

Abstract
HIV infection is persistent in the CNS, to evaluate the compartmentalization of the CNS immune response to HIV, we compared soluble markers of cellular immunity in the blood and CSF among HIV- (n=19) and HIV+ (n=68), as well as among HIV participants with or without CSF pleocytosis. Dysfunction of the blood cerebrospinal fluid barrier (BCSFB) was common in HIV participants. CSF levels of TNFα, IFNγ, IL-2, IL-6, IL-7, IL-10, IP-10, MIP-1α, MIP-1β, and RANTES were significantly higher in participants with CSF pleocytosis (p<0.05); serum levels of these biomarkers were comparable. The CNS immune response is compartmentalized, and remains so despite the BCSFB dysfunction during HIV infection; it is markedly reduced by virology suppression, although BCSFB dysfunction persists on this subgroup.
Keywords: biomarkers, inflammation, HIV-associated neurocognitive disorders, HIV-1, subtype, cerebrospinal fluid, blood-CSF barrier
Graphical Abstract
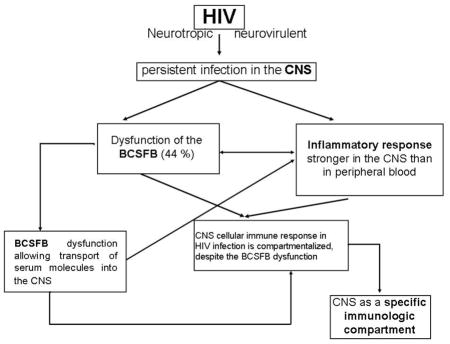
1. Introduction
The CNS barriers, i.e. blood-brain barrier (BBB) and the blood-CSF barrier (BCSFB) are highly selective physiological barriers that restrict the passage of macromolecules from circulating blood to the CNS. These barriers protect neurons from constant changes in the concentration of blood constituents without restricting the transport of nutrients and metabolic products to and from the brain, respectively, as well as the bidirectional communication with the immune system (de Vries et al., 1997; Ballabh et al., 2004; Hamilton et al., 2007; Stamatovic et al., 2008; Abbott et al., 2010; Banks, 2015). Reductions in CSF production, release, and flow rate impair the BCSFB function, thereby increasing the ratio of CSF albumin to serum albumin (albumin quotient QAlb) (Kuehne et al., 2013).
In recent years, there has been a resurgence of interest in the physiology of the brain barriers, which may play critical roles in a wide range of neurological disorders, including those associated with HIV infection (Saunders et al., 2008). Studies conducted prior to the development of highly active anti-retroviral therapy demonstrated that dysfunction of the BCSFB was common in HIV participants, particularly in those with increased CSF white blood cell (WBC) count (pleocytosis) (Marshall et al., 1988; Petito and Cash, 1992). More recent studies also found evidence of BCSFB dysfunction (Andersson et al., 2001; Calcagno et al., 2014), which can occur early in infection, and persist even with highly active anti-retroviral therapy (Calcagno et al., 2015).
HIV is a systemic infection, of which the nervous system is an important component (Brew et al., 1997). Indeed, HIV is highly neurotropic and neurovirulent, and causes persistent infection in the CNS. Invasion of the CNS during acute HIV infection (Resnick et al., 1988; Spudich et al., 2011; Valcour et al., 2013) initiates an inflammatory cascade that eventually compartmentalizes the CNS immune response even with anti-retroviral therapy. Thus we hypothesize that the inflammatory response in HIV infection is stronger in CNS than peripheral blood, characterizing compartmentalization of CNS cellular immune response; despite the dysfunction of the BCSFB during HIV infection. By compartmentalization of immune responses we mean evidence of immune activity, including production of signaling molecules such as chemokines and interleukins, that is CNS specific and would be missed by studying only plasma or serum.
To evaluate the compartmentalization of the CNS immune response to HIV, we compared soluble markers of cellular immunity in the blood and CSF among uninfected and HIV-infected individuals, as well as among HIV participants with or without CSF pleocytosis. We selected biomarkers on the basis of their roles in cellular immunity, neuroinflammation, chemotaxis, and CNS HIV pathobiology (Nath, 1999; Genis et al., 1992; King et al., 2006; Campbell et al., 2007a; Abbas and Herbein, 2013). We also investigated the BCSFB function, and identified the origin of CSF biomarkers.
We anticipated that the data would provide important pathophysiological insights into the neurological impact of HIV and the specific immune response in the CNS. This study extends previous studies (Price et al., 2013; Peterson et al., 2014) by examining cellular immunity in the CSF and serum of participants infected with non-B HIV subtypes, noting that HIV-infected samples we tested were collected from the same geographic area. Indeed, previous studies focused almost exclusively on the effects of HIV-1 subtype B on inflammatory and chemotaxis biomarkers (Genis et al., 1992; Cinque et al., 2007; Brew and Letendre, 2008; Hagberg et al., 2010; Yuan et al., 2013).
2. Materials and methods
This study was a cross-sectional survey of stored CSF and serum samples, and was approved by institutional review boards at University of California San Diego, Hospital de Clínicas-UFPR in Brazil, and Brazil National IRB (CONEP). Participants and methods were described previously (de Almeida et al., 2013; de Almeida et al., 2016).
2.1 Subjects
A total of 87 paired CSF and serum samples were analyzed. Demographic characteristics, HIV status, and co-infections, if any, are summarized in Table 1 for HIV-positive (n = 68) and HIV-negative (n = 19) volunteers.
Table 1.
Demographics, HIV infection characteristics, and co-morbidities of HIV participants and uninfected volunteers
| HIV+ (n = 68) | HIV− (n = 19) | P | |
|---|---|---|---|
| Demographics | |||
| Age, years | 43 (35; 48) | 41 (38; 50) | 0.91 |
|
| |||
| Education, years | 8 (5; 11) | 12 (11; 15) | 0.0001 |
|
| |||
| Gender, n male (%) | 33 (49) | 14 (74) | 0.07 |
|
| |||
| HIV status and treatment | |||
| AIDS, n (%) | 55 (81) | - | - |
|
| |||
| Duration of infection, months | 99 (29; 140) | - | - |
|
| |||
| Current CD4 | 369 (201; 534) | - | - |
|
| |||
| Nadir CD4 | 92 (37; 267) | - | - |
|
| |||
| Log Plasma HIV RNA1 | 1.7 (1.7; 3.5) | - | - |
|
| |||
| Plasma HIV RNA < 50 copies/mL, n (%) | 38 (56) | - | - |
|
| |||
| Log CSF HIV RNA | 1.7 (1.7; 2.8) | - | - |
|
| |||
| CSF HIV RNA < 50 copies/mL, n (%) | 36 (53) | - | - |
|
| |||
| on CART2, n (%) | 55 (81) | - | - |
|
| |||
| CPE3 | 8 (6; 9) | - | - |
|
| |||
| Adherence4, n (%) | 51 (93) | - | - |
|
| |||
| Co-morbidities | |||
| HCV5, n (%) | 12 (18) | 0 | - |
|
| |||
| Log Plasma HCV RNA, n | 2.9 (1.7; 5.9) | 0 | - |
Plasma viral load, log10 c/mL;
CART, combination anti-retroviral therapy;
CPE, anti-retroviral CNS penetration effectiveness (Letendre et al., 2010);
Anti-retroviral treatment adherence was evaluated using AIDS clinical trial (ACTG) adherence questionnaire (4-day recall);
Hepatitis C virus (HCV) status was assessed by antibody testing (Abbott-Architect). Participants co-infected with HCV were not on treatment with interferon-gamma. Data are median (IQR) or number of cases (%).
2.1.1. HIV participants
HIV participants were recruited at Hospital de Clínicas, Universidade Federal do Paraná, Curitiba, Paraná, Brazil. Individuals with opportunistic CNS infections were excluded. All volunteers provided blood and CSF samples, and underwent serological testing to confirm HIV status before enrollment, in accordance with guidelines published by the Brazilian Ministry of Health (2009). For participants with clinically resistant infection, the infecting HIV strain was genotyped using pol sequences, while env sequences were used for all other participants. Genotyping indicated that 27 individuals were infected with HIV subtype B, and 40 with non-B HIV subtypes (C-26, BF-10, BC-1, CF-1, and F-2). In one participant, the HIV subtype could not be determined.
2.1.2. Uninfected controls
As lumbar punctures could not be performed in uninfected volunteers in Brazil, we recruited a control group of 19 age-matched HIV-negative individuals at HIV Neurobehavioral Research Center, University of California San Diego. These volunteers were free of neurological co-morbidities, and tested negative on serological tests for hepatitis C virus and syphilis. The neurochemical criteria for inclusion into the control group was CSF WBC count ≤ 5 cells/mm3, CSF total protein ≤ 45 mg/dL, and CSF glucosis ≥ 55 mg/dL.
2.2. Laboratory Methods
Lumbar punctures were performed aseptically using an atraumatic spinal needle. CSF total protein and glucose were quantified by benzethonium chloride and hexoquinase/G-6-PDH (Architect-Abbott, IL), respectively. Total WBC/mm3 was determined in fresh, uncentrifuged CSF by manual counting in a Fuchs-Rosenthal chamber. CSF pleocytosis was defined as WBC > 5 cells/mm3. For differential leukocyte counts, CSF samples were concentrated by Shandon Cytospin (Pittsburgh, USA), mounted, and stained by May-Grünwald-Giemsa technique. Aliquots of CSF and serum were refrozen and stored at -80°C for subsequent batch testing for cytokines and chemokines.
2.2.1. Clinical laboratory parameters
HIV RNA levels in serum and CSF were quantified by branched DNA assay (Siemens) with nominal limit of detection 50 copies/mL. CD4 counts were quantified by flow cytometry (FACSCalibur-Multitest), while nadir CD4 was extracted from medical records.
2.2.1.1. CSF and serum biomarkers
RANTES was quantified by high-sensitivity enzyme-linked immunosorbent assay (R&D Systems, Minneapolis, MN). TNFα, IFNγ, IL-1β, IL-2, IL-4, IL-6, IL-7, IL-10, MCP-1, MIP-1α, MIP-1β, and IP-10 were quantified by multiplex bead suspension array immunoassays (EMD Millipore, Billerica, MA), in which primary antibodies are immobilized to dyed fluorescent microspheres. The analytic sensitivity of these assays ranged between 0.1 to 10.1 pg/mL. All samples were assayed concurrently in duplicate according to manufacturers’ instructions.
2.2.1.2. Blood-CSF barrier function
CSF and serum albumin were quantified by the nephelometric method (Dade Behring BNII, Deerfield, IL). The functional integrity of the blood-CSF barrier was then assessed by the CSF albumin/serum albumin quotient, which was calculated as QAlb = AlbCSF/Albserum. The upper limit of the reference range for QAlb is age-dependent, was calculated for each participant according to the equation: 4+ age in years/15 (Reiber and Peter, 2001). The albumin and IgG hyperbolic function was calculated using the Reibergram plot (Reiber, 1995). Albumin leakage across the blood-brain barrier over 24 hrs was also calculated (Tourtellotte et al., 1989).
2.3. Data analyses
Demographic data, HIV disease characteristics, and CSF biochemical, cytological, and virological measures were compared between groups using independent samples t-tests for continuous variables and Fisher’s exact test for binary and categorical variables (gender, AIDS diagnostic, ART, HCV serostatus, HIV RNA in plasma and CSF). The demographic data and CSF biochemical and cytological measures were compared between the HIV-positive and the HIV-negative control groups using similar methods. The distribution of plasma HIV viral loads was highly skewed so the Wilcoxon rank-sum test was used.
In order to detect the source of the biomarkers in CSF was calculated the coefficient of variation (CV) for each biomarker in CSF and serum in the normal control group; to the evaluation of the inter-individual variation. The comparison of biological variation of molecule concentration can be used to compare functionally connected compartments of the same group; if we could expect variation of propagation for molecules due to the passage from one compartment to the other (Kuehne et al., 2013).
The CSF and serum biomarker values were log10-transformed to normalize their distributions, and presented in terms of mean (SD), since they are approximately normal on the log scale.
The biomarkers were compared between the HIV-positive and HIV-negative control groups as well as between the HIV-positive samples with pleocytosis in the CSF and HIV-positive samples with a normal CSF WBC count. We used a multivariable linear regression (adjusted analysis), controlling for plasma HIV viral load suppression, and nadir CD4 count, which has been shown in previous studies to be associated with increased soluble biomarkers of inflammation and chemotaxis in HIV (Gisslen et al., 1994; Mooney et al, 2015; Noel et al., 2014). In a second moment results were adjusted for QAlb for the comparison of groups HIV+ and HIV-; and QAlb in addition to suppressed plasma and CSF viral load, and CD4 nadir for the comparison of the groups with and without CSF pleocytosis.
Correlations between variables were calculated using Spearman’s rank-order correlation. Results were considered statistically significant at the 5% alpha level. Cohen’s d effect sizes (and 95% confidence intervals) were reported for differences between groups.
3. RESULTS
As shown in Table 1, ART was prescribed to 55 (81%) of the HIV+ participants. The most frequent ART regimen (33 participants; 60%) contained a ritonavir-boosted HIV protease inhibitor (PI) plus two nucleoside/nucleotide reverse transcriptase inhibitors (NRTI; NtRTI); 18 participants (33%) received a non-nucleoside RT inhibitor (NNRTI) with 2 NRTIs, and 4 (7%) participants received other regimen types. Darunavir was not available in Brazil during the time that these samples were collected. Because of this the Brazilian National HIV Therapy protocol recommended that patients with resistance receive a regimen of an NNRTI combined with 2 NRTIs and ritonavir-boosted lopinavir. The number of participants on ARV and plasma HIV RNA <50 copies/mL was 37 (67%).
3.1. CSF profile and HIV RNA levels
The biochemical, cytological, and virological characteristics of the CSF in uninfected and HIV participants are summarized in Table 2. CSF pleocytosis was detected exclusively in 20/68 (29.4 %, P < 0.0001) among HIV participants. CSF pleocytosis can be attributed solely to HIV infection, since we excluded other opportunistic or co-infections by standard etiological investigation methods. All participants in this study were ambulatory volunteers without signs and symptoms of active opportunistic disease or co-infections. In addition, all CSF samples with pleocytosis underwent standard microbiological tests to exclude coinfections including gram smear and culture for bacterial infection; direct CSF smear, culture and latex agglutination tests for C. neoformans capsular antigen; Ziehl smear, culture and PCR for M. tuberculosis; and PCR for HSV, enterovírus, VZV, CMV, HHV6, HHV7, EBV and JC virus.
Table 2.
Biochemical, cytological, and virological characteristics of the CSF in HIV-positive and uninfected volunteers. Significant differences are highlighted in bold
| HIV+ (n = 68) | HIV− (n = 19) | P | |
|---|---|---|---|
| WBC, cells/mm3 | 2.1 (0.6; 7.2) | 2 (1;2.5) | 0.4024 |
| WBC count > 5 cells/mm3 | 20 (29 %) | 0 | < 0.0001 |
| Glucose, mg/dL | 57 (53; 62) | 63 (59; 71) | 0.0003 |
| Total protein, mg/dL | 40 (32; 46) | 30 (26; 38) | 0.0026 |
| Total protein > 45 mg/dL | 20 (29 %) | 0 | 0.0048 |
| Albumin, mg/dL | 22.4 (16.4; 28.9) | 18 (15; 24) | 0.019 |
| Albumin quotient, QAlb | 0.0064 (0.0049; 0.0097) | 0.005 (0.004; 0.006) | 0.0023 |
| Lactic acid, mmol/L | 1.6 (1.5; 1.8) | - | - |
| RBC, cells/mm3 | 0.5 (0; 7.5) | 2.0 (1.0, 4.0) | 0.3993 |
| Log CSF HIV RNA | 1.7 (1.7; 2.8) | - | - |
| Viral load < 50 | 35 | - | - |
| HIV RNA CSF > blood | 12 (18 %) | - | - |
CSF glucose was significantly higher in HIV-negative volunteers than in HIV participants, although levels in both groups were below reference range. Data are median (IQR) or number of cases (%).
3.2. Inflammatory biomarkers
3.2.1. Comparison between HIV-positive and HIV-negative groups
Cytokine and chemokine levels are listed in Table 3. In CSF, 9 of 13 cytokines and chemokines were significantly elevated (P < 0.05) in HIV participants than in uninfected controls. In contrast, only 4 of 13 markers were significantly elevated in serum (Table 3).
Table 3.
HIV-positive (n=68) and HIV-negative (n=19) levels of cellular chemotaxis and inflammatory biomarkers in cerebrospinal fluid and serum. Significant differences are in bold typeface.
| CSF | SERUM | CSF/serum | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HIV (+) | HIV (−) | Cohen’s d (95%CI) (b) | P (a) | HIV (+) | HIV (−) | Cohen’s d (95%CI) (b) | P (a) | HIV (+) | HIV (−) | Cohen’s d (95%CI) (b) | P (a) | |
| Cytokines | ||||||||||||
| TNFα | 0.33 (0.48) | 0.04 (0.24) | 0.66 (0.3,1.02) | 0.0006 | 0.94 (0.21) | 0.79 (0.18) | 0.72 (0.2,1.23) | 0.0071 | −0.61(0.47) | − 0.75(0.26) | 0.33 (− 0.19,0.84) | 0.2100 |
| IFN γ | 0.27 (0.43) | 0.07 (0.29) | 0.50 (− 0.02,1.02) | 0.0230 | 0.27 (0.45) | 0.56 (0.47) | −0.62 (− 1.13, −0.1) | 0.0200 | 0.001(0.63) | − 0.48(0.53) | 0.79 (0.28,1.31) | 0.0029 |
| IL1 β | −0.11 (0.03) | −0.1 (0.0) | −0.31 (− 0.58, −0.04) | 0.0270 | − 0.09(0.07) | − 0.02(0.18) | −0.60 (− 1.44,0.23) | 0.1500 | −0.02(0.08) | − 0.07(0.18) | 0.5 (− 0.31,1.32) | 0.2100 |
| IL2 | 0.02 (0.09) | 0 (0.0) | 0.27 (0,0.54) | 0.0540 | 0.06 (0.31) | 0.08 (0.19) | −0.07 (− 0.58,0.45) | 0.7500 | −0.04(0.33) | − 0.08(0.19) | 0.13 (− 0.38,0.65) | 0.6100 |
| IL4 | 0.65 (0) | 0.65 (0) | 0 (0,0) | 1.0000 | 0.69 (0.16) | 0.77 (0.39) | −0.35 (− 1.18,0.48) | 0.3900 | −0.03(0.16) | − 0.11(0.39) | 0.35 (− 0.48,1.18) | 0.3900 |
| IL6 | 0.16 (0.34) | 0.09 (0.27) | 0.22 (− 0.3,0.73) | 0.4100 | 0.07 (0.29) | 0.08(0.26) | −0.04 (− 0.56,0.47) | 0.8700 | 0.09(0.45) | 0.01(0.35) | 0.19 (− 0.32,0.71) | 0.4600 |
| IL7 | 0.28 (0.20) | 0.17 (0.14) | 0.60 (0.09,1.12) | 0.0220 | 0.64 (0.37) | 0.65 (0.28) | −0.04 (− 0.55,0.48) | 0.8800 | −0.36(0.41) | − 0.49(0.33) | 0.32 (− 0.2,0.84) | 0.2200 |
| IL10 | 0.25 (0.41) | 0.04 (0.0) | 0.56 (0.29,0.84) | 0.0001 | 0.31(0.45) | 0.30(0.40) | 0.01 (− 0.51,0.53) | 0.9700 | −0.06(0.58) | − 0.26(0.40) | 0.37 (− 0.15,0.88) | 0.1600 |
| Chemokines | ||||||||||||
| IP10 | 3.26 (0.75) | 2.98 (0.20) | 0.42 (0.12,0.72) | 0.0067 | 2.98 (0.35) | 2.57(0.2) | 1.27 (0.75,1.78) | <0.0001 | 0.29(0.67) | 0.41(0.20) | −0.21 (− 0.52,0.1) | 0.1900 |
| MCP1 | 3.16 (0.19) | 3.05 (0.12) | 0.62 (0.1,1.13) | 0.0200 | 2.73 (0.21) | 2.63(0.22) | 0.46 (− 0.05,0.98) | 0.0770 | 0.43(0.27) | 0.42(0.28) | 0.04 (− 0.47,0.56) | 0.8800 |
| MIP1α | 1.10 (0.14) | 1.01 (0.06) | 0.66 (0.31,1.01) | 0.0004 | 0.61 (0.29) | 0.64(0.29) | −0.11 (− 0.63,0.4) | 0.6600 | 0.49(0.34) | 0.37(0.27) | 0.37 (− 0.15,0.88) | 0.1600 |
| MIP1β | 1.09 (0.28) | 0.95 (0.30) | 0.49 (− 0.03,1) | 0.0650 | 1.71 (0.27) | 1.72(0.19) | −0.05 (− 0.56,0.47) | 0.8500 | −0.62(0.33) | − 0.77(0.24) | 0.48 (− 0.04,0.99) | 0.0690 |
| RANTES | 0.58 (0.62) | 0.37 (0.57) | 0.34 (− 0.18,0.85) | 0.2000 | 4.48 (0.26) | 4.75(0.2) | −1.09 (− 1.6, −0.57) | <0.0001 | −3.9(0.67) | − 4.38(0.57) | 0.73 (0.22,1.25) | 0.0060 |
Values are log transformed and presented in mean (SD). (a) unadjusted analysis. Adjusted analysis for CSF/serum albumin quotient (Q alb.) there was no significant difference between the groups for all cytokines and chemokines measured (all p>0.05); for CSF/serum ratio after adjustment for Q alb. there was significant difference for CSF/serum ratio of IFNγ, IL-1β and IP-10 (p= 0.035; 0.027 and 0.0009 respectively). (b) Groups differences presented as Cohen’s d effect sizes and 95% confidence intervals.
In unadjusted regression models, CSF levels of the cytokines TNFα, IFNγ, IL-1β, IL-2, IL-7, and IL-10 were significantly elevated in HIV participants than in uninfected individuals. In contrast, only TNFα and IFNγ were significantly elevated in serum. CSF levels of the chemokines IP-10, MCP-1, and MIP-1α were also significantly elevated in HIV participants than in uninfected controls, while IP-10 and RANTES were significantly elevated in serum. Standardized differences in CSF between HIV-positive and HIV-negative subjects (effect sizes) were larger, on average, than differences in serum between the two groups (Table 3). Adjusted analysis for CSF/serum albumin quotient (Q alb.) there was no significant difference between the groups for all cytokines and chemokines measured (p>0.05).
Collectively, the data indicate that the cellular immune response differs qualitatively between CSF and serum, as more types of cytokines and chemokines were stimulated in CSF than in serum. Also levels of some cytokines and chemokines were also higher in CSF than in serum, clearly indicating intrathecal synthesis (Table 3). Hence, we conclude that the immunological response is stronger in CSF than in serum. In a subgroup analysis of plasma virologically suppressed individuals (n=37), MIP-1β was significantly elevated compared to HIV negative subjects (p=0.027).
3.2.2. Comparison between HIV participants with or without CSF pleocytosis
CSF and serum cytokines and chemokines in HIV participants with or without CSF pleocytosis are summarized in Table 4. After adjusting for CD4 nadir and viral load suppression in plasma and CSF, most cytokines, including TNFα, IFNγ, IL1β, IL-2, IL-6, IL-7, and IL-10, were elevated, along with the chemokines IP10, MIP-1α, MIP-1β and RANTES, in the CSF of HIV participants with CSF pleocytosis than in participants without pleocytosis. Additionally, CSF levels of MCP-1 tended to be higher in participants with pleocytosis, although differences were not statistically significant. In contrast, serum cytokines and chemokines were comparable. These findings are consistent with the idea that the immune response to HIV differs between blood and CNS.
Table 4.
Comparison of cerebrospinal fluid (CSF) and serum chemotaxis and inflammatory biomarkers in HIV-positive samples with an increased CSF WBC count (n=20), and normal CSF WBC count (n=48). Significant differences are in bold typeface
| CSF | SER UM | CSF/se rum | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WBC> 5 | WBC <5 | Cohen’ s d (95%C I) (b) | (a) p | WBC >5 | WB C<5 | Cohen’ s d (95%C I) (b) | (a) p | WBC> 5 | WBC <5 | Diff (95% CI) (b) | (a)p | |
| Cytokines | ||||||||||||
| TNFα | 0.81 (0.56) | 0.13 (0.26) | 1.65 (1.06, 2.23) | <0.0001 | 1.03 (0.19) | 0.90 (0.21) | 0.5 (− 0.08, 1.09) | 0. 09 | −0.22 (0.54) | −0.77 (0.33) | 1.24 (0.65, 1.82) | <0.0001 |
| IFN γ | 0.52 (0.54) | 0.17 (0.33) | 0.63 (0.04, 1.21) | 0.0360 | 0.34 (0.57) | 0.25 (0.40) | 0.35 (− 0.23, 0.94) | 0.23 | 0.19 (0.84) | −0.08 (0.51) | 0.14 (− 0.44,0.72) | 0.6300 |
| IL1β | − 0.11(0. 05) | − 0.1(0. 02) | −0.37 (− 0.96,0.21) | 0.210 | −0.07 (0.14) | −0.1 (0) | 0.34 (− 0.24, 0.93) | 0.25 | −0.05 (0.14) | −0.01 (0.02) | −0.48 (− 1.06,0.11) | 0.1100 |
| IL2 | 0.08 (0.16) | 0 (0) | 0.86 (0.28, 1.45) | 0.0044 | 0.02 (0.12) | 0.08 (0.36) | −0.15 (− 0.73, 0.44) | 0. 62 | 0.06 (0.22) | −0.08 (0.36) | 0.36 (− 0.22,0.95) | 0.2200 |
| IL4 | 0.65 (0) | 0.65 (0) | −0.07 (− 0.66,0.51) | 0.8100 | 0.72 (0.22) | 0.67 (0.14) | 0.3 (− 0.29, 0.88) | 0. 32 | −0.06 (0.22) | −0.02 (0.14) | −0.3 (− 0.88,0.29) | 0.3200 |
| IL6 | 0.35 (0.41) | 0.08 (0.28) | 0.75 (0.17, 1.34) | 0.0120 | 0.07 (0.26) | 0.06 (0.31) | −0.09 (− 0.67, 0.49) | 0. 76 | 0.28 (0.45) | 0.02(0.44) | 0.61 (0.03, 1.2) | 0.0400 |
| IL7 | 0.41 (0.17) | 0.23 (0.19) | 0.72 (0.14, 1.31) | 0.0160 | 0.53 (0.36) | 0.69 (0.37) | −0.28 (− 0.86, 0.31) | 0. 35 | −0.13 (0.41) | −0.46 (0.38) | 0.63 (0.05, 1.22) | 0.0340 |
| IL10 | 0.62 (0.56) | 0.09 (0.18) | 1.58 (1,2.17) | <0.0001 | 0.48 (0.45) | 0.23 (0.43) | 0.57 (− 0.01, 1.16) | 0. 06 | 0.14(0. 75) | −0.14 (0.49) | 0.49 (− 0.1,1. 07) | 0.1000 |
| Chemokines | ||||||||||||
| IP10 | 4.0 (0.95) | 2.97 (0.37) | 1.53 (0.94, 2.11) | <0.0001 | 3.16 (0.35) | 2.90 (0.32) | 0.46 (− 0.12, 1.05) | 0. 12 | 0.82 (0.89) | 0.07 (0.38) | 1.33 (0.75, 1.92) | <0.0001 |
| MCP1 | 3.24(0. 22) | 3.12 (0.16) | 0.48 (− 0.1,1. 07) | 0.1000 | 2.71 (0.19) | 2.74 (0.21) | 0.23 (− 0.36, 0.81) | 0. 44 | 0.53 (0.27) | 0.39 (0.26) | 0.18 (− 0.4,0. 76) | 0.5400 |
| MIP1α | 1.21 (0.2) | 1.05 (0.07) | 1.13 (0.55, 1.72) | 0.0003 | 0.63 (0.35) | 0.59 (0.27) | 0.38 (− 0.21, 0.96) | 0. 20 | 0.57 (0.43) | 0.46 (0.29) | 0.09 (− 0.49,0.68) | 0.7600 |
| MIP1β | 1.21 (0.25) | 1.04 (0.28) | 0.97 (0.38, 1.55) | 0.0016 | 1.64 (0.3) | 1.74 (0.26) | 0.07 (− 0.52, 0.65) | 0. 82 | − 0.43(0. 33) | −0.70 (0.31) | 0.75 (0.17,1. 34) | 0.0130 |
| RANTES | 1.14 (0.79) | 0.34 (0.32) | 1.43 (0.85, 2.02) | <0.0001 | 4.51 (0.23) | 4.47 (0.27) | 0.23 (− 0.35, 0.82) | 0. 43 | −3.36 (0.84) | −4.13 (0.41) | 1.17 (0.59,1. 76) | 0.0002 |
Values are log transformed, presented in mean (SD). (a) Adjusted for plasma and HIV viral load suppression, and CD4 nadir.
Unadjusted and Adjusted analysis adding CSF/serum albumin quotient (Q alb.) to plasma and HIV viral load suppression, and CD4 nadir in the multivariable model are not shown. After adding Q alb. in the adjustment multivariable model, persisted significance for TNFα, IL-10, IP-10, MIP-1α, MIP-1β and RANTES (p <0.0001; <0.0001; <0.0001; 0.0069; 0.014; 0.00015 respectively) and for the CSF/serum TNFα, IL-7, IP-10, MIP-1β, MIP-1β and RANTES (p= 0.002; 0.045; 0.0007; 0.029; 0.0016 respectively) for all the other results p>0.05.
(b) Groups differences presented as Cohen’s d effect sizes and 95% confidence intervals.
We note that in participants with pleocytosis, the cytokines IFNγ, IL-1β, IL-2, IL-6, and IL-10, as well as the chemokines IP-10, MCP-1, and MIP-1α, were higher in CSF than in serum, indicating intrathecal synthesis (Table 4).
Finally, statistically significant differences in TNFα, IL-10, IP-10 (all P < 0.0001), MIP-1α (P = 0.0069), MIP-1β (P = 0.014), and RANTES (P = 0.00015) persisted after adjusting for QAlb, CD4 nadir, and viral load suppression in plasma and CSF.
3.3. Origin of CSF biomarkers
In uninfected controls, the coefficients of variation for IFNγ, IL-7, and MCP-1 were much smaller in CSF (0.81, 0.32, and 0.24, respectively) than in serum (1.25, 0.57, and 0.49, respectively), suggesting that these molecules are not derived from blood, but from the brain or the leptomeninges (Kuehne et al., 2013). In contrast, we found that the coefficient of variation for albumin was higher in CSF (0.299) than in serum (0.063), in line with published data (Kuehne et al., 2013). Indeed, a molecule that passes from blood to CSF should have a larger inter-individual variation in CSF than in blood due to variability in CSF flow and transport across barriers (Reiber et al, 2012). Thus, albumin can be used as positive control for this analysis, because CSF albumin is exclusively blood-derived. For all other biomarkers (IL-6, TNFα, IP-10, MIP-1α, MIP-1β, and RANTES) the variability was higher in CSF than in serum. These results, however, are inconclusive, and may indicate either transport from blood or limited regulatory control of release from the brain (Reiber et al, 2012). For IL-1β, IL-2, IL-4, and IL-10, the coefficient of variation is smaller in CSF than in blood, although levels were below the assay detection limits in the uninfected volunteers.
In HIV participants, the levels of the cytokines IL-6 and the chemokines IP-10, MCP-1, and MIP-1α were higher in CSF than in serum, also indicating intrathecal synthesis (Table 3).
3.4. Blood-CSF Barrier function
QAlb was significantly higher in HIV participants than in uninfected controls, and was measured to be 6.4 × 10−3 (4.9 × 1−3; 9.7 × 1−3) in the former and 5 × 1−3 (4 × 1−3; 6 × 1−3) in the latter (Table 2; Fig. 1; P = 0.0023). In HIV participants with and without CSF pleocytosis, QAlb was determined to be 7.9 × 1−3 (6.3 × 1−3; 11.6 × 1−3) and 6.1 × 1−3 (4.5 × 1−3; 9.2 × 1−3), respectively (P = 0.014). Based on age-dependent reference values, there was BCSFB dysfunction in 30 (44 %) HIV-positive individuals, but not in uninfected controls (P = 0.0002). BCSFB dysfunction was observed in 13 (65 %) HIV participants with CSF pleocytosis, as well as in 17 (35 %) participants with normal white blood cell count in the CSF (P = 0.033). QAlb and albumin Tourtellotte’s formula are summarized in Fig. 1A and B, respectively; the Reibergram plot is shown in Fig. 2. In a subgroup analysis of plasma virologically suppressed individuals (n=37), QAlb and albumin Tourtellotte’s formula remained significantly elevated compared to HIV negative subjects (p=0.001 and 0.002).
Figure 1. Albumin quotient, QAlb (A) and albumin Tourtellotte’s formula (B) in HIV positive and uninfected volunteers; and in HIV positive participants with or without CSF pleocytosis (WBC>5 cell/mm3).

QAlb and albumin Tourtellotte’s formula were strongly and positively correlated (rs = 0.99; P < 0.0001), in accordance with Syndulko et al. (1993).
Figure 2. Hyperbolic function (Reibergram plot) of blood-CSF barrier function and intrathecal IgG synthesis in HIV positive participants.

Hyperbolic functions are a consequence of nonlinear interaction between molecular flux and CSF flow rate as derived from the laws of diffusion (Reiber, 1995). Filled circles are participants HIV+ with CSF WBC > 5 cell/mm3, while open squares are participants HIV+ with CSF WBC < 5 cell/mm3. The age-corrected normal range of QAlb was 7 × 10−3, as indicated in the graphic. BCSFB dysfunction (area with vertical stripes) was observed in 12 (60 %) participants with white blood cell count > 5 cell/mm3, as well as in 20 (41.67 %) participants with white blood cell count < 5 cell/mm3 (χ2 P = 0.192). Intrathecal IgG synthesis was present in nine cases in both groups, at a prevalence of 45 % and 18.75 % respectively (χ2 P = 0.05). BCSFB barrier dysfunction was noted in five of these participants, all of whom presented pleocytosis. The plot was generated with CSF Research software.
In HIV participants, the CSF WBC count was weakly but positively correlated with QAlb (rs = 0.27; P = 0.0275), indicating that CSF WBC is not a marker of BCSFB function. However, there was strong correlation between CSF total protein and QAlb (rs = 0.8; P < 0.0001).
3.5. Correlation of CSF inflammatory biomarkers with CSF flow rate; CSF or plasma HIV RNA
QAlb, a measure of CSF flow rate, was moderately and positively correlated with CSF for the majotity of cytokines and chemokines studied on the HIV+ participants, rs and P values ranged from rs= 0.289 (P = 0.017) to rs= 0.483 (P < 0.0001), for IL-7 and TNFα respectively. The correlation was not significant for IL-1B, IL-4, and IL-10. Similar results were obtained when biomarkers were tested for correlation with albumin Tourtellotte’s formula. These correlations may explain why incorporation of QAlb into regression models diminishes the significance of a majority of biomarkers. Hence, we conclude that BCSFB dysfunction critically affects and accounts for levels of inflammatory biomarkers in CSF.
CSF and plasma viral load are important as HIV antigen load may drive cytokine responses. Indeed, CSF HIV RNA was moderately and positively correlated with levels of CSF IFNγ (rs= 0.472, P<0.0001); IL-10 (rs= 0.380, P=0.0013); IL-6 (rs= 0.415, P= 0.0004); IL-7 (rs= 0.518, P<0.0001); TNFα (rs= 0.618, p<0.0001); IP-10 (rs = 0.562, P p<0.0001); MCP-1 (rs= 0.333, P=0.0055); MIP-1α (rs= 0.488, P<0.0001); and RANTES (rs= 0.458, P<0.0001). Plasma HIV RNA was moderately and positively correlated with CSF IFNγ (rs= 0.369, P=0.002); IL-6 (rs= 0.263, P= 0.031); IL-7 (rs= 0.464, P<0.0001); TNFα (rs= 0.418, p=0.0004); IP-10 (rs= 0.474, P<0.0001); MIP-1α (rs= 0.397, P=0.0008); RANTES (rs= 0.284, P=0.019). The majority of cases (26; 81%) with detectable HIV CSF viral load also had detectable in HIV RNA in blood. To account for this our statistical analysis adjusted for blood viral load.
4. DISCUSSION
In this cross-sectional analysis, unadjusted regression models detected differences in biomarkers in CSF, but not in serum, between HIV-positive and HIV-negative volunteers. This result strongly suggested compartmentalization of the CNS inflammatory response to HIV. In addition, we observed dysfunction of the BCSFB in HIV participants, especially among participants with CSF pleocytosis. The functional integrity of this barrier is generally defined by diffusion and CSF flow rate, the latter being a function of CSF production and release, and is typically assessed by measuring CSF albumin. Dysfunction of the BCSFB is generally due to a decrease in CSF flow rate, rather than structural disruption (Reiber et al, 2012).
In HIV participants, CSF levels of a majority of inflammation biomarkers were positively correlated with CSF/serum albumin ratio. Consequently, incorporation of this ratio into regression models diminished the differences between HIV-positive and HIV-negative CSF, and between HIV participants with or without CSF pleocytosis. These observations indicate that CSF flow rate plays a critical role in CNS inflammation, presumably by facilitating influx of inflammatory molecules, white blood cells, HIV particles, and HIV proteins from peripheral blood, and thereby exacerbating and sustaining cellular immunity and inflammation in the CNS. If the BCSFB dysfunction is cause or consequence of the inflammation can’t be answered by this study. Presumably probably both process are involved, this observation is shared by others authors (Saunders et al., 2015). CNS barriers actively regulate brain homeostasis, and specifically respond to events in peripheral tissues and in the brain parenchyma. Indeed, these activities should be considered to fully characterize CNS diseases. Consequently, there has been a resurgence of interest in the possibility that barrier dysfunction may be involved in different neurological disorders (Saunders et al., 2015).
Our data indicate that in HIV participants, cytokine levels were altered more drastically in the CSF than in the serum. Although this phenomenon was observed for both pro-inflammatory (TNFα and IFNγ) and anti-inflammatory (IL-10) cytokines, the effect was stronger in the former. Levels of IL-7, an important inducer of autoimmune reactions that elicit a strong response to IL-17, i.e., Th-17 response were also elevated in CSF. In contrast, only pro-inflammatory cytokines (TNFα and IFNγ) were altered in the serum. Taken together, the data support the notion that the CNS is a primary site of HIV infection, and are consistent with the hypothesis that the CNS reacts immunologically when stimulated appropriately, although the system is considered immunologically privileged (Barker and Billingham 1977; Kreutzberg, 1996).
Although CNS HIV infection originates from viral and immune components transported into the CNS, the infection eventually becomes “compartmentalized” and acquires characteristics distinct from those observed in the blood. Thus, viral load and levels of immune components may be determined by independent processes in the CSF and blood (Price et al., 2013). Accordingly, a previous study demonstrated that inflammatory reactions in the CSF and serum are comparable during primary HIV infection, but eventually diverge as the infection progresses and as HIV-associated neurocognitive disorders develop. In addition, persistent CSF and blood inflammation despite HIV suppression suggests incomplete control of systemic and CNS infection (Peterson et al., 2014).
We note that all chemokines stimulated by HIV infection are induced via IFNγ, for instance, IP-10 and MCP-1. Similarly, RANTES, was almost twice as high in the CSF in HIV participants, although this difference was not statistically significant. MIP-1α is also elevated, although this chemokine is down-regulated by IFNγ in vitro (Sherry et al., 1998). In addition, IP-10 and RANTES levels were elevated in the serum of HIV participants.
The origins of most CSF biomarkers tested in this study are unknown, and tracing such origins may help elucidate the dynamics of these biomarkers. CSF molecules can be derived exclusively from blood, brain parenchyma or from the leptomeninges (Kuehne et al., 2013; Reiber, 2001; Reiber et al., 2012). Our data indicate that IL-6, IL-7, IFNγ, MCP-1, MIP-1α, and IP-10 are derived from intrathecal synthesis, either because the concentration was higher in CSF than in serum (IL-6, IP-10, MCP-1, and MIP-1α), or because the coefficient of variation was smaller in CSF than in serum (IFNγ, IL-7, and MCP-1). The latter result suggests that inter-individual variations in CSF biomarkers do not depend on serum concentration. Thus, we believe that CSF/serum quotients for these biomarkers primarily derived from the brain are not informative (Reiber, 2001, 2003). However, for proteins derived primarily from blood, the CSF/serum quotients may indeed be informative. We note that coefficients of variation for molecules derived from blood increase with transport into CSF because of variability in barrier traffic and CSF flow rate (Kuehne et al., 2013).
Further, we note that several proteins we analyzed are of small size, including IFNγ (17 kDa), IL-1β (17.5 kDa), IL-2 (15 kDa), IL-4 (17 kDa), IL-6 (25 kDa), IL-7 (17 kDa), IL-10 (20 kDa), TNFα (18 kDa), IP-10 (8.7 kDa), MCP-1 (8 kDa), MIP-1α (20 kDa), MIP-1β (8–14 kDa), and RANTES (7.8 kDa). Hence, these proteins may also cross the blood-CSF barrier independent of its integrity, as diffusion through an intact barrier may occur along a concentration gradient (Reiber, 2001, 2003; Reiber et al., 2012), although such diffusion maybe affected by changes in CSF flow rate. Our data are in line with these observations, as CSF levels of all cytokines and chemokines tested, with the exception of IL-1B, IL-4, and IL-10, were positively correlated with QAlb.
This paper has several positive points; we believe that the sample size was sufficiently large, and consisted of 87 samples, of which 68 were obtained from HIV participants, and 19 from uninfected controls. Nevertheless, we detected significant medium- and large-effect sizes between infected and uninfected volunteers, and between HIV participants with or without CSF pleocytosis. These effects are expressed in terms of Cohen’s d and 95 % con dence intervals. Notably, Cohen’s effect sizes were mostly large in comparisons between HIV participants with or without pleocytosis. Furthermore, the samples included participants infected with non-B HIV subtypes, in contrast to previously published papers. Although HIV-1 subtypes differences on biomarkers was not the aim of this study, comparisons on the same panel of inflammatory biomarkers between HIV-1 subtype B and C were published on a previous paper, we note that inflammatory biomarkers were found to be comparable between between participants infected with these subtypes (de Almeida et al., 2016).
Finally, previous papers did not fully integrate multiple results from studies of the BCSFB during HIV infection, and did not reach the conclusion that a dysfunctional barrier helps to maintain and increase the divergence of the immune response in CSF and blood. Nevertheless, this study is not without limitations, and we emphasize that results are cross-sectional, and not longitudinal.
As conclusion, our findings indicate that despite dysfunction of the the BCSFB, cellular immune response remain highly compartmentalized. Thus markers of cellular immune responses to HIV in CSF are distinct from those in serum. Furthermore, chemokines and cytokines were frequently elevated in the CSF of HIV participants, in comparison to uninfected controls. CSF chemokines and cytokines correlated with CSF and plasma HIV RNA, suggesting that HIV antigen load may drive cytokine responses. These results support the notion that the CNS behaves as a separate immunologic compartment. This compartmentalized immune response is markedly reduced by virology suppression, although BCSFB dysfunction persists on this subgroup. Finally, we found that dysfunction of the BCSFB critically contributes to the strong CNS inflammatory response to HIV, presumably by allowing transport of WBC, serum molecules, HIV particles, and HIV proteins into the CNS.
Highlights.
Soluble biomarkers reflecting cellular immune response were different in CSF than serum among HIV+ participants.
CSF chemokines and cytokines correlated with CSF and plasma HIV RNA, suggesting that HIV antigen load may drive cytokine responses.
The data indicates that CNS cellular immune response in HIV infection is compartmentalized, and remains so despite the BCSFB dysfunction during HIV infection.
The compartmentalized immune response is markedly reduced by virology suppression, although BCSFB dysfunction persists on this subgroup.
These results support the notion that the CNS behaves as a separate immunologic compartment.
Acknowledgments
Part of this work was previously presented at 13th International Symposium on NeuroVirology and 2015 Conference on HIV in the Nervous System, June 2–6, 2015, in San Diego, California, USA.
The authors would like to thank Professor Hansotto Reiber for careful and critical reading of the manuscript, especially of details regarding the blood-CSF barrier and CSF flow rate.
FUNDING
This work was supported by the following grants: National Institute of Health, NIH R21 MH76651 (Ellis, Ronald J; Almeida, Sergio M.), NIH R01 MH83552 (Smith, David M.), S10 RR31646 (Letendre, Scott), K24 MH097673(Letendre, Scott); University of California, San Diego, Center for AIDS Research (CFAR), an NIH-funded program (P30 AI036214), which is supported by the following NIH Institutes and Centers: NIAID, NCI, NIMH, NIDA, NICHD, NHLBI, NIA, NIGMS, and NIDDK; Ministério da Ciência e Tecnologia/Conselho Nacional de Desenvolvimento Científico e Tecnológico, MCT/CNPq-Universal 014/2008, Brazil (Almeida, Sergio M.).
The HIV Neurobehavioral Research Center (HNRC) is supported by Center award P30MH062512 from NIMH. The San Diego HIV Neurobehavioral Research Center [HNRC] group is affiliated with the University of California, San Diego, the Naval Hospital, San Diego, and the Veterans Affairs San Diego Healthcare System, and includes: Director: Robert K. Heaton, Ph.D., Co-Director: Igor Grant, M.D.; Associate Directors: J. Hampton Atkinson, M.D., Ronald J. Ellis, M.D., Ph.D., and Scott Letendre, M.D.; Center Manager: Thomas D. Marcotte, Ph.D.; Jennifer Marquie-Beck, M.P.H.; Melanie Sherman; Neuromedical Component: Ronald J. Ellis, M.D., Ph.D. (P.I.), Scott Letendre, M.D., J. Allen McCutchan, M.D., Brookie Best, Pharm.D., Rachel Schrier, Ph.D., Debra Rosario, M.P.H.; Neurobehavioral Component: Robert K. Heaton, Ph.D. (P.I.), J. Hampton Atkinson, M.D., Steven Paul Woods, Psy.D., Thomas D. Marcotte, Ph.D., Mariana Cherner, Ph.D., David J. Moore, Ph.D., Matthew Dawson; Neuroimaging Component: Christine Fennema-Notestine, Ph.D. (P.I.), Monte S. Buchsbaum, M.D., John Hesselink, M.D., Sarah L. Archibald, M.A., Gregory Brown, Ph.D., Richard Buxton, Ph.D., Anders Dale, Ph.D., Thomas Liu, Ph.D.; Neurobiology Component: Eliezer Masliah, M.D. (P.I.), Cristian Achim, M.D., Ph.D.; Neurovirology Component: David M. Smith, M.D. (P.I.), Douglas Richman, M.D.; International Component: J. Allen McCutchan, M.D., (P.I.), Mariana Cherner, Ph.D.; Developmental Component: Cristian Achim, M.D., Ph.D.; (P.I.), Stuart Lipton, M.D., Ph.D.; Participant Accrual and Retention Unit: J. Hampton Atkinson, M.D. (P.I.), Jennifer Marquie-Beck, M.P.H.; Data Management and Information Systems Unit: Anthony C. Gamst, Ph.D. (P.I.), Clint Cushman; Statistics Unit: Ian Abramson, Ph.D. (P.I.), Florin Vaida, Ph.D. (Co-PI), Reena Deutsch, Ph.D., Anya Umlauf, M.S.
ABBREVIATIONS
- BBB
blood-brain barrier
- BCSFB
blood-CSF barrier
- CART
combination anti-retroviral therapy
- CPE
CNS anti-retroviral penetration effectiveness
- IFN
interferon
- IL
interleukin
- IP-10
Interferon gamma-induced protein 10
- QAlb
albumin quotient
- MCP
monocyte chemoattractant protein
- MIP
macrophage inflammatory protein
- RANTES
Regulated on activation, normal T cell expressed and secreted
- TNF
Tumor necrosis factor
- WBC
white blood cell
Footnotes
The views expressed in this article are those of the authors and do not reflect the official policy or position of the Department of the Navy, Department of Defense, nor the United States Government.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbas W, Herbein G. T-Cell Signaling in HIV-1 Infection. Open Virol J. 2013;26:57–71. doi: 10.2174/1874357920130621001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Andersson M, Alvarez-Cermeño J, Bernardi G, Cogato I, Fredman P, Frederiksen J, et al. Cerebrospinal fluid in the diagnosis of multiple sclerosis: a consensus report. J Neurol Neurosurg Psych. 1994;57:897–902. doi: 10.1136/jnnp.57.8.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson LM, Hagberg L, Fuchs D, Svennerholm B, Gisslen M. Increased blood–brain barrier permeability in neuro-asymptomatic HIV-1-infected individuals, correlation with cerebrospinal fluid HIV-1 RNA and neopterin levels. J NeuroVirol. 2001;7:542–547. doi: 10.1080/135502801753248123. [DOI] [PubMed] [Google Scholar]
- Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Banks WA. The blood-brain barrier in neuroimmunology: tales of separation and assimilation. Brain Behav Immun. 2015;44:1–8. doi: 10.1016/j.bbi.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker CF, Billingham RE. Immunologically privileged sites. Adv Immunol. 1977;25:1–54. [PubMed] [Google Scholar]
- Brasil: Ministério da Saúde. Programa Nacional de DST/AIDS. 2009 http://www.aids.gov.br/assistencia/manualdst/item12.htm.
- Brew BJ, Pemberton L, Cunninghan P, Law MG. Levels of human immunodeficiency virus type 1 RNA in cerebrospinal fluid correlate with AIDS dementia stage. J Infect Dis. 1997;175:963–6. doi: 10.1086/514001. [DOI] [PubMed] [Google Scholar]
- Brew BJ, Letendre SL. Biomarkers of HIV related central nervous system disease. Int Rev Psych. 2008;20:73–88. doi: 10.1080/09540260701878082. [DOI] [PubMed] [Google Scholar]
- Calcagno A, Alberione MC, Romito A, Imperiale D, Ghisetti V, Audagnotto S, Lipani F, Raviolo S, Di Perri G, Bonora S. Prevalence and predictors of blood–brain barrier damage in the HAART era. J Neurovirol. 2014;20:521–5. doi: 10.1007/s13365-014-0266-2. [DOI] [PubMed] [Google Scholar]
- Calcagno A, Motta I, Ghisetti V, Lo Re S, Allice T, Marinaro L, Milia MG, Tettoni MC, Trentini L, Orofino G, Salassa B, Di Perri G, Bonora S. HIV-1 Very Low Level Viremia is Associated with Virological Failure in Highly Active Antiretroviral Treatment-Treated Patients. AIDS Res Hum Retrov. 2015;31:999–1008. doi: 10.1089/AID.2015.0102. [DOI] [PubMed] [Google Scholar]
- Campbell GR, Watkins JD, Singh KK, Loret EP, Spector SA. Human immunodeficiency virus type 1 subtype C Tat fails to induce intracellular calcium flux and induces reduced tumor necrosis factor production from monocytes. J Virol. 2007;81:5919–5928. doi: 10.1128/JVI.01938-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cinque P, Brew BJ, Gisslen M, Hagberg L, Price RW. Cerebrospinal fluid markers in central nervous system HIV infection and AIDS dementia complex. Handb Clin Neurol. 2007;85:261–300. doi: 10.1016/S0072-9752(07)85017-2. [DOI] [PubMed] [Google Scholar]
- de Almeida SM, Ribeiro CE, de Pereira AP, Badiee J, Cherner M, Smith D, et al. Neurocognitive impairment in HIV-1 clade C- versus B-infected individuals in Southern Brazil. J Neurovirol. 2013;19:550–6. doi: 10.1007/s13365-013-0215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida SM, Rotta I, Jiang Y, Li X, Raboni SM, Ribeiro CE, et al. Biomarkers of chemotaxis and inflammation in cerebrospinal fluid and serum in individuals with HIV-1 Subtype C versus B. J Neurovirol. 2016 doi: 10.1007/s13365-016-0437-4. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries HE, Kuiper J, de Boer AG, Van Berkel TJC, Breimer DD. The blood-brain barrier in neuroinflammatory diseases. Pharmacol Rev. 1997;49:143–156. [PubMed] [Google Scholar]
- Genis P, Jett M, Bernton EW, Boyle T, Gelbard HA, Dzenko K, et al. Cytokines and arachidonic metabolites produced during human immunodeficiency virus (HIV)-infected macrophage-astroglia interactions: implications for the neuropathogenesis of HIV disease. J Exp Med. 1992;176:1703–18. doi: 10.1084/jem.176.6.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gisslen M, Chiodi F, Fuchs D, Norkrans G, Svennerholm B, Wachter H, Hagberg L. Markers of immune stimulation in the cerebrospinal fluid during HIV infection: a longitudinal study. Scand J Infect Dis. 1994;26:523–533. doi: 10.3109/00365549409011810. [DOI] [PubMed] [Google Scholar]
- Hagberg L, Cinque P, Gisslen M, Brew BJ, Spudich S, Bestetti A, et al. Cerebrospinal fluid neopterin: an informative biomarker of central nervous system immune activation in HIV-1 infection. AIDS Res Ther. 2010;7:15. doi: 10.1186/1742-6405-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton RD, Foss AJ, Leach L. Establishment of a human in vitro model of the outer blood-retinal barrier. J Anat. 2007;211:707–16. doi: 10.1111/j.1469-7580.2007.00812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King JE, Eugenin EA, Buckner CM, Berman JW. HIV Tat and neurotoxicity. Microb Infect. 2006;8:1347–1357. doi: 10.1016/j.micinf.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–8. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- Kuehne LK, Reiber H, Bechter K, Hagberg L, Fuchs D. Cerebrospinal fluid neopterin is brain-derived and not associated with blood-CSF barrier dysfunction in non-inflammatory affective and schizophrenic spectrum disorders. J Psych Res. 2013;47:1417–22. doi: 10.1016/j.jpsychires.2013.05.027. [DOI] [PubMed] [Google Scholar]
- Letendre S, Ellis R, Deutsch R, Clifford D, Marra C, McCutchan A, et al. Correlates of time-to-loss-of-viral response in CSF and plasma in the CHARTER cohort. Program and Abstracts of the 17th Conference on Retroviruses and Opportunistic Infections; San Francisco, CA. 2010. (poster 430) [Google Scholar]
- Marshall DW, Brey RL, Cahill WT, Houk RW, Zajac RA, Boswell RN. Spectrum of cerebrospinal fluid in various stages of human immunodeficiency virus infection. Arch Neurol. 1988;45:954–958. doi: 10.1001/archneur.1988.00520330032007. [DOI] [PubMed] [Google Scholar]
- Mooney S, Tracy R, Osler T, Grace C. Elevated Biomarkers of Inflammation and Coagulation in Patients with HIV Are Associated with Higher Framingham and VACS Risk Index Scores. PLoS One. 2015;10(12):e0144312. doi: 10.1371/journal.pone.0144312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A. Pathobiology of human immunodeficiency virus dementia. Semin Neurol. 1999;19:113–27. doi: 10.1055/s-2008-1040830. [DOI] [PubMed] [Google Scholar]
- Noel N, Boufassa F, Lecuroux C, Saez-Cirion A, Bourgeois C, Dunyach-Remy C, Goujard C, Rouzioux C, Meyer L, Pancino G, Venet A, Lambotte O ANRS C021 CODEX Study Group. Elevated IP10 levels are associated with immune activation and low CD4(+) T-cellcounts in HIV controller patients. Aids. 2014;28:467–476. doi: 10.1097/qad.0000000000000174. [DOI] [PubMed] [Google Scholar]
- Peterson J, Keating S, Fuchs D, Norris P, Zetterberg H, Shacklett B, et al. Differences between cerebrospinal fluid and blood biomarkers of inflammation in HIV infection. Conference on Retroviruses and Opportunistic Infections; Boston, MA. 2014. (abstract 484) [Google Scholar]
- Petito CK, Cash KS. Blood-brain barrier abnormalities in the acquired immunodeficiency syndrome: immunohistochemical localization of serum proteins in postmortem brain. Ann Neurol. 1992;32:658–66. doi: 10.1002/ana.410320509. [DOI] [PubMed] [Google Scholar]
- Price RW, Peterson J, Fuchs D, Angel TE, Zetterberg H, Hagberg L, et al. Approach to cerebrospinal fluid (CSF) biomarker discovery and evaluation in HIV Infection. J Neuroim Pharmacol. 2013;8:1147–58. doi: 10.1007/s11481-013-9491-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiber H. External quality assessment in clinical neurochemistry: survey of analysis for cerebrospinal fluid (CSF) proteins based on CSF/serum quotients. Clin Chem. 1995;41:256–63. [PubMed] [Google Scholar]
- Reiber H. Dynamics of brain-derived proteins in cerebrospinal fluid. Clin Chim Acta. 2001;310:173–186. doi: 10.1016/s0009-8981(01)00573-3. [DOI] [PubMed] [Google Scholar]
- Reiber H. Proteins in cerebrospinal fluid and blood: barriers, CSF flow rate and source-related dynamics. Restor Neurol Neurosci. 2003;21:79–96. [PubMed] [Google Scholar]
- Reiber H, Peter JB. Cerebrospinal fluid analysis: disease-related data patterns and evaluation programs. J Neurol Sci. 2001;184:101–22. doi: 10.1016/s0022-510x(00)00501-3. [DOI] [PubMed] [Google Scholar]
- Reiber H, Padilla-Docal B, Jensenius JC, Dorta-Contreras AJ. Mannan-binding lectin in cerebrospinal fluid: a leptomeningeal protein. Fluids Barriers CNS. 2012;9:17. doi: 10.1186/2045-8118-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick L, Berger JR, Shapshak P, Tourtellotte WW. Early penetration of the blood-brain-barrier by HIV. Neurology. 1988;38:9–14. doi: 10.1212/wnl.38.1.9. [DOI] [PubMed] [Google Scholar]
- Saunders NR, Ek CJ, Habgood MD, Dziegielewska KM. Barriers in the brain: a renaissance? Trends Neurosci. 2008;31:279–86. doi: 10.1016/j.tins.2008.03.003. [DOI] [PubMed] [Google Scholar]
- Sherry B, Espinoza M, Manogue KR, Cerami A. Induction of the chemokine beta peptides, MIP-1 alpha and MIP-1 beta, by lipopolysaccharide is differentially regulated by immunomodulatory cytokines gamma-IFN, IL-10, IL-4, and TGF-beta. Mol Med. 1998;4:648–57. [PMC free article] [PubMed] [Google Scholar]
- Spudich S, Gisslen M, Hagberg L, Lee E, Liegler T, Brew B, et al. Central nervous system immune activation characterizes primary human immunodeficiency virus 1 infection even in participants with minimal cerebrospinal fluid viral burden. J Infect Dis. 2011;204:753–60. doi: 10.1093/infdis/jir387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syndulko K, Tourtellotte WW, Conrad AJ, Izquierdo G. Trans-blood-brain-barrier albumin leakage and comparisons of intrathecal IgG synthesis calculations in multiple sclerosis patients. Multiple Sclerosis Study Group, Alpha Interferon Study Group, and Azathioprine Study Group. J Neuroimmunol. 1993;46:185–92. doi: 10.1016/0165-5728(93)90248-w. [DOI] [PubMed] [Google Scholar]
- Stamatovic SM, Keep RF, Andjelkovic AV. Brain endothelial cell-cell junctions: how to open the blood brain barrier. Curr Neuropharmacol. 2008;6:179–92. doi: 10.2174/157015908785777210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourtellotte WW, Shapshak P, Osborne MA, Rubinshtein G, Lee M, Staugaitis SM. New formula to calculate the rate of albumin blood-brain-barrier leakage. Ann Neurol. 1989;26:176. [Google Scholar]
- Valcour V, Chalermchai T, Sailasuta N, Marovich M, Lerdlum S, Suttichom D, et al. Central nervous system viral invasion and inflammation during acute HIV infection. J Infect Dis. 2013;206:275–82. doi: 10.1093/infdis/jis326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Qiao L, Wei F, Yin J, Liu L, Ji Y, et al. Cytokines in CSF correlate with HIV-associated neurocognitive disorders in the post-HAART era in China. J Neurovirol. 2013;19:144–9. doi: 10.1007/s13365-013-0150-5. [DOI] [PMC free article] [PubMed] [Google Scholar]