

Conspectus
Porphobilinogen synthase (PBGS), also known as 5-aminolevulinate dehydratase, is an essential enzyme in the biosynthesis of all tetrapyrroles, which function in respiration, photosynthesis, and methanogenesis. Throughout evolution, PBGS adapted to a diversity of cellular niches and evolved to use an unusual variety of metal ions both for catalytic function and to control protein multimerization. With regard to the active site, some PBGS require Zn2+; a subset of those, including human PBGS, contain a constellation of cysteine residues that acts as a sink for the environmental toxin Pb2+. PBGS that do not require the soft metal ion Zn2+ at the active site, instead are suspected of using the hard metal, Mg2+.
The most unexpected property of the PBGS family of enzymes is a dissociative allosteric mechanism that utilizes an equilibrium of architecturally and functionally distinct protein assemblies. The high-activity assembly is an octamer in which inter-subunit interactions modulate active-site lid motion. This octamer can dissociate to dimer, the dimer can undergo a hinge twist, and the twisted dimer can assemble to a low-activity hexamer. The hexamer does not have the intersubunit interactions required to stabilize a closed conformation of the active site lid. PBGS active site chemistry benefits from a closed lid because porphobilinogen biosynthesis includes Schiff base formation, which requires deprotonated lysine amino groups. N-terminal and C-terminal sequence extensions dictate whether or not a specific species of PBGS can sample the hexameric assembly. The bulk of species (nearly all except animals and yeasts) use Mg2+ as an allosteric activator. Mg2+ functions allosterically by binding to an intersubunit interface that is present in the octamer but absent in the hexamer. This conformational selection allosteric mechanism is purported to be essential to avoid the untimely accumulation of phototoxic chlorophyll precursors in plants. For those PBGS that do not use the allosteric Mg2+, there is a spatially equivalent arginine derived guanidium group. Deprotonation of this residue promotes formation of the hexamer and accounts for the basic arm of the bell-shaped pH vs. activity profile of human PBGS.
A human inborn error of metabolism, known as ALAD porphyria, is attributed to PBGS variants that favor the hexameric assembly. The existence of one such variant, F12L, which dramatically stabilizes the human PBGS hexamer, allowed crystal structure determination for the hexamer. Without this crystal structure, and octameric PBGS structures containing the allosteric Mg2+, it would have been difficult to decipher the structural basis for PBGS allostery. The requirement for multimer dissociation as an intermediate step in PBGS allostery was established by monitoring subunit disproportionation during the turnover-dependent transition of heteromeric PBGS (comprised of human wild type and F12L) from hexamer to octamer. One outcome of these studies was the definition of the dissociative morpheein model of protein allostery. The phylogenetically variable timescales for PBGS multimer interconversion result in atypical kinetic and biophysical behaviors. These behaviors can serve to identify other proteins that use the morpheein model of protein allostery.
Graphical Abstract

Introduction
Porphobilinogen is the fundamental biological pyrrole precursor to a rich spectrum of tetrapyrrole pigments (e.g. porphyrins, corrins, chlorins). Tetrapyrroles participate in respiration, photosynthesis, and methanogenesis and are essential for nearly all cellular life. Only one enzyme is known to synthesize porphobilinogen; originally named δ-aminolevulinic acid dehydra(ta)se (ALAD)1, the IUPAC-IUB rules of the 1970’s suggested the name porphobilinogen synthase (PBGS) (EC 4.2.1.24).2 Both names persist; PBGS is used herein. PBGS catalyzes the first of three common steps in tetrapyrrole biosynthesis, after which there is extensive pathway diversity.3 The enzyme catalyzed reaction is an asymmetric condensation of two molecules of 5-aminolevulinic acid (ALA) 4, which is biosynthesized either from succinyl-CoA and glycine or from glutamic acid.5 The PBGS substrates are identified as P-side ALA and A-side ALA to indicate their fate in the product (described in Fig. 1). The requirement for Schiff base formation between ALA and active-site lysine residues requires a highly basic active site environment. The enzyme gates active site access to isolate the required chemistry from the cellular environment.6 Indeed direct-detect 13C and 15N NMR studies of isotopically labeled enzyme-bound P-side Schiff base intermediate and enzyme-bound product reveal chemical shifts like these molecules in aqueous solution at pH ~11, consistent with a very basic active site environment .7 This article provides an overview of our understanding of the PBGS family of enzymes and traces the path of discovery for the enzyme’s most unexpected properties.
Figure 1. The PBGS catalyzed reaction.

The chemically identical substrates 5-aminolevulinic acid (ALA) are named for their fate in the product porphobilinogen. A-side ALA yields the amino nitrogen and the acetic acid side chain. P-side ALA yields the pyrrole nitrogen and the propionyl side chain.
The evolution of PBGS resulted in an unusual quaternary structure equilibrium linked to a rich variation in metal ion usage
Throughout evolution, PBGS adapted to diverse biological niches and varied cellular localizations (e.g. cytosol, chloroplast, apicoplast). This is reflected in variations in metal ion usage and in quaternary structure equilibria. However, the overall domain structure and sequence identity is substantial (≥35% for any two species).
PBGS domains and quaternary structure interactions
The PBGS monomer (≥330 residues) consists of two or three domains (Fig 2a). The N-terminal “arm” (≥25 residues) is an extended structure essential for multimerization. The central (αβ)8-barrel domain is ~300 residues and contains all the components of the enzyme active site, including a flexible active-site lid. A short C-terminal extension (~12 – 20 residues), present in PBGS from a small number of species, limits quaternary structure mobility.
Figure 2. PBGS structure.

(a) The PBGS domain structure, numbered for human PBGS. The asterisks mark the two active site lysine residues. (b) The human PBGS quaternary structure equilibrium (two subunits colored as part a, transitions labeled as in Scheme 1). The octamer-specific subunit-subunit interface is in the rectangular box. The hexamer-specific surface is depicted by a circle. (c) The pro-octamer dimer of T. gondii PBGS (PDB id 1ohl) includes the C-terminal extension, which disallows the hinge motion required for dimer interconversion. (d) Three subunits of the yeast PBGS octamer (PDB id 1ohl) illustrates the hugging interaction between subunits 1 & 3 facilitated by the longer N-terminal domain.
All known PBGS are homo-multimers and various multimers differ in quaternary structure interactions that govern enzymatic activity by impinging upon active-site lid motion. Some PBGS multimers readily dissociate to a conformationally flexible dimer; this allows PBGS to sample an equilibrium of architecturally distinct assemblies.8 Scheme 1 describes the equilibrium of octamer, dimers, and hexamer, illustrated in Fig 2b, which is well established for both human and plant PBGS. Dissociation to dimer occurs along a large, highly conserved, and hydrophilic subunit-subunit interface; residues that bind inter-subunit water molecules are highly conserved. The dimers constitute a low mole fraction (~0.5%) of the total population for human PBGS.9
Scheme 1.

The equilibrium of PBGS quaternary structures.
The PBGS active site contains two universally conserved lysine residues, located ~53 residues apart in sequence (see Fig 2a), but arranged side-by-side in structure. The active site lid, which is variable in both length and sequence, begins and ends between the active site lysine residues. The PBGS quaternary structure impacts active site lid conformation due to interactions between the C-terminal end of the lid of one subunit and the N-terminal arm of an adjacent subunit (within the rectangle in Fig 2b) (e.g. Cys223-Phe12 in Fig 3a). In the octameric assembly, these interactions stabilize “closed” conformations of the lid. Lid-closed conformations can also involve interactions between basic lid residues and the carboxylate group of A-side ALA (Fig 3a). Consistent with this, A-side ALA (or product) binding draws the PBGS quaternary structure equilibrium toward the octamer.10 A-side ALA is the second substrate to bind and its binding determines the Km. Hence, the octamer has a low Km (≤150 µM, which approximates the physiological concentration of ALA). The PBGS hexamer and the PBGS dimers do not have quaternary structure interactions that can stabilize “closed” conformations of the active-site lid (see Fig 2b). The PBGS hexamer crystal structure shows the lid to be disordered.8 The hexamer has a high Km (ranging from ~ 1 mM to >10 mM).8,9,11
Figure 3. Maintenance of the human PBGS structure.

(a) The important players in human PBGS octamer stabilization. Each interaction is essential for maintenance of the octamer. P-side ALA is shown as the Schiff base intermediate. A-side ALA is shown as a bidentate ligand to the essential active-site Zn2+. (b) Native PAGE illustrates how chelation of Zn2+ destabilizes the product-bound human PBGS octamer and hexamer accumulates with time.
C-terminal and N-terminal extensions impact the PBGS quaternary structure equilibrium. The C-terminal extension, when present, forms a domain-swapped β-sheet that prevents reaction II of Scheme 1. For example, Fig 2c shows PBGS from the apicomplexan parasite Toxoplasma gondii where the C-terminal extension secures the hinge between N-terminal and αβ-barrel domains and thus prevents equilibration between dimers.11 T. gondii PBGS participates in an octamer ⬄ pro-octamer dimer equilibrium.12 Fig 2d shows a yeast PBGS where the (~35 residue) N-terminal arm has extensive interactions with two neighboring subunits, thus effectively preventing octamer dissociation (reaction I in Scheme 1).6 Yeast PBGS appears to function as a stable octamer.
It is possible that the regulation of the PBGS quaternary structure equilibrium in the mammalian red blood cell, or the microbial world, is related to one or more of the protein’s reported moonlighting functions as a proteasome inhibitor and/or as a co-chaperone.13,14 In support of the importance of potential non-catalytic PBGS moonlighting functions, there is estimated to be an order of magnitude more PBGS in humans than is required for heme biosynthesis.15 Evidence for PBGS moonlighting functions is growing.16
Variations in PBGS metal ion usage
Two significant sequence variations exists in the αβ-barrel domain that dictate differences in metal ion binding sites.17 The first is a striking sequence variation in the enzyme active site that dictates differences in the requirement for a catalytically essential metal ion.17,18 The unusual nature of this variation is a toggle between PBGS requiring the soft transition metal Zn2+, with its plastic coordination sphere, and the hard alkaline earth metal Mg2+, with its rigid octahedral coordination preference. PBGS from archaea, metazoan, fungi and many bacteria all require an active site Zn2+ that is essential for binding A-side ALA (Fig 3a).19 Crystal structures of product-bound human, yeast, and E. coli PBGS show this Zn2+ directly ligated to the amino nitrogen of porphobilinogen (PDB ids 1e51, 1ohl, 5ic2). The catalytic Zn2+ binds to the underlined cysteine residues in the sequence DXCXCX(Y/F)X3G(H/Q)CG.6 This cysteine-rich site is peculiar for a catalytic Zn2+-binding site.20 Fig 4 describes the PBGS phylogenetic variation with regard to the cysteine-rich Zn2+-binding site. Because Zn2+ is absolutely required for catalysis by these enzymes, PBGS with an alternate active-site metal-binding sequence likely proceed via a different enzyme-catalyzed reaction mechanism.21 With few exceptions, those PBGS that do not require Zn2+ have an alternate cysteine-free sequence, DXALDX(F/Y)X3G(H/Q)DG, that was originally proposed to bind a catalytically essential Mg2+.18 This more “acidic” sequence is present in PBGS from many bacteria as well as all eukaryotes that are not metazoan or fungi (e.g. plants). The crystal structure of one such PBGS (T. gondii) shows no metal ion in the vicinity of enzyme-bound porphobilinogen (PDB id 3obk). Although most crystal structures of PBGS that contain the acidic sequence do not show metal bound, structures are documented that contain Mg2+ in the enzyme active site.22 It is unknown whether the catalytic metal ion dissociates turning enzymatic turnover.
Figure 4. Phylogenetic variation in PBGS active site and allosteric metal ion usage.

Yellow vs. red reports respectively on the presence or absence of the cysteine-rich active-site Zn2+-binding sequence. Blue vs. white reports the presence of absence of the allosteric Mg2+-binding site. The key depicts how the colors are mixed.17
A full description of metal binding to the PBGS active site requires addressing a subtle sequence variation that affects the susceptibility of Zn2+-requiring PBGS to inhibition by the environmental toxin Pb2+. PBGS from many metazoa and fungi also contain a second Zn2+-binding site that involves a cysteine residue at the C-terminal end of the active site lid. The constellation of cysteine residues in these two sites allows for formation of a tight binding site for Pb2+.23,24 Consequently, human PBGS is the primary reservoir for blood Pb2+, which can serve as a clinical measure of lead exposure.
The second important PBGS sequence variation dictates the presence or absence of an allosteric Mg2+-binding site, which is present in nearly all PBGS except metazoan, fungi, and a small number of bacterial species (see Fig 4). Unfortunately, allosteric Mg2+ action serves to confound the interpretation of enzyme kinetic data with regard to the essential nature of an active-site Mg2+. The sequence determinants for the allosteric Mg2+-binding site are a specific arginine in the N-terminal domain and a specific glutamic acid on the outer rim of the αβ-barrel domain, depicted for E. coli PBGS in Fig 5a. The allosteric Mg2+-binding site straddles the octamer-specific subunit interface (rectangle in Fig 2b), stabilizes the octamer, and thus impacts the equilibrium between PBGS octamer and hexamer, causing activation. Removal of Mg2+ from this metal-binding site of PBGS can result in a decrease in the mole fraction of octamer and an increase in the mole fraction of hexamer and/or dimer (Fig 6a,b).8 Fig 6b also shows that Mg2+ (and/or substrate) addition favors accumulation of the E. coli PBGS octamer. Fig 6c shows Mg2+ stabilization of the P. aeruginosa PBGS octamer; in this case purification without Mg2+ resulted in isolation of a dimeric PBGS.
Figure 5. The allosteric Mg2+ binding site, when it is present and when it is not.43.

(a) The E. coli PBGS (PDB id 1l6s) allosteric Mg2+ binding site. The dotted yellow oval highlights an interaction between the N-terminal arm of one subunit and the αβ-barrel of a neighboring subunit, detailed in the image below. (b) The spatially equivalent location of the guanidinium group of Arg240 of human PBGS (PDB id 1e51). (c) No such quaternary structure interaction exists in hexameric human PBGS variant F12L (PDB d 1pv8), where Ser5 of the neighboring subunit is distant from Arg240.
Figure 6. Native PAGE illustrates how the allosteric Mg2+ modulates PBGS quaternary structure.

(a) A plant PBGS (magenta quadrant, Fig 4), purified in the presence of Mg2+ is predominantly octamer. Hexamer accumulates upon EDTA treatment.8 (b) E. coli PBGS (green quadrant, Fig 4), purified in the presence of Zn2+, but not Mg2+, shows a mixture of alternate multimers 36. Addition of EDTA increases the population of dimer. Addition of Mg2+, substrate, or both increase the population of octamer. Substrate-induced transition to octamer can occur within the gel matrix.36 (c) Pseudomonas aeruginosa PBGS (magenta quadrant, Fig 4), purified in the absence of Mg2+ appears dimeric. Dialysis against Mg2+ dramatically stabilizes the octamer.
PBGS lacking the allosteric Mg2+-binding site contain a spatially equivalent arginine-derived guanidinium group (e.g. Arg240 of human PBGS is shown in Fig 5b). In contrast, the human PBGS hexamer, shown in Fig 5c does not contain this multimer-stabilizing interaction (see also Fig 2b). To illustrate the importance of Arg240, the human PBGS variant R240A purifies predominantly as hexamer. However, enzyme turnover promotes a slow, reversible, and pH dependent equilibrium shift toward octamer (Fig 7). This behavior exemplifies the surprisingly metastable nature of the human PBGS quaternary structure equilibrium.
Figure 7. The slow reversible equilibration of human PBGS hexamer and octamer.44.

(a) Human PBGS variant R240A, which purifies predominantly as hexamer, transitions to octamer upon dialysis against substrate (pH 7, open circles; pH 9, open squares). Filled symbols denote a second dialysis without ALA. (b) There is a time-dependent increase in R240A specific activity (10 mM ALA) corresponding to the transition from hexamerlow-activity to octamerhigh-active.
At various evolutionary junctures, additional metal ions have been co-opted to serve in the control of the PBGS quaternary structure equilibrium and thus in the control of PBGS activity. 25 For example, an insect PBGS was discovered to house an inhibitory Zn2+ site that stabilizes the closed-lid conformation.26 Monovalent K+ has also been demonstrated to modulate PBGS activity and can be seen to bind at a subunit-subunit interface that secures a PBGS quaternary structure assembly.
The physiological rationale for the evolution of metal ion usage in PBGS (both catalytic and allosteric) remains largely unknown. We have speculated that the allosteric Mg2+ of plant PBGS plays a role in chloroplasts to protect the plant from accumulation of photoreactive chlorophyll precursors.17 In the 1980’s ALA was considered as a potential herbicide.27 Sprayed on corn fields at dusk, ALA was preferentially taken up by the weeds, turned into photoreactive chlorophyll precursors during the dark hours, and the weeds burned up when the sun emerged in the morning. The analogous accumulation of phototoxic heme precursors serves similarly when ALA is used for photodynamic detection and photodynamic therapy for some human cancers.28
PBGS quaternary structure dynamics impinges upon enzyme kinetic observations
There are myriad kinetic phenomena that arise from the equilibrium of PBGS assemblies; these are dependent upon on the rate of multimer equilibration relative to the timescale of measurements. For example, PBGS in the magenta and red quadrants of Fig 4 equilibrate sufficiently rapidly to show a protein concentration dependence to the enzyme specific activity below ~1 µM subunit (Fig 8a). Below ~30 nM subunit concentration, the quaternary structure equilibrium is dominated by the high Km and low Vmax dimer/hexamer species; above ~1 µM, the protein exists almost completely as the low Km and high Vmax octamer. For PBGS whose multimer equilibration is slower (e.g. human PBGS) one can observe a rate vs. [substrate] relationship that is the sum of the slowly exchanging low Km, high Vmax octamer and the high Km, low Vmax hexamer. At [substrate] below and through the low Km, the rate is dominated by the octamer. As [substrate] approaches the high Km, catalysis by the hexamer contributes to the observed rate. This data is appropriately fit to the sum of the two hyperbolic equations where the apparent Vmax values are relative to the proportion of the low Km and high Km species.29 For human PBGS, the ratio of octamer to hexamer is very sensitive to the protein sequence; all of the porphyria-associated protein variants favor hexamer more than wild-type.30 In addition, deprotonation of octamer-stabilizing basic residues (e.g. Arg240 at the octamer-specific interface, Arg221 on the active site lid) manifest as a pH dependence to the octamer-hexamer equilibrium. At neutral pH, wild-type human PBGS is predominantly octameric; at pH 9, wild-type human PBGS is predominantly hexameric.9 The pH dependence of the human PBGS quaternary structure equilibrium correlates with the basic arm of the human PBGS pH rate profile (see below). The interaction of enzyme concentration, assay pH, and both monovalent and divalent metal ion binding can yield complex PBGS activity profiles, one of which is illustrated in Fig 8b.31
Figure 8. Kinetic phenomena that can arise from the PBGS quaternary structure equilibrium.
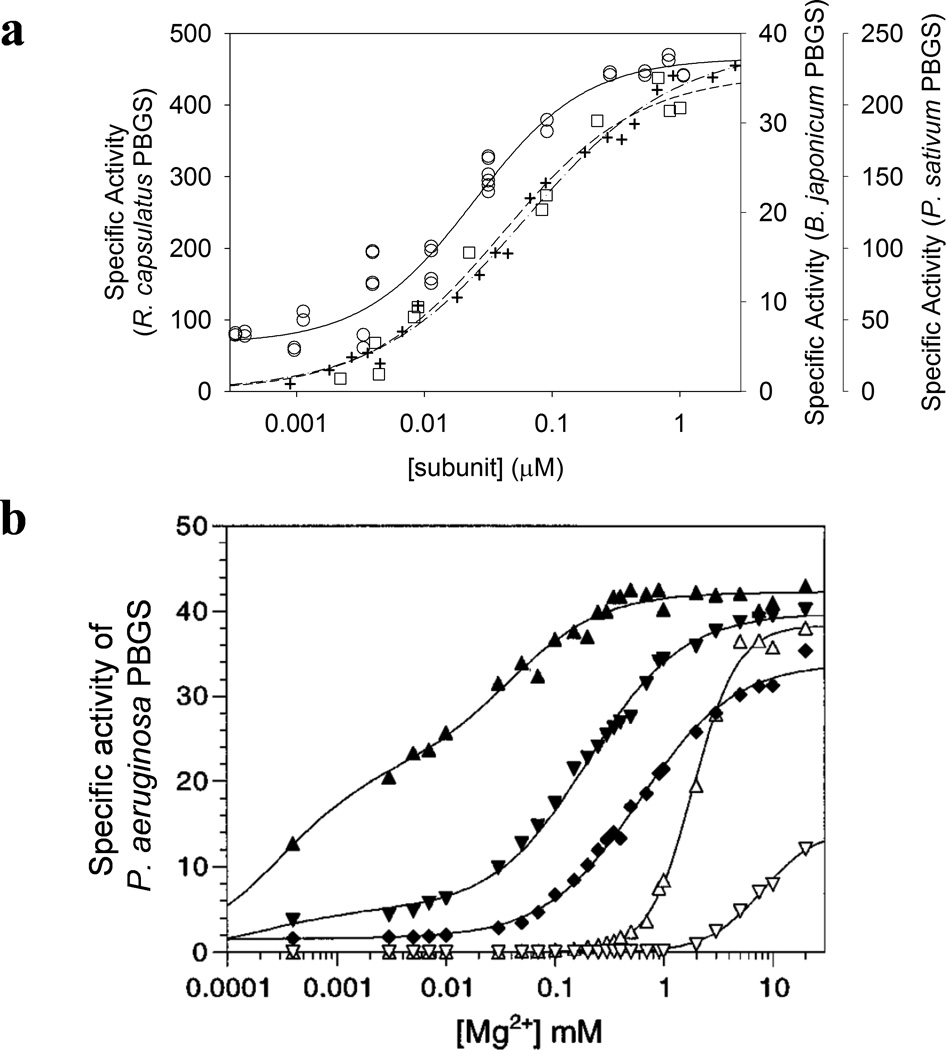
(a) PBGS from organisms in the magenta and red quadrants show a protein concentration dependence to the specific activity (determined at 10 mM ALA). Rhodobacter capsulatus PBGS is in the red quadrant. Pisum sativum and Bradyrhizobium japonicum PBGS are in the magenta quadrant. (b) The dependence of B. japonicum PBGS specific activity (at 10 mM ALA, in Bis-Tris propane-HCl) on pH (pH 6.5, diamonds; 6.9, downward triangles; 8.2, upward triangles) and on the presence (closed symbols) or absence (open symbols) of 0.1 M K+ , as a function of Mg2+ concentration.31
The PBGS-catalyzed reaction mechanism
Investigations into the PBGS-catalyzed reaction mechanism focus on the order of bond formation and bond breakage; and most mechanistic work assumed a phylogenetically conserved reaction mechanism.21 In fact, much of our early work presumed that all PBGS required a catalytically essential Zn2+ and that all PBGS would be inhibited by Pb2+; both these assumptions turned out to be invalid. However, the common aspects of the PBGS catalyzed reaction include metal ion-independent binding of P-side ALA followed by Schiff base formation to the more C-terminal active-site lysine residue (Fig 3a). A-side ALA binds next; and can require coordination to the essential active site Zn2+. Crystal structures with bi-substrate analogs suggest that A-side ALA may form a Schiff base intermediate with the more N-terminal active site lysine residue.32 In all cases, A-side ALA binding favors active site lid closure after which the remaining chemistry proceeds isolated from bulk solvent. Catalysis stalls after the formation of a tightly bound product molecule, which has been observed both by direct detect 13C and 15N NMR and in the above-mentioned crystal structures.7 For some PBGS, product release appears to be rate limiting (e.g. mammalian PBGS has a maximal turnover number of ~0.6/sec at 37 °C); this may be related to the phenomenon of half-of-the-sites reactivity.
Significant data suggests that metazoan and some bacterial PBGS experience half-of-the-sites reactivity 19,26,33,34, which implies a ratcheting mechanism wherein substrate must bind to one hemisphere of the PBGS multimer before product can be released from the other hemisphere. The pathway for PBGS intersubunit communication required for half-of-the-sites reactivity remains an outstanding question, though half-of-the-sites ligand binding is seen in both octameric and hexameric human PBGS crystal structures (e.g. PDB ids 1e51, 1pv8, 5hnr) and P. aeruginosa PBGS (PDB id 1gzg). We have considered that half-of-the-sites may be characteristic of PBGS that lack the octamer-stabilizing C-terminal extension or the longer “hugging” N-terminal arm (Figs 2c and 2d). For these, enzyme-bound product secures the active site lid, which secures the neighboring N-terminal arm and maintains the protein as an active octamer. Using human PBGS as the example, Fig 3a shows all of the interactions that are necessary to maintain a stable octamer. Without any one of these stabilizing interactions, the protein favors (or slowly reverts to) the hexameric assembly. For example, treatment of the enzyme-product complex of human PBGS with a Zn2+-chelator results in accumulation of hexamer (Fig 3b).
Discovery of the PBGS octamer hexamer equilibrium
The discovery of the PBGS quaternary structure equilibrium was facilitated by the natural occurrence of the F12L variant, which dramatically shifts the equilibrium toward the low-activity hexamer. An asymptomatic child was discovered to have 12% PBGS activity and was heterozygous for genes encoding WT and F12L.35 Phe12 is a phylogenetically variable residue, located in the middle of the N-terminal arm, whose backbone is within van der Waals contact of Cys223. A native Western blot published with the identification of F12L showed that the amino acid substitution limits the conformational space available to PBGS (Fig 9); however, identification of the alternate conformers was not addressed.35 With 20/20 hindsight, Fig 9 revealed the human PBGS quaternary structure equilibrium between octamer and hexamer as a natural property of the wild type protein and that this equilibrium is shifted for disease-associated PBGS variants. At that time however, knowing that the PBGS crystal structures (to date) were octameric, it was far from obvious that the F12L variant would be a hexamer. Despite our extensive investigation summarized below, it remains unclear why this particular Phe-to-Leu substitution has such a dramatic effect on the PBGS quaternary structure equilibrium.
Figure 9.

The original native PAGE Western blot of human PBGS variants expressed in CHO cells.35
The characterization of human PBGS WT, F12L, and WT+F12L hetero-multimers used a classic (not-tagged) purification scheme during which we achieved anion exchange resolution of alternate assemblies. The resolution of alternate metastable assemblies was an essential component of the discovery process.8 We initially found that the neutral substitution of Leu for Phe caused a dramatic change in the protein’s retention on anion exchange resin and on the enzyme’s pH activity profile (10 mM ALA) (Fig 10a and 10c). Coexpression yielded alternate multimers, one of which co-eluted from IEC with WT and the other co-eluted with F12L. Mass spectral analysis of a tryptic digest proved that the IEC resolved WT+F12L multimers were both heteromeric, and the pH activity profiles of the hetero-multimers were similar (though not identical) to the homo-multimers of WT and F12L. Native PAGE showed that the heteromultimers were very stable once resolved from each other, but the faster migrating band (hexamer) slowly transitioned to the slower migrating band (octamer) during catalytic turnover. Furthermore, 2D native PAGE established that the hexamer to octamer transition during turnover could occur within the native gel matrix. Later studies of the temperature dependence of the human PBGS WT+F12L hetero-hexamer morphing to the hetero-octamer during turnover established that the rate determining step is the protein hinge motion that must occur when pro-hexamer dimer morphs to pro-octamer dimer (Scheme 1, reaction II).9 Although our initial analytical ultracentrifugation data suggested that F12L is a hexamer, we resisted this interpretation as it made no sense within the context of the known octameric PBGS crystal structures. It wasn’t until we solved the crystal structure of the F12L hexamer (PDB id 1pv8) that the pieces of the puzzle snapped together. Had we not already known a great deal about the phylogenetic variation in PBGS behaviors, we might have dismissed the human PBGS hexamer as a crystallographic artifact.
Figure 10. Human PBGS F12L compared to wild type (N59 allele)8.
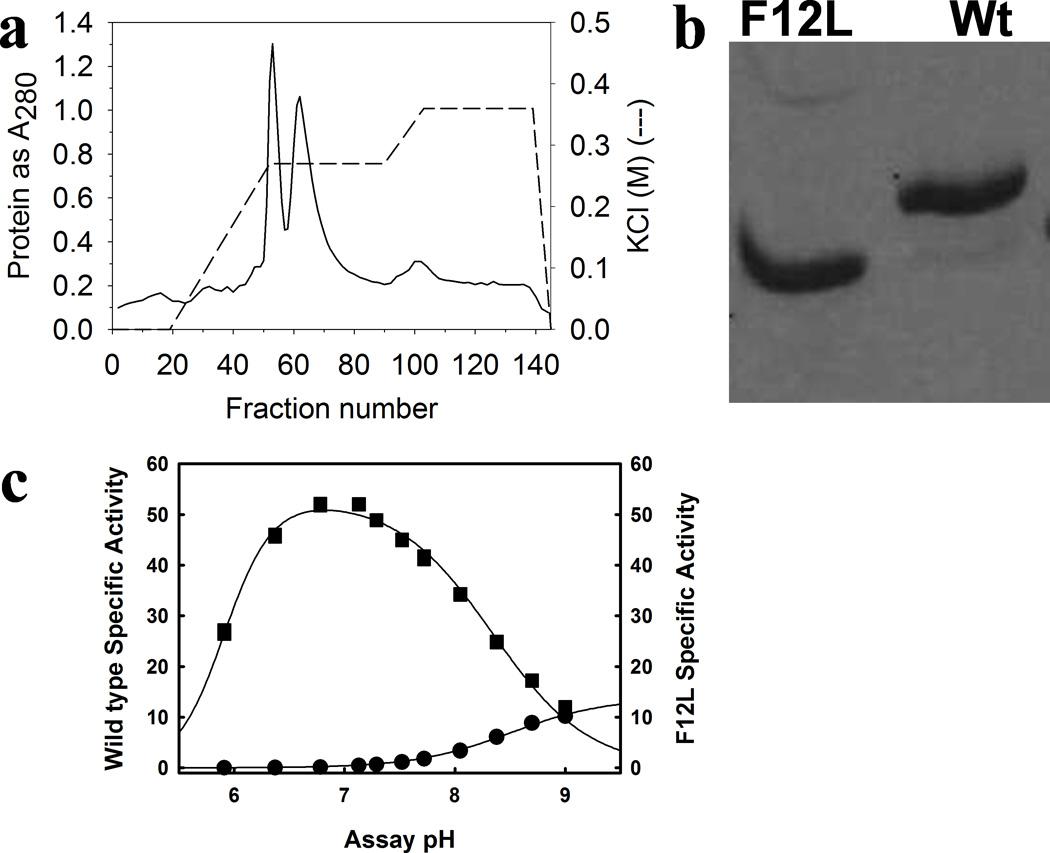
(a) The anion exchange separation of PBGS hexamer (early eluting peak, exemplified by F12L) and the PBGS octamer (later eluting peak, as exemplified by the wild type allele N59). (b) Native PAGE (Phastgel) behavior of purified F12L, shown by crystallography to be a hexamer, and wild-type human PBGS, shown by crystal structure to be an octamer. (c) The pH activity profile for wild type human PBGS (squares) vs. the F12L variant (circles), at 10 mM ALA, in 0.1 M bis-tris propane. Later work9 established that at pH 9, wild type human PBGS is predominantly hexamer.
The crystal structure of F12L defined the architecture of the PBGS hexamer.8 The hexamer’s absent inter-subunit interface (rectangle in Fig 2b) correlated with the binding site for the allosteric Mg2+ of the Zn2+-requiring E. coli PBGS (PDB id 1i8j) (Fig 5a) and the Mg2+-dependent Pseudomonas aeruginosa PBGS (PDB id 1gzg).32,34 E. coli PBGS had been purified without Mg2+; the purified protein had a high Km (~1mM) and separated into various species on native PAGE (Fig 6b).36 Addition of Mg2+ to this protein yielded a species with a low Km, and all of the multimers transitioned to a single larger assembly (Fig 6b). A Mg2+-dependent plant PBGS was treated with EDTA, which caused a change in a quaternary structure equilibrium between two species similar in mobility on native PAGE to the separation of human PBGS WT and F12L (Figs 6a).8,37 Together these provided the necessary insight to propose that an octamer ⬄ hexamer equilibrium could provide the basis for allosteric regulation of PBGS. The possible inclusion of an octamer ⬄ dimer component to the PBGS multimeric equilibrium arose from various observations of smaller components on native PAGE (e.g. Figs 6b, 6c). However, it took us a while to realize that the probable physiologically relevant pro-octamer dimer was distinct from the “hugging”-dimer that comprised the asymmetric units of some octameric crystal structures of PBGS. The dissociative nature of the octamer ⬄ hexamer equilibrium was suggested by the fact that one could not push a PBGS dimer into the hexamer or pull a PBGS dimer out of the octamer without destroying the assembly symmetry. The original model of the predominant PBGS quaternary structure equilibrium was refined to consist of the octamer ⬄ pro-octamer dimer ⬄ pro-hexamer dimer ⬄ hexamer, in part or in whole (Fig 2b). The structures of the minor species that appear to run on native PAGE at a mobility predicted to be tetrameric remain unknown.
The key to proving the dissociative nature of the multimer interconversion process benefited from isolation of the metastable WT+F12L hexamer followed by catalytic turnover to stimulate the transition from hexamer to octamer, which was readily monitored by native PAGE.10 The heteromeric transition was stopped at a point that yields a mixture of hetero-hexamer and hetero-octamers. This mixture is resolved using anion exchange chromatography. Analysis by mass spec then showed that the WT chains accumulate in the resulting octamer while the F12L chains are enriched in the remaining hexamer (Table 1). This disproportionation of chains establishes that the multimers must dissociate during the multimeric reequilibration. Specific activity assays at both pH 7 and pH 9 of the various heteromultimer pools before and after multimeric transition are included in Table 1.
Table 1.
Disproportionation of WT and F12L chains during dialysis vs. ALA
| Sample | pH 7 Specific Activity (10 mM ALA) |
pH 9 Specific Activity (10 mM ALA) |
Phe:Leu |
|---|---|---|---|
| F12L homo-hexamer | 00.3 | 14.5 | 000:100 |
| WT homo-octamer | 56.9 | 13.1 | 100:000 |
| Initial WT+F12L hetero-hexamer | 07.9 | 06.6 | 32:68 |
| Post dialysis isolated hetero-hexamer | 00.5 | 05.8 | 13:87 |
| Post dialysis isolated hetero-octamer | 11.5 | 06.8 | 51:49 |
| Initial WT+F12L hetero-octamer | 21.8 | 05.4 | 67:33 |
| Post dialysis isolated hetero-hexamer | 00.2 | 01.6 | 14:84 |
| Post-dialysis isolated hetero-octamer | 17.6 | 05.7 | 81:19 |
Conclusion
The story describing experimental observations that revealed the PBGS quaternary structure equilibrium reveals the many ways that our (e.g. my) traditional view of protein structure/function relationships was flawed. The first indications of the quaternary structure repertoire available to PBGS occurred ~fifteen years ago, when there were few precedents for allosteric agents that bound to a multimer-specific subunit interface and modulated the equilibrium between architecturally and functionally distinct assemblies. Even fewer examples proved that the alternate assemblies could not interconvert in the absence of multimer dissociation. The physiologic significance of dissociative allostery had been considered previously,38,39 but the existence of alternate finite multimers was largely considered artifactual, irreversible, or associated with protein “misfolding”. The novelty of the PBGS quaternary structure equilibrium caused us to define the dissociative morpheein model of protein allostery, whose defining characteristic is a conformational change in a dissociated state followed by multimer reassembly. However, this concept languished for lack of other compelling examples. Recently reports of other proteins with morpheein-like properties suggest that a metastable mixture of functionally distinct multimers may be a common phenomenon whereby Nature extracts broadened functionality from a limited set of protein transcripts. The best example to date is the three structurally and functionally distinct assemblies of the Ebola virus VP40 protein (dimer, hexamer, octamer), each of which is essential for some part of the viral life cycle.40 Although the structural repertoire of VP40 might be considered a special property of viral proteins, because viruses must do the most with a limited genome, the physical chemistry that governs the behavior of a viral protein is no different from the physical chemistry that governs the behavior of any other protein. For example, very recent work defines the alternate functions of dimeric vs. hexameric activities of the α-subunit of mammalian ribonucleotide reductase.41 An equilibrium of structurally and functionally distinct protein assemblies opens new avenues for targeting multimer-specific binding sites for drug discovery,42 though this aspect of our work with PBGS is not described herein.
Biography
Eileen K. Jaffe was an NSF graduate fellow at University of Pennsylvania where she obtained her Ph.D. with the late Mildred Cohn, focused on 31P NMR as a probe of nucleoside phosphorothioates in kinases. An NIH post-doctoral fellowship with the late Jeremy R. Knowles at Harvard University employed isotope effects to probe chorismate mutase function. Prof. Jaffe’s independent research career, mostly as an NIH funded PI, has been in Philadelphia, where serendipity introduced her to PBGS. Through a circuitous route, this lead to her current focus on the role of protein quaternary structure dynamics in human health and disease.
References
- 1.Gibson KD, Neuberger A, Scott JJ. The enzymic conversion of delta-aminolaevulic acid to porphobilinogen. Biochem. J. 1954;58:xli–xlii. [PubMed] [Google Scholar]
- 2.Nomenclature of multiple forms of enzymes. Recommendations(1976) IUPAC-IUB Commission on Biochemical Nomenclature (CBN) J. Biol. Chem. 1977;252:5939–5941. [PubMed] [Google Scholar]
- 3.Yin L, Bauer CE. Controlling the delicate balance of tetrapyrrole biosynthesis. Philos. Trans. R. Soc. Lond. B. 2013;368:20120262. doi: 10.1098/rstb.2012.0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shemin D, Russell CS. Delta-aminolevulinic acid, its role in the biosynthesis of porphyrins and purines. J. Am. Chem. Soc. 1953;75:4873. [Google Scholar]
- 5.Beale SI. Biosynthesis of 5-aminolevulinic acid. Adv. Photosynth. Respir. 2006;25:147–158. [Google Scholar]
- 6.Erskine PT, Senior N, Awan S, Lambert R, Lewis G, Tickle LJ, Sarwar M, Spencer P, Thomas P, Warren MJ, Shoolingin-Jordan PM, Wood SP, Cooper JB. X-ray structure of 5-aminolaevulinate dehydratase, a hybrid aldolase. Nat. Struct. Biol. 1997;4:1025–1031. doi: 10.1038/nsb1297-1025. [DOI] [PubMed] [Google Scholar]
- 7.Jaffe EK, Markham GD, Rajagopalan JS. N-15 and C-13 Nmr-Studies of Ligands Bound to the 280000-Dalton Protein Porphobilinogen Synthase Elucidate the Structures of Enzyme-Bound Product and a Schiff-Base Intermediate. Biochemistry. 1990;29:8345–8350. doi: 10.1021/bi00488a021. [DOI] [PubMed] [Google Scholar]
- 8.Breinig S, Kervinen J, Stith L, Wasson AS, Fairman R, Wlodawer A, Zdanov A, Jaffe EK. Control of tetrapyrrole biosynthesis by alternate quaternary forms of porphobilinogen synthase. Nat. Struct. Biol. 2003;10:757–763. doi: 10.1038/nsb963. [DOI] [PubMed] [Google Scholar]
- 9.Selwood T, Tang L, Lawrence SH, Anokhina Y, Jaffe EK. Kinetics and thermodynamics of the interchange of the morpheein forms of human porphobilinogen synthase. Biochemistry. 2008;47:3245–3257. doi: 10.1021/bi702113z. [DOI] [PubMed] [Google Scholar]
- 10.Tang L, Stith L, Jaffe EK. Substrate-induced interconversion of protein quaternary structure isoforms. J. Biol. Chem. 2005;280:15786–15793. doi: 10.1074/jbc.M500218200. [DOI] [PubMed] [Google Scholar]
- 11.Jaffe EK, Shanmugam D, Gardberg A, Dieterich S, Sankaran B, Stewart LJ, Myler PJ, Roos DS. Crystal Structure of Toxoplasma gondii Porphobilinogen Synthase: Insights on octameric structure and porphobilinogen formation. J. Biol. Chem. 2011;286:15298–15307. doi: 10.1074/jbc.M111.226225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shanmugam D, Wu B, Ramirez U, Jaffe EK, Roos DS. Plastid-associated Porphobilinogen Synthase from Toxoplasma gondii - Kinetic and structural properties validate therapeutic potential. J. Biol. Chem. 2010;285:22122–22131. doi: 10.1074/jbc.M110.107243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guo GG, Gu M, Etlinger JD. 240-kDa proteasome inhibitor (CF-2) is identical to delta-aminolevulinic acid dehydratase. J. Biol. Chem. 1994;269:12399–12402. [PubMed] [Google Scholar]
- 14.Gross M, Hessefort S, Olin A. Purification of a 38-kDa protein from rabbit reticulocyte lysate which promotes protein renaturation by heat shock protein 70 and its identification as delta-aminolevulinic acid dehydratase and as a putative DnaJ protein. J. Biol. Chem. 1999;274:3125–3134. doi: 10.1074/jbc.274.5.3125. [DOI] [PubMed] [Google Scholar]
- 15.Shemin D. In: The Enzymes. 3rd. Boyer P, editor. VII. New York: Academic Press; 1972. pp. 232–237. [Google Scholar]
- 16.Bardag-Gorce F, French SW. Delta-aminolevulinic dehydratase is a proteasome interacting protein. Exp. Mol. Patho.l. 2011;91:485–489. doi: 10.1016/j.yexmp.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaffe EK. An unusual phylogenetic variation in the metal ion binding sites of porphobilinogen synthase. Chem. Biol. 2003;10:25–34. doi: 10.1016/s1074-5521(02)00296-x. [DOI] [PubMed] [Google Scholar]
- 18.Boese QF, Spano AJ, Li JM, Timko MP. Aminolevulinic acid dehydratase in pea (Pisum sativum L.). Identification of an unusual metal-binding domain in the plant enzyme. J. Biol. Chem. 1991;266:17060–17066. [PubMed] [Google Scholar]
- 19.Jaffe EK, Hanes D. Dissection of the early steps in the porphobilinogen synthase catalyzed reaction. Requirements for Schiff's base formation. J. Biol. Chem. 1986;261:9348–9353. [PubMed] [Google Scholar]
- 20.Berg JM, Godwin HA. Lessons from zinc-binding peptides. Annu. Rev. Biophys. Biomol. Struct. 1997;26:357–371. doi: 10.1146/annurev.biophys.26.1.357. [DOI] [PubMed] [Google Scholar]
- 21.Jaffe EK. The porphobilinogen synthase catalyzed reaction mechanism. Bioorg. Chem. 2004;32:316–325. doi: 10.1016/j.bioorg.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 22.Heinemann IU, Schulz C, Schubert WD, Heinz DW, Wang YG, Kobayashi Y, Awa Y, Wachi M, Jahn D, Jahn M. Structure of the heme biosynthetic Pseudomonas aeruginosa porphobilinogen synthase in complex with the antibiotic alaremycin. Antimicrob. Agents. Chemother. 2010;54:267–272. doi: 10.1128/AAC.00553-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaffe EK, Martins J, Li J, Kervinen J, Dunbrack RL. The molecular mechanism of lead inhibition of human porphobilinogen synthase. J.Biol. Chem. 2001;276:1531–1537. doi: 10.1074/jbc.M007663200. [DOI] [PubMed] [Google Scholar]
- 24.Warren MJ, Cooper JB, Wood SP, Shoolingin-Jordan PM. Lead poisoning, haem synthesis and 5-aminolaevulinic acid dehydratase. Trends Biochem. Sci. 1998;23:217–221. doi: 10.1016/s0968-0004(98)01219-5. [DOI] [PubMed] [Google Scholar]
- 25.Jaffe EK, Lawrence SH. The dance of porphobilinogen synthase in the control of tetrapyrrole biosynthesis. In: Ferreira GC, editor. Handbook of Porphyrin Science. Vol. 26. World Scientific Publishing Co. Pte. Ltd.; 2014. pp. 79–128. [Google Scholar]
- 26.Kundrat L, Martins J, Stith L, Dunbrack RL, Jaffe EK. A structural basis for half-of-the-sites metal binding revealed in Drosophila melanogaster porphobilinogen synthase. J. Biol. Chem. 2003;278:31325–31330. doi: 10.1074/jbc.M304124200. [DOI] [PubMed] [Google Scholar]
- 27.Rebeiz CA, Montazerzouhoor A, Hopen HJ, Wu SM. Photodynamic Herbicides .1. Concept And Phenomenology. Enzyme Microb. Technol. 1984;6:390–401. [Google Scholar]
- 28.Fukuda H, Casas A, Batlle A. Aminolevulinic acid: from its unique biological function to its star role in photodynamic therapy. Int. J. Biochem. Cell Biol. 2005;37:272–276. doi: 10.1016/j.biocel.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 29.Lawrence SH, Jaffe EK. Expanding the Concepts in Protein Structure-Function Relationships and Enzyme Kinetics: Teaching using Morpheeins. Biochem. Mol. Biol. Educ. 2008;36:274–283. doi: 10.1002/bmb.20211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaffe EK, Stith L. ALAD porphyria is a conformational disease. Am. J. Hum. Genet. 2007;80:329–337. doi: 10.1086/511444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petrovich RM, Litwin S, Jaffe EK. Bradyrhizobium japonicum porphobilinogen synthase uses two Mg(II) and monovalent cations. J. Biol. Chem. 1996;271:8692–8699. doi: 10.1074/jbc.271.15.8692. [DOI] [PubMed] [Google Scholar]
- 32.Kervinen J, Jaffe EK, Stauffer F, Neier R, Wlodawer A, Zdanov A. Mechanistic basis for suicide inactivation of porphobilinogen synthase by 4,7-dioxosebacic acid, an inhibitor that shows dramatic species selectivity. Biochemistry. 2001;40:8227–8236. doi: 10.1021/bi010656k. [DOI] [PubMed] [Google Scholar]
- 33.Bevan DR, Bodlaender P, Shemin D. Mechanism of porphobilinogen synthase. Requirement of Zn2+ for enzyme activity. J. Biol. Chem. 1980;255:2030–2035. [PubMed] [Google Scholar]
- 34.Frankenberg N, Erskine PT, Cooper JB, Shoolingin-Jordan PM, Jahn D, Heinz DW. High resolution crystal structure of a Mg2+-dependent porphobilinogen synthase. J. Mol. Biol. 1999;289:591–602. doi: 10.1006/jmbi.1999.2808. [DOI] [PubMed] [Google Scholar]
- 35.Akagi R, Yasui Y, Harper P, Sassa S. A novel mutation of delta-aminolaevulinate dehydratase in a healthy child with 12% erythrocyte enzyme activity. Br. J. Haematol. 1999;106:931–937. doi: 10.1046/j.1365-2141.1999.01647.x. [DOI] [PubMed] [Google Scholar]
- 36.Jaffe EK, Ali S, Mitchell LW, Taylor KM, Volin M, Markham GD. Characterization of the Role of the Stimulatory Magnesium of Escherichia-Coli Porphobilinogen Synthase. Biochemistry. 1995;34:244–251. doi: 10.1021/bi00001a029. [DOI] [PubMed] [Google Scholar]
- 37.Kervinen J, Dunbrack RL, Litwin S, Martins J, Scarrow RC, Volin M, Yeung AT, Yoon E, Jaffe EK. Porphobilinogen synthase from pea: Expression from an artificial gene, kinetic characterization, and novel implications for subunit interactions. Biochemistry. 2000;39:9018–9029. doi: 10.1021/bi000620c. [DOI] [PubMed] [Google Scholar]
- 38.Kurganov BI. Allosteric Enzymes. New York: John Wiley & Sons; 1982. [Google Scholar]
- 39.Traut TW. Allosteric regulatory enzymes. New York, NY: Springer; 2008. [Google Scholar]
- 40.Bornholdt ZA, Noda T, Abelson DM, Halfmann P, Wood MR, Kawaoka Y, Saphire EO. Structural rearrangement of ebola virus VP40 begets multiple functions in the virus life cycle. Cell. 2013;154:763–774. doi: 10.1016/j.cell.2013.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wisitpitthaya S, Zhao Y, Long MJ, Li M, Fletcher EA, Blessing WA, Weiss RS, Aye Y. Cladribine and Fludarabine Nucleotides Induce Distinct Hexamers Defining a Common Mode of Reversible RNR Inhibition. ACS Chem. Biol. 2016;11:2021–2032. doi: 10.1021/acschembio.6b00303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jaffe EK. Morpheeins - a New Pathway for Allosteric Drug Discovery. Open Conf. Proc. J. 2010;1:1–6. doi: 10.2174/2210289201001010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jaffe EK, Lawrence SH. Allostery and the dynamic oligomerization of porphobilinogen synthase. Arch. Biochem. Biophys. 2012;519:144–153. doi: 10.1016/j.abb.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang L, Breinig S, Stith L, Mischel A, Tannir J, Kokona B, Fairman R, Jaffe EK. Single amino acid mutations alter the distribution of human porphobilinogen synthase quaternary structure isoforms (morpheeins) J. Biol. Chem. 2006;281:6682–6690. doi: 10.1074/jbc.M511134200. [DOI] [PubMed] [Google Scholar]