

Abstract
Tissue injury can initiate bidirectional signaling between neurons, glia and immune cells that creates and amplifies pain. While the ability for neurotransmitters, neuropeptides, and cytokines to initiate and maintain pain has been extensively studied, recent work has identified a key role for reactive oxygen and nitrogen species (nitroxidative species), including superoxide, peroxynitrite, and hydrogen peroxide. In this review, we describe how nitroxidative species are generated after tissue injury, and the mechanisms by which they enhance neuroexcitability in pain pathways. Finally, we discuss potential therapeutic strategies for normalizing nitroxidative signaling, which may also enhance opioid analgesia, to help to alleviate the enormous burden of pathological pain.
Keywords: NADPH oxidase, mitochondria, sensitization, TRP channels, neuroinflammation, exercise
The link between nitroxidative signaling and pain
Investigation of oxidative processes, such as rusting, began with the “phlogiston theory”, developed by Georg Ernest Stahl during the scientific revolution, which postulated that a fire-like element (phlogiston) is released during combustion. Oxidation was formally linked to biology during the early 20th Century, when it was found to underpin cellular metabolism [1–3]. The connection between reactive oxygen species (ROS) and altered sensory processing was empirically identified around the same time [4]. Since then, research has shown that prolonged, unchecked increases in reactive oxygen and nitrogen (nitroxidative) species after infection or tissue damage can promote cytotoxicity and inflammation. These processes can cause peripheral and central sensitization, which underlie pathological pain (see Glossary) [5,6]. Thus, restoring nitroxidative balance in peripheral and central nervous systems (PNS, CNS) is a possible therapeutic approach for ameliorating neuropathology [6–10].
In this review, we summarize recent research on how nitroxidative species participate in neuroimmune signaling throughout the neuraxis to drive pathological pain. We additionally discuss potential therapeutic strategies for normalizing nitroxidative signaling by activating endogenous antioxidant systems, which may also enhance opioid analgesia. As pathological pain is often intractable to current therapies, new strategies to normalize nitroxidative signaling may help to alleviate the enormous burden of pain [11].
Production of nitroxidative species by neurons, glia, and immune cells
The role of nitroxidative signaling in pain has been studied using rodent experimental models of inflammatory pain (e.g. intraplantar complete Freund’s adjuvant (CFA), formalin) and neuropathic pain (e.g. peripheral nerve injury (PNI), chemotherapy-induced peripheral neuropathy (CIPN), diabetic neuropathy (DN), spinal cord injury (SCI), experimental autoimmune encephalomyelitis (EAE)), which have recently been reviewed elsewhere [12]. There are numerous endogenous sources of ROS and nitric oxide (NO) that are engaged during pain processing [13]. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, NO synthases and mitochondrial respiration are among the best characterized ROS/NO producers, and will be discussed here (Figs. 1 and 2).
Figure 1. Induction of nitroxidative species after tissue injury.
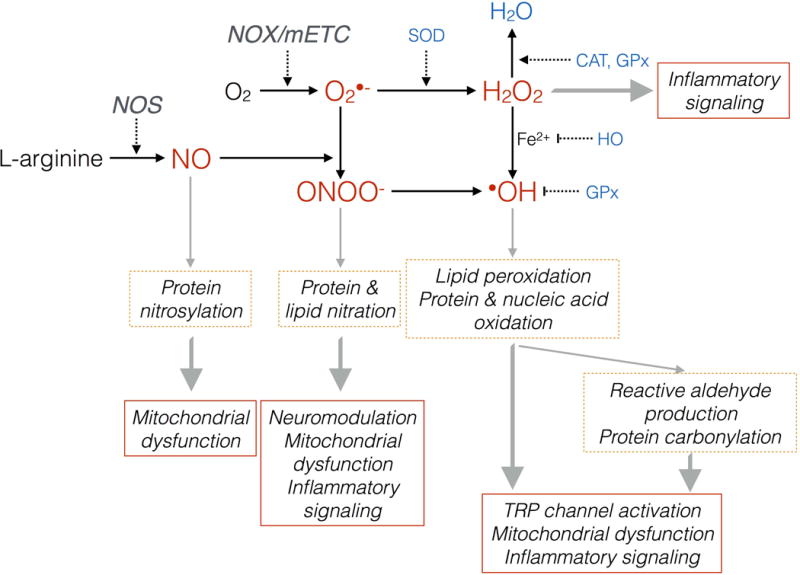
Nitroxidative species can induce posttranslational modifications of proteins and lipids, which subsequently drive pathological pain by modulating nociceptive neurotransmission, activating TRP channels, inducing mitochondrial dysfunction, and induce inflammatory signaling. In healthy cells, endogenous antioxidant systems prevent nitroxidative damage. Cell damage/pathology can perturb this balance, driving accumulation of potentially damaging nitroxidative species. O2: oxygen; NO: nitric oxide; O2•−: superoxide; ONOO−: peroxynitrite; H2O2: hydrogen peroxide; •OH: hydroxyl radical; H2O: water; NOX: NADPH oxidase; NOS: nitric oxide synathse; mETC: mitochondrial electron transport chain; SOD: superoxide dismutase; CAT: catalase; GPx: glutathione; HO: heme oxygenase.
Figure 2. Sources of nitroxidative species after tissue injury.
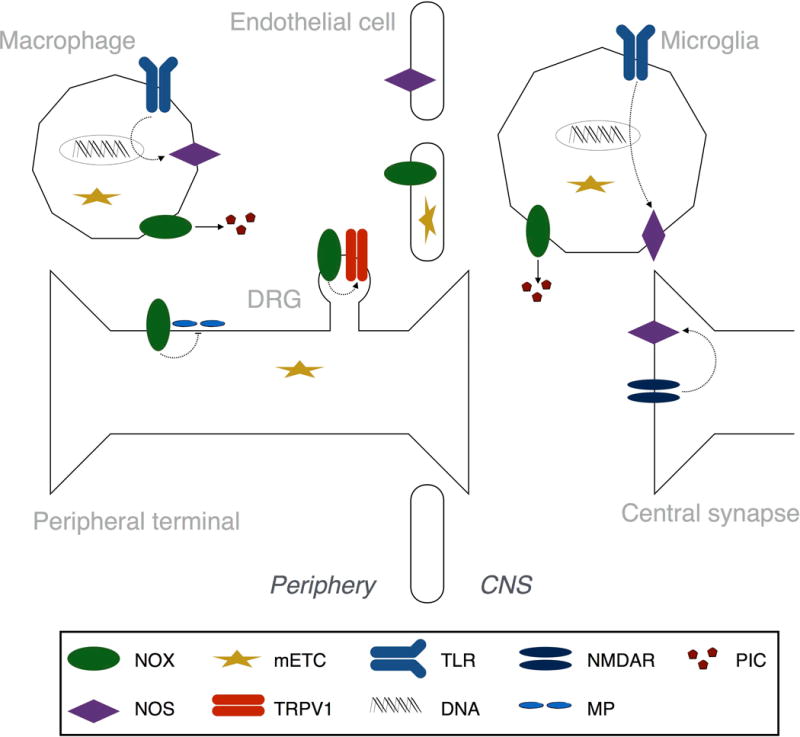
Principal sources of nitroxidative species include NADPH oxidase (NOX), nitric oxide synthase (NOS), and electron leakage from the mitochondrial electron transport chain (mETC). The NOX1, 2, and 4 isoforms are differentially expressed across cell types and tissues after injury. NOX1-derived reactive oxygen species induce enhance Transient Receptor Potential (TRP) V1 activity in dorsal root ganglia (DRG) neurons. NOX2 activity in macrophages and microglia drives mRNA expression of proinflammatory cytokines (PIC) in DRG the spinal dorsal horn. NOX4 expression at the site of peripheral nerve injury decreases expression of myelin proteins (MP). The three NOS isoforms—NOS1 (neuronal), 2 (inducible), and 3 (endothelial)—are also differentially expressed by cell type. In abnormal pain states, N-methyl-D-aspartate receptors (NMDARs) are activated, resulting in calcium influx and activation of NOS1. Transcription of NOS2 is initiated by Toll like receptors (TLRs). These enzymes and processes have a well-established role in pathological pain.
NADPH oxidases
NADPH oxidases (NOX) are membrane-bound enzyme complexes. They transport electrons donated from cytosolic NADPH to generate extracellular or luminal superoxide anions or hydrogen peroxide, that can be transported into the cytosol via aquaporin channels [13,14]. In contrast to other sources of ROS that are generated as a byproduct of catabolism, ROS generation is the primary function of NOX. There are seven members in the NOX family; NOX1, 2, and 4 have been implicated in pathological inflammatory and neuropathic pain models [13,15,16]. NOX1 and 2 are expressed at the cellular membrane, and produce superoxide anions following phosphorylation of cytosolic subunits [17]. NOX4 is expressed on organelles, such as the endoplasmic reticulum, and constitutively produces hydrogen peroxide [17].
NOX1 is inducibly expressed by microglia, neurons, astrocytes, and macrophages in the dorsal root ganglion (DRG) and CNS [17–19]. Nociceptive hypersensitivity induced by the inflammatory stimuli formalin and carrageenan is attenuated in Nox1 deficient mice [18]. NOX1-derived ROS induce translocation of PKCε to the membrane to enhance Transient Receptor Potential (TRP) V1 activity in DRG neurons [18], a change consistent with pain amplification (Fig. 2). In contrast, another study showed that NOX1 mRNA failed to upregulate in the DRG following peripheral nerve injury (PNI) [20]. These results indicate that DRG NOX1 may have a preferential role in inflammatory versus neuropathic pain.
NOX2 is predominantly expressed by phagocytic cells—peripheral macrophages and CNS microglia [13]. PNI induces a rapid upregulation of NOX2 mRNA by DRG macrophages and spinal microglia, which is correlated with increased intracellular superoxide [20,21]. PNI-induced nociceptive hypersensitivity was attenuated in Nox2 deficient mice [20,21]. Nox2 deficiency attenuated TNF, but not IL-1β, mRNA expression, as well as expression of the neuronal injury marker ATF3 in DRG (Fig. 2) [20]. However, Nox2 deficiency did not influence macrophage recruitment to the injured DRG, suggesting a role for NOX2 in macrophage function rather than chemotaxis [20]. Nox2 deficiency attenuated PNI-induced Iba1 expression and the attendant expression of pro-inflammatory cytokines TNF and IL-1β in the spinal dorsal horn [21]. As these studies were performed in global knockouts, it is still unclear whether alterations in the DRG and dorsal horn are subject to NOX-dependent changes in macrophage function at the injury site. In contrast to NOX1, NOX2 activity in monocytes appears to play no role in inflammatory pain [22].
NOX4 is expressed by DRG neurons—both myelinated (A-fibers) and unmyelinated (C-fibers) DRG neurons—and by microglia, astrocytes and macrophages [13,23,24]. Nociceptive hypersensitivity following PNI is attenuated in Nox4 deficient mice, with attenuation of hydrogen peroxide at the sciatic nerve injury site [23]. These results are supported by the absence of NOX4 upregulation in the DRG after PNI [20]. The myelin proteins MPZ and PMP22 are decreased at the sciatic nerve injury site over time in an NOX4-dependent fashion, suggesting that myelin degeneration by hydrogen peroxide may maintain neuropathic pain (Fig. 2). However, attenuated damage at the injury site did not alter expression of the nitroxidative stress and neuroinflammation indices at the spinal dorsal horn or DRG (microglia proliferation, hydrogen peroxide levels) [23]. This contrasts with other studies showing that such processes are dependent on manipulations at the sciatic nerve [25–27]. Finally, a role for NOX4 may be limited to neuropathic, rather than inflammatory pain [23].
Together, these data suggest that NOX1, 2, and 4 isoforms contribute to pathological pain. Future studies could expand the role of various NOX isoforms to other sites in the neuraxis, and well as identifying a role for other NOX isoforms in pain. These data may help to guide development of therapeutics that target the activity of specific NOX isoforms to reduce nitroxidative stress and pain.
Nitric oxide synthases
NO is a diffusible gas mediator that is synthesized from L-arginine by one of three nitric oxide synthase (NOS) isoforms: NOS1 (neuronal), 2 (inducible), and 3 (endothelial). NO and all three NOS isoforms have a well-established role in nociception (Fig. 2) [28]. It easily passes through membranes to directly impact nearby cells.
NOS1 is constitutively expressed in the cytosolic compartment of postsynaptic terminals of neurons, and of stressed Schwann cells, and requires calcium for its activation [29–31]. In abnormal pain states, N-methyl-D-aspartate (NMDA) receptors are activated, resulting in calcium influx and activation of NOS1 [28]. Nociceptive hypersensitivity induced by PNI and CIPN is attenuated by genetic ablation and pharmacological inhibition of NOS1 [32–35].
NOS2 is a cytosolic isoform that is widely expressed in many immune cells and in glia. Transcription of NOS2 is initiated by Toll like receptors (TLRs) and, once translated, is constitutively active—that is, unlike NOS1 and 3, its activity is independent of calcium [28]. NOS2 inhibition attenuates nociceptive hypersensitivity associated with inflammatory and neuropathic pain models [15,36,37].
NOS3 is best known for its expression in the cardiovascular system as a regulator of vascular tone. NOS3 is a membrane-bound enzyme that is constitutively expressed; however, it requires the interaction of calcium and calmodulin for its activation [28]. NOS3 expression is increased in the DRG after subcutaneous administration of CFA, and is correlated with allodynia, suggestive of increased NOS3 activity [38]. CFA-induced inflammatory pain is attenuated by NOS3 inhibition [38].
Cellular respiration
One critical function of mitochondria is energy metabolism. The mitochondrial electron transport chain (mETC) is a series of five molecular complexes through which electrons are transported to synthesize ATP from ADP. Premature electron leakage can occur during cellular respiration, particularly at Complexes I and III, resulting in superoxide production (Fig. 2) [39].
Mitochondrial ROS are elevated in spinal neurons, microglia and astrocytes in neuropathic pain models [21,40,41]. Furthermore, blocking the mETC attenuates hyperalgesia associated with a range of inflammatory and neuropathic pain models [42–45]. However, a direct link between mETC-dependent pain and mitochondrial ROS has yet to be shown. These results suggest that cellular respiration is increased, but is inefficient due to enhanced ROS-generating electron leakage from the mETC, as ATP production by sciatic nerves is impaired during CIPN [46].
Mechanisms of nitroxidative signaling in neuronal hyperexcitability
Injury or disease can provoke intense, repeated, and sustained activity of primary afferent (sensory) neurons. This activity, together with the release of mediators from reactive glia and immune cells, elicits well-characterized changes in neuronal and biochemical processing at peripheral terminals and central synapses [5,47–50]. This is termed ‘sensitization’, and results in nociceptive hypersensitivity. Here, we discuss how nitroxidative signaling engages neurons in pain pathways, leading to peripheral and central sensitization (Figs. 1 and 3).
Figure 3. Nitroxidative mechanisms of neuroexcitability after tissue injury.
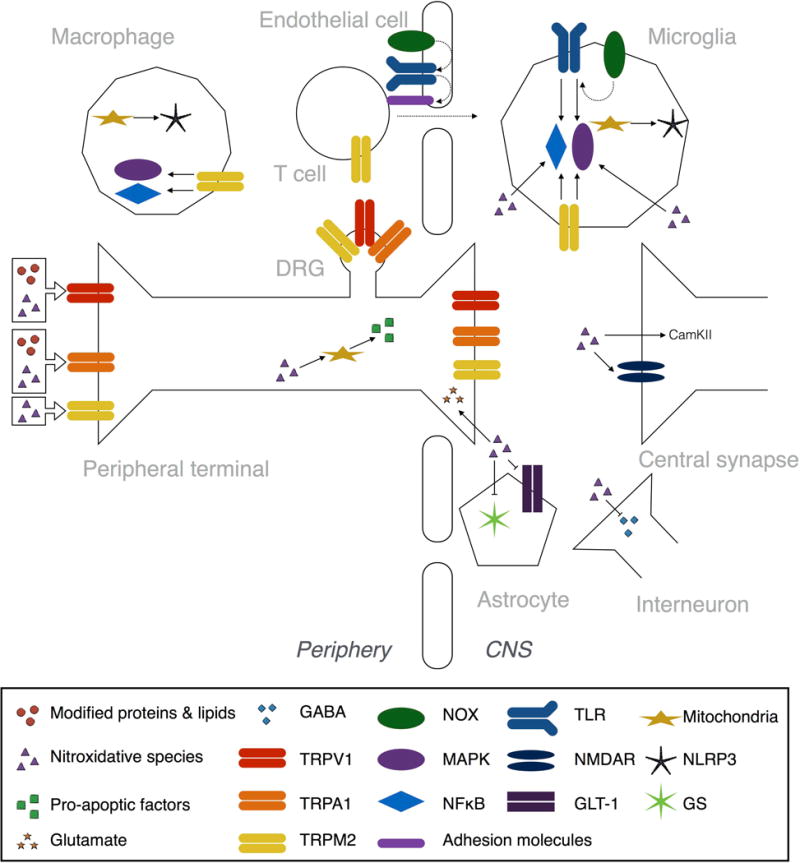
Reactive nitroxidative species, such as hydrogen peroxide and peroxynitrite, and modified proteins and lipids, like carbonylated proteins, peroxidated and nitrated lipids, and reactive aldehydes, all contribute to peripheral and central sensitization after tissue injury. These processes drive pathological pain. Several of the Transient Receptor Potential (TRP) family of nonselective cation channels are activated by nitroxidative species and modified proteins and lipids (see Nitroxidative species activate TRP channels). TRPA1 is expressed by peptidergic C-fibers, and is activated by modified proteins and lipids. TRPM2, which is expressed by neurons, monocytes/macrophages, microglia, and T cells, is directly activated by nitroxidative species. TRPM2 also activates intracellular signaling pathways, including mitogen activated protein kinase (MAPK) and nuclear translocation of nuclear factor κ-light-chain-enhancer of activated B cells (NFκB) pathways. TRPV1 is found on C-fibers and is directly activated by some modified proteins and lipids, as well as being a target of oxidation and nitration events by nitroxidative species that increase responsiveness of the channel. Reactive nitroxidative species can directly modulate neuroexcitibility in central synapses by promoting glutamate release from primary afferent terminals, by activating calcium calmodulin-dependent protein kinase II (CamKII) in glutamatergic spinal neurons, and by inhibiting GABAergic interneurons (see Nitroxidative species as neuromodulators in pain pathways). Nitroxidative species also disrupt glutamate homeostasis by nitration and phosphorylation of NMDA receptor (NMDAR) subunits, as well as inhibiting glutamine synthetase (GS) and the glutamate transporter GLT-1. Mitochondrial DNA is a target of oxidation and nitration, while some nitroxidative species can form adducts with many mitochondrial proteins, which together impairs the structural integrity and function of mitochondria (see Nitroxidative species induce mitochondrial dysfunction). Nitroxidative species can also trigger release of pro-apoptotic factors from mitochondria by disrupting organelle dynamics. Nitroxidative species induce production of proinflammatory mediators, and can activate NFκB and MAPK intracellular signaling pathways (see Nitroxidative species induce neuroinflammatory signaling). Toll like receptors (TLRs) bind a variety of endogenous danger signals, including those released from nitroxidative-damaged mitochondria, to activate NFκB and MAPKs. NOX-derived ROS are second messengers for NFκB- and p38 MAPK-dependent TLR signaling, and TLR expression. The TLR2-NOX1 interaction also upregulates adhesion molecules via CCL3, which facilitates transendothelial cell migration into the CNS. Mitochondria-derived ROS also activate NLRP3 inflammasomes, which are protein complexes responsible for the proteolytic activation of IL-1β.
Nitroxidative species as neuromodulators in pain pathways
Nitroxidative species can directly increase the excitability of nociceptive neurons. Intraplantar administration of superoxide, peroxynitrite, or intrathecal delivery of the ROS donor tert-butyl hydroperoxide (tBOOH) is sufficient to induce nociceptive hypersensitivity in naïve rats [51–54]. These studies demonstrated that ROS activates calcium calmodulin-dependent protein kinase II (CamKII) in glutamatergic spinal neurons, and induced presynaptic inhibition of GABAergic interneurons (disinhibition). Furthermore, hydrogen peroxide enhanced the frequency and amplitude of action potentials of DRG neurons from neuropathic rats (Fig. 3) [55].
In neuropathic pain models, administration of the non-selective ROS scavenger phenyl-N-tert-butylnitrone (PBN), selective small molecule superoxide and peroxynitrite decomposition catalysts such as M40403, FeTMPyP5+ and MnTE-2-PyP5+, or selective peroxynitrite decomposition catalysts such as SRI6 and SRI110 attenuated nociceptive hypersensitivity [15,51,53,54,56–58]. Accordingly, PBN attenuated injury-induced hyperexcitability of spinal dorsal (sensory) horn “pain” responsive neurons and phosphorylation of CamKII [51,57], an effect consistent with pain normalization. Several mechanisms of enhanced excitatory signaling have been identified. Hydrogen peroxide can activate cGKIα, resulting in increased neurotransmitter release from the terminals of primary afferent neurons in the dorsal horn [59,60]. Peroxynitrite and ROS disrupt glutamate homeostasis leading to potentiation of synaptic currents and calcium influx, and ultimately excitotoxicity [56,61]. Mechanisms include nitration and phosphorylation of several NMDA receptor subunits, as well as inhibition of glutamine synthetase and the glutamate transporter GLT-1 that limit the synaptic half-life of glutamate [15,56,62,63]. Nitroxidative products also induce disinhibition after PNI, as PBN normalized the decrease in GAD-67+ GABAergic dorsal horn neurons, and increased GABA release (Fig. 3) [53,64]. Together, these data suggest that nitroxidative species directly enhance neuroexcitability in pain pathways.
Nitroxidative species activate TRP channels
The TRP family of nonselective cation channels plays a vital role in the molecular integration of multiple endogenous and exogenous sensory stimuli [65]. Several of these channels, expressed at the peripheral and central terminals and cells bodies of primary afferent neurons, are activated by nitroxidative species and products. TRP channel activation by nitroxidative species can also initiate neurogenic inflammation— recruitment and activation of immune cells following release of neuropeptides by neurons—which is a key process underlying pathological pain (Fig. 3) [5,66]. Here, we focus on known roles of TRPA1, TRPM2, and TRPV1.
TRPA1 is a chemoreceptor expressed exclusively by peptidergic C-fibers [65]. Nitroxidative species induce protein carbonylation, and membrane phospholipid peroxidation and nitration, and subsequent production of reactive aldehydes such as acrolein (Fig. 1). These products all share the ability to induce nociceptive hypersensitivity by directly activating TRPA1 [67–72]. Acrolein is elevated in the DRG and spinal cord after SCI, and blockade with hydralazine or phenelzine partially attenuated allodynia [73,74]. Moreover, nociceptive hypersensitivity induced by CIPN was abolished in Trpa1 deficient mice, or with a TRPA1 antagonist [75]. In this model, the chemotherapeutic bortezamib did not directly activate TRPA1, suggesting that ROS may act as an intermediate [75].
TRPM2 is expressed by neurons, and abundantly by immune cells, including monocytes/macrophages, neutrophils and T cells, and microglia. This channel is directly activated by hydrogen peroxide, and cytosolic ADP-ribose that is generated after nitroxidative damage to mitochondria [76–81]. Furthermore, TRPM2 activation is critical for activation of spinal microglia and for macrophage infiltration into the spinal cord after PNI [82]. TRPM2 also activates ERK MAPK and induces nuclear translocation of NFκB, resulting in production of proinflammatory cytokines and chemokines [76,77,81,83,84]. Consequently, pharmacological and genetic studies have demonstrated that TRPM2 contributes to inflammatory and neuropathic nociceptive hypersensitivity [77–79,82,85].
TRPV1 is found on unmyelinated, slowly conducting neuronal C-fibers, and is an essential component underlying injury-elicited thermal hyperalgesia and nociceptive hypersensitivity [65]. TRPV1 expression is upregulated by an exogenous ROS donor (tBOOH), and is a target of oxidation and nitration events that increase responsiveness of the channel [18,86–88]. Moreover, linoleic acid metabolites, created during production of eicosanoids, are endogenous TRPV1 agonists when oxidized, and contribute to nociceptive signaling [89,90].
Nitroxidative species induce mitochondrial dysfunction
Mitochondria have pivotal roles in a variety of cellular functions, including energy metabolism, calcium homeostasis, lipid synthesis, and apoptosis. As noted above, cellular respiration can be elevated under neuropathic pain conditions, with an attendant elevation of ROS derived from neuronal and microglial mitochondria [21,40,41]. Together with nitroxidative species derived from NOX and NOS enzymes, these species disrupt mitochondrial homeostasis via several mechanisms, leading to bioenergetic crisis (due to impaired mETC efficiency) and degeneration of primary afferents (Fig. 3) [91].
Mitochondrial DNA is a target of oxidation and nitration, while peroxidated lipid end-products, such as reactive aldehydes, can form covalent modifications (adducts) with an array of mitochondrial proteins, including antioxidants [92,93]. Together, these changes impair the structural integrity and function of mitochondria. Nitroxidative species can also trigger release of pro-apoptotic factors from mitochondria. For example, NO can disrupt mitochondrial dynamics (fission and fusion; responsible for maintaining metabolic homeostasis) that results in translocation of Bcl-2-associated X protein from the cytosol to the organelle membrane, where it activates apoptosis pathways [94–96]. Activation of apoptosis pathways contributes to neuropathic pain, as inhibition of several caspase enzymes attenuates vincristine- and dideoxycytidine-induced nociceptive hypersensitivity [97]. Neuropathic pain is associated with impaired mitochondrial function, and nociceptive hypersensitivity is accordingly attenuated by pharmacologically normalizing mitochondrial dynamics or preventing mitotoxicity [46,98–100].
Nitroxidative species induce neuroinflammatory signaling
Pro-inflammatory mediators released by glial and immune cells increase neuroexcitability in pain pathways after injury (e.g. TNF, IL-1β, BDNF) [5,50,101–103]. Several mechanisms include enhanced glutamate release, increased AMPA receptor expression, phosphorylated NMDA receptor subunits, and downregulated astrocyte glutamate transporters [5]. These proinflammatory mediators can also induce disinhibition of neuronal excitability by attenuating GABA and glycine release from interneurons and inhibitory descending projections, and downregulating KCC2 on postsynaptic terminals [5].
Nitroxidative species regulate the production of proinflammatory mediators during pathological pain. For example, NFκB and p38 MAPK are responsible for the production of a wide array of proinflammatory mediators in immune cells. Nitroxidative products degrade/inhibit IκB and MAPK phosphatases, resulting in activation of NFκB and p38 that both mediate inflammatory and neuropathic pain [52,104–107]. Furthermore, nitroxidative species may promote release of neuron-to-glia signals, such as matrix metalloproteases (MMPs) (Fig. 3) [108].
Nitroxidative species also elicit proinflammatory responses via toll-like receptor (TLR) signaling. TLRs bind a variety of endogenous ligands (danger associated molecular patterns: DAMPs), including DNA and N-formyl peptides from nitroxidatively damaged mitochondria, to trigger innate immune responses that contribute to pathological pain [5,109]. ROS serve a vital role as second messengers for TLR signaling. A rapid (minutes) respiratory burst occurs upon activation of TLR2 and 4, which is mediated by a direct interaction with the intracellular domains of NOX1, 2, and 4 enzymes. This NOX activity is essential for downstream NFκB- and p38 MAPK-dependent cytokine production [110–114]. Furthermore, activation of NOX enzymes by TLR signaling induces transcription of TLRs, and promotes membrane expression in lipid rafts, which is necessary for efficient signaling [111,115,116]. In concert with disruption of blood-brain barrier tight junctions by nitroxidative species, the TLR2-NOX1 interaction also upregulates adhesion molecules via CCL3 to facilitate transendothelial cell migration, which contributes to nociceptive hypersensitivity after PNI (Fig. 3) [102,110,117].
ROS have been implicated in the activation of NLRP3 inflammasomes [118]—protein complexes responsible for the proteolytic activation of IL-1β, a pro-inflammatory cytokine with a well-established role in pathological pain [5,101,119]. Among the various sensor molecules that trigger formation of inflammasomes, NLRP3 has been most widely investigated, and has a recently described role in neuropathic pain [120]. The relative contributions of ROS to the activation versus priming of NLRP3 inflammasomes remains to be elucidated [119]. Mitochondria are key participants in the activation of NLRP3 inflammasomes; they are a source of ROS that can directly activate NLRP3, as well as oxidized mitochondrial DNA that can also activate NLRP3 (Fig. 3) [118,121–123]. Furthermore, TRPM2 activation by nitroxidative species induces a calcium flux that activates the NLRP3 inflammasome [124].
Finally, there is a reciprocal relationship between nitroxidative species and inflammatory signaling. For example, the transcription of NOX and NOS enzymes is upregulated by TLR4 and 9 signaling, and by NFκB and p38 activation [19,125–129]. The purinergic receptor P2X7, which has a documented role in pathological pain, also induces ROS production [5,120,130]. ATP signaling through P2X7R activates NOX2 in a calcium and p38-dependent fashion [131–133].
Endogenous regulators of nitroxidative signaling
Under healthy conditions, nitroxidative species and antioxidants exist in a balanced state, as nitroxidative products play a vital physiological role in cellular processes (e.g. signal transduction, pathogen defense [134–136]). In response to increased production of nitroxidative species during injury or infection, antioxidant and regulatory systems are activated in an attempt to recover homeostasis (Fig. 1) [14].
Antioxidant defense
Transcription of antioxidant genes is a critical step in controlling nitroxidative signaling. One key transcription factor is nuclear factor E2-related factor 2 (Nrf2). Nrf2 is expressed in CNS and PNS neurons, macrophages, Schwann cells, astrocytes, and microglia [137–139]. Under homeostatic conditions, cytosolic Nrf2 is sequestered by the protein Keap1 and ubiquinated for degradation. However, in the presence of oxidants and electrophiles Nrf2 is released from Keap1 and translocates to the nucleus [140]. Nrf2 binds to the antioxidant response element (ARE) promoter region to elicit expression of 200+ antioxidant genes, including superoxide dismutases (SOD1: cytosolic; SOD2: mitochondrial), catalase, glutathione, and heme-oxygenases [140]. Another transcription factor, forkhead box, class O (FoxO), is also responsible for the production of SOD2 and catalase [141]. Many of these antioxidants are ubiquitously expressed, and their catabolic function is summarized in Figure 1 [142].
These endogenous antioxidant systems collaborate to detoxify reactive nitroxidative species (Fig. 1). Evidence is mixed whether neuroinflammatory or traumatic events increase nervous system antioxidant levels [143–152]. This likely reflects a temporally-and injury-specific antioxidant response, and the fact that injury-induced nitroxidative species can negatively regulate antioxidant production [15,76]. Antioxidant system activation can limit pathological pain: deletion of SOD1 exacerbates neuropathic pain, while exogenous antioxidants attenuate nociceptive hypersensitivity in a range of inflammatory and neuropathic pain models [37,108,153–155]. Similarly, hemeoxgenases, which elicit expression of various antioxidants, protects cells and could improve inflammation and neuropathic pain [21]. Therefore, therapies that increase antioxidant systems could resolve neuroinflammation and pain symptoms.
Anti-inflammatory cytokine and adenosine signaling
Cytokines such as IL-10 and TGFβ counter-regulate proinflammatory signaling and contribute to the resolution of neuropathic pain hypersensitivity [5,156,157]. One mechanism of action is regulation of nitroxidative signaling. For example, IL-10 and TGFβ inhibit NOX2 activity and promote antioxidant production [158–160]. This is a reciprocal relationship, as antioxidants can also drive production of anti-inflammatory cytokines [161,162]. Adenosine signaling is also anti-nociceptive in pathological pain models [163–165]. Signaling through A2A and A3 receptors inhibits NOX activity, and drives production of anti-inflammatory cytokines and antioxidants [163,166,167].
Opposition of opioid analgesia by nitroxidative species
Opioid analgesics remain the cornerstone of management of moderate-to-severe pain. However, the clinical utility of opioids is limited by tolerance, which is characterized by dose escalation due to reduced sensitivity to an opioid agonist, as well as hyperalgesia, a paradoxical increase in pain sensitivity due to opioid exposure [168,169]. Recent evidence has identified a role for nitroxidative signaling in these phenomena [6,170].
NOX activity is elevated by morphine, and genetic or pharmacological disruption of these enzymes attenuates tolerance and hyperalgesia [171–173]. Superoxide and peroxynitrite have been implicated as downstream mediators, as decomposition catalysts also attenuate tolerance and hyperalgesia [174–176]. It remains unclear how morphine engages these enzymes, but it may be mediated by classical μ-opioid receptors and/or TLR4 [168]. The pro-nociceptive mechanisms of nitroxidative species, described above, may act as an opponent process of neuronally-mediated opioid analgesia to create tolerance, or may overshadow analgesia to induce hyperalgesia. Therefore, correcting nitroxidative imbalance may improve the clinical profile of opioids [170].
Nitroxidative signaling also disrupts endogenous opioid analgesia in supraspinal sites that is engaged to inhibit spinal nociception via descending projections. For example, induction of peroxynitrite during inflammatory pain results in nitration of met-enkephalin in the rostral ventromedial medulla (RVM), which reduces opioid receptor binding affinity [177]. This may be normalized by intra-RVM microinjections of FeTMPyP5+, which was antinociceptive in inflammatory and neuropathic pain models [177].
Nitroxidative signaling as a therapeutic target for pathological pain
Under pathological conditions, endogenous antioxidant responses can be insufficient, leading to an accumulation of toxic nitroxidative species. As mentioned above, unchecked increases in nitroxidative species can promote cytotoxicity and inflammation via cascading pronociceptive signaling. Therefore, discovering therapeutic treatments that enhance cellular antioxidant capacity could help achieve nitroxidative balance to recover homeostasis.
Initial efforts to combat increases in nitroxidative species in a wide range of neurological disorders used direct antioxidant compounds (e.g. vitamins C and E, co-enzyme Q). The consensus view is that the possible beneficial effects are outweighed by unfavorable pharmacokinetic and pharmacodynamic profiles [13,178,179]. A variety of redox-active therapeutics are being developed to overcome these issues and are effective in in treating cancer-induced bone pain, inflammatory, and neuropathic pain, and can also potentiate opioid analgesia [9,10,180].
Newer approaches have instead aimed to inhibit sources of nitroxidative species, stimulate endogenous antioxidants, and prevent nitroxidative damage [13,178]. To this end, inhibitors of specific NOX and NOS isoforms, and ROS toxifiers such as MPO, are being developed and may prove effective for pain treatment [13,181]. As noted above, A2A and A3 adenosine receptor agonists attenuate spinal NOX activity and promote antioxidant production, with a concomitant decrease in neuropathic pain [163–165]. Another promising approach is the development of small molecules that catalyze the clearance of reactive aldehydes [182].
Indirect antioxidants augment the redox response without being antioxidants themselves. For example, sulforaphane, resveratrol, and curcumin induce nuclear translocation of Nrf2, a transcription factor responsible for the production of a wide array of antioxidants, and attenuate nociceptive hypersensitivity in neuropathic pain models [21,183–186]. Non-pharmacological approaches may also function in this capacity. For example, exercise increases Nrf2 expression and promotes the expression of antioxidants in the CNS as well as peripherally [187–189]. Consequently, voluntary wheel running has been shown to both prevent and reverse neuropathic pain [187,190].
Finally, ROS have a role in normal physiological processes [134–136], and there is some evidence that ROS may have protective effects after injury. For example, inflammation induced by endotoxin is exacerbated in NADPH-impaired mice, relative to their wild-type counterparts [191]. In another study, yeast survival to hydrogen peroxide stress was dependent on superoxide [192]. Further work is required to determine whether reactive oxygen species may also have a protective role after sterile nervous system injury. However, agents have been developed to spare superoxide (e.g. peroxynitrite decomposition catalysts SRI110 and SRI6 [15]), and such approaches may prove to be important for restoring homeostasis after nervous system injury.
Concluding remarks
Nitroxidative species are generated by mitochondria and by NOX and NOS enzymes. They enhance neuroexcitability in pain pathways through direct neuronal interactions, and indirectly by impairing mitochondria and inducing neuroinflammation. Normalizing nitroxidative signaling may be an alternative strategy to help to alleviate the enormous burden of pathological pain, which affects ∼20% of the population, and is poorly treated [11,193,194]. There are several areas of basic science research that may move us towards that goal (see Outstanding Questions).
Despite the extensive research implicating nitroxidative species in pathological pain states, no studies to date have quantified the critical relationships between real-time local cellular creation of nitroxidative species, their concentration at the effect site, or the distribution of their direct effect. This challenge has not been overcome owing to the volatility of these nitroxidative species and hence the very short life-time in vivo and ex vivo. Several new technologies are being developed to address these issues, and are discussed in Box 1.
Box 1. New and emerging tools to study nitroxidative species.
Colorimetric and fluorescent methods for detecting the “shadow” of the presence of nitroxidative species production is well established by the quantification of attendant cellular events (e.g. oxidative stress such as lipid peroxidation (TBARS) [195]; and DNA damage (8-Oxoguanine: 8-OxoG) [196,197]) or the quantification of more stable metabolites (e.g. nitrite/nitrate using Griess reaction [198]). These methods are not only limited in their temporal and spatial resolution, but also due to their insufficient ability to define concentrations and time courses of specific nitroxidative species. Establishing differential regulation of distinct nitroxidative species would be useful, as specific oxygen or nitrogen species have unique outcomes in the neuroinflammatory responses. A recent example demonstrated that specifically targeting peroxynitrite reduced inflammatory progression via NLRP3 inflammasome-dependent IL-1β/IL-18 release following ICH induced inflammatory injury [199]. Thus, new biosensors are required to improve our mechanistic understanding of how nitroxidative species affect the nervous system.
The chemistry of fluorescent probes for specific detection of both ex vivo and in vivo production of nitroxidative species has grown rapidly. A range of approaches and hence biosensors have been created that exploit platform sensing modalities, such as photoinduced electron transfer (PET) and Förster resonance energy transfer (FRET) signalling. Additionally, composite biosensors that incorporate a sensor functionalised to a nanoparticle (gold particles, UCNP and QDots) are used to detect and/or measure ROS/RNS species (detailed in Table 1). Such ROS species probes can quantify hypochlorite [200,201], hydroxyl [202,203], superoxide [204], hydrogen peroxide [205] and singlet oxygen [206]. Biosensors for nitric oxide [207,208], nitroxyl [209–211], peroxynitrite [212] are also being developed.
These probes detect targeted species either in cell-lines, in ex vivo tissue, or in in vivo models of inflammation. However, these biosensor tools require further optimization. Further refining biosensors will help improve the stability of the probe; the brightness of the fluorescing molecule; the specificity to defined species; the sensitivity of detection; and the consumption of the probe in the sensing process. Thus real-time continued visualisation and/or quantification of nitroxidative species within the CNS of a behaving preclinical rodent model of pathological pain remains an elusive goal.
The ultimate nitroxidative species biosensor would have real-time sensing capacity, with signal brightness that detected subcellular localisation of the nitroxidative species; ideally, this probe would not be consumed/bleached in the sensing process allowing for repeated measurements in vivo. Next generation probes will address some of these limitations. For instance, a redox sensitive fluorescent protein (rxRFP1), whose fluorescence intensity is positively related to the extent of oxidation of the probe, can detect varying amounts of oxidative stress within separate cellular compartments [213]. Further refining these tools will enable an improved understanding of how certain species contribute to oxidative or nitrosative stress, and will allow researchers to define how spatiotemporal regulation of nitroxidative activity contributes to pathological pain.
Lessons from the failure of direct antioxidants to improve clinical disease need to be recognized within the pain field; the effects of direct antioxidants on preclinical pain models continue to be reported, despite the strong probability that the results will not translate clinically. Several studies suggest that more robustly engaging antioxidant systems after injury can help alleviate pain: for instance, in animal pain models, increasing action of master antioxidant transcription factors Nrf2 or FoxO, or activating the heme-oxygenase system show promising pain-relieving effects. Future studies could explore whether combinatorial strategies – aimed at boosting multiple antioxidants or targeting both antioxidant and nitroxidative systems simultaneously – dampen inflammation and pain. Nitroxidant dysregulation clearly contributes to neuropathology; thus, discovering new targets and therapies that restore nitroxidative balance could help relieve pathological pain.
Table 1.
Probes able to detect specific ROS and RNS species, in vitro, ex vivo or in vivo
| ROS Species | Probe type | Imaging platform used | Tested in vitro | Tested ex vivo/in vivo | ROS/RNS stimulation | Ref |
|---|---|---|---|---|---|---|
| Hypochlorite | Iridium (III) complex-based two-photon phosphorescent probe. | Two-photon laser scanning fluorescence microscope & Confocal laser microscope | HeLa cells/RAW 264.7 cells | Zebrafish | 10mM NaClO (HeLa) 1mg/mL LPS (RAW 264.7 & Zebrafish) |
[201] |
| Hypochlorite | Rhodamine-based hydrazide protein fluorescent probe | Fluorescence microscope | HeLa cells | 50μM OCl− | [200] | |
| Hydroxyl | Ratiometric fluorescence biosensor (gold particles conjugated with organic fluorophore) | Confocal laser microscope | HeLa cells | 10μg/mL LPS | [203] | |
| Hydroxyl | Ratiometric fluorescence biosensor (upconversion nanaoparticles conjugated with organic fluorophore) | Fluorescence microscope equipped with 980nm laser. | HeLa cells | Mouse liver | 500ng/mL PMA (in vitro) 1–4mg LPS/100g body weight (in vivo) |
[202] |
| Superoxide | Fluorescein protein based fluorescent probe | Confocal laser microscope | HCT116/BV-2/RAW 264.7 cells | Zebrafish | 500ng/mL LPS & 50ng/mL IFN-γ (in vitro) PMA 200 ng/mL or antimycin A 500 nM (in vivo) |
[204] |
| Hydrogen Peroxide | Chemo-selective fluorescent naphthylimide peroxide probe | Two-photon laser scanning fluorescence microscope | RAW 264.7 cells | Mouse lung & skin | 1μg/ml LPS (in vitro) 20μg LPS (in vivo) |
[205] |
| Singlet Oxygen | Far-red silicon-rhodamine based chemical fluorescent probe | Fluorescence microscope with 640nm laser | HeLa cells/RAW 264.7 cells | Photosensitizers: 150μg/mL 5-ALA 5μM TMPyP4 |
[206] | |
| RNS Species | ||||||
| Nitric oxide | Chemo-selective copper(II) based fluorescence probe | Confocal laser microscope | HeLa cells/RAW 264.7 cells | 50–200μM DEA-NONOate (HeLa) 200ng/mL LPS (RAW 264.7) |
[208] | |
| Nitric oxide | Far-red two-photon chemical fluorescent probe | Two-photon laser scanning fluorescence microscope | HeLa cells/RAW 264.7 cells | Mouse liver | 25μM NOC-9 (HeLa) 20μg/mL LPS, 200U/mL IFN-γ and 0.5mg/mL L- arginine (RAW 264.7) 1–4mg/ml LPS (in vivo) |
[207] |
| Nitroxyl | FRET-based ratiometric chemical fluorescent probe | Confocal laser microscope | HeLa cells | 100μM AS | [211] | |
| Nitroxyl | Near infra-red chemical fluorescent probe | Confocal laser microscope & In Vivo Imaging System | RAW 264.7 cells | Mouse (in vivo) | 100μM AS (RAW 264.7) 500μM AS (i.p. Mouse) |
[210] |
| Nitroxyl | Lysosome-targetable near infra-red chemical fluorescent probe | Confocal laser microscope & In Vivo Imaging System | RAW 264.7 cells | Mouse (in vivo) | 200μM AS (RAW 264.7) 1mM AS (i.p. Mouse) |
[209] |
| Peroxynitrite | Boronate-based chemical fluorescent probe | Confocal laser microscope | HeLa cells/RAW 264.7 cells | 5 & 20μM Peroxynitrite solution (HeLa) 1μg/mL LPS, 50ng/ml IFN-γ, 2.5ng/ml PMA (RAW 264.7) |
[212] |
LPS: Lipopolysaccharide (produces endogenous ROS/RNS); PMA: phorbol 12-myristate-13-acetate (activates protein kinase C in vivo and in vitro); IFN-γ: Interferon gamma (produces endogenous ROS/RNS); Antimycin A: Produces endogenous ROS/RNS by driving apoptosis; 5-ALA: 5-Aminolevulinic acid (drug used in photodynamic therapy, known to produce singlet oxygen); TMPyP4: 5, 10, 15, 20-tetra-(N-methyl-4-pyridyl)porphyrin (drug used in photodynamic therapy, known to produce singlet oxygen); DEA-NONOate: 2-(N,N-Diethylamino)-diazenolate 2-oxide (Nitric Oxide donor); NOC-9: 6-(2-Hydroxy-1-methyl-2-nitrosohydrazino)-N-methyl-1-hexanamine (Nitric Oxide donor); AS: Angeli’s salt (Nitroxyl donor)
Outstanding Questions.
How ubiquitous are nitroxidative signaling mechanisms within the neuraxis, beyond the classical sites already tested (peripheral nerve injury site, DRG, spinal cord)?
Are nitroxidative signaling mechanisms common or different between different preclinical pain models?
What is the relationship between the antioxidant and anti-inflammatory cytokine systems?
Do indirect antioxidants have improved translational potential for treatment of pathological pain?
Trends.
Nitroxidative species (reactive nitrogen and oxygen species, and their products) contribute to peripheral and central sensitization after tissue injury, which leads to pathological pain.
There is a reciprocal relationship between nitroxidative and inflammatory signaling that drives peripheral and central sensitization.
New approaches to restoring nitroxidative balance may reveal effective strategies to treat pathological pain
The development of new tools may enhance our understanding of the critical relationships between real-time local creation of nitroxidative species, their concentration at the effect site, and the distribution of their direct effects.
Acknowledgments
This work was supported by an Australian Postgraduate Research scholarship (V.S.), an Australian Research Council Research Fellowship (DP110100297; M.R.H.), funding from the ARC Centre of Excellence for Nanoscale Biophotonics (CE140100003; M.R.H.), NIH grant DE021966 (L.R.W.), and a Wings for Life Project Grant (L.R.W./A.D.G.).
Glossary
- Neuroimmune signaling
bidirectional communication between leukocytes, glia and neurons
- Pathological pain
maladaptive pain that serves no useful purpose
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Krebs HA, Johnson WA. The role of citric acid in intermediate metabolism in animal tissues. Enzymologia. 1937;4:148–156. doi: 10.4159/harvard.9780674366701.c143. [DOI] [PubMed] [Google Scholar]
- 2.Gozsy B, Szent-Gyorgyi A. On the mechanism of primary respiration in pigeon breast muscle. Hoppe Seylers Z Physiol Chem. 1934;224:1–10. [Google Scholar]
- 3.Harden A, Young WJ. The Alcoholic Ferment of Yeast-Juice. Proc R Soc Lond B Biol Sci. 1906;77:405–420. [Google Scholar]
- 4.Maass O, Hatcher WH. The properties of pure hydrogen peroxide. I. J Am Chem Soc. 1920;42:2548–69. [Google Scholar]
- 5.Grace PM, et al. Pathological pain and the neuroimmune interface. Nat Rev Immunol. 2014;14:217–231. doi: 10.1038/nri3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salvemini D, et al. Roles of reactive oxygen and nitrogen species in pain. Free Radic Biol Med. 2011;51:951–966. doi: 10.1016/j.freeradbiomed.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Janes K, et al. Anti-superoxide and anti-peroxynitrite strategies in pain suppression. Biochim Biophys Acta. 2012;1822:815–821. doi: 10.1016/j.bbadis.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Little JW, et al. Reactive nitroxidative species and nociceptive processing: determining the roles for nitric oxide, superoxide, and peroxynitrite in pain. Amino Acids. 2012;42:75–94. doi: 10.1007/s00726-010-0633-0. [DOI] [PubMed] [Google Scholar]
- 9.Symons-Liguori A, et al. The Contribution of Nitroxidative Stress to Pathophysiological Pain and Opioid Analgesic Failure. In: Batinic-Haberle I, et al., editors. Redox-Active Therapeutics. Springer International Publishing; 2016. pp. 563–95. [Google Scholar]
- 10.Batinic-Haberle I, et al. Mn Porphyrin-Based Redox-Active Therapeutics. In: Batinic-Haberle I, et al., editors. Redox-Active Therapeutics. Springer International Publishing; 2016. pp. 165–211. [Google Scholar]
- 11.Pizzo PA, Clark NM. Alleviating suffering 101–pain relief in the United States. N Engl J Med. 2012;366:197–199. doi: 10.1056/NEJMp1109084. [DOI] [PubMed] [Google Scholar]
- 12.Burma NE, et al. Animal models of chronic pain: Advances and challenges for clinical translation. J Neurosci Res. 2016 doi: 10.1002/jnr.23768. [DOI] [PubMed] [Google Scholar]
- 13.Casas AI, et al. Reactive Oxygen-Related Diseases: Therapeutic Targets and Emerging Clinical Indications. Antioxid Redox Signal. 2015;23:1171–1185. doi: 10.1089/ars.2015.6433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15:411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- 15.Doyle T, et al. Targeting the Overproduction of Peroxynitrite for the Prevention and Reversal of Paclitaxel-Induced Neuropathic Pain. J Neurosci. 2012;32:6149–6160. doi: 10.1523/JNEUROSCI.6343-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hassler SN, et al. Reactive oxygen species and lipid peroxidation inhibitors reduce mechanical sensitivity in a chronic neuropathic pain model of spinal cord injury in rats. J Neurochem. 2014;131:413–417. doi: 10.1111/jnc.12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nayernia Z, et al. New insights on NOX enzymes in the central nervous system. Antioxid Redox Signal. 2014;20:2815–2837. doi: 10.1089/ars.2013.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ibi M, et al. Reactive oxygen species derived from NOX1/NADPH oxidase enhance inflammatory pain. J Neurosci Off J Soc Neurosci. 2008;28:9486–9494. doi: 10.1523/JNEUROSCI.1857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JS, et al. Glycogen synthase kinase 3beta and beta-catenin pathway is involved in toll-like receptor 4-mediated NADPH oxidase 1 expression in macrophages. FEBS J. 2010;277:2830–2837. doi: 10.1111/j.1742-4658.2010.07700.x. [DOI] [PubMed] [Google Scholar]
- 20.Kallenborn-Gerhardt W, et al. Nox2-dependent signaling between macrophages and sensory neurons contributes to neuropathic pain hypersensitivity. Pain. 2014;155:2161–2170. doi: 10.1016/j.pain.2014.08.013. [DOI] [PubMed] [Google Scholar]
- 21.Kim D, et al. NADPH oxidase 2-derived reactive oxygen species in spinal cord microglia contribute to peripheral nerve injury-induced neuropathic pain. Proc Natl Acad Sci U S A. 2010;107:14851–14856. doi: 10.1073/pnas.1009926107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hackel D, et al. The connection of monocytes and reactive oxygen species in pain. PloS One. 2013;8:e63564. doi: 10.1371/journal.pone.0063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kallenborn-Gerhardt W, et al. NADPH oxidase-4 maintains neuropathic pain after peripheral nerve injury. J Neurosci Off J Soc Neurosci. 2012;32:10136–10145. doi: 10.1523/JNEUROSCI.6227-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CF, et al. Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death. Circ Res. 2010;106:1489–1497. doi: 10.1161/CIRCRESAHA.109.215392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grace PM, et al. A novel animal model of graded neuropathic pain: utility to investigate mechanisms of population heterogeneity. J Neurosci Methods. 2010;193:47–53. doi: 10.1016/j.jneumeth.2010.08.025. [DOI] [PubMed] [Google Scholar]
- 26.Wen YR, et al. Nerve conduction blockade in the sciatic nerve prevents but does not reverse the activation of p38 mitogen-activated protein kinase in spinal microglia in the rat spared nerve injury model. Anesthesiology. 2007;107:312–321. doi: 10.1097/01.anes.0000270759.11086.e7. [DOI] [PubMed] [Google Scholar]
- 27.Xie W, et al. Early blockade of injured primary sensory afferents reduces glial cell activation in two rat neuropathic pain models. Neuroscience. 2009;160:847–857. doi: 10.1016/j.neuroscience.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cury Y, et al. Pain and analgesia: The dual effect of nitric oxide in the nociceptive system. Nitric Oxide Biol Chem Off J Nitric Oxide Soc. 2011;25:243–254. doi: 10.1016/j.niox.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Bredt DS, et al. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347:768–770. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- 30.Cao J, et al. The PSD95-nNOS interface: a target for inhibition of excitotoxic p38 stress-activated protein kinase activation and cell death. J Cell Biol. 2005;168:117–126. doi: 10.1083/jcb.200407024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Askwith T, et al. Taurine reduces nitrosative stress and nitric oxide synthase expression in high glucose-exposed human Schwann cells. Exp Neurol. 2012;233:154–162. doi: 10.1016/j.expneurol.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Annedi SC, et al. Discovery of N-(3-(1-methyl-1,2,3,6-tetrahydropyridin-4-yl)-1H-indol-6-yl) thiophene-2-carboximidamide as a selective inhibitor of human neuronal nitric oxide synthase (nNOS) for the treatment of pain. J Med Chem. 2011;54:7408–7416. doi: 10.1021/jm201063u. [DOI] [PubMed] [Google Scholar]
- 33.Keilhoff G, et al. Time-course of neuropathic pain in mice deficient in neuronal or inducible nitric oxide synthase. Neurosci Res. 2013;77:215–221. doi: 10.1016/j.neures.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 34.Mihara Y, et al. Involvement of spinal NR2B-containing NMDA receptors in oxaliplatin-induced mechanical allodynia in rats. Mol Pain. 2011;7:8. doi: 10.1186/1744-8069-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mukherjee P, et al. Development of nitric oxide synthase inhibitors for neurodegeneration and neuropathic pain. Chem Soc Rev. 2014;43:6814–6838. doi: 10.1039/c3cs60467e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonnefous C, et al. Discovery of inducible nitric oxide synthase (iNOS) inhibitor development candidate KD7332, part 1: Identification of a novel, potent, and selective series of quinolinone iNOS dimerization inhibitors that are orally active in rodent pain models. J Med Chem. 2009;52:3047–3062. doi: 10.1021/jm900173b. [DOI] [PubMed] [Google Scholar]
- 37.Tanabe M, et al. Pharmacological assessments of nitric oxide synthase isoforms and downstream diversity of NO signaling in the maintenance of thermal and mechanical hypersensitivity after peripheral nerve injury in mice. Neuropharmacology. 2009;56:702–708. doi: 10.1016/j.neuropharm.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 38.Borsani E, et al. Endothelial nitric oxide synthase in dorsal root ganglia during chronic inflammatory nociception. Cells Tissues Organs. 2013;197:159–168. doi: 10.1159/000342518. [DOI] [PubMed] [Google Scholar]
- 39.Galluzzi L, et al. Mitochondria: master regulators of danger signalling. Nat Rev Mol Cell Biol. 2012;13:780–788. doi: 10.1038/nrm3479. [DOI] [PubMed] [Google Scholar]
- 40.Schwartz ES, et al. Oxidative stress in the spinal cord is an important contributor in capsaicin-induced mechanical secondary hyperalgesia in mice. Pain. 2008;138:514–524. doi: 10.1016/j.pain.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwartz ES, et al. Persistent pain is dependent on spinal mitochondrial antioxidant levels. J Neurosci Off J Soc Neurosci. 2009;29:159–168. doi: 10.1523/JNEUROSCI.3792-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chu C, et al. Mitochondrial dependence of nerve growth factor-induced mechanical hyperalgesia. Pain. 2011;152:1832–1837. doi: 10.1016/j.pain.2011.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferrari LF, Levine JD. Alcohol consumption enhances antiretroviral painful peripheral neuropathy by mitochondrial mechanisms. Eur J Neurosci. 2010;32:811–818. doi: 10.1111/j.1460-9568.2010.07355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Joseph EK, Levine JD. Mitochondrial electron transport in models of neuropathic and inflammatory pain. Pain. 2006;121:105–114. doi: 10.1016/j.pain.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 45.Joseph EK, Levine JD. Multiple PKCε-dependent mechanisms mediating mechanical hyperalgesia. Pain. 2010;150:17–21. doi: 10.1016/j.pain.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng H, et al. Functional deficits in peripheral nerve mitochondria in rats with paclitaxel- and oxaliplatin-evoked painful peripheral neuropathy. Exp Neurol. 2011;232:154–161. doi: 10.1016/j.expneurol.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Basbaum AI, et al. Cellular and Molecular Mechanisms of Pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med. 2010;16:1248–1257. doi: 10.1038/nm.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain Off J Am Pain Soc. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ren K, Dubner R. Interactions between the immune and nervous systems in pain. Nat Med. 2010;16:1267–1276. doi: 10.1038/nm.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gwak YS, et al. Reactive oxygen species contribute to neuropathic pain and locomotor dysfunction via activation of CamKII in remote segments following spinal cord contusion injury in rats. Pain. 2013;154:1699–1708. doi: 10.1016/j.pain.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 52.Ndengele MM, et al. Cyclooxygenases 1 and 2 contribute to peroxynitrite-mediated inflammatory pain hypersensitivity. FASEB J Off Publ Fed Am Soc Exp Biol. 2008;22:3154–3164. doi: 10.1096/fj.08-108159. [DOI] [PubMed] [Google Scholar]
- 53.Yowtak J, et al. Reactive oxygen species contribute to neuropathic pain by reducing spinal GABA release. Pain. 2011;152:844–852. doi: 10.1016/j.pain.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang ZQ, et al. A newly identified role for superoxide in inflammatory pain. J Pharmacol Exp Ther. 2004;309:869–878. doi: 10.1124/jpet.103.064154. [DOI] [PubMed] [Google Scholar]
- 55.Sözbir E, Nazıroğlu M. Diabetes enhances oxidative stress-induced TRPM2 channel activity and its control by N-acetylcysteine in rat dorsal root ganglion and brain. Metab Brain Dis. 2016;31:385–393. doi: 10.1007/s11011-015-9769-7. [DOI] [PubMed] [Google Scholar]
- 56.Chen Z, et al. NMDA-receptor activation and nitroxidative regulation of the glutamatergic pathway during nociceptive processing. Pain. 2010;149:100–106. doi: 10.1016/j.pain.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee I, et al. The role of reactive oxygen species in capsaicin-induced mechanical hyperalgesia and in the activities of dorsal horn neurons. Pain. 2007;133:9–17. doi: 10.1016/j.pain.2007.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rausaria S, et al. Retooling manganese(III) porphyrin-based peroxynitrite decomposition catalysts for selectivity and oral activity: a potential new strategy for treating chronic pain. J Med Chem. 2011;54:8658–8669. doi: 10.1021/jm201233r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lorenz JE, et al. Oxidant-induced activation of cGMP-dependent protein kinase Iα mediates neuropathic pain after peripheral nerve injury. Antioxid Redox Signal. 2014;21:1504–1515. doi: 10.1089/ars.2013.5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luo C, et al. Presynaptically localized cyclic GMP-dependent protein kinase 1 is a key determinant of spinal synaptic potentiation and pain hypersensitivity. PLoS Biol. 2012;10:e1001283. doi: 10.1371/journal.pbio.1001283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Muscoli C, et al. Posttranslational nitration of tyrosine residues modulates glutamate transmission and contributes to N-methyl-D-aspartate-mediated thermal hyperalgesia. Mediators Inflamm. 2013;2013:950947. doi: 10.1155/2013/950947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao X, et al. Reactive oxygen species (ROS) are involved in enhancement of NMDA-receptor phosphorylation in animal models of pain. Pain. 2007;131:262–271. doi: 10.1016/j.pain.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zanelli SA, et al. Nitration is a mechanism of regulation of the NMDA receptor function during hypoxia. Neuroscience. 2002;112:869–877. doi: 10.1016/s0306-4522(02)00141-0. [DOI] [PubMed] [Google Scholar]
- 64.Yowtak J, et al. Effect of antioxidant treatment on spinal GABA neurons in a neuropathic pain model in the mouse. Pain. 2013;154:2469–2476. doi: 10.1016/j.pain.2013.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Julius D. TRP channels and pain. Annu Rev Cell Dev Biol. 2013;29:355–384. doi: 10.1146/annurev-cellbio-101011-155833. [DOI] [PubMed] [Google Scholar]
- 66.Xanthos DN, Sandkühler J. Neurogenic neuroinflammation: inflammatory CNS reactions in response to neuronal activity. Nat Rev Neurosci. 2014;15:43–53. doi: 10.1038/nrn3617. [DOI] [PubMed] [Google Scholar]
- 67.Andersson DA, et al. Streptozotocin Stimulates the Ion Channel TRPA1 Directly: INVOLVEMENT OF PEROXYNITRITE. J Biol Chem. 2015;290:15185–15196. doi: 10.1074/jbc.M115.644476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bautista DM, et al. TRPA1 Mediates the Inflammatory Actions of Environmental Irritants and Proalgesic Agents. Cell. 2006;124:1269–1282. doi: 10.1016/j.cell.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 69.Bessac BF, et al. Transient receptor potential ankyrin 1 antagonists block the noxious effects of toxic industrial isocyanates and tear gases. FASEB J. 2009;23:1102–1114. doi: 10.1096/fj.08-117812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Taylor-Clark TE, Undem BJ. Ozone activates airway nerves via the selective stimulation of TRPA1 ion channels. J Physiol. 2010;588:423–433. doi: 10.1113/jphysiol.2009.183301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Taylor-Clark TE, et al. Nitrooleic Acid, an Endogenous Product of Nitrative Stress, Activates Nociceptive Sensory Nerves via the Direct Activation of TRPA1. Mol Pharmacol. 2009;75:820–829. doi: 10.1124/mol.108.054445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trevisani M, et al. 4-Hydroxynonenal, an endogenous aldehyde, causes pain and neurogenic inflammation through activation of the irritant receptor TRPA1. Proc Natl Acad Sci. 2007;104:13519–13524. doi: 10.1073/pnas.0705923104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Due MR, et al. Acrolein involvement in sensory and behavioral hypersensitivity following spinal cord injury in the rat. J Neurochem. 2014;128:776–786. doi: 10.1111/jnc.12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen Z, et al. Mitigation of sensory and motor deficits by acrolein scavenger phenelzine in a rat model of spinal cord contusive injury. J Neurochem. 2016;138:328–338. doi: 10.1111/jnc.13639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Trevisan G, et al. Novel therapeutic strategy to prevent chemotherapy-induced persistent sensory neuropathy by TRPA1 blockade. Cancer Res. 2013;73:3120–3131. doi: 10.1158/0008-5472.CAN-12-4370. [DOI] [PubMed] [Google Scholar]
- 76.Chen S, et al. Role of TRPM2 in cell proliferation and susceptibility to oxidative stress. Am J Physiol Cell Physiol. 2013;304:C548–560. doi: 10.1152/ajpcell.00069.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Haraguchi K, et al. TRPM2 Contributes to Inflammatory and Neuropathic Pain through the Aggravation of Pronociceptive Inflammatory Responses in Mice. J Neurosci. 2012;32:3931–3941. doi: 10.1523/JNEUROSCI.4703-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Naziroğlu M, et al. Role of TRPM2 cation channels in dorsal root ganglion of rats after experimental spinal cord injury. Muscle Nerve. 2013;48:945–950. doi: 10.1002/mus.23844. [DOI] [PubMed] [Google Scholar]
- 79.Özdemir ÜS, et al. Hypericum perforatum Attenuates Spinal Cord Injury-Induced Oxidative Stress and Apoptosis in the Dorsal Root Ganglion of Rats: Involvement of TRPM2 and TRPV1 Channels. Mol Neurobiol. 2015;53:3540–3551. doi: 10.1007/s12035-015-9292-1. [DOI] [PubMed] [Google Scholar]
- 80.Perraud AL, et al. Accumulation of Free ADP-ribose from Mitochondria Mediates Oxidative Stress-induced Gating of TRPM2 Cation Channels. J Biol Chem. 2005;280:6138–6148. doi: 10.1074/jbc.M411446200. [DOI] [PubMed] [Google Scholar]
- 81.Yamamoto S, et al. TRPM2-mediated Ca2+ influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med. 2008;14:738–747. doi: 10.1038/nm1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Isami K, et al. Involvement of TRPM2 in Peripheral Nerve Injury-Induced Infiltration of Peripheral Immune Cells into the Spinal Cord in Mouse Neuropathic Pain Model. PLOS ONE. 2013;8:e66410. doi: 10.1371/journal.pone.0066410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chung MK, et al. The role of TRPM2 in hydrogen peroxide-induced expression of inflammatory cytokine and chemokine in rat trigeminal ganglia. Neuroscience. 2015;297:160–169. doi: 10.1016/j.neuroscience.2015.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nazıroğlu M, et al. Modulation of oxidative stress and Ca(2+) mobilization through TRPM2 channels in rat dorsal root ganglion neuron by Hypericum perforatum. Neuroscience. 2014;263:27–35. doi: 10.1016/j.neuroscience.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 85.So K, et al. Involvement of TRPM2 in a wide range of inflammatory and neuropathic pain mouse models. J Pharmacol Sci. 2015;127:237–243. doi: 10.1016/j.jphs.2014.10.003. [DOI] [PubMed] [Google Scholar]
- 86.Chuang H, Lin S. Oxidative challenges sensitize the capsaicin receptor by covalent cysteine modification. Proc Natl Acad Sci U S A. 2009;106:20097–20102. doi: 10.1073/pnas.0902675106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Susankova K, et al. Reducing and oxidizing agents sensitize heat-activated vanilloid receptor (TRPV1) current. Mol Pharmacol. 2006;70:383–394. doi: 10.1124/mol.106.023069. [DOI] [PubMed] [Google Scholar]
- 88.Westlund KN, et al. Impact of Central and Peripheral TRPV1 and ROS Levels on Proinflammatory Mediators and Nociceptive Behavior. Mol Pain. 2010;6:1744–8069. 6–46. doi: 10.1186/1744-8069-6-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Patwardhan AM, et al. Activation of TRPV1 in the spinal cord by oxidized linoleic acid metabolites contributes to inflammatory hyperalgesia. Proc Natl Acad Sci U S A. 2009;106:18820–18824. doi: 10.1073/pnas.0905415106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Patwardhan AM, et al. Heat generates oxidized linoleic acid metabolites that activate TRPV1 and produce pain in rodents. J Clin Invest. 2010;120:1617–1626. doi: 10.1172/JCI41678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bennett GJ, et al. Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nat Rev Neurol. 2014;10:326–336. doi: 10.1038/nrneurol.2014.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.MacMillan-Crow LA, Thompson JA. Tyrosine modifications and inactivation of active site manganese superoxide dismutase mutant (Y34F) by peroxynitrite. Arch Biochem Biophys. 1999;366:82–88. doi: 10.1006/abbi.1999.1202. [DOI] [PubMed] [Google Scholar]
- 93.Zarkovic N, et al. Pathophysiological relevance of aldehydic protein modifications. J Proteomics. 2013;92:239–247. doi: 10.1016/j.jprot.2013.02.004. [DOI] [PubMed] [Google Scholar]
- 94.Sinha K, et al. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87:1157–1180. doi: 10.1007/s00204-013-1034-4. [DOI] [PubMed] [Google Scholar]
- 95.Yuan H, et al. Mitochondrial fission is an upstream and required event for bax foci formation in response to nitric oxide in cortical neurons. Cell Death Differ. 2006;14:462–471. doi: 10.1038/sj.cdd.4402046. [DOI] [PubMed] [Google Scholar]
- 96.Scheving R, et al. Protein S-nitrosylation and denitrosylation in the mouse spinal cord upon injury of the sciatic nerve. J Proteomics. 2012;75:3987–4004. doi: 10.1016/j.jprot.2012.05.006. [DOI] [PubMed] [Google Scholar]
- 97.Joseph EK, Levine JD. Caspase signalling in neuropathic and inflammatory pain in the rat. Eur J Neurosci. 2004;20:2896–2902. doi: 10.1111/j.1460-9568.2004.03750.x. [DOI] [PubMed] [Google Scholar]
- 98.Ferrari LF, et al. Role of Drp1, a key mitochondrial fission protein, in neuropathic pain. J Neurosci Off J Soc Neurosci. 2011;31:11404–11410. doi: 10.1523/JNEUROSCI.2223-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Janes K, et al. Bioenergetic deficits in peripheral nerve sensory axons during chemotherapy-induced neuropathic pain resulting from peroxynitrite-mediated post-translational nitration of mitochondrial superoxide dismutase. Pain. 2013;154:2432–2440. doi: 10.1016/j.pain.2013.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lim TKY, et al. Mitochondrial and bioenergetic dysfunction in trauma-induced painful peripheral neuropathy. Mol Pain. 2015;11:58. doi: 10.1186/s12990-015-0057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Austin PJ, Moalem-Taylor G. The neuro-immune balance in neuropathic pain: involvement of inflammatory immune cells, immune-like glial cells and cytokines. J Neuroimmunol. 2010;229:26–50. doi: 10.1016/j.jneuroim.2010.08.013. [DOI] [PubMed] [Google Scholar]
- 102.Grace PM, et al. Peripheral immune contributions to the maintenance of central glial activation underlying neuropathic pain. Brain Behav Immun. 2011;25:1322–1332. doi: 10.1016/j.bbi.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 103.Gaudet AD, et al. Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J Neuroinflammation. 2011;8:110. doi: 10.1186/1742-2094-8-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Genovese T, et al. Effects of a metalloporphyrinic peroxynitrite decomposition catalyst, ww-85, in a mouse model of spinal cord injury. Free Radic Res. 2009;43:631–645. doi: 10.1080/10715760902954126. [DOI] [PubMed] [Google Scholar]
- 105.Kamata H, et al. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 106.Matata BM, Galiñanes M. Peroxynitrite is an essential component of cytokines production mechanism in human monocytes through modulation of nuclear factor-kappa B DNA binding activity. J Biol Chem. 2002;277:2330–2335. doi: 10.1074/jbc.M106393200. [DOI] [PubMed] [Google Scholar]
- 107.Zhou H, et al. CD11b/CD18 (Mac-1) is a novel surface receptor for extracellular double-stranded RNA to mediate cellular inflammatory responses. J Immunol Baltim Md 1950. 2013;190:115–125. doi: 10.4049/jimmunol.1202136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li J, et al. N-acetyl-cysteine attenuates neuropathic pain by suppressing matrix metalloproteinases. Pain. 2016;157:1711–1723. doi: 10.1097/j.pain.0000000000000575. [DOI] [PubMed] [Google Scholar]
- 109.Zhang Q, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–107. doi: 10.1038/nature08780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lee JH, et al. Interaction of NADPH oxidase 1 with Toll-like receptor 2 induces migration of smooth muscle cells. Cardiovasc Res. 2013;99:483–493. doi: 10.1093/cvr/cvt107. [DOI] [PubMed] [Google Scholar]
- 111.Nakahira K, et al. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med. 2006;203:2377–2389. doi: 10.1084/jem.20060845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Park HS, et al. Cutting Edge: Direct Interaction of TLR4 with NAD(P)H Oxidase 4 Isozyme Is Essential for Lipopolysaccharide-Induced Production of Reactive Oxygen Species and Activation of NF-κB. J Immunol. 2004;173:3589–3593. doi: 10.4049/jimmunol.173.6.3589. [DOI] [PubMed] [Google Scholar]
- 113.Yang CS, et al. ASK1-p38 MAPK-p47phox activation is essential for inflammatory responses during tuberculosis via TLR2-ROS signalling. Cell Microbiol. 2008;10:741–754. doi: 10.1111/j.1462-5822.2007.01081.x. [DOI] [PubMed] [Google Scholar]
- 114.Yang CS, et al. NADPH Oxidase 2 Interaction with TLR2 Is Required for Efficient Innate Immune Responses to Mycobacteria via Cathelicidin Expression. J Immunol. 2009;182:3696–3705. doi: 10.4049/jimmunol.0802217. [DOI] [PubMed] [Google Scholar]
- 115.Matsuzawa A, et al. ROS-dependent activation of the TRAF6-ASK1-p38 pathway is selectively required for TLR4-mediated innate immunity. Nat Immunol. 2005;6:587–592. doi: 10.1038/ni1200. [DOI] [PubMed] [Google Scholar]
- 116.Wong SW, et al. Fatty acids modulate Toll-like receptor 4 activation through regulation of receptor dimerization and recruitment into lipid rafts in a reactive oxygen species-dependent manner. J Biol Chem. 2009;284:27384–27392. doi: 10.1074/jbc.M109.044065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lochhead JJ, et al. Tempol modulates changes in xenobiotic permeability and occludin oligomeric assemblies at the blood-brain barrier during inflammatory pain. Am J Physiol Heart Circ Physiol. 2012;302:H582–593. doi: 10.1152/ajpheart.00889.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhou R, et al. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 119.Latz E, et al. Activation and regulation of the inflammasomes. Nat Rev Immunol. 2013;13:397–411. doi: 10.1038/nri3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Grace PM, et al. Morphine paradoxically prolongs neuropathic pain in rats by amplifying spinal NLRP3 inflammasome activation. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1602070113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Nakahira K, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shimada K, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhou R, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 124.Zhong Z, et al. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun. 2013;4:1611. doi: 10.1038/ncomms2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Anrather J, et al. NF-kappaB regulates phagocytic NADPH oxidase by inducing the expression of gp91phox. J Biol Chem. 2006;281:5657–5667. doi: 10.1074/jbc.M506172200. [DOI] [PubMed] [Google Scholar]
- 126.Guo Z, et al. Identification of a classic cytokine-induced enhancer upstream in the human iNOS promoter. FASEB J Off Publ Fed Am Soc Exp Biol. 2007;21:535–542. doi: 10.1096/fj.06-6739com. [DOI] [PubMed] [Google Scholar]
- 127.Lee JG, et al. A combination of Lox-1 and Nox1 regulates TLR9-mediated foam cell formation. Cell Signal. 2008;20:2266–2275. doi: 10.1016/j.cellsig.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 128.Li Y, et al. Regulation of neuronal nitric oxide synthase exon 1f gene expression by nuclear factor-kappaB acetylation in human neuroblastoma cells. J Neurochem. 2007;101:1194–1204. doi: 10.1111/j.1471-4159.2006.04407.x. [DOI] [PubMed] [Google Scholar]
- 129.Yoo BK, et al. Activation of p38 MAPK induced peroxynitrite generation in LPS plus IFN-gamma-stimulated rat primary astrocytes via activation of iNOS and NADPH oxidase. Neurochem Int. 2008;52:1188–1197. doi: 10.1016/j.neuint.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 130.Sorge RE, et al. Genetically determined P2X7 receptor pore formation regulates variability in chronic pain sensitivity. Nat Med. 2012;18:595–599. doi: 10.1038/nm.2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Apolloni S, et al. The NADPH oxidase pathway is dysregulated by the P2X7 receptor in the SOD1-G93A microglia model of amyotrophic lateral sclerosis. J Immunol Baltim Md 1950. 2013;190:5187–5195. doi: 10.4049/jimmunol.1203262. [DOI] [PubMed] [Google Scholar]
- 132.Kim SY, et al. ATP released from beta-amyloid-stimulated microglia induces reactive oxygen species production in an autocrine fashion. Exp Mol Med. 2007;39:820–827. doi: 10.1038/emm.2007.89. [DOI] [PubMed] [Google Scholar]
- 133.Parvathenani LK, et al. P2X7 Mediates Superoxide Production in Primary Microglia and Is Up-regulated in a Transgenic Mouse Model of Alzheimer’s Disease. J Biol Chem. 2003;278:13309–13317. doi: 10.1074/jbc.M209478200. [DOI] [PubMed] [Google Scholar]
- 134.Sena LA, Chandel NS. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zuo L, et al. Biological and physiological role of reactive oxygen species – the good, the bad and the ugly. Acta Physiol. 2015;214:329–348. doi: 10.1111/apha.12515. [DOI] [PubMed] [Google Scholar]
- 136.Massaad CA, Klann E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid Redox Signal. 2011;14:2013–2054. doi: 10.1089/ars.2010.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Dang J, et al. Nrf2 expression by neurons, astroglia, and microglia in the cerebral cortical penumbra of ischemic rats. J Mol Neurosci MN. 2012;46:578–584. doi: 10.1007/s12031-011-9645-9. [DOI] [PubMed] [Google Scholar]
- 138.Ishii T, et al. Oxidative stress-inducible proteins in macrophages. Free Radic Res. 1999;31:351–355. doi: 10.1080/10715769900300921. [DOI] [PubMed] [Google Scholar]
- 139.Vincent AM, et al. Sensory neurons and schwann cells respond to oxidative stress by increasing antioxidant defense mechanisms. Antioxid Redox Signal. 2009;11:425–438. doi: 10.1089/ars.2008.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39:199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 141.Klotz LO, et al. Redox regulation of FoxO transcription factors. Redox Biol. 2015;6:51–72. doi: 10.1016/j.redox.2015.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Vilhardt F, et al. Microglia antioxidant systems and redox signaling. Br J Pharmacol. 2016 doi: 10.1111/bph.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Goecks CSB, et al. Assessment of oxidative parameters in rat spinal cord after chronic constriction of the sciatic nerve. Neurochem Res. 2012;37:1952–1958. doi: 10.1007/s11064-012-0815-0. [DOI] [PubMed] [Google Scholar]
- 144.Guedes RP, et al. Neuropathic pain modifies antioxidant activity in rat spinal cord. Neurochem Res. 2006;31:603–609. doi: 10.1007/s11064-006-9058-2. [DOI] [PubMed] [Google Scholar]
- 145.Guedes RP, et al. Sciatic nerve transection increases gluthatione antioxidant system activity and neuronal nitric oxide synthase expression in the spinal cord. Brain Res Bull. 2009;80:422–427. doi: 10.1016/j.brainresbull.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 146.Hu P, et al. Secoisolariciresinol diglycoside, a flaxseed lignan, exerts analgesic effects in a mouse model of type 1 diabetes: Engagement of antioxidant mechanism. Eur J Pharmacol. 2015;767:183–192. doi: 10.1016/j.ejphar.2015.10.024. [DOI] [PubMed] [Google Scholar]
- 147.Kallenborn-Gerhardt W, et al. Antioxidant activity of sestrin 2 controls neuropathic pain after peripheral nerve injury. Antioxid Redox Signal. 2013;19:2013–2023. doi: 10.1089/ars.2012.4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Liu X, et al. Spinal Heme Oxygenase-1 (HO-1) Exerts Antinociceptive Effects Against Neuropathic Pain in a Mouse Model of L5 Spinal Nerve Ligation. Pain Med Malden Mass. 2015 doi: 10.1111/pme.12906. [DOI] [PubMed] [Google Scholar]
- 149.Naik AK, et al. Role of oxidative stress in pathophysiology of peripheral neuropathy and modulation by N-acetyl-L-cysteine in rats. Eur J Pain Lond Engl. 2006;10:573–579. doi: 10.1016/j.ejpain.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 150.Pabreja K, et al. Minocycline attenuates the development of diabetic neuropathic pain: possible anti-inflammatory and anti-oxidant mechanisms. Eur J Pharmacol. 2011;661:15–21. doi: 10.1016/j.ejphar.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 151.Pathak NN, et al. Atorvastatin attenuates neuropathic pain in rat neuropathy model by down-regulating oxidative damage at peripheral, spinal and supraspinal levels. Neurochem Int. 2014;68:1–9. doi: 10.1016/j.neuint.2014.01.014. [DOI] [PubMed] [Google Scholar]
- 152.Scheid T, et al. Sciatic nerve transection modulates oxidative parameters in spinal and supraspinal regions. Neurochem Res. 2013;38:935–942. doi: 10.1007/s11064-013-1000-9. [DOI] [PubMed] [Google Scholar]
- 153.Berger JV, et al. Enhanced neuroinflammation and pain hypersensitivity after peripheral nerve injury in rats expressing mutated superoxide dismutase 1. J Neuroinflammation. 2011;8:33. doi: 10.1186/1742-2094-8-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kong Q, et al. Heme oxygenase-1 inhibits neuropathic pain in rats with diabetic mellitus. Neural Regen Res. 2012;7:2305–2311. doi: 10.3969/j.issn.1673-5374.2012.29.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Shen Y, et al. Exogenous induction of HO-1 alleviates vincristine-induced neuropathic pain by reducing spinal glial activation in mice. Neurobiol Dis. 2015;79:100–110. doi: 10.1016/j.nbd.2015.04.012. [DOI] [PubMed] [Google Scholar]
- 156.Chen G, et al. Intrathecal bone marrow stromal cells inhibit neuropathic pain via TGF-β secretion. J Clin Invest. 2015;125:3226–3240. doi: 10.1172/JCI80883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kwilasz AJ, et al. The therapeutic potential of interleukin-10 in neuroimmune diseases. Neuropharmacology. 2015;96:55–69. doi: 10.1016/j.neuropharm.2014.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lee TS, Chau LY. Heme oxygenase-1 mediates the anti-inflammatory effect of interleukin-10 in mice. Nat Med. 2002;8:240–246. doi: 10.1038/nm0302-240. [DOI] [PubMed] [Google Scholar]
- 159.Qian L, et al. Interleukin-10 protects lipopolysaccharide-induced neurotoxicity in primary midbrain cultures by inhibiting the function of NADPH oxidase. J Pharmacol Exp Ther. 2006;319:44–52. doi: 10.1124/jpet.106.106351. [DOI] [PubMed] [Google Scholar]
- 160.Qian L, et al. Potent anti-inflammatory and neuroprotective effects of TGF-beta1 are mediated through the inhibition of ERK and p47phox-Ser345 phosphorylation and translocation in microglia. J Immunol Baltim Md 1950. 2008;181:660–668. doi: 10.4049/jimmunol.181.1.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jadhav A, et al. The heme oxygenase system selectively enhances the anti-inflammatory macrophage-M2 phenotype, reduces pericardial adiposity, and ameliorated cardiac injury in diabetic cardiomyopathy in Zucker diabetic fatty rats. J Pharmacol Exp Ther. 2013;345:239–249. doi: 10.1124/jpet.112.200808. [DOI] [PubMed] [Google Scholar]
- 162.Piantadosi CA, et al. Heme Oxygenase-1 Couples Activation of Mitochondrial Biogenesis to Anti-inflammatory Cytokine Expression. J Biol Chem. 2011;286:16374–16385. doi: 10.1074/jbc.M110.207738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Janes K, et al. A3 adenosine receptor agonist prevents the development of paclitaxel-induced neuropathic pain by modulating spinal glial-restricted redox-dependent signaling pathways. Pain. 2014;155:2560–2567. doi: 10.1016/j.pain.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Little JW, et al. Endogenous adenosine A3 receptor activation selectively alleviates persistent pain states. Brain. 2015;138:28–35. doi: 10.1093/brain/awu330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Loram LC, et al. Enduring reversal of neuropathic pain by a single intrathecal injection of adenosine 2A receptor agonists: a novel therapy for neuropathic pain. J Neurosci Off J Soc Neurosci. 2009;29:14015–14025. doi: 10.1523/JNEUROSCI.3447-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sun W, et al. Pharmacologic characterization of novel adenosine A2A receptor agonists in equine neutrophils. Am J Vet Res. 2007;68:981–987. doi: 10.2460/ajvr.68.9.981. [DOI] [PubMed] [Google Scholar]
- 167.Sun W, et al. Effects of stimulation of adenosine A2A receptors on lipopolysaccharide-induced production of reactive oxygen species by equine neutrophils. Am J Vet Res. 2007;68:649–656. doi: 10.2460/ajvr.68.6.649. [DOI] [PubMed] [Google Scholar]
- 168.Grace PM, et al. Opioid-induced central immune signaling: implications for opioid analgesia. Headache. 2015;55:475–489. doi: 10.1111/head.12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Hutchinson MR, et al. Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol Rev. 2011;63:772–810. doi: 10.1124/pr.110.004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Salvemini D, Neumann WL. Peroxynitrite: a strategic linchpin of opioid analgesic tolerance. Trends Pharmacol Sci. 2009;30:194–202. doi: 10.1016/j.tips.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 171.Doyle T, et al. Spinal NADPH oxidase is a source of superoxide in the development of morphine-induced hyperalgesia and antinociceptive tolerance. Neurosci Lett. 2010;483:85–89. doi: 10.1016/j.neulet.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Doyle T, et al. NADPH-oxidase 2 activation promotes opioid-induced antinociceptive tolerance in mice. Neuroscience. 2013;241:1–9. doi: 10.1016/j.neuroscience.2013.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ibi M, et al. Involvement of NOX1/NADPH oxidase in morphine-induced analgesia and tolerance. J Neurosci Off J Soc Neurosci. 2011;31:18094–18103. doi: 10.1523/JNEUROSCI.4136-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Little JW, et al. Spinal mitochondrial-derived peroxynitrite enhances neuroimmune activation during morphine hyperalgesia and antinociceptive tolerance. Pain. 2013;154:978–986. doi: 10.1016/j.pain.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Muscoli C, et al. Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. J Clin Invest. 2007;117:3530–3539. doi: 10.1172/JCI32420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Batinić-Haberle I, et al. Lipophilicity is a critical parameter that dominates the efficacy of metalloporphyrins in blocking the development of morphine antinociceptive tolerance through peroxynitrite-mediated pathways. Free Radic Biol Med. 2009;46:212–219. doi: 10.1016/j.freeradbiomed.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Little JW, et al. Supraspinal peroxynitrite modulates pain signaling by suppressing the endogenous opioid pathway. J Neurosci Off J Soc Neurosci. 2012;32:10797–10808. doi: 10.1523/JNEUROSCI.6345-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Schmidt HHHW, et al. Antioxidants in Translational Medicine. Antioxid Redox Signal. 2015;23:1130–1143. doi: 10.1089/ars.2015.6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Salvemini D, et al. SOD mimetics are coming of age. Nat Rev Drug Discov. 2002;1:367–374. doi: 10.1038/nrd796. [DOI] [PubMed] [Google Scholar]
- 180.Slosky LM, et al. The Cystine/Glutamate Antiporter System xc- Drives Breast Tumor Cell Glutamate Release and Cancer-Induced Bone Pain. Pain. 2016 doi: 10.1097/j.pain.0000000000000681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Dao VTV, et al. Pharmacology and Clinical Drug Candidates in Redox Medicine. Antioxid Redox Signal. 2015;23:1113–1129. doi: 10.1089/ars.2015.6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Zambelli VO, et al. Aldehyde dehydrogenase-2 regulates nociception in rodent models of acute inflammatory pain. Sci Transl Med. 2014;6:251ra118. doi: 10.1126/scitranslmed.3009539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Fattori V, et al. Curcumin inhibits superoxide anion-induced pain-like behavior and leukocyte recruitment by increasing Nrf2 expression and reducing NF-κB activation. Inflamm Res Off J Eur Histamine Res Soc Al. 2015;64:993–1003. doi: 10.1007/s00011-015-0885-y. [DOI] [PubMed] [Google Scholar]
- 184.Juan SH, et al. Mechanism of concentration-dependent induction of heme oxygenase-1 by resveratrol in human aortic smooth muscle cells. Biochem Pharmacol. 2005;69:41–48. doi: 10.1016/j.bcp.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 185.Negi G, et al. Nrf2 and NF-κB modulation by sulforaphane counteracts multiple manifestations of diabetic neuropathy in rats and high glucose-induced changes. Curr Neurovasc Res. 2011;8:294–304. doi: 10.2174/156720211798120972. [DOI] [PubMed] [Google Scholar]
- 186.Yang Y, et al. Resveratrol suppresses glial activation and alleviates trigeminal neuralgia via activation of AMPK. J Neuroinflammation. 2016;13:84. doi: 10.1186/s12974-016-0550-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Benson C, et al. Voluntary wheel running delays disease onset and reduces pain hypersensitivity in early experimental autoimmune encephalomyelitis (EAE) Exp Neurol. 2015;271:279–290. doi: 10.1016/j.expneurol.2015.05.017. [DOI] [PubMed] [Google Scholar]
- 188.Navarro A, et al. Beneficial effects of moderate exercise on mice aging: survival, behavior, oxidative stress, and mitochondrial electron transfer. Am J Physiol Regul Integr Comp Physiol. 2004;286:R505–511. doi: 10.1152/ajpregu.00208.2003. [DOI] [PubMed] [Google Scholar]
- 189.Souza PS, et al. Physical Exercise Attenuates Experimental Autoimmune Encephalomyelitis by Inhibiting Peripheral Immune Response and Blood-Brain Barrier Disruption. Mol Neurobiol. 2016 doi: 10.1007/s12035-016-0014-0. [DOI] [PubMed] [Google Scholar]
- 190.Grace PM, et al. Prior voluntary wheel running attenuates neuropathic pain. Pain. 2016;157:2012–2023. doi: 10.1097/j.pain.0000000000000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Deng J, et al. Protective role of reactive oxygen species in endotoxin-induced lung inflammation through modulation of IL-10 expression. J Immunol Baltim Md 1950. 2012;188:5734–5740. doi: 10.4049/jimmunol.1101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Thorpe GW, et al. Superoxide radicals have a protective role during H2O2 stress. Mol Biol Cell. 2013;24:2876–2884. doi: 10.1091/mbc.E13-01-0052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Blyth FM, et al. Chronic pain in Australia: a prevalence study. Pain. 2001;89:127–134. doi: 10.1016/s0304-3959(00)00355-9. [DOI] [PubMed] [Google Scholar]
- 194.Breivik H, et al. Survey of chronic pain in Europe: Prevalence, impact on daily life, and treatment. Eur J Pain. 2006;10:287–333. doi: 10.1016/j.ejpain.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 195.Patton S, Kurtz GW. 2-Thiobarbituric Acid as a Reagent for Detecting Milk Fat Oxidation1. J Dairy Sci. 1951;34:669–674. [Google Scholar]
- 196.Bespalov IA, et al. Fabs specific for 8-oxoguanine: control of DNA binding. J Mol Biol. 1999;293:1085–1095. doi: 10.1006/jmbi.1999.3214. [DOI] [PubMed] [Google Scholar]
- 197.Soultanakis RP, et al. Fluorescence detection of 8-oxoguanine in nuclear and mitochondrial DNA of cultured cells using a recombinant Fab and confocal scanning laser microscopy. Free Radic Biol Med. 2000;28:987–998. doi: 10.1016/s0891-5849(00)00185-4. [DOI] [PubMed] [Google Scholar]
- 198.Griess P. Bemerkungen zu der Abhandlung der HH. Weselsky und Benedikt „Ueber einige Azoverbindungen”. Berichte Dtsch Chem Ges. 1879;12:426–428. [Google Scholar]
- 199.Feng L, et al. P2X7R blockade prevents NLRP3 inflammasome activation and brain injury in a rat model of intracerebral hemorrhage: involvement of peroxynitrite. J Neuroinflammation. 2015;12:190. doi: 10.1186/s12974-015-0409-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Goswami S, et al. Nanomolar Detection of Hypochlorite by a Rhodamine-Based Chiral Hydrazide in Absolute Aqueous Media: Application in Tap Water Analysis with Live-Cell Imaging. Anal Chem. 2014;86:6315–6322. doi: 10.1021/ac500418k. [DOI] [PubMed] [Google Scholar]
- 201.Li G, et al. A mitochondrial targeted two-photon iridium(III) phosphorescent probe for selective detection of hypochlorite in live cells and in vivo. Biomaterials. 2015;53:285–295. doi: 10.1016/j.biomaterials.2015.02.106. [DOI] [PubMed] [Google Scholar]
- 202.Li Z, et al. A Rationally Designed Upconversion Nanoprobe for in Vivo Detection of Hydroxyl Radical. J Am Chem Soc. 2015;137:11179–11185. doi: 10.1021/jacs.5b06972. [DOI] [PubMed] [Google Scholar]
- 203.Zhuang M, et al. Ratiometric Fluorescence Probe for Monitoring Hydroxyl Radical in Live Cells Based on Gold Nanoclusters. Anal Chem. 2014;86:1829–1836. doi: 10.1021/ac403810g. [DOI] [PubMed] [Google Scholar]
- 204.Hu JJ, et al. Fluorescent Probe HKSOX-1 for Imaging and Detection of Endogenous Superoxide in Live Cells and In Vivo. J Am Chem Soc. 2015;137:6837–6843. doi: 10.1021/jacs.5b01881. [DOI] [PubMed] [Google Scholar]
- 205.Rong L, et al. Hydrogen peroxide detection with high specificity in living cells and inflamed tissues. Regen Biomater. 2016 doi: 10.1093/rb/rbw022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kim S, et al. Far-red fluorescence probe for monitoring singlet oxygen during photodynamic therapy. J Am Chem Soc. 2014;136:11707–11715. doi: 10.1021/ja504279r. [DOI] [PubMed] [Google Scholar]
- 207.Mao Z, et al. NIR in, far-red out: developing a two-photon fluorescent probe for tracking nitric oxide in deep tissue. Chem Sci. 2016;7:5230–5235. doi: 10.1039/c6sc01313a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sun X, et al. A Water-Soluble Copper(II) Complex for the Selective Fluorescence Detection of Nitric Oxide/Nitroxyl and Imaging in Living Cells. ChemPlusChem. 2016;81:30–34. doi: 10.1002/cplu.201500436. [DOI] [PubMed] [Google Scholar]
- 209.Jing X, et al. Visualization of nitroxyl (HNO) in vivo via a lysosome-targetable near-infrared fluorescent probe. Chem Commun Camb Engl. 2014;50:14253–14256. doi: 10.1039/c4cc07561g. [DOI] [PubMed] [Google Scholar]
- 210.Liu P, et al. A near-infrared fluorescent probe for the selective detection of HNO in living cells and in vivo. Analyst. 2015;140:4576–4583. doi: 10.1039/c5an00759c. [DOI] [PubMed] [Google Scholar]
- 211.Zhang H, et al. A FRET-based ratiometric fluorescent probe for nitroxyl detection in living cells. ACS Appl Mater Interfaces. 2015;7:5438–5443. doi: 10.1021/am508987v. [DOI] [PubMed] [Google Scholar]
- 212.Sun X, et al. A water-soluble boronate-based fluorescent probe for the selective detection of peroxynitrite and imaging in living cells. Chem Sci. 2014;5:3368–3373. [Google Scholar]
- 213.Fan Y, Ai H. Development of redox-sensitive red fluorescent proteins for imaging redox dynamics in cellular compartments. Anal Bioanal Chem. 2016;408:2901–2911. doi: 10.1007/s00216-015-9280-3. [DOI] [PubMed] [Google Scholar]