

Abstract
Natural products serve important roles as drug candidates and as tools for chemical biology. However, traditional natural product discovery, largely based on bioassay-guided approaches, is biased towards abundant compounds and rediscovery rates are high. Orthogonal methods to facilitate discovery of new natural products are thus needed, and herein we describe an isotope tag-based expansion of reactivity-based natural product screening to address these shortcomings. Reactivity-based screening is a directed discovery approach in which a specific reactive handle on the natural product is targeted by a chemoselective probe to enable its detection by mass spectrometry. In this study, we have developed an aminooxy-containing probe to guide the discovery of aldehyde- and ketone-containing natural products. To facilitate the detection of labeling events, the probe was dibrominated, imparting a unique isotopic signature to distinguish labeled metabolites from spectral noise. As a proof of concept, the probe was then utilized to screen a collection of bacterial extracts, leading to the identification of a new analog of antipain, deimino-antipain. The bacterial producer of deimino-antipain was sequenced and the responsible biosynthetic gene cluster was identified by bioinformatic analysis and heterologous expression. These data reveal the previously undetermined genetic basis for a well-known family of aldehyde-containing, peptidic protease inhibitors, including antipain, chymostatin, leupeptin, elastatinal, and microbial alkaline protease inhibitor (MAPI), which have been widely used for over 40 years.
Graphical Abstract
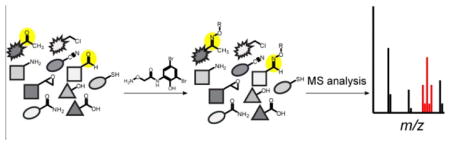
Introduction
Natural products (NPs) have historically been a valuable source of important drugs and drug leads, as well as serving as the inspiration for generations of synthetic organic chemists.1–5 While new NPs are still being discovered, rapid determination of structural novelty, a process referred to as dereplication, has become increasingly challenging.6,7 This is especially true for traditional bioassay-guided isolation approaches, which are strongly biased toward highly active/abundant compounds and most often result in the rediscovery of NPs commonly produced by many species, such as streptomycin.8 Rediscovery is further exacerbated by a focus on screening easily cultivable bacterial strains often already thoroughly combed over by industry and academia during the last 75 years. However, even in these heavily investigated strains, many NPs are easily missed by traditional screening due to sub-detection threshold production levels, and retrospective genomic analysis has demonstrated that many more complex small molecules are encoded in bacterial and fungal genomes than are currently known.2 A number of inventive methods to facilitate NP discovery that circumvent the issues surrounding bioactivity-based screening have been advanced, including new cultivation techniques, bioinformatics-guided discovery, transcriptional activation of silent gene clusters, and chemoselective enrichment.7,9–13 Similar to chemoselective enrichment, another strategy for the rapid identification of NPs, termed reactivity-based screening (RBS), involves targeting a specific functional group present on the NP. This orthogonal, chemistry-based approach is agnostic to the bioactivity of a NP, and when interfaced with genomic knowledge, becomes a powerful NP discovery platform. Typically, RBS is performed on exported metabolites without cell lysis, meaning the chemoselective probes used in this method do not have to contend with functional groups present on cytosolic compounds. Recent examples of this approach have utilized thiol-based probes to successfully target electron-deficient alkenes, β-lactams, β-lactones, and epoxides by nucleophilic addition, resulting in the discovery of several novel natural products.14,15,16 Here we expand the scope of RBS to target additional functional groups as well as introduce a method for the straightforward identification of labeled compounds based on a unique isotopic signature (Figure 1).
Figure 1.
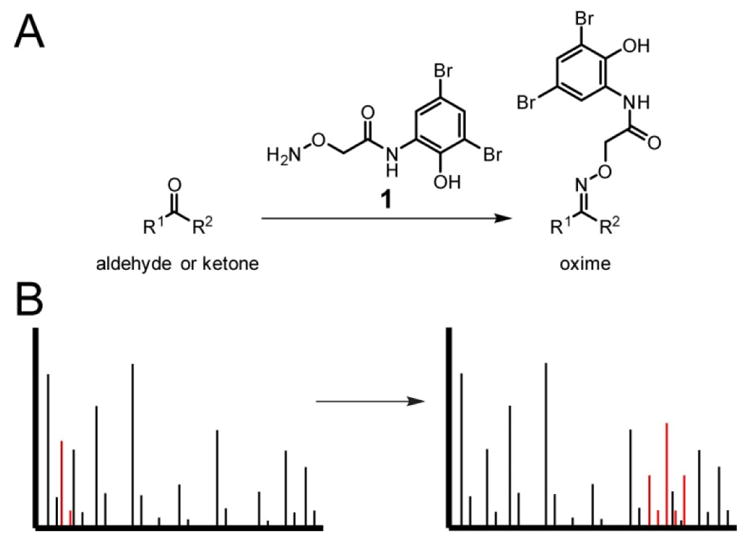
Overview of reactivity-based screening strategy. (A) Reaction scheme for labeling aldehydes and ketones with dibrominated aminooxy probe 1. (B) Labeled compounds show a mass shift accompanied by a distinctive isotopic distribution by mass spectrometry, allowing straightforward identification of peaks of interest.
Given the presence of aldehydes and ketones in many clinically important drugs and biological tools derived from NPs (Figure 2), we believed that these moieties would be attractive targets for RBS. Aldehydes in particular are often the active warhead on covalent inhibitors (e.g. protease inhibitors such as leupeptin, Figure 2), but they can also be found on drugs acting through non-covalent mechanisms (e.g. streptomycin, Figure 2).17,18 Aldehydes and ketones are found on a diverse variety of NPs, especially those produced by polyketide synthases (PKS).19 For example, the ketone moiety of erythromycin is formed due to the absence of a ketoreductase domain in the third module of the PKS; a parallel strategy applies to many other PKS pathways.19–21 Aldehyde biosynthesis remains underexplored for many NPs, although two distinct strategies have been described. In the first, the aldehyde is formed through oxidation of a hydroxyl group by cytochrome P450-type enzymes, as in the polyketides tylosin and rosamicin (Figure 2).22,23 The second pathway is unique to non-ribosomal peptides (NRPs) and results from reductive release from the non-ribosomal peptide synthetase (NRPS, e.g. flavopeptin, Figure 2).24 Due to their reactivity towards nucleophiles and biological rarity, aldehydes have been exploited for bioconjugation25,26 with numerous examples with oligonucleotides27,28 and glycoproteins.29,30 These bioconjugations are typically carried out with aminooxy or hydrazide groups to afford oxime or hydrazone linkages, respectively.25 Although oxime formation can be sluggish for some ketone substrates at neutral pH, sufficient reaction progress for detection of labeling can be achieved for aldehydes and more electrophilic ketones under mild conditions within a few hours.31,32 We therefore reasoned that the aminooxy group could likewise be utilized in a probe for the discovery of aldehyde- and sterically unencumbered ketone-containing natural products.
Figure 2.

Structures of representative NPs containing aldehyde and/or ketone functional groups. Such groups can be either critical or dispensable for the bioactivity of the NP.
A major hurdle of NP discovery is the often low quantity of compound produced under laboratory conditions.2 Coupled with a complicated metabolic background, reactivity-based labeling events could be easily overlooked in mass spectra for low abundance or less reactive compounds. We postulated that introduction of a unique isotopic signature to the reactive probe would help ameliorate this issue. This concept was recently demonstrated by Bertozzi and coworkers, who leveraged the naturally occurring 1:1 ratio of 79Br to 81Br to provide a readily detectable isotopic pattern by mass spectrometry (MS).33 Dibromination results in a symmetrical triplet with major peaks at M, M+2, and M+4. Based on this distribution, a mass pattern prediction program, IsoStamp,33 was developed and successfully used for glycoproteome profiling.34,35 By including two bromines within an aminooxy-based probe, we aimed to facilitate the discovery of lower abundance NPs through reactivity-based screening, especially in impure, complex settings (Figure 1).
Results and Discussion
Probe design and validation
Probe 1 was designed with aminooxy functionality for the specific labeling of aldehydes and ketones. While oxime formation can require hours or even days to reach completion at room temperature and neutral pH for poorly reactive electrophiles, complete labeling is not unnecessary for RBS and is actually undesirable, as the simultaneous presence of labeled and unlabeled NP aids in hit identification. We hypothesized that 1 would be sufficient for labeling target NPs as oximes36 in 1–2 h under mild conditions.31 Probe 1 is readily synthesized, requiring only a standard amide bond coupling followed by an acid-mediated deprotection (Figure 3A).
Figure 3.

Synthesis and validation of aminooxy probe 1. (A) Scheme for the synthesis of 1: a, EDC, HOBt, THF (63%); b, 4 M HCl in dioxane (55%). (B) MALDI-TOF MS of the water-soluble, excreted metabolites from Streptomyces griseus WC-3480 either unreacted (top) or labeled with 1 (bottom). The labeled streptomycin peak is magnified to display the unique dibromine isotopic pattern. (C) MALDI-TOF MS of Streptomyces bikiniensis subsp. bikiniensis ISP-5582 extract, either unreacted (top) or labeled with 1 (bottom).
With 1 in hand, we sought to validate probe reactivity toward known aldehyde- and ketone-containing NPs. We selected a small panel of commercially available compounds for this purpose. As oxime formation on aldehydes is considerably more rapid than on most ketones,32,37 we first tested labeling on streptomycin (Figure 2). As an aminoglycoside antibiotic produced by a number of actinomycetes, including Streptomyces griseus, streptomycin contains an aldehyde group installed during the penultimate biosynthetic step.38 Commercially obtained streptomycin was reacted with 1 in water for 2 h at room temperature and the crude mixture was subjected to matrix assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) analysis along with an unreacted standard. Near complete conversion of streptomycin (m/z 582.3 Da) was observed along with a new peak containing the expected dibromine isotope signature that corresponded to labeled streptomycin (m/z 902.2 Da, Figure S1). To determine if 1 would function in the context of a bacterial extract, a streptomycin producer, Streptomyces griseus WC-3480, was grown on solid media, and the exported metabolites from several colonies were extracted with water. As with commercial streptomycin, efficient labeling was readily observed (Figure 3). The labeling reaction was also successful in a range of solvents, including MeOH, n-BuOH, MeCN, EtOAc, and CHCl3, indicating that the labeling reaction could be performed on extracts directly without further sample handling. To determine the limit of detection for streptomycin, we diluted streptomycin into the organic extracts of Streptomyces lividans and performed the labeling reaction on a series of 10-fold dilutions. These samples were then analyzed by MALDI-TOF MS for evidence of labeling. The limit of detection was determined to be between 100 nM and 1 μM with the actual amount of labeled streptomycin in the 1 μM sample for MS being 1 pmol (Figure S2). We expect the limit of detection of this method to vary considerably due to differences in ionization efficiency of the natural product, the extent of labeling, and the presence of other compounds in the extract. Therefore, caution is warranted in extrapolating this limit of detection to other natural products. Interestingly, compounds labeled with 1 usually displayed more intense signals by MS than their unlabeled counterparts from the same initial sample throughout this study. An example of this can be seen in Figure 3B and C. It has been noted previously that analyte detection by MALDI-TOF MS can be greatly enhanced by covalent attachment to a chromophore capable of absorbing the MALDI UV laser energy, which may account for the signal enhancement seen herein upon labeling with 1.39,40
As aryl aldehydes are known to form oximes more slowly than alkyl aldehydes,32 the labeling reaction was also performed on anisaldehyde (Figure S3A). Although the unreacted compound could not be detected using the parameters employed, a peak with the dibromine isotopic signature was observed at the expected mass of the labeled product (m/z 456.9 Da, Figure S3B). In addition to confirming that 1 is effective at labeling aryl aldehydes, this result also supports the utility of this labeling strategy for the detection of very small molecules that are below the standard mass range of MALDI-TOF MS. Without the dibromine tag, the appearance of new peaks at low m/z ratios would have been difficult to interpret due to the unreacted NP never being observed.
Reactions with 1 were next evaluated with ketone-containing NPs to determine if oxime formation would proceed rapidly enough to be useful. Three NPs within chemically distinct ketones—daunomycin, tacrolimus, and virginiamycin S1 (Figure 2)—were evaluated for suitability with RBS-based NP discovery (Figure S4–S6). Daunomycin, containing an unhindered methyl ketone, and tacrolimus, containing both an α-oxoamide and a dialkyl ketone, both labeled to a relatively small but sufficient extent. Despite containing two ketones, only a single labeling event was observed on tacrolimus, which was localized to the dialkyl ketone by 1H NMR (Figure S7). In contrast, virginiamycin S1, which contains a piperidone, appeared to fully label under the mild reaction conditions employed. As expected, these data demonstrate that the chemical context of the ketone will influence the extent of labeling. If more robust labeling were desired while specifically targeting less reactive ketones, a number of methods for accelerating oxime formation have been reported and could be utilized in labeling reactions, including numerous organic catalysts,41–43 pH adjustments,44 and freeze-thaw cycling.45
While aminooxy groups are primarily used for oxime formation reactions with aldehydes and ketones, reactions with other electrophilic functional groups are also possible and could lead to off-target reactivity. However, these reactions typically occur either with highly reactive moieties that would be unlikely to persist in a bacterial extract (e.g. acyl chlorides) or require additional reagents and/or very long reaction times (e.g. epoxides, alkyl chlorides).46 Thus, it is unlikely that off-target reactivity would pose a major problem during aminooxy-based RBS, and indeed, several natural products containing epoxide or alkyl chloride groups were submitted to standard labeling conditions and did not react to any detectable degree (Figure S8).
Screening of bacterial extracts
A random collection of 348 actinomycetes extracts grown on solid media were next screened with probe 1. From this extract collection, 36 contained metabolites that clearly underwent labeling in the initial screen. Several strains produced more than one labeled compound, which could result from either the presence of multiple aldehyde- or ketone-containing NPs (an example of which is given below) or from MS artifacts (e.g. daunomycin degrades during MS analysis, Figure S4). Eleven of these strains were prioritized for further examination based on the presence of unique masses (many hits within the original 36 extracts contained redundant masses). Six of these 11 strains reproducibly generated the metabolite resulting in labeling.
The extract from Streptomyces bikiniensis subsp. bikiniensis ISP-5582 contained two compounds that underwent complete labeling (m/z 582.3 and 744.3, Figure 3C). A literature search revealed the organism to be a known producer of streptomycin and mannosidostreptomycin (Figure S9).47 Additionally, the genome of strain ISP-5582 contains a biosynthetic gene cluster (BGC) with identical gene architecture to the canonical streptomycin BGC from S. griseus.48 The ISP-5582 BGC resides within nucleotide positions 22,768 to 54,261 of NCBI Reference Sequence: WP_030220122.1. High resolution (HR) MS and MS/MS analysis supported the assignment of (mannosido)streptomycin (Figure S9), and thus these RBS-identified NPs were considered a further validation of the method. The required presence of an aldehyde or ketone group for labeling on an unknown NP should facilitate dereplication in future cases as well. Combined with a HR mass, NP databases can be quickly searched for known compounds that fit both the mass and reactivity criteria. While this does not guarantee that the detected compound is a novel NP, it does guide prioritization toward such compounds. Accordingly, another hit was detected in Streptomyces albulus NRRL B-3066 (unlabeled m/z 606.3 Da, Figure 4A) and chosen for follow up because its HR mass did not correspond to any known structures (Figure S10).
Figure 4.

Deimino-antipain from S. albulus NRRL B-3066. (A) MALDI-TOF MS of bacterial extract, either unreacted (top) or labeled with 1 (bottom). The region containing the labeled peak is shown magnified and each isotope peak is colored red to display the unique isotopic pattern. The intensity of peak m/z 655 Da (unknown) was reduced in the labeled spectrum, resulting in other background peaks appearing more intense. (B) Structure of deimino-antipain with carbon numbering shown. (C) Structures of previously known antipain family members.
Isolation and structure determination of deimino-antipain
Despite numerous attempts to purify the unlabeled (native) compound from S. albulus NRRL B-3066, various chromatographic methods were all found to be insufficient. Chromatographic difficulties were exacerbated by the compound bearing a weak chromophore, rendering UV-based monitoring inconvenient. Therefore, the entire extract from S. albulus NRRL B-3066 was reacted with probe 1, resulting in complete conversion to the oxime-derivatized metabolite that now harbored an easily-monitored UV chromophore (Figure S11). This derivatization endowed the compound with chemical attributes that gave significantly improved chromatographic behavior. Analysis of these HPLC traces revealed that the RBS-identified compound was actually a collection of two major and two minor species that were isobaric (Figure S11). One of the major isomers was isolated in sufficient quantity to allow structural characterization by NMR.
Using HRMS, the molecular mass of the oxime-derivatized, protonated species was determined to be m/z 926.2156 Da, and the collision-induced dissociation (CID) spectrum suggested the presence of phenylalanine (Figure S10). These data, in conjunction with NMR spectroscopy, established the peptidic nature and connectivity of the labeled NP. NMR assignments, 1H-1H COSY, 1H-1H TOCSY, 1H-1H NOESY, and 1H-13C HMBC are provided in the supplemental data (Figures S12–S13, Table S1). Interestingly, a ureido group was found between 2-C and 9-C. Additionally, while NMR analysis initially suggested two Arg may be present, further analysis incorporating MS/MS data indicated that the residue at 9-C was in fact citrulline (Cit), an amino acid rare in small-molecule NPs (only 9 entries in the Dictionary of Natural Products [DNP] contain an unmodified Cit, Figure S14). Comparison of the determined structure to NP databases revealed that the compound was similar to the known NP antipain49,50 but differed by the substitution of Cit for an Arg (Figure 4). This new NP was thus named deimino-antipain. Chiral amino acid using the standard Marfey method established all amino acids within deimino-antipain to be L, in accord with antipain.
Antipain is produced by several actinomycetes, including Streptomyces yokosukanensis and Streptomyces michiganensis,49 and is also a commercially available Ser and Cys protease inhibitor. An analog, antipain Y, contains Tyr in place of Phe (Figure 4C).51 Antipain exists as a mixture of the D- and L-arginal, with the aldehyde primarily masked in either a cyclic hemiaminal formed from the ε-nitrogen or in a hydrate.52 As two stereoisomers result from formation of the cyclic hemiaminal, antipain exists as a mixture of eight isoforms: four cyclic hemiaminals, two hydrates, and two free aldehydes (Figure S15).52 This complexity, as well as rapid conversion between the various isoforms, likely contributed to the chromatographic difficulties encountered previously. The presence of both D- and L-arginal may account for two of the detected isobaric species (Figure S11). In the reported isolations of antipain and antipain Y, cation exchange chromatography took advantage of the net positive charge imparted by the two Arg at neutral pH (Figure 4C). Replacement of one Arg with Cit would require highly acidic conditions to purify deimino-antipain by this method. Thus, the four isobaric isomers of deimino-antipain were purified as oxime-labeled derivatives, which also facilitated their UV-based detection. Nevertheless, after obtaining sufficient quantities of deimino-antipain, the oxime derivatization was reversed by acid treatment in the presence of acetone to quench the released aminooxy probe. A final round of HPLC purification yielded pure deimino-antipain. Upon assessing protease inhibitory activity, deimino-antipain was found to behave similarly to antipain (Figure S16).
During the purification of deimino-antipain, we discovered an additional, low-abundance compound labeled with 1 that was shifted +14 Da from deimino-antipain. While the material was insufficient for NMR characterization, HRMS/MS analysis supports the assignment of this species as a methylated analog of deimino-antipain (HRMS m/z calculated, 620.3515; experimental 620.3509; ppm error, 0.97; Figure S17). A probable structure would replace Val with either Leu or Ile (Figure S17). Such substitutions are known for branched, hydrophobic amino acids in NRPs, with the alternative residues being loaded at lower efficiencies than the preferred substrate;53 for instance, the antipain-like NP chymostatin is produced with internal Val/Leu/Ile substitutions. While it is unclear at this time if Leu, Ile, or both, are incorporated, this would account for the lower abundance of this analog of deimino-antipain.
Genetic characterization of deimino-antipain
Deimino-antipain belongs to a small family of peptide aldehyde protease inhibitors which includes chymostatin, elastatinal, microbial alkaline protease inhibitor (MAPI), etc. (Figure 5A). These peptides are related by their relatively low molecular weight, hydrophobic character, presence of a C-terminal aldehyde, and an internal ureido linkage; their high structural similarity implies a common biosynthetic pathway, and thus, evolutionarily related BGCs. While this family of protease inhibitors has been known for over 40 years and appears in commercial protease inhibitor cocktails,49,54,55 the genetic origin of these compounds has not yet been reported. We were thus interested in locating the deimino-antipain BCG and other peptide aldehyde protease inhibitors. Toward this goal, the genome of S. albulus NRRL B-3066 was sequenced and deposited in GenBank (accession number LWBU00000000).
Figure 5.

BGCs for deimino-antipain and related NPs. (A) Structures of peptide aldehyde protease inhibitors with the variable side chains shown in red. (B) S. albulus NRRL B-3066 BGC for deimino-antipain. A, adenylation. C, condensation. PCP, peptide carrier protein/thiolation. R, reduction. (C) Proposed proteins involved in biosynthesis of C-terminal aldehyde and capreomycidine ring of antipain-like NPs.
Numerous peptidic NPs are known to contain ureido-modified α-amino groups, including the anabaenopeptins, pacidamycins, and syringolins (Figure S18). Several of these have been demonstrated to be of non-ribosomal origin,56–58 suggesting that deimino-antipain may also be a NRP. Further, it is known that during syringolin biosynthesis, the first adenylation (A) domain installs both amino acids adjacent to the ureido group, as well as the linkage itself, resulting in one fewer A domain than would be predicted based on the number of amino acids.59 The prediction programs antiSMASH60 and PRISM61 were used to locate possible NRPS clusters; a single BGC containing the expected three A domains was identified in the S. albulus NRRL B-3066 genome (termed anpA–H, Figure 5B, Table 1). Genes anpC–G constitute the NRPS, with anpD, anpE, and anpF containing A domains predicted to install Arg, Phe, and Val, respectively, indicating that the cluster is not classically co-linear. The absence of a fourth A domain to install Arg (or Cit) suggests that either the Phe loading module (AnpE) also installs Arg/Cit, analogous to syringolin biosynthesis59 or that the Arg-specific module (AnpD) acts twice in a non-consecutive manner. The installation of two dissimilar amino acids by a single A domain in deimino-antipain would constitute a first for NRP biosynthesis (e.g. Val is installed twice for syringolin). The alternative scenario, in which AnpD installs Cit (or Arg with subsequent deimination to Cit) followed by Arg in a specific, nonconsecutive manner, would also be highly unusual. Of the remaining NRPS genes, anpC solely contains a condensation (C) domain while anpG contains thiolation and peptide carrier protein (PCP) and C domains, as well as a putative NAD-binding reductive (R) domain (instead of a traditional thioesterase) that is likely responsible for release of the product to yield the C-terminal aldehyde.24 Outside of the NRPS genes, anpA encodes a putative hydrolase that could potentially play a role in Cit formation, either pre- or post-installation of Arg. The most similar protein to AnpA occurs in Streptomyces exfoliatus (81% aa sequence identity, 88% similarity). However, the gene is not located near a NRPS or PKS BGC, nor are any other highly similar homologs. Finally, AnpB belongs to the major facilitator superfamily (MFS) and thus is assigned as an exporter, while AnpH is a predicted histidine kinase and proposed to serve a regulatory role.
Table 1.
Description of genes from S. albulus NRRL B-3066 involved in deimino-antipain biosynthesis.
| ORF1 | Product | NCBI Accession | Length (aa) | Functional Assignment | Top BLAST-P match (GenBank, NR2 database) | NCBI Accession | Length (aa) | Identity/Similarity (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | AnpA | OAL11431 | 364 | hydrolase | hydrolase [Streptomyces exfoliatus] | WP_030550770 | 350 | 81/88 |
| 2 | AnpB | OAL11432 | 438 | transporter (major facilitator superfamily) | MFS transporter [Streptomyces sp. WM4235] | WP_063839671 | 462 | 75/83 |
| 3 | AnpC | OAL11433 | 472 | NRPS; C domain | NRPS [Streptomyces sp. FxanaC1] | WP_018091965 | 431 | 67/72 |
| 4 | AnpD | OAL11435 | 1119 | NRPS; C, A, T domains | NRPS [Streptomyces auratus] | WP_006607925 | 1062 | 76/81 |
| 5 | AnpE | OAL11436 | 645 | NRPS; A, T domains | NRPS/PKS hybrid [Streptomyces sp. NRRL S-1813] | WP_030982618 | 645 | 76/81 |
| 6 | AnpF | OAL11475 | 626 | NRPS; A, T domains | NRPS [Streptomyces sp. NRRL S-1813] | WP_051817706 | 622 | 80/86 |
| 7 | AnpG | OAL11476 | 980 | NRPS; C, T domains; NAD-binding domain R | NRPS/PKS hybrid [Streptomyces decoyicus] | WP_053208647 | 951 | 71/78 |
| 8 | AnpH | OAL11437 | 158 | regulator | regulator [Streptomyces puniciscabiei] | WP_055706148 | 159 | 65/74 |
ORF, open reading frame;
NR, non-redundant
Heterologous expression of deimino-antipain
To determine if the anp gene cluster was sufficient for deimino-antipain production, a fosmid containing the putative BGC was generated and transformed into Streptomyces lividans for heterologous expression. Growth in liquid media for 10 d, however, resulted in the production of antipain, as determined by HRMS and MS/MS of the aminooxy-labeled product (Figure S19). Deimino-antipain was not detected, suggesting that Cit installation requires genes distal to the BGC that are either not present or are non-functional in S. lividans. Conversely, the native producer, S. albulus NRRL B-3066, grown under identical conditions, formed deimino-antipain with no detectable antipain (Figure S20). Interestingly, multiple other species with masses similar to antipain were also labeled with probe 1 during heterologous S. lividans expression that were not present in S. albulus or in control cultures of wild type S. lividans (Figure S20). The identity of the minor species could not be determined; however, the exact mass and MS/MS of one major component were consistent with β-MAPI, an antipain analog with a C-terminal Phe (HRMS m/z of the labeled compound, calculated, 916.1987; experimental 916.1991; ppm error, 0.44; Figure S21). The presence of these additional analogs may be due to a relaxed NRPS substrate specificity in S. lividans. Nevertheless, the heterologous production of the anp BGC demonstrates these genes are sufficient for antipain biosynthesis.
Bioinformatic survey of peptide aldehyde BGCs
We sought to identify peptide aldehyde BGCs in all available genomes to provide bioinformatic context for the deimino-antipain BGC. We thus identified highly similar homologs of AnpA–H using BLAST, aligned them, and generated profile hidden Markov models (pHMMs) of each. Next, we annotated the local genomic region of all AnpG homologs identified within GenBank.62–64 AnpG was selected for this analysis as it is putatively responsible for the installation of the aldehyde, and we believed it may be conserved in other aldehyde-containing NRPSs. Seventy-two unique AnpG proteins from 115 strains were part of similar BGCs with high individual protein sequence identities to the BGC from S. albulus B-3066 (Table S2). Given the sequence variability found among the homologs (Table S2), these are likely to represent BGCs for compounds related to but distinct from antipain. Several BGCs appear on the edge of whole-genome sequencing contigs or are missing several proteins; therefore, these cases are of indeterminate architecture. However, 91 are intact, and these were subjected to genome neighborhood analysis. Anp-like BGCs form three general architectures, but all maintain a consistent anpB–G gene direction and order (Figure 6B). The principal difference between the three BGC types is the presence or absence of an acyl-CoA dehydrogenase (present in 63/91, 69%). The additional acyl-CoA dehydrogenase gene anpI commonly occurs between anpD and anpE, but in four cases, this gene instead appears after anpG. In the latter instances, the AnpI homolog is more divergent (29% identity/43% similarity in clade A, vs. 75–100/83–100% identity/similarity for those in clade B and C, Figure 6A) and may have a different function. The MFS exporter AnpB appears in 79/91 BGCs (87%). Interestingly, none of the identified BGCs contain homologs of AnpA, the putative hydrolase from the S. albulus deimino-antipain cluster. However, homologs of AnpH appear in 4/91 cases (4%). Several additional protein families commonly co-occur in these BGCs, including regulators (Pfam identifier: PF00440; 59/91, 65%), hydrolases (PF00561, 51/91, 56%), FAD-binding domains (PF01494; 10/91, 11%), acetyltransferases (PF00583 and PF13302; 14/91, 15%), amidases (PF01425; 45/91, 49%), and methyltransferases (PF08241; 8/91, 9%), among others (Figure 6C). A phylogenetic tree annotated with homolog co-occurrence revealed that the BGCs form clades consistent with gene organization as well as protein co-occurrences (Figure 6C), and a sequence similarity network (SSN) of AnpG proteins showed grouping principally on the basis of the dehydrogenase presence. SSNs were also generated for the other NRPS proteins (AnpC-F) and these displayed the same network topology as with AnpG, again segregating into groups coinciding with the presence of the dehydrogenase (Figure S22).
Figure 6.

Analysis of antipain-like BGCs. (A) Sequence similarity network (SSN) of AnpG homologs. The network is visualized with an edge cutoff score of 10−214. Node color indicates which gene architecture shown in (B) is represented by the BGC, consisting either a deimino-antipain-like BGC lacking an acyl-CoA dehydrogenase (grey), or a BGC with an acyl-CoA dehydrogenase located either in the center (orange) or edge (purple) of the BGC. Node shapes indicate select MALDI-TOF MS screening hits (MAPI or chymostatin) upon oxime labeling. BGCs containing the internal dehydrogenase give rise to products 2 Da lighter than BGCs without it. (B) Antipain-like BGCs adopt three architectures, with NRPS domains indicated. Abbreviations: A, adenylation. C, condensation. PCP, peptide carrier protein. R, reduction. (C) Phylogenetic tree of AnpG homologs from bioinformatically identified whole (i.e non-cutoff) BGCs. The proteins form clades consistent with their groupings in the SSN in (A). The tree is annotated with the presence of the dehydrogenase and the most commonly co-occurring Pfams within 10 genes of the AnpG homolog in each BGC.
To corroborate our bioinformatic survey, we carried out a targeted screen of several strains encoding a peptide aldehyde BGC using probe 1. We noticed a series of labeled peaks of either m/z 914 or 916 Da [M+H]+ in many of the extracts (594 or 596 Da unlabeled). The presence of the heavier peaks corresponded to genomes with BGCs lacking the AnpI dehydrogenase; the -2 Da peaks entirely co-occurred with AnpI. We found that the extract from Streptomyces sp. S-98 abundantly produced a 596 Da species (916 Da labeled), as well as a 427 Da species (747 Da labeled). HR-MS/MS analysis revealed a fragmentation pattern for the 596 Da species consistent with the structure of β-MAPI (dihydrochymostatin B) (Figure S23). Additionally, HR-MS/MS showed the 427 Da species to be leupeptin (Figure S24). Interestingly, the BGC in S. sp S-98 contains an acetyltransferase (PF13302; WP_030828785) immediately downstream of its AnpG, as do several other peptide aldehyde BGCs. Thus, given the structural similarity of leupeptin and elastatinal to antipain, chymostatin, and MAPI (Figure 5A), as well as the promiscuity of the antipain BGC observed upon heterologous expression, it is most probable that these NPs share a common biosynthetic origin. We also detected peaks consistent with elastatinal A (m/z 513 Da; S. monomycini B-24309 and S. albus subsp. albus F-4371) and chymostatin B (m/z 594 Da; S. lavendulae subsp. lavendulae B-2275 and S. rimosus subsp. rimosus B-8076).
Within the peptide aldehyde protease inhibitor family, a few (e.g. elastatinal and chymostatin) are adorned with a 6-membered, capreomycidine ring (Figure 5), which is putatively formed via the dehydrogenative cyclization of Arg. The biosynthetic basis for this internal Arg cyclization is not known, given that the BGCs are elusive for all members of the family despite the common use of chymostatin and leupeptin in commercially popular protease inhibitor cocktails. Amongst known NPs, capreomycidine is relatively rare, being found in less than 50 NPs, including its eponym, capreomycin (Figure S25). Along with the AnpI/mass spectrometry correlation data, we noted that AnpI is similar (56% sequence identity) to Mur22 (ADZ45334.1) involved in the biosynthesis of muraymycin, which also displays a capreomycidine adjacent to a ureido linkage.65 Sequence analysis predicts that Mur22 and AnpI are acyl-CoA dehydrogenases while Mur15/16 (ADZ45327.1/ADZ45328.1), which were previously implicated in capreomycidine installation,65 are homologs of known transcription factors and hydroxylases. Additionally, genomic analysis of S. lavendulae, a known producer of chymostatin B, revealed no homologs of Mur15/Mur16. During capreomycin and viomycin biosynthesis, however, capreomycidine is pre-formed before NRPS elongation via β-hydroxylation of Arg followed by elimination and conjugate addition.66,67 The proteins that perform this chemistry, CmnCD/VmnCD, (ABR67746.1/ABR67747.1 and AAP92493.1/AAP92494.1) share no demonstrable sequence similarity to AnpI/Mur22 nor to Mur15/Mur16, indicating that there at least two routes for capreomycidine biosynthesis. For the peptide aldehydes NRPs, we tentatively assign AnpI as responsible for capreomycidine installation. Confirmation of this prediction will require future experimentation.
Conclusion and outlook
In this study, we have developed a new NP discovery tool that utilizes oxime formation as the basis for compound identification. Reactivity-based (aminooxy-functionalized) probe 1 selectively labels aldehydes and ketones under mild conditions. To extend the technique to less reactive ketones, previously reported catalysts41–45 could be employed to widen the scope of the reaction. Free aldehydes are generally rare among exported primary metabolites, so hits identified from mild reaction conditions have a high probability of being potentially interesting NPs. The lack of off-target reactivity also allows large excesses of probe to be used without the need for accurate stoichiometric calculations. The two bromines within probe 1 provided a unique isotopic distribution that allowed the unambiguous identification of labeling events even amidst complex or noisy data. Oxime derivatization was validated by labeling several aldehyde- or ketone-containing NPs, including streptomycin, daunomycin, and virginiamycin S1. Screening of a random collection of 348 bacterial extracts led to the discovery of a new NP, deimino-antipain, in addition to a methylated congener. Labeling of deimino-antipain with probe 1 facilitated purification, assessment of bioactivity, and structural elucidation by MS and NMR. The genome of the deimino-antipain producer, S. albulus NRRL B-3066, was sequenced, and the BGC for deimino-antipain was identified and validated by heterologous expression in S. lividans. Bioinformatic analysis of genome neighborhoods revealed 115 peptide aldehyde BGCs among sequenced genomes, providing the first genomic insights into this protease inhibitor family that includes the long-known compounds β-MAPI, chymostatin, and elastatinal. This work contributes to the collective knowledge of genetic pathways to aldehyde formation and lays the foundation to guide future screening efforts. Many aldehydes within NPs serve critical functional roles, and notable examples exist on approved drugs. Therefore, oxime-based strategies are well-poised to locate additional NPs that have evaded previous, bioassay-guided isolation efforts.
Supplementary Material
Acknowledgments
We would like to thank W. Metcalf, J. Doroghazi, R. Haines, K. Tchalukov, and M. Goettge for donating bacterial extracts, molecular biology reagents, and for technical assistance with fosmid generation. We are grateful to P. Blair for HRMS assistance. This research was supported in part by the NIH Director’s New Innovator Award Program (DP2 OD008463, to D.A.M.) and the David and Lucile Packard Fellowship for Science and Engineering (to D.A.M.). T.M. was supported in part by fellowships from the Department of Chemistry at the University of Illinois at Urbana-Champaign and the NIH Chemical Biology Interface Training Program (T32 GM070421). J.I.T. was supported in part by the Robert C. and Carolyn J. Springborn Endowment, an ACS Division of Medicinal Chemistry Predoctoral Fellowship, and the NIH Chemical Biology Interface Training Program (T32 GM070421). The Bruker Ultra-fleXtreme MALDI TOF/TOF mass spectrometer was purchased in part with a grant from the National Center for Research Resources, National Institutes of Health (S10 RR027109 A).
Footnotes
The authors declare no competing financial interest.
Additional experimental procedures, structural characterization details, and supporting figures as mentioned in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Dias DA, Urban S, Roessner U. Metabolites. 2012;2:303. doi: 10.3390/metabo2020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harvey AL, Edrada-Ebel R, Quinn RJ. Nat Rev Drug Discovery. 2015;14:111. doi: 10.1038/nrd4510. [DOI] [PubMed] [Google Scholar]
- 3.Newman DJ, Cragg GM. J Nat Prod. 2016;79:629. doi: 10.1021/acs.jnatprod.5b01055. [DOI] [PubMed] [Google Scholar]
- 4.Carlson EE. ACS Chem Biol. 2010;5:639. doi: 10.1021/cb100105c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crews CM, Splittgerber U. Trends Biochem Sci. 1999;24:317. doi: 10.1016/s0968-0004(99)01425-5. [DOI] [PubMed] [Google Scholar]
- 6.Sashidhara KV, Rosaiah JN. Nat Prod Commun. 2007;2:193. [Google Scholar]
- 7.Lewis K. Nat Rev Drug Discov. 2013;12:371. doi: 10.1038/nrd3975. [DOI] [PubMed] [Google Scholar]
- 8.Baltz RH. J Ind Microbiol Biotechnol. 2006;33:507. doi: 10.1007/s10295-005-0077-9. [DOI] [PubMed] [Google Scholar]
- 9.Luo Y, Cobb RE, Zhao H. Curr Opin Biotechnol. 2014;30:230. doi: 10.1016/j.copbio.2014.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doroghazi JR, Albright JC, Goering AW, Ju KS, Haines RR, Tchalukov KA, Labeda DP, Kelleher NL, Metcalf WW. Nat Chem Biol. 2014;10:963. doi: 10.1038/nchembio.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scherlach K, Hertweck C. Org Biomol Chem. 2009;7:1753. doi: 10.1039/b821578b. [DOI] [PubMed] [Google Scholar]
- 12.Odendaal AY, Trader DJ, Carlson EE. Chem Sci. 2011;2:760. doi: 10.1039/C0SC00620C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goering AW, McClure RA, Doroghazi JR, Albright JC, Haverland NA, Zhang Y, Ju KS, Thomson RJ, Metcalf WW, Kelleher NL. ACS Cent Sci. 2016;2:99. doi: 10.1021/acscentsci.5b00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox CL, Tietz JI, Sokolowski K, Melby JO, Doroghazi JR, Mitchell DA. ACS Chem Biol. 2014;9:2014. doi: 10.1021/cb500324n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molloy EM, Tietz JI, Blair PM, Mitchell DA. Bioorg Med Chem. 2016 doi: 10.1016/j.bmc.2016.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Castro-Falcon G, Hahn D, Reimer D, Hughes CC. ACS Chem Biol. 2016;11:2328. doi: 10.1021/acschembio.5b00924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carter AP, Clemons WM, Brodersen DE, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. Nature. 2000;407:340. doi: 10.1038/35030019. [DOI] [PubMed] [Google Scholar]
- 18.Demirci H, Murphy Ft, Murphy E, Gregory ST, Dahlberg AE, Jogl G. Nat Commun. 2013;4:1355. doi: 10.1038/ncomms2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Staunton J, Weissman KJ. Nat Prod Rep. 2001;18:380. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 20.Staunton J, Wilkinson B. Chem Rev. 1997;97:2611. doi: 10.1021/cr9600316. [DOI] [PubMed] [Google Scholar]
- 21.Khosla C, Herschlag D, Cane DE, Walsh CT. Biochemistry. 2014;53:2875. doi: 10.1021/bi500290t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiou KC. Thesis, Doctor of Philosophy. University of Michigan; Ann Arbor, MI: 2013. [Google Scholar]
- 23.Iizaka Y, Higashi N, Ishida M, Oiwa R, Ichikawa Y, Takeda M, Anzai Y, Kato F. Antimicrob Agents Chemother. 2013;57:1529. doi: 10.1128/AAC.02092-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, McClure RA, Zheng Y, Thomson RJ, Kelleher NL. J Am Chem Soc. 2013;135:10449. doi: 10.1021/ja4031193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalia J, Raines RT. Curr Org Chem. 2010;14:138. doi: 10.2174/138527210790069839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shieh P, Bertozzi CR. Org Biomol Chem. 2014;12:9307. doi: 10.1039/c4ob01632g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zatsepin TS, Stetsenko DA, Gait MJ, Oretskaya TS. Bioconjugate Chem. 2005;16:471. doi: 10.1021/bc049712v. [DOI] [PubMed] [Google Scholar]
- 28.Kojima N, Takebayashi T, Mikami A, Ohtsuka E, Komatsu Y. Nucleic Acids Symp Ser. 2009;53:45. doi: 10.1093/nass/nrp023. [DOI] [PubMed] [Google Scholar]
- 29.Zeng Y, Ramya TN, Dirksen A, Dawson PE, Paulson JC. Nat Methods. 2009;6:207. doi: 10.1038/nmeth.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bayer EA, Benhur H, Wilchek M. Anal Biochem. 1988;170:271. doi: 10.1016/0003-2697(88)90631-8. [DOI] [PubMed] [Google Scholar]
- 31.Kool ET, Crisalli P, Chan KM. Org Lett. 2014;16:1454. doi: 10.1021/ol500262y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kool ET, Park DH, Crisalli P. J Am Chem Soc. 2013;135:17663. doi: 10.1021/ja407407h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palaniappan KK, Pitcher AA, Smart BP, Spiciarich DR, Iavarone AT, Bertozzi CR. ACS Chem Biol. 2011;6:829. doi: 10.1021/cb100338x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Breidenbach MA, Palaniappan KK, Pitcher AA, Bertozzi CR. Mol Cell Proteomics. 2012;11:M111 015339. doi: 10.1074/mcp.M111.015339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Woo CM, Iavarone AT, Spiciarich DR, Palaniappan KK, Bertozzi CR. Nat Methods. 2015;12:561. doi: 10.1038/nmeth.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kalia J, Raines RT. Angew Chem Int Ed. 2008;47:7523. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bure C, Marceau P, Meudal H, Delmas AF. J Pept Sci. 2012;18:147. doi: 10.1002/psc.1429. [DOI] [PubMed] [Google Scholar]
- 38.Maier S, Grisebach H. Biochim Biophys Acta. 1979;586:231. doi: 10.1016/0304-4165(79)90095-3. [DOI] [PubMed] [Google Scholar]
- 39.Cabrera-Pardo JR, Chai DI, Liu S, Mrksich M, Kozmin SA. Nat Chem. 2013;5:423. doi: 10.1038/nchem.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mandal A, Das AK, Basak A. RSC Advances. 2015;5:106912. [Google Scholar]
- 41.Rashidian M, Mahmoodi MM, Shah R, Dozier JK, Wagner CR, Distefano MD. Bioconjugate Chem. 2013;24:333. doi: 10.1021/bc3004167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Larsen D, Pittelkow M, Karmakar S, Kool ET. Org Lett. 2015;17:274. doi: 10.1021/ol503372j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wendeler M, Grinberg L, Wang X, Dawson PE, Baca M. Bioconjugate Chem. 2014;25:93. doi: 10.1021/bc400380f. [DOI] [PubMed] [Google Scholar]
- 44.Córdova T, Peraza AJ, Calzadilla M, Malpica A. J Phys Org Chem. 2001;15:48. [Google Scholar]
- 45.Agten SM, Suylen DP, Hackeng TM. Bioconjugate Chem. 2016;27:42. doi: 10.1021/acs.bioconjchem.5b00611. [DOI] [PubMed] [Google Scholar]
- 46.Laulhe S. Thesis, Doctor of Philosophy. University of Kentucky; Louisville, KT: 2013. [Google Scholar]
- 47.Johnstone DB, Waksman SA. J Bacteriol. 1948;55:317. doi: 10.1128/jb.55.3.317-326.1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Distler J, Mansouri K, Mayer G, Stockmann M, Piepersberg W. Gene. 1992;115:105. doi: 10.1016/0378-1119(92)90547-3. [DOI] [PubMed] [Google Scholar]
- 49.Suda H, Takeuchi T, Umezawa H, Aoyagi T, Hamada M. J Antibiot. 1972;25:263. doi: 10.7164/antibiotics.25.263. [DOI] [PubMed] [Google Scholar]
- 50.Umezawa S, Tatsuta K, Fujimoto K, Tsuchiya T, Umezawa H. J Antibiot. 1972;25:267. doi: 10.7164/antibiotics.25.267. [DOI] [PubMed] [Google Scholar]
- 51.Nakae K, Kojima F, Sawa R, Kubota Y, Igarashi M, Kinoshita N, Adachi H, Nishimura Y, Akamatsu Y. J Antibiot. 2010;63:41. doi: 10.1038/ja.2009.109. [DOI] [PubMed] [Google Scholar]
- 52.Hoebeke J, Busattosamsoen C, Davoust D, Lebrun E. Magn Reson Chem. 1994;32:220. [Google Scholar]
- 53.Challis GL, Ravel J, Townsend CA. Chem Biol. 2000;7:211. doi: 10.1016/s1074-5521(00)00091-0. [DOI] [PubMed] [Google Scholar]
- 54.Umezawa H, Aoyagi T, Morishima H, Kunimoto S, Matsuzaki M. J Antibiot. 1970;23:425. doi: 10.7164/antibiotics.23.425. [DOI] [PubMed] [Google Scholar]
- 55.Umezawa H, Aoyagi T, Okura A, Morishima H, Takeuchi T. J Antibiot. 1973;26:787. doi: 10.7164/antibiotics.26.787. [DOI] [PubMed] [Google Scholar]
- 56.Rounge TB, Rohrlack T, Nederbragt AJ, Kristensen T, Jakobsen KS. BMC Genomics. 2009;10:396. doi: 10.1186/1471-2164-10-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang W, Ostash B, Walsh CT. Proc Natl Acad Sci U S A. 2010;107:16828. doi: 10.1073/pnas.1011557107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amrein H, Makart S, Granado J, Shakya R, Schneider-Pokorny J, Dudler R. Mol Plant Microbe Interact. 2004;17:90. doi: 10.1094/MPMI.2004.17.1.90. [DOI] [PubMed] [Google Scholar]
- 59.Imker HJ, Walsh CT, Wuest WM. J Am Chem Soc. 2009;131:18263. doi: 10.1021/ja909170u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weber T, Blin K, Duddela S, Krug D, Kim HU, Bruccoleri R, Lee SY, Fischbach MA, Muller R, Wohlleben W, Breitling R, Takano E, Medema MH. Nucleic Acids Res. 2015;43:W237. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Skinnider MA, Dejong CA, Rees PN, Johnston CW, Li H, Webster AL, Wyatt MA, Magarvey NA. Nucleic Acids Res. 2015;43:9645. doi: 10.1093/nar/gkv1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. Nucleic Acids Res. 2016;44:D67. doi: 10.1093/nar/gkv1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, Salazar GA, Tate J, Bateman A. Nucleic Acids Res. 2016;44:D279. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eddy SR. PLoS Comput Biol. 2011;7:e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cheng L, Chen W, Zhai L, Xu D, Huang T, Lin S, Zhou X, Deng Z. Mol Biosyst. 2011;7:920. doi: 10.1039/c0mb00237b. [DOI] [PubMed] [Google Scholar]
- 66.Ju J, Ozanick SG, Shen B, Thomas MG. ChemBioChem. 2004;5:1281. doi: 10.1002/cbic.200400136. [DOI] [PubMed] [Google Scholar]
- 67.Felnagle EA, Rondon MR, Berti AD, Crosby HA, Thomas MG. Appl Environ Microbiol. 2007;73:4162. doi: 10.1128/AEM.00485-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.