10.1158/0008-5472.CAN-06-4149. [DOI] [PubMed] [Google Scholar]
- Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U S A. 2005;102:8204–8209. doi: 10.1073/pnas.0502857102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florez JC. The new type 2 diabetes gene TCF7L2. Curr Opin Clin Nutr Metab Care. 2007;10:391–396. doi: 10.1097/MCO.0b013e3281e2c9be. [DOI] [PubMed] [Google Scholar]
- Gaulton KJ, Willer CJ, Li Y, Scott LJ, Conneely KN, Jackson AU, Duren WL, Chines PS, Narisu N, Bonnycastle LL, et al. Comprehensive association study of type 2 diabetes and related quantitative traits with 222 candidate genes. Diabetes. 2008;57:3136–3144. doi: 10.2337/db07-1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloria-Bottini F, Banci M, Saccucci P, Magrini A, Bottini E. Is there a role of p53 codon 72 polymorphism in the susceptibility to type 2 diabetes in overweight subjects? A study in patients with cardiovascular diseases. Diabetes Res Clin Pract. 2011;91:e64–67. doi: 10.1016/j.diabres.2010.11.031. [DOI] [PubMed] [Google Scholar]
- GoDarts, Group UDPS, Wellcome Trust Case Control C. Zhou K, Bellenguez C, Spencer CC, Bennett AJ, Coleman RL, Tavendale R, Hawley SA, et al. Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat Genet. 2011;43:117–120. doi: 10.1038/ng.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein I, Ezra O, Rivlin N, Molchadsky A, Madar S, Goldfinger N, Rotter V. p53, a novel regulator of lipid metabolism pathways. J Hepatol. 2012;56:656–662. doi: 10.1016/j.jhep.2011.08.022. [DOI] [PubMed] [Google Scholar]
- Goldstein I, Rotter V. Regulation of lipid metabolism by p53 - fighting two villains with one sword. Trends Endocrinol Metab. 2012;23:567–575. doi: 10.1016/j.tem.2012.06.007. [DOI] [PubMed] [Google Scholar]
- Goldstein I, Yizhak K, Madar S, Goldfinger N, Ruppin E, Rotter V. p53 promotes the expression of gluconeogenesis-related genes and enhances hepatic glucose production. Cancer Metab. 2013;1:9. doi: 10.1186/2049-3002-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38:320–323. doi: 10.1038/ng1732. [DOI] [PubMed] [Google Scholar]
- Gruben N, Shiri-Sverdlov R, Koonen DP, Hofker MH. Nonalcoholic fatty liver disease: A main driver of insulin resistance or a dangerous liaison? Biochim Biophys Acta. 2014;1842:2329–2343. doi: 10.1016/j.bbadis.2014.08.004. [DOI] [PubMed] [Google Scholar]
- Gurel Z, Zaro BW, Pratt MR, Sheibani N. Identification of O-GlcNAc modification targets in mouse retinal pericytes: implication of p53 in pathogenesis of diabetic retinopathy. PLoS One. 2014;9:e95561. doi: 10.1371/journal.pone.0095561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinault C, Kawamori D, Liew CW, Maier B, Hu J, Keller SR, Mirmira RG, Scrable H, Kulkarni RN. Delta40 Isoform of p53 controls beta-cell proliferation and glucose homeostasis in mice. Diabetes. 2011;60:1210–1222. doi: 10.2337/db09-1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayounfar R, Jeddi-Tehrani M, Cheraghpour M, Ghorbani A, Zand H. Relationship of p53 accumulation in peripheral tissues of high-fat diet-induced obese rats with decrease in metabolic and oncogenic signaling of insulin. Gen Comp Endocrinol. 2015;214:134–139. doi: 10.1016/j.ygcen.2014.06.029. [DOI] [PubMed] [Google Scholar]
- Hoshino A, Ariyoshi M, Okawa Y, Kaimoto S, Uchihashi M, Fukai K, Iwai-Kanai E, Ikeda K, Ueyama T, Ogata T, et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proc Natl Acad Sci U S A. 2014;111:3116–3121. doi: 10.1073/pnas.1318951111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Zhang C, Wu R, Sun Y, Levine A, Feng Z. Glutaminase 2, a novel p53 target gene regulating energy metabolism and antioxidant function. Proc Natl Acad Sci U S A. 2010;107:7455–7460. doi: 10.1073/pnas.1001006107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ide T, Brown-Endres L, Chu K, Ongusaha PP, Ohtsuka T, El-Deiry WS, Aaronson SA, Lee SW. GAMT, a p53-inducible modulator of apoptosis, is critical for the adaptive response to nutrient stress. Mol Cell. 2009;36:379–392. doi: 10.1016/j.molcel.2009.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Inoue N, Yahagi N, Yamamoto T, Ishikawa M, Watanabe K, Matsuzaka T, Nakagawa Y, Takeuchi Y, Kobayashi K, Takahashi A, et al. Cyclin-dependent kinase inhibitor, p21WAF1/CIP1, is involved in adipocyte differentiation and hypertrophy, linking to obesity, and insulin resistance. J Biol Chem. 2008;283:21220–21229. doi: 10.1074/jbc.M801824200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazayeri L, Callaghan MJ, Grogan RH, Hamou CD, Thanik V, Ingraham CR, Capell BC, Pelo CR, Gurtner GC. Diabetes increases p53-mediated apoptosis following ischemia. Plast Reconstr Surg. 2008;121:1135–1143. doi: 10.1097/01.prs.0000302499.18738.c2. [DOI] [PubMed] [Google Scholar]
- Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol. 2011;13:310–316. doi: 10.1038/ncb2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, Plas DR, Kubek S, Buzzai M, Mu J, Xu Y, Birnbaum MJ, Thompson CB. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18:283–293. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- Karunakaran U, Kim HJ, Kim JY, Lee IK. Guards and culprits in the endoplasmic reticulum: glucolipotoxicity and beta-cell failure in type II diabetes. Exp Diabetes Res. 2012;2012:639762. doi: 10.1155/2012/639762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi K, Araki K, Tobiume K, Tanaka N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat Cell Biol. 2008;10:611–618. doi: 10.1038/ncb1724. [DOI] [PubMed] [Google Scholar]
- Kim WH, Lee JW, Gao B, Jung MH. Synergistic activation of JNK/SAPK induced by TNF-alpha and IFN-gamma: apoptosis of pancreatic beta-cells via the p53 and ROS pathway. Cell Signal. 2005;17:1516–1532. doi: 10.1016/j.cellsig.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J. 2001;358:69–77. doi: 10.1042/0264-6021:3580069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65:177–185. [PubMed] [Google Scholar]
- Koshkin V, Wang X, Scherer PE, Chan CB, Wheeler MB. Mitochondrial functional state in clonal pancreatic beta-cells exposed to free fatty acids. J Biol Chem. 2003;278:19709–19715. doi: 10.1074/jbc.M209709200. [DOI] [PubMed] [Google Scholar]
- Kubota T, Kubota N, Kumagai H, Yamaguchi S, Kozono H, Takahashi T, Inoue M, Itoh S, Takamoto I, Sasako T, et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011;13:294–307. doi: 10.1016/j.cmet.2011.01.018. [DOI] [PubMed] [Google Scholar]
- Kung CP, Budina A, Balaburski G, Bergenstock MK, Murphy M. Autophagy in tumor suppression and cancer therapy. Crit Rev Eukaryot Gene Expr. 2011;21:71–100. doi: 10.1615/critreveukargeneexpr.v21.i1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung CP, Khaku S, Jennis M, Zhou Y, Murphy ME. Identification of TRIML2, a novel p53 target, that enhances p53 SUMOylation and regulates the transactivation of proapoptotic genes. Mol Cancer Res. 2015;13:250–262. doi: 10.1158/1541-7786.MCR-14-0385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung CP, Leu JI, Basu S, Khaku S, Anokye-Danso F, Liu Q, George DL, Ahima RS, Murphy ME. The P72R Polymorphism of p53 Predisposes to Obesity and Metabolic Dysfunction. Cell Rep. 2016;14:2413–2425. doi: 10.1016/j.celrep.2016.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Las G, Shirihai OS. The role of autophagy in beta-cell lipotoxicity and type 2 diabetes. Diabetes Obes Metab. 2010;12(Suppl 2):15–19. doi: 10.1111/j.1463-1326.2010.01268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer. 2009;9:749–758. doi: 10.1038/nrc2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Tomasini R, McKeon FD, Mak TW, Melino G. The p53 family: guardians of maternal reproduction. Nat Rev Mol Cell Biol. 2011;12:259–265. doi: 10.1038/nrm3086. [DOI] [PubMed] [Google Scholar]
- Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R, Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149:1269–1283. doi: 10.1016/j.cell.2012.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Liu J, Feng Z. The regulation of cellular metabolism by tumor suppressor p53. Cell Biosci. 2013;3:9. doi: 10.1186/2045-3701-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovis P, Roggli E, Laybutt DR, Gattesco S, Yang JY, Widmann C, Abderrahmani A, Regazzi R. Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes. 2008;57:2728–2736. doi: 10.2337/db07-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan E, Gogna R, Bhatt M, Pati U, Kuppusamy P, Mahdi AA. Regulation of glucose metabolism by p53: emerging new roles for the tumor suppressor. Oncotarget. 2011;2:948–957. doi: 10.18632/oncotarget.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathupala SP, Heese C, Pedersen PL. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J Biol Chem. 1997;272:22776–22780. doi: 10.1074/jbc.272.36.22776. [DOI] [PubMed] [Google Scholar]
- Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM. p53 regulates mitochondrial respiration. Science. 2006;312:1650–1653. doi: 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci U S A. 2001;98:11598–11603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez D, Shatz M, Resnick MA. Interactions between the tumor suppressor p53 and immune responses. Curr Opin Oncol. 2013;25:85–92. doi: 10.1097/CCO.0b013e32835b6386. [DOI] [PubMed] [Google Scholar]
- Minamino T, Orimo M, Shimizu I, Kunieda T, Yokoyama M, Ito T, Nojima A, Nabetani A, Oike Y, Matsubara H, et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 2009;15:1082–1087. doi: 10.1038/nm.2014. [DOI] [PubMed] [Google Scholar]
- Molchadsky A, Ezra O, Amendola PG, Krantz D, Kogan-Sakin I, Buganim Y, Rivlin N, Goldfinger N, Folgiero V, Falcioni R, et al. p53 is required for brown adipogenic differentiation and has a protective role against diet-induced obesity. Cell Death Differ. 2013;20:774–783. doi: 10.1038/cdd.2013.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto Y, Bando YK, Shigeta T, Monji A, Murohara T. Atorvastatin prevents ischemic limb loss in type 2 diabetes: role of p53. J Atheroscler Thromb. 2011;18:200–208. doi: 10.5551/jat.6437. [DOI] [PubMed] [Google Scholar]
- Moyers JS, Bilan PJ, Reynet C, Kahn CR. Overexpression of Rad inhibits glucose uptake in cultured muscle and fat cells. J Biol Chem. 1996;271:23111–23116. doi: 10.1074/jbc.271.38.23111. [DOI] [PubMed] [Google Scholar]
- Muris DM, Houben AJ, Schram MT, Stehouwer CD. Microvascular dysfunction is associated with a higher incidence of type 2 diabetes mellitus: a systematic review and meta-analysis. Arterioscler Thromb Vasc Biol. 2012;32:3082–3094. doi: 10.1161/ATVBAHA.112.300291. [DOI] [PubMed] [Google Scholar]
- Nakatsuka A, Wada J, Hida K, Hida A, Eguchi J, Teshigawara S, Murakami K, Kanzaki M, Inoue K, Terami T, et al. RXR antagonism induces G0/G1 cell cycle arrest and ameliorates obesity by up-regulating the p53-p21(Cip1) pathway in adipocytes. J Pathol. 2012;226:784–795. doi: 10.1002/path.3001. [DOI] [PubMed] [Google Scholar]
- Nguyen PD, Tutela JP, Thanik VD, Knobel D, Allen RJ, Jr, Chang CC, Levine JP, Warren SM, Saadeh PB. Improved diabetic wound healing through topical silencing of p53 is associated with augmented vasculogenic mediators. Wound Repair Regen. 2010;18:553–559. doi: 10.1111/j.1524-475X.2010.00638.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orimo M, Minamino T, Miyauchi H, Tateno K, Okada S, Moriya J, Komuro I. Protective role of SIRT1 in diabetic vascular dysfunction. Arterioscler Thromb Vasc Biol. 2009;29:889–894. doi: 10.1161/ATVBAHA.109.185694. [DOI] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Panasiuk A, Dzieciol J, Panasiuk B, Prokopowicz D. Expression of p53, Bax and Bcl-2 proteins in hepatocytes in non-alcoholic fatty liver disease. World J Gastroenterol. 2006;12:6198–6202. doi: 10.3748/wjg.v12.i38.6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietsch EC, Sykes SM, McMahon SB, Murphy ME. The p53 family and programmed cell death. Oncogene. 2008;27:6507–6521. doi: 10.1038/onc.2008.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polonsky KS. The past 200 years in diabetes. N Engl J Med. 2012;367:1332–1340. doi: 10.1056/NEJMra1110560. [DOI] [PubMed] [Google Scholar]
- Porteiro B, Diaz-Ruiz A, Martinez G, Senra A, Vidal A, Serrano M, Gualillo O, Lopez M, Malagon MM, Dieguez C, et al. Ghrelin requires p53 to stimulate lipid storage in fat and liver. Endocrinology. 2013;154:3671–3679. doi: 10.1210/en.2013-1176. [DOI] [PubMed] [Google Scholar]
- Poyurovsky MV, Prives C. P53 and aging: A fresh look at an old paradigm. Aging (Albany NY) 2010;2:380–382. doi: 10.18632/aging.100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puzio-Kuter AM. The Role of p53 in Metabolic Regulation. Genes Cancer. 2011;2:385–391. doi: 10.1177/1947601911409738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Z, He J, Zhang Y, Shao Y, Ding S. Exercise training attenuates oxidative stress and decreases p53 protein content in skeletal muscle of type 2 diabetic Goto-Kakizaki rats. Free Radic Biol Med. 2011;50:794–800. doi: 10.1016/j.freeradbiomed.2010.12.022. [DOI] [PubMed] [Google Scholar]
- Qu L, He B, Pan Y, Xu Y, Zhu C, Tang Z, Bao Q, Tian F, Wang S. Association between polymorphisms in RAPGEF1, TP53, NRF1 and type 2 diabetes in Chinese Han population. Diabetes Res Clin Pract. 2011;91:171–176. doi: 10.1016/j.diabres.2010.11.019. [DOI] [PubMed] [Google Scholar]
- Reiling E, Lyssenko V, Boer JM, Imholz S, Verschuren WM, Isomaa B, Tuomi T, Groop L, Dolle ME. Codon 72 polymorphism (rs1042522) of TP53 is associated with changes in diastolic blood pressure over time. Eur J Hum Genet. 2012;20:696–700. doi: 10.1038/ejhg.2011.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynet C, Kahn CR. Rad: a member of the Ras family overexpressed in muscle of type II diabetic humans. Science. 1993;262:1441–1444. doi: 10.1126/science.8248782. [DOI] [PubMed] [Google Scholar]
- Rother K, Johne C, Spiesbach K, Haugwitz U, Tschop K, Wasner M, Klein-Hitpass L, Moroy T, Mossner J, Engeland K. Identification of Tcf-4 as a transcriptional target of p53 signalling. Oncogene. 2004;23:3376–3384. doi: 10.1038/sj.onc.1207464. [DOI] [PubMed] [Google Scholar]
- Ruiz-Lozano P, Hixon ML, Wagner MW, Flores AI, Ikawa S, Baldwin AS, Jr, Chien KR, Gualberto A. p53 is a transcriptional activator of the muscle-specific phosphoglycerate mutase gene and contributes in vivo to the control of its cardiac expression. Cell Growth Differ. 1999;10:295–306. [PubMed] [Google Scholar]
- Samarakoon R, Overstreet JM, Higgins SP, Higgins PJ. TGF-beta1 –> SMAD/p53/USF2 –> PAI-1 transcriptional axis in ureteral obstruction-induced renal fibrosis. Cell Tissue Res. 2012;347:117–128. doi: 10.1007/s00441-011-1181-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148:852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446:444–448. doi: 10.1038/nature05602. [DOI] [PubMed] [Google Scholar]
- Schwartzenberg-Bar-Yoseph F, Armoni M, Karnieli E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res. 2004;64:2627–2633. doi: 10.1158/0008-5472.can-03-0846. [DOI] [PubMed] [Google Scholar]
- Secchiero P, Toffoli B, Melloni E, Agnoletto C, Monasta L, Zauli G. The MDM2 inhibitor Nutlin-3 attenuates streptozotocin-induced diabetes mellitus and increases serum level of IL-12p40. Acta Diabetol. 2013;50:899–906. doi: 10.1007/s00592-013-0476-8. [DOI] [PubMed] [Google Scholar]
- Sen N, Satija YK, Das S. PGC-1alpha, a key modulator of p53, promotes cell survival upon metabolic stress. Mol Cell. 2011;44:621–634. doi: 10.1016/j.molcel.2011.08.044. [DOI] [PubMed] [Google Scholar]
- Shang X, Vasudevan SA, Yu Y, Ge N, Ludwig AD, Wesson CL, Wang K, Burlingame SM, Zhao YJ, Rao PH, et al. Dual-specificity phosphatase 26 is a novel p53 phosphatase and inhibits p53 tumor suppressor functions in human neuroblastoma. Oncogene. 2010;29:4938–4946. doi: 10.1038/onc.2010.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- Shepherd PR, Kahn BB. Glucose transporters and insulin action–implications for insulin resistance and diabetes mellitus. N Engl J Med. 1999;341:248–257. doi: 10.1056/NEJM199907223410406. [DOI] [PubMed] [Google Scholar]
- Shimizu I, Yoshida Y, Katsuno T, Tateno K, Okada S, Moriya J, Yokoyama M, Nojima A, Ito T, Zechner R, et al. p53-induced adipose tissue inflammation is critically involved in the development of insulin resistance in heart failure. Cell Metab. 2012;15:51–64. doi: 10.1016/j.cmet.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Shimizu I, Yoshida Y, Moriya J, Nojima A, Uemura A, Kobayashi Y, Minamino T. Semaphorin3E-induced inflammation contributes to insulin resistance in dietary obesity. Cell Metab. 2013;18:491–504. doi: 10.1016/j.cmet.2013.09.001. [DOI] [PubMed] [Google Scholar]
- Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Lango Allen H, Lindgren CM, Luan J, Magi R, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitsina EV, Iakunina N, Chudakova DA, Nikitin AG, Svetlova GN, Soluianova TN, Strokov IA, Nosikov VV. Association of polymorphous markers Pro72Arg and C(−594)CC OF TP53 gene with diabetic polyneuropathy in patients with type 1 diabetes mellitus living in Moscow. Mol Biol (Mosk) 2007;41:989–993. [PubMed] [Google Scholar]
- Stavridi ES, Halazonetis TD. p53 and stress in the ER. Genes Dev. 2004;18:241–244. doi: 10.1101/gad.1181704. [DOI] [PubMed] [Google Scholar]
- Szoke D, Molnar B, Solymosi N, Racz K, Gergics P, Blasko B, Vasarhelyi B, Vannay A, Mandy Y, Klausz G, et al. Polymorphisms of the ApoE, HSD3B1, IL-1beta and p53 genes are associated with the development of early uremic complications in diabetic patients: results of a DNA resequencing array study. Int J Mol Med. 2009;23:217–227. [PubMed] [Google Scholar]
- Takagi M, Tsuchida R, Oguchi K, Shigeta T, Nakada S, Shimizu K, Ohki M, Delia D, Chessa L, Taya Y, et al. Identification and characterization of polymorphic variations of the ataxia telangiectasia mutated (ATM) gene in childhood Hodgkin disease. Blood. 2004;103:283–290. doi: 10.1182/blood-2003-01-0094. [DOI] [PubMed] [Google Scholar]
- Tavana O, Puebla-Osorio N, Sang M, Zhu C. Absence of p53-dependent apoptosis combined with nonhomologous end-joining deficiency leads to a severe diabetic phenotype in mice. Diabetes. 2010;59:135–142. doi: 10.2337/db09-0792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita K, Teratani T, Suzuki T, Oshikawa T, Yokoyama H, Shimamura K, Nishiyama K, Mataki N, Irie R, Minamino T, et al. p53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J Hepatol. 2012;57:837–843. doi: 10.1016/j.jhep.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Tornovsky-Babeay S, Dadon D, Ziv O, Tzipilevich E, Kadosh T, Schyr-Ben Haroush R, Hija A, Stolovich-Rain M, Furth-Lavi J, Granot Z, et al. Type 2 diabetes and congenital hyperinsulinism cause DNA double-strand breaks and p53 activity in beta cells. Cell Metab. 2014;19:109–121. doi: 10.1016/j.cmet.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasquez DA, Martinez G, Romero A, Vazquez MJ, Boit KD, Dopeso-Reyes IG, Lopez M, Vidal A, Nogueiras R, Dieguez C. The central Sirtuin 1/p53 pathway is essential for the orexigenic action of ghrelin. Diabetes. 2011;60:1177–1185. doi: 10.2337/db10-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Ryan KM. p53 and metabolism. Nat Rev Cancer. 2009;9:691–700. doi: 10.1038/nrc2715. [DOI] [PubMed] [Google Scholar]
- Wang SJ, Yu G, Jiang L, Li T, Lin Q, Tang Y, Gu W. p53-Dependent regulation of metabolic function through transcriptional activation of pantothenate kinase-1 gene. Cell Cycle. 2013;12:753–761. doi: 10.4161/cc.23597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Rimm EB, Stampfer MJ, Willett WC, Hu FB. Comparison of abdominal adiposity and overall obesity in predicting risk of type 2 diabetes among men. Am J Clin Nutr. 2005;81:555–563. doi: 10.1093/ajcn/81.3.555. [DOI] [PubMed] [Google Scholar]
- Webster NJ, Resnik JL, Reichart DB, Strauss B, Haas M, Seely BL. Repression of the insulin receptor promoter by the tumor suppressor gene product p53: a possible mechanism for receptor overexpression in breast cancer. Cancer Res. 1996;56:2781–2788. [PubMed] [Google Scholar]
- Whibley C, Pharoah PD, Hollstein M. p53 polymorphisms: cancer implications. Nat Rev Cancer. 2009;9:95–107. doi: 10.1038/nrc2584. [DOI] [PubMed] [Google Scholar]
- Williams KH, Shackel NA, Gorrell MD, McLennan SV, Twigg SM. Diabetes and nonalcoholic Fatty liver disease: a pathogenic duo. Endocr Rev. 2013;34:84–129. doi: 10.1210/er.2012-1009. [DOI] [PubMed] [Google Scholar]
- Wrede CE, Dickson LM, Lingohr MK, Briaud I, Rhodes CJ. Protein kinase B/Akt prevents fatty acid-induced apoptosis in pancreatic beta-cells (INS-1) J Biol Chem. 2002;277:49676–49684. doi: 10.1074/jbc.M208756200. [DOI] [PubMed] [Google Scholar]
- Yahagi N, Shimano H, Matsuzaka T, Najima Y, Sekiya M, Nakagawa Y, Ide T, Tomita S, Okazaki H, Tamura Y, et al. p53 Activation in adipocytes of obese mice. J Biol Chem. 2003;278:25395–25400. doi: 10.1074/jbc.M302364200. [DOI] [PubMed] [Google Scholar]
- Yahagi N, Shimano H, Matsuzaka T, Sekiya M, Najima Y, Okazaki S, Okazaki H, Tamura Y, Iizuka Y, Inoue N, et al. p53 involvement in the pathogenesis of fatty liver disease. J Biol Chem. 2004;279:20571–20575. doi: 10.1074/jbc.M400884200. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Waki H, Kamon J, Murakami K, Motojima K, Komeda K, Miki H, Kubota N, Terauchi Y, Tsuchida A, et al. Inhibition of RXR and PPARgamma ameliorates diet-induced obesity and type 2 diabetes. J Clin Invest. 2001;108:1001–1013. doi: 10.1172/JCI12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama M, Okada S, Nakagomi A, Moriya J, Shimizu I, Nojima A, Yoshida Y, Ichimiya H, Kamimura N, Kobayashi Y, et al. Inhibition of endothelial p53 improves metabolic abnormalities related to dietary obesity. Cell Rep. 2014;7:1691–1703. doi: 10.1016/j.celrep.2014.04.046. [DOI] [PubMed] [Google Scholar]
- Yuan H, Zhang X, Huang X, Lu Y, Tang W, Man Y, Wang S, Xi J, Li J. NADPH oxidase 2-derived reactive oxygen species mediate FFAs-induced dysfunction and apoptosis of beta-cells via JNK, p38 MAPK and p53 pathways. PLoS One. 2010;5:e15726. doi: 10.1371/journal.pone.0015726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci U S A. 2011;108:16259–16264. doi: 10.1073/pnas.1113884108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Liu J, Liang Y, Wu R, Zhao Y, Hong X, Lin M, Yu H, Liu L, Levine AJ, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935. doi: 10.1038/ncomms3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Liu J, Wu R, Liang Y, Lin M, Liu J, Chan CS, Hu W, Feng Z. Tumor suppressor p53 negatively regulates glycolysis stimulated by hypoxia through its target RRAD. Oncotarget. 2014;5:5535–5546. doi: 10.18632/oncotarget.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Tan Y, Dai J, Li B, Guo L, Cui J, Wang G, Shi X, Zhang X, Mellen N, et al. Exacerbation of diabetes-induced testicular apoptosis by zinc deficiency is most likely associated with oxidative stress, p38 MAPK activation, and p53 activation in mice. Toxicol Lett. 2011;200:100–106. doi: 10.1016/j.toxlet.2010.11.001. [DOI] [PubMed] [Google Scholar]
- Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001;3:973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhang E, Berggreen C, Jing X, Osmark P, Lang S, Cilio CM, Goransson O, Groop L, Renstrom E, et al. Survival of pancreatic beta cells is partly controlled by a TCF7L2-p53-p53INP1-dependent pathway. Hum Mol Genet. 2012;21:196–207. doi: 10.1093/hmg/ddr454. [DOI] [PubMed] [Google Scholar]