

Abstract
Biased ligands (also known as functionally selective ligands) of G protein-coupled receptors are valuable tools for dissecting the roles of G protein-dependent and independent signaling pathways in health and disease. Biased ligands have also been increasingly pursued by the biomedical community as promising therapeutics with improved efficacy and reduced side effects compared with unbiased ligands. We previously discovered first-in-class β-arrestin-biased agonists of dopamine D2 receptor (D2R) by extensively exploring multiple regions of aripiprazole, a balanced D2R agonist. In our continuing efforts to identify biased agonists of D2R, we unexpectedly discovered a G protein-biased agonist of D2R, compound 1, which is the first G protein-biased D2R agonist from the aripiprazole scaffold. We designed and synthesized novel analogs to explore two regions of 1 and conducted structure–functional selectivity relationship (SFSR) studies. Here we report the discovery of 1, findings from our SFSR studies, and characterization of novel G protein-biased D2R agonists.
Graphical abstract

INTRODUCTION
The D2 dopamine receptor (D2R) remains a main target for the development of anti-Parkinson1 and antipsychotic agents.2 Although all five dopamine receptors (D1R - D5R) are expressed in brain,3 D2R is by far the most highly studied4 and is involved in many neuropsychiatric diseases5 including attention-deficit disorders6 and schizophrenia.7 It is therefore not surprising that all of the current FDA-approved drugs for the treatment of schizophrenia have direct action at D2R.8 Despite the prevalence of drugs targeting D2R, early typical antipsychotic drugs are known to have debilitating motor side-effects including extrapyramidal symptoms (EPS),9 often leading to permanent symptoms such as tardive dyskinesia.10 Thus, safer and more effective drugs that target D2R are needed not only to treat the positive and negative symptoms of schizophrenia,11 but also to minimize the motor side-effects commonly observed with chronic antipsychotic treatment.12 Newer generation antipsychotics such as aripiprazole13 possess partial agonist actions at D2R14 and have less propensity to produce motor side-effects,15 but mounting evidence in the last decade has suggested that aripiprazole, which is a balanced D2R agonist,16 can activate a plethora of downstream signaling pathways.17–18
The growing realization of the complexity of G protein coupled receptor (GPCR) mediated signal transduction pathways, specifically D2R mediated signaling pathways, has provided a theoretical framework for the development of functionally selective or biased ligands,4, 19 which refer to a ligand’s ability to differentially modulate canonical (e.g., G protein-dependent) versus non-canonical (e.g., G protein-independent, β-arrestin-mediated) signaling pathways.20 We previously discovered novel β-arrestin-biased agonists of D2R by extensively exploring multiple regions of the aripiprazole scaffold.16, 21 Using these β-arrestin-biased D2R agonists, which were tested in a battery of in vitro and in vivo assays, we found that the β-arrestin bias appears to be important to exert superior therapeutic utility in animal models of schizophrenia and elicit a lower level of catalepsy compared to the D2R antagonist haloperidol.16, 22 Others have recently reported the discovery of multiple functionally selective D2R ligands based on the potential antipsychotic agent cariprazine and other D2R targeting compounds.23–25,26 In addition, the first G protein-biased agonist of D2R was recently reported.25,27 Biased D2R ligands are thought to stabilize different conformations of D2R leading to the preference for either G protein-dependent or G protein-independent signaling,28–30 usually through recruitment or inhibition of β-arrestin.31–34 Future characterization of biochemical and behavioral effects of biased D2R ligands and optimization of their drug-like properties could ultimately lead to improved antipsychotic drugs that are safer and more effective than existing antipsychotics. While a number of β-arrestin-biased D2R agonists have been generated and progressed towards clinical development, there is only one reported G protein-biased D2R agonist.27
To develop G protein-biased D2R agonists as useful tools for elucidating the role of G protein-mediated D2R signaling in physiology and pathophysiology, we extended our structure–functional selectivity relationship (SFSR) studies based on the aripiprazole scaffold. From these studies, we discovered compound 1, a benzothiazole containing D2R agonist with an unexpected bias for G protein signaling. We then explored two regions of this lead and uncovered structural features that affect ligand bias. By combining preferred structural motifs, novel G protein-biased D2R agonists devoid of any measurable β-arrestin recruitment were discovered.
RESULTS AND DISCUSSION
Discovery of Compound 1
We previously identified UNC9994 (Figure 1) as a β-arrestin-biased D2R agonist that lacks Gαi/o activity.21 A close analog of this compound with the middle piperidine replaced by piperazine, UNC9995 (Figure 1), retained the β-arrestin-biased feature for D2R with slightly decreased potency.21 Adding a simple methyl group at the 2 position of the benzothiazole ring resulted in compound 2 (Figure 1), which is a balanced D2R agonist with similar efficacy and potency in both β-arrestin and Gi/o pathways. Surprisingly, extending the middle linker from propoxy (compound 2) to butoxy (compound 1, Figure 1) resulted in the first G protein-biased D2R ligand from the aripiprazole scaffold, which exhibited D2R Gi/o partial agonist activity (EC50 = 15 nM, Emax = 50%) with weak efficacy for β-arrestin recruitment (EC50 = 13 nM, Emax = 23%).
Figure 1. Discovery of compound 1, a G protein-biased D2R partial agonist.

Benzothiazole substitution and linker modification of a β-arrestin-biased D2R agonist led to compound 1, which is a D2R Gi/o partial agonist with weak efficacy for β-arrestin recruitment.
To develop more G protein-biased D2R agonists, we conducted structure–functional selectivity relationship (SFSR) studies based on this newly identified G protein-biased lead. We designed, synthesized and evaluated derivatives of compound 1 to determine which structural modifications would favor Gi/o activation over β-arrestin recruitment. We previously found that conformationally-constrained central linkers could lead to a significant bias for β-arrestin recruitment over Gi/o signaling.21 We and others also found that the butoxy was preferred compared to propoxy (Figure 1) or pentoxy35 as a central linker. Based on these findings, we kept the butoxy constant as the central linker for the SFSR studies. We then focused our exploration on two regions of compound 1, namely the left hand side (LHS) phenylpiperazine and right hand side (RHS) benzothiazole moieties.
SFSR Studies of the Benzothiazole Moiety
To determine the effects of substituents at the 2-position of the benzothiazole moiety on ligand bias, we designed the analogs of compound 1 outlined in Table 1. The synthesis of these compounds is summarized in Scheme 1. Generally, our previously well-established two-step alkylation sequence21 was employed to prepare these compounds. Commercially available 2-methylbenzo[d]thiazol-5-ol, 2-methylbenzo[d]thiazol-6-ol, 2-methylbenzo[d]oxazol-5-ol, 2-chlorobenzo[d]thiazol-5-ol, and 5-hydroxybenzo[d]thiazol-2(3H)-one were reacted with 1,4-dibromobutane to give the bromo intermediates, which were then reacted with 1-(2,3-dichlorophenyl)piperazine (5) to afford the target compounds 1, 3, 4, 6, and 7. The 2-trifluoromethyl benzo[d]thiazole-5-ol (10) was synthesized from commercially available 3-methoxyaniline. Trifluoroacetic acid was reacted with 3-methoxyaniline to give intermediate 8, which was treated with NaSH˙H2O and then cyclized to yield intermediate 9.36 Subsequent dimethylation reaction afforded intermediate 10,37 which underwent the two-step alkylation sequence to furnish compound 11. The 2-ethyl, 2-isopropyl, 2-methylamine and 2-dimethylamine substituted benzothiazole building blocks were synthesized starting from commercially available 1-isothiocyanoto-3-methoxybenzene. Typical Grignard reaction procedures with ethylmagnesium chloride and isopropylmagnesium chloride yielded the corresponding thioamide compounds 12 and 13, which were then cyclized to give intermediates 14 and 15.38 Removal of the methyl group followed by the two-step alkylation reactions afforded compounds 18 and 19. In addition, 1-isothiocyanato-3-methoxybenzene was treated with methanamine or dimethylamine in the presence of PhCH2N(CH3)3Br3 to give aminobenzothiazoles 21 and 22,39 which were subjected the deprotection and two-step alkylation sequence to furnish the desired compounds 24 and 25.
Table 1.
SFSR of the RHS Benzothiazole Moiety.a
 | |||||
|---|---|---|---|---|---|
| Compound | RHS Group | β-arrestin | cAMP | ||
| Emax (%) | EC50 (nM) | Emax (%) | EC50 (nM) | ||
| 1 | 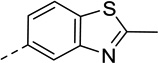 |
23 | 13 | 50 | 15 |
| 3 | 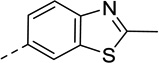 |
22 | 76 | 42 | 29 |
| 4 | 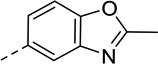 |
25 | 24 | 36 | 21 |
| 6 | 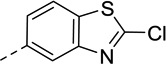 |
40 | 48 | 50 | 43 |
| 7 | 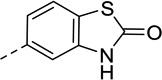 |
59 | 2.6 | 67 | 5.2 |
| 11 | 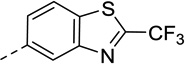 |
40 | 160 | 32 | 140 |
| 18 | 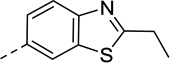 |
<20 | N/C | 37 | 98 |
| 19 | 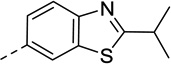 |
<20 | N/C | 33 | 72 |
| 24 | 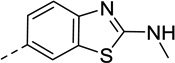 |
<20 | N/C | 40 | 24 |
| 25 | 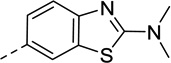 |
N/A | N/A | N/A | N/A |
EC50 and Emax values are the average of at least 3 independent experiments performed in triplicate. Standard error of the mean (SEM): < ± 20%.
N/C. not calculated. N/A. no activity.
Scheme 1.

Synthesis of compound 1 analogs to explore the RHS benzothiazole moiety.a
a Reagents and Conditions: (a) K2CO3, EtOH, reflux, 6 h, 30–80% yield; (b) NaI/K2CO3, CH3CN, reflux, 6 h, 50–70% yield; (c) (i) Ph3P, Et3N, CCl4, TFA, 0 °C, 10 min; (ii) 3-methoxyaniline/CCl4, reflux, 3h, 86% yield; (d) NaSH˙xH2O, PdCl2, DMSO, 50–110 °C, 3 h, 85% yield; (e) HI, reflux, 5 h, 83–90%; (f) RMgBr, THF, −10 °C, 90 min, ~100% yield; (g) (i) K3Fe(CN)6, NaOH, H2O, CH3OH, 60 °C, 2 h, (ii) K2CO3, 60 °C, 1 h, 82–87% yield; (h) NHR’R”, PhCH2N(CH3)3Br3, THF, r.t., overnight, 93–96% yield.
All the newly synthesized compounds were evaluated in: (1) D2R-mediated cAMP accumulation assay, which measures inhibition of isoproterenol-stimulated cAMP production;40 and (2) D2R-mediated β-arrestin-2 recruitment Tango assay to determine potency and efficacy for β-arrestin recruitment.41 Quinpirole, a full agonist of D2R,42 was used as the positive control in both cAMP inhibition and β-arrestin-2 recruitment Tango assays.
As summarized in Table 1, by reversing the positions of nitrogen and sulfur in the thiazole structure, compound 3 retained similar efficacy in both β-arrestin and Gi/o pathways but showed decreased potencies. The potency in β-arrestin recruitment decreased more than that in Gi/o assay (5-fold versus 2-fold), thus favoring G protein bias. By replacing sulfur in the thiazole ring with oxygen, the bioisostere compound 4 exhibited similar potency and efficacy in β-arrestin pathway but its efficacy in Gi/o pathway decreased, resulting in a less G protein-biased compound compared with compound 1. With 2-Cl substitution, compound 6 showed similar efficacy in Gi/o pathway compared to compound 1 whereas its efficacy in β-arrestin recruitment was similar to Gi/o activity, exhibiting less G protein bias. Interestingly, the 2-OH substitution, tautomerized to 2-keto, in compound 7 reversed the bias for G protein, demonstrating balanced signaling for both Gi/o signaling and β-arrestin recruitment with high potencies. Incorporation of a trifluoromethyl group, in most instances, in place of a methyl group, has been a commonly pursued modification to lead compounds in medicinal chemistry.43 However, changing the methyl group (compound 1) to trifluoromethyl group (compound 11) suffered from reduced potency in both assays, and its efficacy in the β-arrestin pathway increased whereas its Gi/o activity decreased, making it a balanced D2R agonist like compounds 6 and 7, albeit with much weaker potency in both pathways. Replacing the methyl group with ethyl, isopropyl, or methylamine group (in compounds 18, 19 and 24, respectively) resulted in very weak efficacy in β-arrestin recruitment (Emax < 20%) but maintained low to moderate Gi/o efficacy (Emax = 43%, 33% and 40%, respectively). Although the potency of compound 18 and 19 in the Gi/o pathway was reduced by 5–7 fold, compound 24 was able to maintain similar potency as compound 1 in Gi/o pathway (EC50 = 24 nM). Surprisingly, with the dimethylamine substitution, compound 25 did not show any activity in both pathways. Taken together, these results suggest that D2R G protein bias is sensitive to subtle structural modifications to the 2-position of the benzothiazole moiety and only limited substitutions are tolerated in order to achieve G protein-biased compounds. 2-Methyl, 2-ethyl, 2-isopropyl, and 2-methylamine are possible RHS benzothizole substitutions that may favor Gi/o signaling over β-arrestin recruitment.
SFSR Studies of the Phenylpiperazine Moiety
We next investigated the LHS phenylpiperazine moiety of compound 1. We previously reported that this moiety was very tolerant to modifications, but most single substitutions on the phenyl ring appeared to generate balanced compounds with similar efficacy in both β-arrestin and Gi/o pathways.21 Therefore, we designed most compounds with 2,3-disubstitutions, including cyclized 2,3-disubstitutions on the phenyl ring. 2-Methoxy substitution on the phenyl ring was found to increase the compound’s binding affinity towards D2R as well as the potency in both signaling pathways,21 therefore 1-(2-methoxyphenyl)piperazine was selected as the only single substituted phenylpiperazine. Because 7-(piperazin-1-yl)benzo[d]oxazol-2(3H)-one is present in the structure of bifeprunox,44 which exhibited extraordinary potency and efficacy in both signaling pathways in our assays, we included this moiety in our design to enhance the comparatively low efficacy and low potency of compound 1. In addition, piperazine was replaced by piperidine, homopiperazine, or substituted piperazine to explore whether any one of these ring replacements would favor Gi/o signaling over β-arrestin recruitment. The synthesis of these compounds is outlined in Scheme 2.
Scheme 2.

Synthesis of compound 1 analogs to explore the LHS phenylpiperazine moiety.a
a Reagents and Conditions: (a) NaI/K2CO3, CH3CN, reflux, 6 h, 50–70% yield; (b) Pd2(dba)3, BINAP, t-BuONa, toluene, sealed tube, 100 °C, overnight, 52–89% yield; (c) TFA, CH2Cl2, r.t., 1 h, 92–96% yield; (d) Pd2(dba)3, BINAP, t-BuONa, toluene, sealed tube, 100 °C, 4h, 56–67% yield; (e) Pd(PPh3)4, K2CO3, dioxane, H2O, microwave, 93% yield.
4-(2,3-Dichlorophenyl)piperidine (27)21 and 1-(2,3-dichlorophenyl)-1,4-diazepane (28)16 were prepared according to the previously published procedures. Compounds 30, 31, and 32 were prepared by the nucleophilic displacement of 5-(4-bromobutoxy)-2-methylbenzo[d]thiazole (26) with corresponding starting materials 27, 28, and commercially available 1-(2-methoxyphenyl)piperazine (29). Buchwald–Hartwig amination between 1-bromo-2,3-dichlorobenzene and Boc-protected methylpiperazine,45 followed by the removal of the Boc protecting group gave intermediate 33, which underwent the nucleophilic displacement reaction with bromide 26 to furnish compound 34. Similarly, 8-bromo-2-methylquinoline underwent the Buchwald–Hartwig amination reaction with unprotected piperazine yielded intermediate 35. It was necessary to reduce the reaction time from overnight to 4 hours to minimize the over-reacted byproduct. Likewise, intermediates 37 and 38 were synthesized by the reaction of piperazine with 8-bromo-3,4-dihydro-2H-benzo[b][1,4]oxazine and 5-bromo-2,3-dihydrobenzo[b][1,4]dioxine. Nucleophilic displacement of bromide 26 with intermediates 35, 37, and 38 afforded compounds 36, 39 and 40, respectively. Overnight Buchwald–Hartwig amination of 1,3-dibromo-2-methylbenzene with Boc-protected piperazine yielded intermediate 41, which was reacted with phenylboronic acid under typical Suzuki coupling reaction conditions to afford intermediate 42, Removal of the Boc protecting group, followed by the nucleophilic displacement reaction with bromide 26, furnished compound 44. Compound 45 was prepared from the nucleophilic displacement reaction of 7-(piperazin-1-yl)benzo[d]oxazol-2(3H)-one44 with bromide 26.
Results of these compounds in β-arrestin-2 recruitment Tango and inhibition of cAMP accumulation assays are summarized in Table 2. Replacing the piperazine group in compound 1 with piperidine group in compound 30 resulted in a similar bias for G protein with slightly higher efficacy but slightly lower potency in both Gi/o and β-arrestin pathways. By contrast, replacing the piperazine group (compound 1) with homopiperazine (compound 31) led to significantly decreased efficacy in both Gi/o and β-arrestin signaling, resulting in a highly potent low efficacy partial agonist at activating Gi/o-mediated cAMP inhibition (EC50 = 1.9 nM, Emax = 22%), which was inactive for β-arrestin recruitment. With 1-(2-methoxyphenyl)piperazine as the LHS group, compound 32 exhibited slight improvement in efficacy in both assays, but the potency in Gi/o pathway dropped around 5-fold compared to compound 1, resulting in less bias for G protein signaling. Surprisingly, the methyl substitution in the middle piperazine region (compound 34) reduced the potency and efficacy drastically in both pathways, and led to no appreciable activity in either Gi/o signaling or β-arrestin recruitment. Interestingly, by replacing the 2,3-dichlorophenyl in compound 1 with the 2-methylquinoline, compound 36 displayed considerably reduced efficacy in both Gi/o and β-arrestin signaling, making compound 36 a potent G protein-biased partial agonist with low efficacy (EC50 = 15 nM, Emax = 25%). Similarly, compound 39, with the dihydrobenzooxazine as the LHS moiety, did not recruit β-arrestin at all, but had improved Gi/o potency yet decreased efficacy compared to compound 1 (EC50 = 5.2 nM, Emax = 29%). Interestingly, the close analog, compound 40, with dihydrobenzodioxine as the LHS moiety, displayed markedly improved potency (EC50 = 0.64 nM) and similar efficacy (Emax = 50%) for activating Gi/o signaling compared to compound 1. Although its efficacy and potency in β-arrestin recruitment also improved, 40 exhibited a similar bias for G protein as 1 and 30. By replacing the 2,3-dichloro substitutions (compound 1) with the 2-methyl-3-phenyl substitutions, compound 44 was found to be completely inactive in both Gi/o and β-arrestin assays. With the benzooxazolone as the LHS moiety, compound 45 displayed significantly improved efficacy and potency in both Gi/o and β-arrestin signaling pathways, making it a potent, balanced full agonist of D2R. In summary, although this region is generally more tolerant to modifications than the RHS benzothiazole region, subtle structural changes can still lead to a very significant impact on ligand bias. Our SFSR studies on this region resulted in the identification of several additional motifs including 2-methylquinoline, dihydrobenzooxazine, and dihydrobenzodioxine that favor Gi/o signaling over β-arrestin recruitment.
Table 2.
SFSR of the LHS Phenylpiperazine Moiety.a
 | |||||
|---|---|---|---|---|---|
| Compound | LHS Group | β-arrestin | cAMP | ||
| Emax (%) | EC50 (nM) | Emax (%) | EC50 (nM) | ||
| 1 | 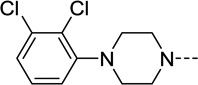 |
23 | 13 | 50 | 15 |
| 30 | 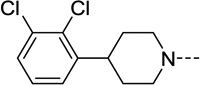 |
31 | 49 | 66 | 76 |
| 31 | 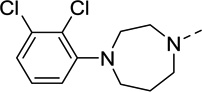 |
<20 | N/C | 22 | 1.9 |
| 32 | 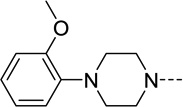 |
25 | 8.5 | 61 | 68 |
| 34 | 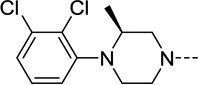 |
N/A | N/A | N/A | N/A |
| 36 |  |
<20 | N/C | 25 | 15 |
| 39 | 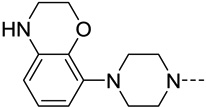 |
<20 | N/C | 29 | 5.2 |
| 40 | 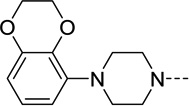 |
35 | 3.6 | 50 | 0.64 |
| 44 | 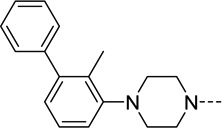 |
N/A | N/A | N/A | N/A |
| 45 | 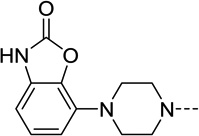 |
88 | 0.36 | 90 | 0.27 |
EC50 and Emax values are the average of at least 3 independent experiments performed in triplicate. SEM: < ± 20%.
N/C. not calculated. N/A. no activity.
SFSR Studies of Combination Compounds
We next designed and synthesized a number of combination compounds (outlined in Scheme 3 and Table 3), which incorporate some of the preferred RHS and LHS structural motifs we identified from the above studies. We selected 2-ethyl, 2-isopropyl, 2-methylamino and 2-dimethylaminobenzothiazol-5-yl and 2-methylbenzothiazol-6-yl as the RHS moiety, and 4-(2,3-dichlorophenyl)piperidine, 2-methylquinoline piperazine, dihydrobenzooxazine piperazine, and dihydrobenzodioxine piperazine as the LHS moiety. The synthetic routes for these combination compounds are summarized in Scheme 3. These compounds (46 – 58) were prepared following the same synthetic approach developed for compounds 1, 3 and 4 using the corresponding LHS and RHS intermediates.
Scheme 3.

Synthesis of combination compounds.a
a Reagents and Conditions: (a) K2CO3, EtOH, reflux, 6 h, 30–80% yield; (b) NaI/K2CO3, CH3CN, reflux, 6 h, 50–70% yield.
Table 3.
SFSR of combination compounds.a
| Cmpd | Structure | β-arrestin | cAMP | ||
|---|---|---|---|---|---|
| Emax (%) | EC50 (nM) | Emax (%) | EC50 (nM) | ||
| 46 | 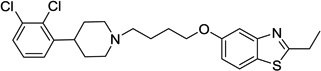 |
<20 | N/C | 43 | 98 |
| 47 | 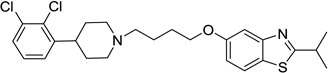 |
<20 | N/C | 33 | 72 |
| 48 | 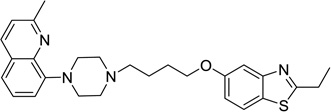 |
<20 | N/C | 33 | 62 |
| 49 | 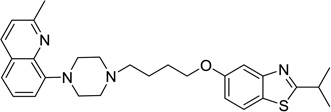 |
<20 | N/C | 32 | 8.3 |
| 50 | 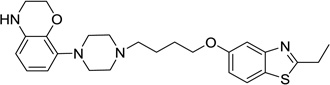 |
33 | 6.4 | 41 | 1.0 |
| 51 | 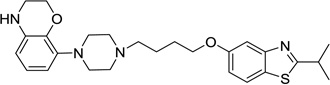 |
36 | 16 | 48 | 2.8 |
| 52 | 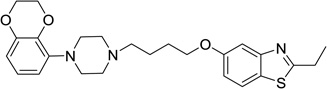 |
N/A | N/A | N/A | N/A |
| 53 | 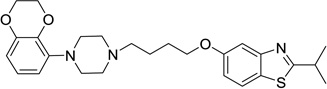 |
<20 | N/C | 37 | 5.5 |
| 54 | 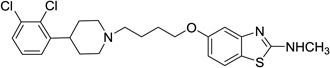 |
<20 | N/C | 47 | 50 |
| 55 | 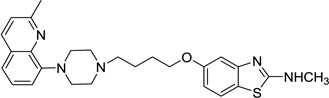 |
<20 | N/C | 31 | 28 |
| 56 | 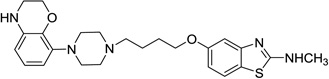 |
29 | 6.5 | 49 | 1.1 |
| 57 | 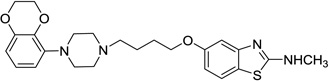 |
N/A | N/A | N/A | N/A |
| 58 | 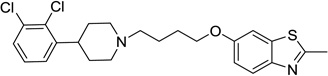 |
24 | 460 | 56 | 88 |
EC50 and Emax values are the average of at least three independent experiments performed in triplicate. SEM: < ± 20%.
N/C. not calculated. N/A. no activity.
We then evaluated compounds 46 – 58 in the D2R β-arrestin-2 recruitment Tango and Gi/o-mediated cAMP inhibition assays (results are summarized in Table 3). Interestingly, seven out of the 13 combination compounds (46 – 49 and 53 – 55) were significantly biased for G protein over β-arrestin recruitment with no activity in β-arrestin recruitment, and four other compounds (50, 51, 56 and 58) exhibited some extent of Gi/o bias with improved potency and/or efficacy in the Gi/o pathway. Surprisingly, two of the combination compounds, 52 and 57, with dihydrobenzodioxinylpiperazine as the LHS moiety and 2-ethyl or 2-methylamino benzothiazole as the RHS moiety, were inactive in both assays. All seven extremely G protein-biased compounds (46 – 49 and 53 – 55) were agonists at Gi/o signaling with low to moderate efficacy and moderate to high potency, but did not display appreciable β-arrestin-2 recruitment activity (<20% of quinpirole). Additionally, we also tested these compounds in cells not expressing D2R to rule out nonspecific cAMP inhibition not related to D2R, and observed no significant cAMP inhibition at concentrations lower than 10 µM (Figure S1). Most notably, as illustrated in Figure 2, compounds 46 and 54, similar to compounds 18 and 24, were more efficacious for Gi/o signaling with Emax 40–50% of quinpirole, and compounds 49 and 53 exhibited high Gi/o potencies (EC50 < 10 nM) but lower efficacies (Emax = 30 – 40%). A bias plot46 comparing relative concentration responses in the Gi/o GloSensor assay versus Tango β-arrestin-2 recruitment activity reveals that quinpirole is equi-efficacious in both assays (noted by the dotted line), but that compounds 1, 24, 46, 49, 53, and 54 deviate from quinpirole and cluster toward G protein activation (Figure 2C). These ligands, which have unique bias profiles, are potentially useful tools for elucidating signaling pathways that may contribute to antipsychotic efficacy and/or side-effects.
Figure 2. Compounds 1, 24, 46, 49, 53, and 54 are G protein-biased D2R partial agonists.

(A) Activity of compounds 1, 24, 46, 49, 53, 54, and quinpirole in the D2R-mediated Gi/o coupled isoproterenol-stimulated cAMP production assay using HEK293T cells expressing D2R and GloSensor-22F. All six tested compounds were partial agonists compared to quinpirole (EC50 = 1.7 nM), which was used as a positive control. (B) Activity of compounds 1, 24, 46, 49, 53, 54, and quinpirole in the D2R-mediated β-arrestin-2 translocation Tango assay using HTLA cells transfected with a D2V2-TCS-tTA construct. 1 recruited β-arrestin weakly and other five compounds did not substantially recruit β-arrestin compared to the full agonist quinpirole (EC50 = 0.8 nM). Data are representative of at least three independent experiments performed in triplicate. (C) Bias plot of compounds 1, 24, 46, 49, 53, 54, and quinpirole in the D2R-mediated cAMP inhibition GloSensor assay versus the D2R-mediated β-arrestin-2 translocation Tango assay. Quinpirole shows equal activity in both assays, but compounds 1, 24, 46, 49, 53, and 54 cluster toward G protein activation.
From these SFSR studies, we observed the following general trends: (1) 2-substitution at the RHS benzothiazole moiety is a critical contributor to bias for Gi/o signaling. 2-Methyl, 2-ethyl, 2-isopropyl, and 2-methylamino are preferred substituents that lead to bias for the G protein signaling over β-arrestin recruitment; (2) the LHS 2,3-dichlorophenyl can be replaced with 2-methylquinoline, dihydrobenzooxazine, and dihydrobenzodioxine, resulting to significant bias for the Gi/o signaling pathway over β-arrestin recruitment; (3) a small substituent such as methyl at the middle piperazine ring can completely abolish agonist activity in both pathways and a bulky substituent such as phenyl at the LHS ring can also completely diminish agonist activity in both pathways; and (4) subtle ligand structural changes can result in major changes in ligand bias, which is consistent with our previous findings.21
Further Characterization of G protein-biased D2R Agonists
Considering that the determination of ligand bias can be problematic due to either system or observational bias,46 which is dependent on cell background (e.g., HEK, CHO cell lines) and signal amplification (i.e., receptor reserve, time-dependence),47 we wanted to confirm the D2R G protein bias of these ligands by orthologous measures of β-arrestin-2 recruitment and G protein activity using bioluminescence resonance energy transfer (BRET)-based assays. Aripiprazole has been previously reported to be either a D2R antagonist31 or a partial agonist48 in the BRET β-arrestin-2 recruitment assay dependent on GRK2 co-expression, and has been observed to act as either a D2R antagonist or partial agonist in G protein-activation assays dependent on receptor expression.49 Although we have quantified the extent of D2R expression comparing D2R expressed in the GloSensor assay versus D2R expressed in the Tango assay and found similar expression levels (Table S1), time-dependent or assay kinetic differences may still interfere with the interpretation of ligand bias.
Therefore, we chose to use the D2R β-arrestin-2 recruitment BRET assay, where D2R arrestin recruitment is measured after only 5 minutes, and compared compound 1 to aripiprazole and the previously reported D2R G protein-biased ligand, MLS1547 (59)27. In HEK293T cells co-expressing D2R C-terminal-tagged renilla luciferase (Rluc), a Venus-tagged β-arrestin-2, and G protein–coupled receptor kinase 2 (GRK2), both aripiprazole and compound 59 displayed partial agonist activity for D2R-mediated β-arrestin-2 recruitment (Figure 3A, Emax = 26% and 68% of quinpirole, respectively) while compound 1 exhibited no activity up to 1 µM (Figure 3A). In fact, 59 also showed robust β-arrestin recruitment in the Tango assay (Figure 3B), albeit with moderate potency in both Tango and Gi/o assays (EC50 = 102 and 46 nM) (Figures 3B and 3C). To address the discrepant partial agonist activity of 59 in the β-arrestin-2 recruitment BRET assay compared to the previously published lack of β-arrestin-2 recruitment as measured using DiscoverX,27 we further evaluated compounds 1, 53, and 59 relative to quinpirole using the D2R DiscoverX β-arrestin recruitment assay and confirmed lack of β-arrestin-2 recruitment activity for these compounds (Figure 3D). The lack of robust β-arrestin-2 recruitment activity by 59 using DiscoverX assays may reflect cell type differences (DiscoverX uses CHO cells versus BRET and Tango use HEK cell background), but also likely reflect the lack of GRK2 co-expression, where GRK2 co-expression has been shown to be necessary for robust β-arrestin-2 recruitment for the mu-opioid receptor using the DiscoverX system.50 Importantly, compound 53 shows no activity in all three measures of D2R β-arrestin-2 recruitment.
Figure 3. Compounds 1, 24, 46, 49, 53, and 54 do not activate β-arrestin recruitment in the D2R-mediated BRET assay while compound 59 is a partial agonist in the D2R β-arrestin-2 recruitment Tango assay and D2R Gi/o-mediated cAMP inhibition assay.

(A) Activity of aripiprazole, compound 1, compound 59 and quinpirole in the D2R-mediated BRET-based β-arrestin-2 recruitment assay using HEK293 cells expressing GRK2. Aripiprazole and compound 59 were partial agonists that promote β-arrestin recruitment to D2R while compound 1 was not active in the BRET assay. Quinpirole (EC50 = 7.0 nM) was used as a positive control. (B) Activity of compound 59 and quinpirole in the D2R-mediated β-arrestin-2 translocation Tango assay using HTLA cells transfected with a D2V2-TCS-tTA construct. Compound 59 displayed robust β-arrestin recruitment in the Tango assay (EC50 = 46 nM, Emax = 67%) compared to the full agonist quinpirole (EC50 = 2.3 nM). (C) Activity of compound 59 and quinpirole in the D2R-mediated Gi/o coupled isoproterenol-stimulated cAMP production assay using HEK293T cells expressing D2R and GloSensor-22F. Compound 59 was a partial agonist (EC50 = 102 nM, Emax = 63%) compared to quinpirole (EC50 = 1.2 nM), which was used as a positive control. (D) D2R β-arrestin recruitment activity by compounds 1, 53, and 59 as measured by DiscoverX in D2R-expressing CHO cells. Compounds 1, 53, and 59 showed no activity up to 10 µM. Quinpirole (EC50 = 28 nM) was used as a positive control. (E) Compound 1 (IC50 = 18 nM) blocked dopamine (DA, EC50 = 11 nM) stimulated β-arrestin recruitment in the BRET assay. (F) Activity of compounds 24, 46, 49, 53 and 54 in the BRET assay. All compounds were not active in the BRET-based β-arrestin-2 recruitment assay while quinpirole (EC50 = 12 nM) was used as a positive control. (G) Activity of aripiprazole and compounds 1 and 53 in the D2R Gαi1-Gγ2 dissociation BRET assay. Aripiprazole (EC50 = 0.99 nM), compound 1 (EC50 = 3.6 nM), and 53 (EC50 = 4.6 nM), all displayed partial agonist activity (78%, 57%, and 47%, respectively) relative to quinpirole. Quinpirole (EC50 = 1.7 nM) was used as a positive control. Except for the DiscoverX assay (n=1, performed in duplicate), data are representative of at least three independent experiments performed in triplicate.
Despite the lack of agonist activity by 1 in the BRET assay, 1 displayed full antagonist activity by blocking dopamine-stimulated β-arrestin-2 recruitment (Figure 3E). In contrast, 1 showed partial inhibition of dopamine-stimulated D2R G protein activation as measured by cAMP inhibition (Figure S2). Similarly, as observed with compound 1, compounds 24, 46, 49, 53, and 54 showed no β-arrestin recruitment up to 1 µM as measured by the BRET assay with GRK2 co-expressed (Figure 3F). Finally, to confirm D2R G protein activation using an orthologous assay, we tested compounds 1 and 53 relative to aripiprazole and quinpirole using a Gαi1-Gγ2 dissociation BRET-based assay (Figure 3G). In this assay, compounds 1 and 53 still exhibited potent partial agonist activity (57% and 47% of quinpirole, respectively) compared to aripiprazole (78%). Therefore, using BRET-based orthologous assay platforms for either G protein activation or arrestin recruitment, we confirmed that compounds 1, 24, 46, 49, 53, and 54 are G protein-biased D2R agonists. As illustrated in Figures 2 – 3, these ligands are superior in their D2R G protein-bias profile compared to the previously reported compound 59.
Finally, we determined binding affinity of our G protein-biased D2R agonists 1, 24, 46, 49, 53, and 54 against a panel of aminergic GPCRs and neurotransmitter transporters (Table 4). Compound 1 exhibited ten-fold higher binding affinity towards D3R than D2R whereas all other five compounds displayed similar binding affinity to D3R compared to D2R. With the exception of compounds 1 and 53, which had similar D4R and D2R binding affinity, all compounds showed low or no affinity for other dopamine receptors (i.e., D1R, D4R and D5R). At serotonin [also known as 5-hydroxytryptamine (5-HT or 5HT)] receptors, all tested compounds displayed moderate to high binding affinities towards 5-HT1A, 5-HT2A, 5-HT2B, 5-HT2C and 5-HT7. All six compounds displayed similar or higher binding affinities for 5-HT1A and 5-HT2B as comparing to D2R. Notably, compound 53 showed very high affinity for 5-HT1A (Ki = 1.4 nM). High affinities were also observed for compound 49 towards 5-HT1A as well as compounds 24 and 54 towards 5-HT2B (Ki < 20 nM). All compounds exhibited low or no affinities towards dopamine transporter (DAT), serotonin transporter (SERT) and H2-histamine receptor with the exception of compound 54 towards SERT. For H1-histamine receptor, compounds 1, 24, 53, and 54 displayed moderate binding affinities while compound 46 showed low affinity and compound 49 did not exhibit appreciable affinity.
Table 4.
Radioligand binding affinity of compounds 1, 24, 46, 49, 53 and 54 at select GPCRs.a
| Receptor | Binding affinity Ki (nM) | |||||
|---|---|---|---|---|---|---|
| 1 | 24 | 46 | 49 | 53 | 54 | |
| D2R | 150 | 60 | 170 | 45 | 30 | 99 |
| D1R | N/A | 1100 | 1700 | 2000 | 2000 | 1100 |
| D3R | 13 | 35 | 100 | 64 | 31 | 69 |
| D4R | 82 | 280 | 1800 | 7100 | 77 | 430 |
| D5R | N/A | 1500 | N/A | 1500 | 1100 | 1800 |
| 5-HT1A | 86 | 55 | 100 | 14 | 1.4 | 66 |
| 5-HT1B | 1700 | 1400 | N/A | 1600 | 420 | 2600 |
| 5-HT1D | 320 | 390 | 820 | 94 | 100 | 410 |
| 5-HT2A | 120 | 72 | 140 | 110 | 29 | 70 |
| 5-HT2B | 38 | 12 | 71 | 43 | 32 | 18 |
| 5-HT2C | 160 | 58 | 210 | 140 | 92 | 73 |
| 5-HT3 | 130 | 330 | 1500 | N/A | 2600 | 370 |
| 5-HT5A | N/A | 1300 | 1600 | 1400 | 330 | 810 |
| 5-HT6 | 720 | 350 | 960 | 2200 | 2300 | 410 |
| 5-HT7 | 140 | 100 | 160 | 69 | 31 | 95 |
| DAT | 690 | 2800 | 4000 | N/A | 2500 | 2100 |
| SERT | 340 | 450 | 580 | 480 | 330 | 62 |
| H1 | 57 | 56 | 690 | N/A | 52 | 28 |
| H2 | 680 | 310 | 2100 | 430 | 240 | 330 |
Ki values are the average of at least 2 duplicate experiments. SEM: < ± 20%.
N/A: not active.
CONCLUSION
In summary, by introducing 2-methyl to the RHS benzothiazole and changing the middle linker from propoxy to butoxy, we unexpectedly converted a β-arrestin-biased D2R agonist to a G protein-biased D2R agonist, compound 1. We conducted structure–functional selectivity studies that focus on exploring two regions of the scaffold represented by this lead. These studies revealed a number of interesting general trends and identified multiple structural motifs that contribute to significant bias for the Gi/o signaling over β-arrestin recruitment. We discovered multiple G protein-biased D2R partial agonists including compounds 24, 46, 49, 53, and 54, which were completely inactive in two measures of β-arrestin recruitment, and in the case of compound 53 inactive in all three measures of β-arrestin recruitment. Based on our assay results, these newly identified ligands are superior G protein-biased D2R partial agonists compared with the previously reported G protein-biased D2R agonist, compound 59. Among the G protein-biased D2R agonists we have identified, compounds 24, 46 and 54 are most efficacious for the Gi/o signaling with Emax around 40–50% and compounds 49 and 53 exhibit the most potent Gi/o activity (EC50 < 10 nM). The unique bias profiles of these ligands make them a set of potentially useful tools for elucidating the role of G protein-mediated D2R signaling in health and disease. Our studies and results will also help identify the next generation of D2R G protein-biased and β-arrestin-biased ligands.
EXPERIMENTAL SECTION
Chemistry General Procedures
Unless stated to the contrary, where applicable, the following conditions apply: All commercial grade reagents were used without further purification. MeCN and CH2Cl2 were distilled from CaH2 under a N2 atmosphere before use; THF was distilled from Na/benzophenone under N2. All other dry solvents were of anhydrous quality purchased from Sigma-Aldrich. Brine (NaCl), NaHCO3, and NH4Cl refer to saturated aqueous (sat aq.) solutions. Column chromatography was performed on silica gel G (200–300 mesh) with reagent grade solvents. Melting points were uncorrected. NMR spectra were recorded on a Varian spectrometer (400 MHz for 1H NMR and 100 MHz for 13C NMR, respectively) at ambient temperature. All 1H and 13C chemical shifts are reported in ppm (δ) relative to CDCl3 (7.26 and 77.16, respectively) or CD3OD (3.30 and 49.00, respectively).51 HPLC data for all compounds were acquired using an Agilent 6110 series system with a UV detector set to 220 nm. Samples were injected (<10 µL) onto an Agilent Eclipse Plus 4.6 × 50 mm, 1.8 µm, C18 column at room temperature (rt) at a flow rate of 1.0 mL/min. A linear gradient from 10% to 100% (vol/vol) B over 5.0 min followed by 2.0 min at 100% B with a mobile phase of (A) H2O + 0.1% acetic acid and (B) MeOH + 0.1% acetic acid was used. Mass spectra (MS) data were acquired in positive ion mode using an Agilent 6110 series single quadrupole mass spectrometer with an electrospray ionization (ESI) source. High-resolution (positive ion) mass spectra (HRMS) were acquired using a Shimadzu LCMS-IT-Tof time-of-flight mass spectrometer. HPLC was used to establish the purity of targeted compounds. All compounds that were evaluated in biological assays had >95% purity using the HPLC methods described above.
5-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butoxy)-2-methylbenzo[d]thiazole (1)
A mixture of 2-methylbenzo[d]thiazol-5-ol (202.7 mg, 1.23 mmol), 1,4-dibromobutane (990.8 mg, 4.91 mmol) and anhydrous K2CO3 (203.5 mg, 1.47 mmol) was dissolved in EtOH (5 mL) and the solution was heated to reflux for 6 hours. The solution was diluted with water and extracted with EtOAc. The combined organic layers were washed with saturated aq NaHCO3, brine, dried over anhydrous Na2SO4, concentrated in vacuo and purified by flash chromatography on silica gel column to give 5-(4-bromobutoxy)-2-methylbenzo[d]thiazole (25, 251.1 mg, 68% yield) as a white solid. Compound 25 (89 mg, 0.30 mmol) was re-dissolved in CH3CN. To this mixture was added NaI (63.2 mg, 0.42 mmol) and the reaction mixture was heated to reflux for 30 min and then cooled to rt. The commercially available compound 4 (70.8 mg, 0.31 mmol) and anhydrous K2CO3 (56.5 mg, 0.41 mmol) were added to the mixture. The resulting mixture was heated to reflux and stirred for 6 h. Precipitated crystals were filtered off and the filtrate was evaporated under reduced pressure. The residue was extracted with EtOAc. The combined EtOAc layers was washed with brine, dried over anhydrous Na2SO4, concentrated in vacuo and purified by flash chromatography on silica gel column (elution with DCM/MeOH = 50:1) to give 5-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butoxy)-2-methylbenzo[d]thiazole (1) as white solid (87.8 mg, yield 65%). 1H NMR (400 MHz, CDCl3): δ 7.64 (d, J = 8.8 Hz, 1H), 7.44 (d, J = 2.4 Hz, 1H), 7.17 – 7.10 (m, 2H), 6.98 (dd, J = 8.8, 2.5 Hz, 1H), 6.95 (dd, J = 6.8, 2.8 Hz, 1H), 4.07 (t, J = 6.3 Hz, 2H), 3.08 (bs, 4H), 2.80 (s, 3H), 2.68 (bs, 4H), 2.55 – 2.48 (m, 2H), 1.93 – 1.83 (m, 2H), 1.75 (ddd, J = 15.1, 8.8, 6.0 Hz, 2H); 13C NMR (101 MHz, CDCl3): δ 168.26, 158.32, 154.75, 151.36, 134.13, 127.63, 127.56, 127.40, 124.70, 121.69, 118.75, 115.15, 106.04, 68.19, 58.30, 53.40, 51.34, 27.32, 23.51, 20.30; HPLC: 99%, RT 4.819 min; MS (ESI) m/z 450.2 [M + H]+. HRMS m/z [M + H]+ calcd for C22H26Cl2N3OS 450.1174, found 450.1168.
5-(3-(4-(2,3-Dichlorophenyl)piperazin-1-yl)propoxy)-2-methylbenzo[d]thiazole (2)
Compound 2 (73 mg) was prepared as white solid by the same procedure as preparing 1 starting from 2-methylbenzo[d]thiazol-5-ol and 1,3-dibromopropane, yield 64%. 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 8.8 Hz, 1H), 7.46 (d, J = 2.4 Hz, 1H), 7.18 – 7.11 (m, 2H), 7.00 (dd, J = 8.8, 2.4 Hz, 1H), 6.96 (dd, J = 6.4, 3.2 Hz, 1H), 4.12 (t, J = 6.3 Hz, 2H), 3.08 (bs, 4H), 2.81 (s, 3H), 2.65 (dd, J = 15.6, 8.4 Hz, 6H), 2.10 – 1.99 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 168.27, 158.33, 154.78, 151.46, 134.15, 127.66, 127.56, 127.47, 124.66, 121.70, 118.75, 115.15, 106.16, 66.76, 55.25, 53.49, 51.50, 26.89, 20.33; HPLC 99%, RT 4.766 min; MS (ESI) m/z 436.1 [M + H]+.
6-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butoxy)-2-methylbenzo[d]thiazole (3)
Compound 3 (63 mg) was prepared as white solid by the same procedure as preparing 1 starting from 2-methylbenzo[d]thiazol-6-ol and 1,4-dibromobutane, yield 67%. 1H NMR (400 MHz, CDCl3): δ 7.80 (d, J = 8.9 Hz, 1H), 7.27 (d, J = 2.5 Hz, 1H), 7.18 – 7.10 (m, 2H), 7.02 (dd, J = 8.9, 2.5 Hz, 1H), 6.95 (dd, J = 7.2, 2.3 Hz, 1H), 4.04 (t, J = 6.0 Hz, 2H), 3.14 (t, J = 4.3 Hz, 4H), 2.77 (bs, 7H), 2.64 – 2.56 (m, 2H), 1.94 – 1.75 (m, 4H); 13C NMR (101 MHz, CDCl3): δ 164.42, 156.76, 150.97, 147.94, 136.95, 134.15, 127.63, 124.93, 122.86, 118.82, 115.47, 105.09, 68.29, 58.17, 53.26, 50.85, 27.27, 23.14, 20.07; HPLC 99%, RT 4.857 min; MS (ESI) m/z 450.2[M + H]+.
6-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butoxy)-2-methylbenzo[d]oxazole (4)
Starting from 2-methylbenzo[d]oxazol-5-ol and 1,4-dibromobutane, compound 4 (58 mg) was prepared as white solid by the same procedure as preparing compound 1, yield 62%. 1H NMR (400 MHz, CDCl3): δ 7.32 (d, J = 8.8 Hz, 1H), 7.14 (dt, J = 6.9, 4.1 Hz, 3H), 6.96 (dd, J = 7.2, 2.3 Hz, 1H), 6.86 (dd, J = 8.8, 2.5 Hz, 1H), 4.02 (t, J = 5.6 Hz, 2H), 3.18 (bs, 4H), 2.82 (bs, 4H), 2.64 (t, J = 6.9 Hz, 2H), 2.59 (s, 3H), 1.93 – 1.77 (m, 4H); 13C NMR (101 MHz, CD3OD): δ 164.73, 156.38, 150.82, 145.71, 142.44, 134.14, 127.64, 127.61, 125.02, 118.87, 113.23, 110.38, 103.69, 68.43, 58.14, 53.17, 50.61, 27.25, 23.00, 14.72; HPLC 99%, RT 4.681 min; MS (ESI) m/z 434.1 [M + H]+.
2-Chloro-5-(4-(4-(2,3-dichlorophenyl)piperazin-1-yl)butoxy)benzo[d]thiazole (6)
Starting from 2-chlorobenzo[d]thiazol-5-ol and 1,4-dibromobutane, compound 6 (48 mg) was prepared as white solid by the same procedure as preparing compound 1, yield 57%. 1H NMR (400 MHz, CD3OD): δ 7.83 (d, J = 8.9 Hz, 1H), 7.50 (d, J = 2.4 Hz, 1H), 7.38 – 7.29 (m, 2H), 7.18 (ddd, J = 11.4, 8.0, 2.5 Hz, 2H), 4.20 (dd, J = 7.6, 3.9 Hz, 2H), 3.74 (d, J = 11.7 Hz, 2H), 3.59 (d, J = 13.6 Hz, 2H), 3.43 – 3.35 (m, 4H), 3.15 (t, J = 12.4 Hz, 2H), 2.11 – 1.94 (m, 4H); 13C NMR (101 MHz, CD3OD): δ 160.06, 155.72, 153.32, 150.65, 135.16, 129.18, 128.65, 123.26, 123.11, 120.51, 117.15, 107.26, 107.13, 68.58, 57.84, 53.50, 29.16, 27.31, 22.23; HPLC 99%, RT 5.164 min; MS (ESI) m/z 470.1 [M + H]+.
5-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butoxy)benzo[d]thiazol-2(3H)-one (7)
Starting from 5-hydroxybenzo[d]thiazol-2(3H)-one and 1,4-dibromobutane, compound 7 (51 mg) was prepared as white solid by the same procedure as preparing compound 1, yield 53%. 1H NMR (400 MHz, CD3OD): δ 10.40 (s, 1H), 7.23 (dd, J = 8.1, 1.5 Hz, 1H), 7.17 (t, J = 8.0 Hz, 1H), 7.11 (d, J = 8.6 Hz, 1H), 6.98 (dd, J = 8.0, 1.5 Hz, 1H), 6.67 (d, J = 2.2 Hz, 1H), 6.61 (dd, J = 8.6, 2.3 Hz, 1H), 3.94 (dd, J = 13.3, 7.7 Hz, 2H), 3.82 (d, J = 11.4 Hz, 2H), 3.42 (d, J = 12.6 Hz, 2H), 3.37 – 3.18 (m, 4H), 3.09 (t, J = 10.3 Hz, 2H), 2.11 – 1.98 (m, 2H), 1.90 – 1.78 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 173.39, 157.98, 149.06, 136.78, 134.39, 128.02, 127.77, 126.30, 122.98, 119.24, 115.34, 110.40, 98.47, 67.17, 57.19, 52.58, 48.49, 26.26, 21.05; HPLC 99%, RT 4.773 min; MS (ESI) m/z 452.1 [M + H]+.
(Z)-2,2,2-Trifluoro-N-(3-methoxyphenyl)acetohydrazonoyl chloride (8)
To a 3-necked flask was charged with Ph3P (17.25 g, 66 mmol), Et3N (3.65 mL, 26.5 mmol), CCl4 (10.55 mL, 110 mmol) and TFA (1.7 mL, 22 mmol). After the solution was stirred at 0°C for about 10 min, 3-methoxyaniline (3.24 g, 26.5 mmol) in CCl4 (10.55 mL, 110 mmol) was added. The mixture was then refluxed under stirring for 3 h. Solvent was removed under reduced pressure, and the residue was diluted with hexane and filtered. Residual solid was washed with hexane several times. The filtrate was concentrated and purified by flash chromatography on silica gel column to give compound 8 as white solid. (4.7792 g, 86%) 1H NMR (400 MHz, CDCl3) δ 7.34 (t, J = 8.1 Hz, 1H), 6.85 (ddd, J = 8.4, 2.5, 0.8 Hz, 1H), 6.68 (ddd, J = 7.9, 1.9, 0.8 Hz, 1H), 6.62 (t, J = 2.2 Hz, 1H), 3.83 (s, 3H); HPLC: 99%, RT 4.881 min; MS (ESI) m/z 253.0 [M + H]+.
5-Methoxy-2-(trifluoromethyl)benzo[d]thiazole (9)
To a solution of compound 8 (242.0 mg, 1.02 mmol) in DMSO (3 mL) was added NaSH˙xH2O (81.5 mg, 1.02 mmol). The resulting mixture was then stirred at 50°C for 30 min. PdCl2 (9.0 mg, 0.05 mmol) was added and the temperature was increased to 110°C. After stirring for 3 h at 110°C, H2O was added and the reaction mixture was cooled, and extracted with EtOAc. Organic layers were combined and concentrated to give the residue which was purified on silica gel column to give compound 9 as white solid (202.2 mg, 85%). 1H NMR (400 MHz, CDCl3) δ 7.78 (d, J = 9.0 Hz, 1H), 7.60 (d, J = 2.5 Hz, 1H), 7.17 (dd, J = 8.9, 2.5 Hz, 1H), 3.88 (s, 3H); HPLC: 99%, RT 5.855 min; MS (ESI) m/z 234.0 [M + H]+.
2-(Trifluoromethyl)benzo[d]thiazol-5-ol (10)
To the flask containing compound 9 (202.2 mg, 0.87 mmol) was added 47% HI (3 mL). The resulting mixture was refluxed while stirring for 5 h. After basifying with NaOH (1N), the mixture was extracted with EtOAc. Organic layers were combined and concentrated to give the residue which was purified on silica gel column to give compound 10 as white solid (174.8 mg, 92%). 1H NMR (400 MHz, CDCl3) δ 7.81 (dd, J = 8.8, 0.9 Hz, 1H), 7.61 (d, J = 2.0 Hz, 1H), 7.16 (dd, J = 8.8, 2.3 Hz, 1H), 6.33 (bs, 1H); HPLC: 99%, RT 5.429 min; MS (ESI) m/z 220.0 [M + H]+.
5-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butoxy)-2-(trifluoromethyl)benzo[d]thiazole (11)
Starting from 2-(trifluoromethyl)benzo[d]thiazol-5-ol (10) and 1,4-dibromobutane, compound 11 (68 mg) was prepared as white solid by the same procedure as preparing compound 1, yield 63%. 1H NMR (400 MHz, CDCl3): δ 7.81 (d, J = 8.9 Hz, 1H), 7.63 (d, J = 2.4 Hz, 1H), 7.19 (dd, J = 8.9, 2.5 Hz, 1H), 7.17 – 7.09 (m, 2H), 6.95 (dd, J = 6.8, 2.7 Hz, 1H), 4.10 (t, J = 6.3 Hz, 2H), 3.10 (s, 4H), 2.70 (s, 4H), 2.60 – 2.48 (m, 2H), 1.97 – 1.86 (m, 2H), 1.78 (dt, J = 14.7, 7.4 Hz, 2H); 13C NMR (101 MHz, CDCl3): δ 159.23, 156.87 (d, J = 40.3 Hz), 153.69, 151.28, 134.14, 127.63, 127.59, 126.91, 124.77, 122.34 (d, J = 4.7 Hz), 119.93 (q, J = 272 Hz), 118.76, 118.57, 107.15 (d, J = 6.4 Hz), 68.30, 58.21, 53.39, 51.27, 27.15, 23.39; HPLC 99%, RT 5.298 min; MS (ESI) m/z 504.1 [M + H]+.
2-Ethyl-5-methoxybenzo[d]thiazole (14)
To a solution of 1-isothiocyanato-3-methoxybenzene (2.9475 g, 17.84 mmol) in THF (12 mL) at −10°C was added ethylmagnesium chloride (2.0 M in THF, 17.84 mL, 35.68 mmol). The resulting mixture was then stirred at −10°C for 90 min. Saturated NH4Cl was added and the mixture was extracted with EtOAc three times. Organic layers were combined, dried over anhydrous Na2SO4 and concentrated to give crude N-(3-methoxyphenyl)propanethioamide (12) which was used for the next reaction without further purification. To a suspension of K3Fe(CN)6 (19.384 g, 58.87 mmol) in H2O (49 mL) at 60°C was added the mixture of compound 12 (17.84 mmol) and NaOH (5.2806 g, 132.01 mmol) in H2O (81 mL) and CH3OH (6 mL). The resulting mixture was the stirred at 60°C for 2 h before K2CO3 (13.0679 g, 94.55 mmol) was added. The mixture was then stirred at 60°C for another hour. After cooling, the mixture was extracted with Et2O. Organic layers were combined and concentrated to give the residue which was purified on silica gel column to give compound 14 as colorless oil (2.8272 g, 82% yield). 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J = 8.8 Hz, 1H), 7.47 (d, J = 2.4 Hz, 1H), 6.99 (dd, J = 8.8, 2.5 Hz, 1H), 3.88 (s, 3H), 3.13 (q, J = 7.6 Hz, 2H), 1.46 (t, J = 7.6 Hz, 3H); HPLC 99%, RT 5.398 min; MS (ESI) m/z 194.0 [M + H]+.
2-Isopropyl-5-methoxybenzo[d]thiazole (15)
Starting from 1-isothiocyanato-3-methoxybenzene and isopropylmagnesium chloride, compound 15 (2.5738 g, 87%) was prepared as yellow oil via the same procedure as preparing compound 14. 1H NMR (400 MHz, CDCl3) δ 7.67 (d, J = 8.8 Hz, 1H), 7.48 (d, J = 2.5 Hz, 1H), 6.98 (dd, J = 8.8, 2.5 Hz, 1H), 3.86 (s, 3H), 3.39 (hept, J = 6.9 Hz, 1H), 1.47 (d, J = 6.9 Hz, 6H); HPLC 99%, RT 5.689 min; MS (ESI) m/z 208.0 [M + H]+.
2-Ethylbenzo[d]thiazol-5-ol (16)
Using the same procedure as preparing compound 10, compound 16 was prepared from compound 14 as white solid (890 mg, 83% yield). 1H NMR (400 MHz, CD3OD) δ 7.66 (d, J = 8.7 Hz, 1H), 7.29 (d, J = 2.3 Hz, 1H), 6.91 (dd, J = 8.7, 2.4 Hz, 1H), 3.07 (q, J = 7.6 Hz, 2H), 1.40 (t, J = 7.6 Hz, 3H); HPLC 99%, RT 4.852 min; MS (ESI) m/z 180.0 [M + H]+.
2-Isopropylbenzo[d]thiazol-5-ol (17)
Using the same procedure as preparing compound 10, compound 17 was prepared from compound 15 as white solid (1.2270 g, 87% yield). 1H NMR (400 MHz, CD3OD) δ 7.70 (d, J = 8.7 Hz, 1H), 7.30 (d, J = 2.3 Hz, 1H), 6.92 (dd, J = 8.7, 2.4 Hz, 1H), 3.44 – 3.34 (m, 1H), 1.45 (d, J = 6.9 Hz, 6H); HPLC 99%, RT 5.189 min; MS (ESI) m/z 194.0 [M + H]+.
5-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butoxy)-2-ethylbenzo[d]thiazole (18)
Starting from 2-ethylbenzo[d]thiazol-5-ol (16) and 1,4-dibromobutane, compound 18 (59 mg) was prepared as white solid by the same procedure as preparing compound 1, yield 64%. 1H NMR (400 MHz, CDCl3): δ 7.67 (d, J = 8.7 Hz, 1H), 7.46 (d, J = 2.3 Hz, 1H), 7.17 – 7.10 (m, 2H), 6.97 (ddd, J = 9.5, 7.7, 2.6 Hz, 2H), 4.07 (t, J = 6.3 Hz, 2H), 3.18 – 3.02 (m, 6H), 2.67 (bs, 4H), 2.55 – 2.46 (m, 2H), 1.93 – 1.82 (m, 2H), 1.74 (dt, J = 7.3 Hz, 2H), 1.46 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ 174.94, 158.33, 154.66, 151.44, 134.15, 127.66, 127.56, 126.83, 124.67, 121.82, 118.75, 115.22, 106.06, 68.22, 58.33, 53.43, 51.44, 29.85, 27.96, 27.35, 23.57, 13.93; HPLC 99%, RT 4.981 min; MS (ESI) m/z 463.9 [M + H]+. HRMS m/z [M + H]+ calcd for C23H28Cl2N3OS 464.1330, found 464.1321.
5-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butoxy)-2-isopropylbenzo[d]thiazole (19)
Starting from 2-isopropylbenzo[d]thiazol-5-ol (17) and 1,4-dibromobutane, compound 19 (63 mg) was prepared as white solid by the same procedure as preparing compound 1, yield 68%. 1H NMR (400 MHz, CDCl3): δ 7.68 (d, J = 8.8 Hz, 1H), 7.46 (d, J = 2.4 Hz, 1H), 7.22 – 7.12 (m, 2H), 6.98 (dd, J = 9.0, 2.3 Hz, 2H), 4.08 (t, J = 5.6 Hz, 2H), 3.39 (dt, J = 13.8, 6.9 Hz, 1H), 3.28 (bs, 4H), 2.94 (bs, 4H), 2.76 (bs, 2H), 1.92 (bs, 4H), 1.47 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ 180.16, 158.08, 154.54, 134.26, 127.77, 126.72, 125.37, 121.96, 119.04, 115.12, 106.15, 67.86, 58.05, 53.06, 50.11, 34.27, 27.10, 23.03; HPLC 99%, RT 5.106 min; MS (ESI) m/z 478.0 [M + H]+.
5-Methoxy-N-methylbenzo[d]thiazol-2-amine (20)
To a solution of 1-isothiocyanato-3-methoxybenzene (235.8 mg, 1.43 mmol) in THF (5 mL) was added CH3NH2 (0.75 mL, 1.50 mmol). The resulting mixture was stirred at room temperature for 30 min before PhCH2NMe3Br3 (556.5 mg, 1.43 mmol) was added. The reaction mixture was then stirred overnight at room temperature. The reaction was diluted with DCM and neutralized with aqueous NaHCO3. The organic layer was dried and concentrated to give the residue which was purified on silica gel column to give compound 20 as white solid (258.3 mg, yield 93%). 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 8.6 Hz, 1H), 7.08 (d, J = 2.5 Hz, 1H), 6.70 (dd, J = 8.6, 2.5 Hz, 1H), 6.22 (bs, 1H), 3.82 (s, 3H), 3.07 (s, 3H); HPLC 99%, RT 4.194 min; MS (ESI) m/z 195.1 [M + H]+.
5-Methoxy-N,N-dimethylbenzo[d]thiazol-2-amine (21)
Starting from 1-isothiocyanato-3-methoxybenzene and (CH3)2NH, compound 21 (white solid, 2.895 g, 96% yield) was prepared by the same procedure as preparing compound 20. 1H NMR (400 MHz, CDCl3) δ 7.42 (d, J = 8.6 Hz, 1H), 7.13 (d, J = 2.5 Hz, 1H), 6.67 (dd, J = 8.6, 2.5 Hz, 1H), 3.81 (s, 3H), 3.16 (s, 6H); HPLC 99%, RT 4.931 min; MS (ESI) m/z 209.0 [M + H]+.
2-(Methylamino)benzo[d]thiazol-5-ol (22)
Using the same procedure as preparing compound 10, compound 22 was prepared from compound 20 as white solid (2.563 g, 85% yield). 1H NMR (400 MHz, CD3OD) δ 7.33 (d, J = 8.5 Hz, 1H), 6.91 (d, J = 2.4 Hz, 1H), 6.62 – 6.50 (m, 1H), 3.00 (s, 3H); HPLC 99%, RT 2.969 min; MS (ESI) m/z 181.0 [M + H]+.
2-(Dimethylamino)benzo[d]thiazol-5-ol (23)
Using the same procedure as preparing compound 10, compound 23 was prepared from compound 21 as white solid (1.227 g, 89% yield). 1H NMR (400 MHz, CD3OD) δ 7.39 (d, J = 8.5 Hz, 1H), 6.94 (d, J = 2.4 Hz, 1H), 6.58 (dd, J = 8.5, 2.4 Hz, 1H), 3.17 (s, 6H); HPLC 99%, RT 3.880 min; MS (ESI) m/z 195.1 [M + H]+.
5-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butoxy)-N methylbenzo[d]thiazol-2-amine (24)
Starting from 2-(methylamino)benzo[d]thiazol-5-ol (22) and 1,4-dibromobutane, compound 24 (45 mg) was prepared as white solid by the same procedure as preparing compound 1, yield 53%. 1H NMR (400 MHz, CDCl3): δ 7.43 (d, J = 8.6 Hz, 1H), 7.20 – 7.13 (m, 2H), 7.11 (d, J = 2.4 Hz, 1H), 6.98 (dd, J = 7.4, 2.2 Hz, 1H), 6.71 (dd, J = 8.6, 2.5 Hz, 1H), 4.04 (t, J = 5.6 Hz, 2H), 3.22 (bs, 4H), 3.10 (s, 3H), 2.86 (bs, 4H), 2.68 (bs, 2H), 1.91 – 1.81 (m, 4H); 13C NMR (101 MHz, CDCl3): δ 169.61, 158.38, 153.96, 134.23, 127.71, 125.11, 122.37, 121.04, 118.97, 110.64, 110.22, 104.30, 67.91, 58.17, 53.16, 50.19, 31.85, 27.24, 22.83; HPLC 99%, RT 4.522 min; MS (ESI) m/z 465.0 [M + H]+. HRMS m/z [M + H]+ calcd for C22H27Cl2N4OS 465.1283, found 465.1266.
5-(4-(4-(2,3-Dichlorophenyl)piperazin-1-yl)butoxy)-N,N-dimethylbenzo[d]thiazol-2-amine (25)
Starting from 2-(dimethylamino)benzo[d]thiazol-5-ol (23) and 1,4-dibromobutane, compound 25 (53 mg) was prepared as white solid by the same procedure as preparing compound 1, yield 58%. 1H NMR (400 MHz, CDCl3): δ 7.41 (d, J = 8.6 Hz, 1H), 7.17 – 7.08 (m, 3H), 6.94 (dd, J = 6.9, 2.6 Hz, 1H), 6.67 (dd, J = 8.6, 2.4 Hz, 1H), 4.01 (t, J = 6.3 Hz, 2H), 3.17 (s, 6H), 3.08 (bs, 4H), 2.67 (bs, 4H), 2.55 – 2.45 (m, 2H), 1.89 – 1.78 (m, 2H), 1.72 (dt, J = 9.5, 7.1 Hz, 2H); 13C NMR (101 MHz, CDCl3): δ 170.10, 158.43, 154.52, 151.34, 134.07, 127.58, 127.53, 124.63, 122.59, 120.89, 118.73, 110.23, 103.83, 68.04, 58.28, 53.34, 51.29, 40.23, 27.35, 23.48; HPLC 99%, RT 4.747 min; MS (ESI) m/z 479.0 [M + H]+.
5-(4-(4-(2,3-Dichlorophenyl)piperidin-1-yl)butoxy)-2-methylbenzo[d]thiazole (30)
Starting from 4-(2,3-dichlorophenyl)piperidine (27) and 5-(4-bromobutoxy)-2-methylbenzo[d]thiazole (26), compound 30 (53 mg) was prepared as white solid by the same procedure as preparing compound 1, yield 61%. 1H NMR (400 MHz, CDCl3): δ 7.66 (d, J = 8.7 Hz, 1H), 7.41 (d, J = 2.4 Hz, 1H), 7.35 (dd, J = 7.9, 1.6 Hz, 1H), 7.29 (dd, J = 7.9, 1.6 Hz, 1H), 7.20 (t, J = 7.9 Hz, 1H), 6.97 (dd, J = 8.8, 2.5 Hz, 1H), 4.08 (t, J = 5.9 Hz, 2H), 3.55 (d, J = 12.0 Hz, 2H), 3.33 (tt, J = 12.2, 3.3 Hz, 1H), 2.99 – 2.90 (m, 2H), 2.80 (s, 3H), 2.70 (t, J = 11.5 Hz, 2H), 2.38 (q, J = 12.1 Hz, 2H), 2.13 – 2.04 (m, 2H), 2.00 (d, J = 13.9 Hz, 2H), 1.96 – 1.87 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 168.51, 157.92, 154.70, 143.14, 133.37, 131.60, 129.02, 128.03, 127.78, 125.63, 121.85, 114.85, 106.18, 67.58, 57.83, 53.58, 38.09, 29.48, 26.93, 21.94, 20.32; HPLC 99%, RT 4.797 min; MS (ESI) m/z 449.1 [M + H]+.
5-(4-(4-(2,3-Dichlorophenyl)-1,4-diazepan-1-yl)butoxy)-2-methylbenzo[d]thiazole (31)
Compound 31 (58 mg) was prepared as white solid by the same procedure as preparing 1 from the reaction of 1-(2,3-dichlorophenyl)-1,4-diazepane (28) and 5-(4-bromobutoxy)-2-methylbenzo[d]thiazole (26), yield 53%. 1H NMR (400 MHz, CDCl3): δ 7.64 (d, J = 8.8 Hz, 1H), 7.39 (d, J = 2.4 Hz, 1H), 7.19 (dd, J = 8.0, 1.7 Hz, 1H), 7.13 (t, J = 8.0 Hz, 1H), 7.02 (dd, J = 7.9, 1.7 Hz, 1H), 6.96 (dd, J = 8.8, 2.5 Hz, 1H), 4.07 (t, J = 5.8 Hz, 2H), 3.69 – 3.41 (m, 6H), 3.32 – 3.18 (m, 4H), 2.79 (s, 3H), 2.48 (bs, 2H), 2.26 – 2.13 (m, 2H), 1.98 – 1.87 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 168.50, 157.77, 154.65, 151.22, 134.22, 127.97, 127.85, 125.79, 121.86, 120.97, 114.78, 106.16, 4, 67.29, 57.63, 56.25, 52.93, 51.95, 49.92, 26.70, 24.17, 21.53, 20.29; HPLC 99%, RT 4.820 min; MS (ESI) m/z 464.2 [M + H]+.
5-(4-(4-(2-Methoxyphenyl)piperazin-1-yl)butoxy)-2-methylbenzo[d]thiazole (32)
Compound 32 (52 mg) was prepared as white solid by the same procedure as preparing 1 from the reaction of 1-(2-methoxyphenyl)piperazine (29) and 5-(4-bromobutoxy)-2-methylbenzo[d]thiazole (26), yield 62%. 1H NMR (400 MHz, CDCl3): δ 7.64 (d, J = 8.8 Hz, 1H), 7.41 (d, J = 2.4 Hz, 1H), 7.03 – 6.95 (m, 2H), 6.95 – 6.87 (m, 2H), 6.85 (d, J = 7.8 Hz, 1H), 4.05 (t, J = 5.8 Hz, 2H), 3.84 (s, 3H), 3.28 (bs, 4H), 2.95 (bs, 4H), 2.79 (s, 3H), 2.78 – 2.70 (m, 2H), 1.98 – 1.81 (m, 4H); 13C NMR (101 MHz, CDCl3): δ 168.30, 158.06, 154.63, 152.22, 140.36, 127.50, 123.60, 121.70, 121.14, 118.59, 114.94, 111.30, 106.06, 67.84, 57.94, 55.46, 53.04, 49.29, 27.02, 22.51, 20.23; HPLC 99%, RT 4.503 min; MS (ESI) m/z 412.3 [M + H]+.
(S)-1-(2,3-Dichlorophenyl)-2-methylpiperazine (33)
To a solution of 1-bromo-2,3-dichlorobenzene (394.3 mg, 1.75 mmol) in toluene (5 mL) was added tert-butyl (S)-3-methylpiperazine-1-carboxylate (402.0 mg, 2.01 mmol), Pd2(dba)3 (7.99 mg, 0.0087 mmol), BINAP (21.7 mg, 0.035 mmol) and t-BuONa (754.8 mg, 7.85 mmol). The resulting mixture was stirred overnight at 100°C in a sealed tube. After cooling down, the reaction mixture was filtered through a pad of celite and the filtrate was concentrated to give the residue which was purified on silica gel column to give tert-butyl (S)-4-(2,3-dichlorophenyl)-3-methylpiperazine-1-carboxylateas as white solid, which was treated with TFA in DCM at room temperature for 1 h to give crude compound 33 as TFA salt (263.2 mg, 52% yield) after removal of all the solvent. This crude compound 33 was used for the next reaction without further purification.
(S)-5-(4-(4-(2,3-Dichlorophenyl)-3-methylpiperazin-1-yl)butoxy)-2-methylbenzo[d]thiazole (34)
Compound 34 (52 mg) was prepared as white solid by the same procedure as preparing 1 from the reaction of (S)-1-(2,3-dichlorophenyl)-2-methylpiperazine (33) and 5-(4-bromobutoxy)-2-methylbenzo[d]thiazole (26), yield 51%. 1H NMR (400 MHz, CDCl3): δ 7.66 (d, J = 8.8 Hz, 1H), 7.42 (d, J = 2.5 Hz, 1H), 7.30 – 7.26 (m, 1H), 7.19 (t, J = 8.0 Hz, 1H), 7.11 (d, J = 7.7 Hz, 1H), 6.97 (dd, J = 8.7, 2.4 Hz, 1H), 4.07 (t, J = 5.6 Hz, 2H), 3.62 (bs, 1H), 3.42 – 2.84 (m, 7H), 2.81 (s, 3H), 2.52 (s, 1H), 2.04 – 1.83 (m, 4H), 0.93 (d, J = 6.1 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ 168.46, 162.81, 158.02, 154.73, 134.09, 127.73, 127.55, 127.11, 123.08, 121.83, 118.29, 114.95, 106.12, 67.64, 58.86, 57.63, 52.84, 51.76, 39.29, 26.87, 25.56, 21.94, 20.32; HPLC 99%, RT 4.935 min; MS (ESI) m/z 464.2 [M + H]+.
2-Methyl-8-(piperazin-1-yl)quinoline (35)
To a sealed tube was added 8-bromo-2-methylquinoline (213.4 mg, 0.96 mmol), piperazine (95.2 mg, 1.11 mmol), Pd2(dba)3 (8.8 mg, 0.0096 mmol), BINAP (12.0 mg, 0.019 mmol), t-BuONa (230.8 mg, 2.40 mmol) and toluene (2 mL). The mixture was then stirred at 100°C for 4 h. After cooling, the mixture was filtered through a pad of celite and the filtrate was concentrated to give the residue which was purified on silica gel column to give compound 35 as white solid (146.2 mg, 67% yield).1H NMR (400 MHz, CDCl3) δ 7.95 (d, J = 8.4 Hz, 1H), 7.39 – 7.29 (m, 2H), 7.21 (d, J = 8.4 Hz, 1H), 7.07 (dd, J = 6.5, 2.4 Hz, 1H), 3.41 (d, J = 4.5 Hz, 4H), 3.30 – 3.18 (m, 4H), 2.70 (s, 3H); HPLC 99%, RT 2.628 min; MS (ESI) m/z 228.2 [M + H]+.
2-Methyl-5-(4-(4-(2-methylquinolin-8-yl)piperazin-1-yl)butoxy)benzo[d]thiazole (36)
Compound 36 (57 mg) was prepared as white solid by the same procedure as preparing 1 from the reaction of 2-methyl-8-(piperazin-1-yl)quinoline (35) and 5-(4-bromobutoxy)-2-methylbenzo[d]thiazole (26), yield 55%. 1H NMR (400 MHz, CDCl3): δ 7.97 (d, J = 8.4 Hz, 1H), 7.65 (d, J = 8.7 Hz, 1H), 7.44 (d, J = 2.4 Hz, 1H), 7.40 – 7.32 (m, 2H), 7.23 (d, J = 8.4 Hz, 1H), 7.09 (dd, J = 6.0, 2.9 Hz, 1H), 6.99 (dd, J = 8.8, 2.4 Hz, 1H), 4.08 (t, J = 6.1 Hz, 2H), 3.50 (bs, 4H), 2.88 (bs, 4H), 2.80 (s, 3H), 2.72 (s, 3H), 2.63 – 2.57 (m, 2H), 1.96 – 1.77 (m, 4H); 13C NMR (101 MHz, CDCl3): δ 168.22, 158.32, 156.76, 154.74, 148.61, 142.03, 136.71, 127.79, 127.38, 125.86, 121.67, 121.62, 121.51, 115.97, 115.15, 106.06, 68.23, 58.49, 53.45, 51.85, 27.38, 25.96, 23.48, 20.29; HPLC 99%, RT 4.572 min; MS (ESI) m/z 447.2 [M + H]+.
8-(Piperazin-1-yl)-3,4-dihydro-2H-benzo[b][1,4]oxazine (37)
Compound 37 (529.2 mg, 56% yield) was prepared starting from 8-bromo-3,4-dihydro-2H-benzo[b][1,4]oxazine and piperazine via the same procedure as preparing compound 35. 1H NMR (400 MHz, CD3OD) δ 6.66 (t, J = 8.0 Hz, 1H), 6.39 (dd, J = 8.0, 1.4 Hz, 1H), 6.34 (dd, J = 8.0, 1.4 Hz, 1H), 4.25 – 4.18 (m, 2H), 3.37 – 3.32 (m, 2H), 3.15 – 3.09 (m, 4H), 3.09 – 3.01 (m, 4H); HPLC 99%, RT 1.737 min; MS (ESI) m/z 220.2 [M + H]+.
1-(2,3-Dihydrobenzo[b][1,4]dioxin-5-yl)piperazine (38)
Compound 38 (719.0 mg, 59% yield) was prepared starting from 5-bromo-2,3-dihydrobenzo[b][1,4]dioxine and piperazine via the same procedure as preparing compound 35. 1H NMR (400 MHz, CD3OD) δ 6.75 (t, J = 8.1 Hz, 1H), 6.56 (ddd, J = 10.3, 8.1, 1.5 Hz, 2H), 4.30 – 4.25 (m, 2H), 4.25 – 4.20 (m, 2H), 3.15 – 3.06 (m, 8H); HPLC 99%, RT 2.977 min; MS (ESI) m/z 221.2 [M + H]+.
8-(4-(4-((2-Methylbenzo[d]thiazol-5-yl)oxy)butyl)piperazin-1-yl)-3,4-dihydro-2H-benzo[b][1,4]oxazine (39)
Compound 39 (68 mg) was prepared as white solid by the same procedure as preparing 1 from the reaction of 8-(piperazin-1-yl)-3,4-dihydro-2H-benzo[b][1,4]oxazine (37) and 5-(4-bromobutoxy)-2-methylbenzo[d]thiazole (26), yield 56%. 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 8.8 Hz, 1H), 7.42 (d, J = 2.4 Hz, 1H), 6.97 (dd, J = 8.8, 2.4 Hz, 1H), 6.77 (t, J = 8.1 Hz, 1H), 6.59 (d, J = 8.2 Hz, 1H), 6.53 (dd, J = 8.0, 1.0 Hz, 1H), 4.30 (dd, J = 7.4, 3.4 Hz, 2H), 4.24 (dd, J = 7.8, 3.3 Hz, 2H), 4.06 (t, J = 5.6 Hz, 2H), 3.20 (bs, 4H), 2.82 (bs, 4H), 2.80 (s, 3H), 2.63 (t, J = 6.4 Hz, 2H), 1.92 – 1.79 (m, 4H); 13C NMR (101 MHz, CDCl3): δ 168.32, 158.22, 154.73, 144.23, 141.35, 136.55, 127.49, 121.73, 120.85, 115.07, 112.31, 110.96, 106.10, 68.06, 64.49, 64.10, 58.21, 53.24, 50.09, 27.22, 23.06, 20.30; HPLC 99%, RT 4.585 min; MS (ESI) m/z 439.3 [M + H]+.
5-(4-(4-(2,3-Dihydrobenzo[b][1,4]dioxin-5-yl)piperazin-1-yl)butoxy)-2-methylbenzo[d]thiazole (40)
Compound 40 (57 mg) was prepared as white solid by the same procedure as preparing 1 from the reaction of 1-(2,3-dihydrobenzo[b][1,4]dioxin-5-yl)piperazine (38) and 5-(4-bromobutoxy)-2-methylbenzo[d]thiazole (26), yield 61%. 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 8.8 Hz, 1H), 7.41 (d, J = 2.4 Hz, 1H), 6.97 (dd, J = 8.8, 2.4 Hz, 1H), 6.69 (t, J = 8.0 Hz, 1H), 6.35 (d, J = 8.0 Hz, 2H), 4.31 – 4.24 (m, 2H), 4.06 (t, J = 5.9 Hz, 2H), 3.43 – 3.32 (m, 6H), 3.12 (bs, 4H), 2.95 – 2.85 (m, 2H), 2.80 (s, 3H), 2.10 – 1.99 (m, 2H), 1.95 – 1.84 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 168.43, 157.98, 154.68, 139.83, 136.45, 134.34, 127.68, 121.80, 121.17, 114.91, 111.52, 108.75, 106.14, 67.68, 65.41, 57.73, 52.76, 48.56, 40.82, 26.91, 21.97, 20.29; HPLC 99%, RT 4.804 min; MS (ESI) m/z 440.2 [M + H]+.
tert-Butyl 4-(3-bromo-2-methylphenyl)piperazine-1-carboxylate (41)
Compound 41 (1.8375 g, 89% yield) was prepared starting from 1,3-dibromo-2-methylbenzene and tert-butyl piperazine-1-carboxylate via the same procedure as preparing compound 35. HPLC 99%, RT 6.703 min; MS (ESI) m/z 355.1 [M + H]+.
tert-Butyl 4-(2-methyl-[1,1'-biphenyl]-3-yl)piperazine-1-carboxylate (42)
To a solution of tert-Butyl 4-(3-bromo-2-methylphenyl)piperazine-1-carboxylate (41, 1.0664 g, 3.00 mmol) in dioxane (20 mL) and H2O (5 mL) was added phenylboronic acid (732.0 mg, 6.00 mmol), Pd(PPh3)4 (346.9 mg, 0.30 mmol), K2CO3 (829.7 mg, 6.00 mmol). The mixture was then stirred at 110°C under microwave irradiation for 20 min. Solvent was removed and the residue was purified on silica gel column to give compound 42 as yellow oil (983.5 mg, 93% yield). HPLC 99%, RT 6.120 min; MS (ESI) m/z 353.3 [M + H]+.
1-(2-Methyl-[1,1'-biphenyl]-3-yl)piperazine (43)
To a solution of compound 42 (983.5 mg, 2.79 mmol) in DCM (20 mL) was added TFA (3 mL). The resulting mixture was stirred at room temperature for 1 h to give crude compound 43 as TFA salt (off-white solid, 1.0222 g, 100% yield) after removal of all the solvent, which was used for the next reaction without further purification. HPLC 95%, RT 4.910 min; MS (ESI) m/z 253.2 [M + H]+.
2-Methyl-5-(4-(4-(2-methyl-[1,1'-biphenyl]-3-yl)piperazin-1-yl)butoxy)benzo[d]thiazole (44)
Compound 44 (69 mg) was prepared as white solid by the same procedure as preparing 1 from the reaction of 1-(2-methyl-[1,1'-biphenyl]-3-yl)piperazine (43) and 5-(4-bromobutoxy)-2-methylbenzo[d]thiazole (26), yield 66%. 1H NMR (400 MHz, CDCl3): δ 7.82 (d, J = 8.9 Hz, 1H), 7.43 – 7.36 (m, 2H), 7.36 – 7.26 (m, 4H), 7.22 (t, J = 7.8 Hz, 1H), 7.10 – 7.06 (m, 1H), 7.04 (dd, J = 8.9, 2.5 Hz, 1H), 7.00 – 6.97 (m, 1H), 4.06 (t, J = 6.1 Hz, 2H), 3.08 (t, J = 4.7 Hz, 4H), 2.79 (s, 3H), 2.74 (bs, 4H), 2.62 – 2.54 (m, 2H), 2.19 (s, 3H), 1.96 – 1.73 (m, 4H); 13C NMR (101 MHz, CDCl3): δ 164.39, 156.81, 151.77, 147.94, 143.83, 142.57, 136.96, 130.44, 129.40, 128.11, 126.82, 126.18, 125.12, 122.87, 118.21, 115.49, 115.47, 105.13, 105.09, 68.53, 68.36, 62.54, 58.30, 53.73, 51.67, 29.58, 27.35, 25.96, 23.29, 20.07, 15.93; HPLC 99%, RT 5.123 min; MS (ESI) m/z 472.3 [M + H]+.
7-(4-(4-((2-Methylbenzo[d]thiazol-5-yl)oxy)butyl)piperazin-1-yl)benzo[d]oxazol-2(3H)-one (45)
Compound 45 (85 mg) was prepared as white solid by the same procedure as preparing 1 from the reaction of 7-(piperazin-1-yl)benzo[d]oxazol-2(3H)-one and 5-(4-bromobutoxy)-2-methylbenzo[d]thiazole (26), yield 62%. 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 8.8 Hz, 1H), 7.44 (d, J = 2.4 Hz, 1H), 7.04 (t, J = 8.1 Hz, 1H), 6.99 (dd, J = 8.8, 2.5 Hz, 1H), 6.66 – 6.58 (m, 2H), 4.07 (t, J = 6.2 Hz, 2H), 3.36 (bs, 4H), 2.81 (s, 3H), 2.73 (bs, 4H), 2.60 – 2.52 (m, 2H), 1.91 – 1.84 (m, 2H), 1.84 – 1.75 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 168.36, 158.30, 154.72, 154.69, 136.00, 134.25, 130.19, 127.44, 124.85, 121.74, 115.18, 110.75, 110.15, 106.04, 102.54, 68.12, 58.28, 53.12, 48.92, 27.26, 23.28, 20.31; HPLC 99%, RT 4.132 min; MS (ESI) m/z 439.2 [M + H]+.
5-(4-(4-(2,3-Dichlorophenyl)piperidin-1-yl)butoxy)-2-ethylbenzo[d]thiazole (46)
Compound 46 (63 mg) was prepared as white solid via the same procedure as preparing 1 starting from 2-ethylbenzo[d]thiazol-5-ol (16) and 1,4-dibromobutane, yield 65%. 1H NMR (400 MHz, CDCl3): δ 7.66 (d, J = 8.7 Hz, 1H), 7.46 (d, J = 2.4 Hz, 1H), 7.31 (dd, J = 7.5, 1.9 Hz, 1H), 7.22 – 7.12 (m, 2H), 6.98 (dd, J = 8.7, 2.4 Hz, 1H), 4.06 (t, J = 6.3 Hz, 2H), 3.16 – 3.02 (m, 5H), 2.51 – 2.42 (m, 2H), 2.11 (t, J = 11.1 Hz, 2H), 1.90 – 1.81 (m, 4H), 1.78 – 1.69 (m, 4H), 1.46 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ 174.90, 158.35, 154.66, 145.77, 133.20, 131.94, 128.18, 127.50, 126.80, 125.42, 121.80, 115.23, 106.06, 68.25, 58.71, 54.38, 39.95, 32.20, 27.96, 27.47, 23.79, 13.92; HPLC 99%, RT 4.917 min; MS (ESI) m/z 463.0 [M + H]+. HRMS m/z [M + H]+ calcd for C24H29Cl2N2OS 463.1378, found 463.1365.
5-(4-(4-(2,3-Dichlorophenyl)piperidin-1-yl)butoxy)-2-isopropylbenzo[d]thiazole (47)
Compound 47 (68 mg) was prepared as white solid via the same procedure as preparing 1 starting from 2-isopropylbenzo[d]thiazol-5-ol (17) and 1,4-dibromobutane, yield 69%. 1H NMR (400 MHz, CDCl3): δ 7.69 (d, J = 8.7 Hz, 1H), 7.43 (d, J = 2.4 Hz, 1H), 7.39 – 7.30 (m, 2H), 7.22 (t, J = 7.9 Hz, 1H), 6.97 (dd, J = 8.7, 2.5 Hz, 1H), 4.08 (t, J = 5.7 Hz, 2H), 3.75 (d, J = 11.5 Hz, 2H), 3.53 – 3.41 (m, 1H), 3.41 – 3.36 (m, 1H), 3.21 – 3.10 (m, 2H), 2.96 (t, J = 11.6 Hz, 2H), 2.70 (d, J = 11.9 Hz, 2H), 2.30 – 2.18 (m, 2H), 2.07 (d, J = 14.2 Hz, 2H), 1.99 – 1.88 (m, 2H), 1.46 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ 180.35, 157.70, 154.50, 142.16, 133.45, 131.49, 129.36, 128.25, 127.05, 125.72, 122.08, 114.80, 106.27, 67.30, 53.29, 37.38, 34.25, 28.46, 26.72, 23.00, 21.24; HPLC 99%, RT 5.069 min; MS (ESI) m/z 477.0 [M + H]+.
2-Ethyl-5-(4-(4-(2-methylquinolin-8-yl)piperazin-1-yl)butoxy)benzo[d]thiazole (48)
Compound 48 (72 mg) was prepared as white solid via the same procedure as preparing 1 starting from 2-ethylbenzo[d]thiazol-5-ol (16) and 1,4-dibromobutane, yield 63%. 1H NMR (400 MHz, CDCl3): δ 7.98 (d, J = 8.4 Hz, 1H), 7.67 (d, J = 8.7 Hz, 1H), 7.47 (d, J = 2.4 Hz, 1H), 7.39 – 7.35 (m, 2H), 7.24 (d, J = 8.4 Hz, 1H), 7.10 (dd, J = 5.3, 3.6 Hz, 1H), 7.00 (dd, J = 8.7, 2.4 Hz, 1H), 4.08 (t, J = 6.3 Hz, 2H), 3.47 (bs, 4H), 3.12 (q, J = 7.6 Hz, 2H), 2.83 (bs, 4H), 2.73 (s, 3H), 2.61 – 2.50 (m, 2H), 1.96 – 1.85 (m, 2H), 1.85 – 1.74 (m, 2H), 1.46 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ 174.89, 158.38, 156.74, 154.67, 148.85, 142.11, 136.70, 127.82, 126.80, 125.89, 121.80, 121.61, 121.40, 115.91, 115.25, 106.09, 68.33, 58.61, 53.58, 52.16, 27.96, 27.46, 25.99, 23.71, 13.93; HPLC 99%, RT 4.471 min; MS (ESI) m/z 461.1 [M + H]+.
2-Isopropyl-5-(4-(4-(2-methylquinolin-8-yl)piperazin-1-yl)butoxy)benzo[d]thiazole (49)
Compound 49 (76 mg) was prepared as white solid via the same procedure as preparing 1 starting from 2-isopropylbenzo[d]thiazol-5-ol (17) and 1,4-dibromobutane, yield 67%. 1H NMR (400 MHz, CDCl3): δ 7.98 (d, J = 8.4 Hz, 1H), 7.68 (d, J = 8.7 Hz, 1H), 7.48 (s, 1H), 7.41 – 7.32 (m, 2H), 7.28 – 7.20 (m, 1H), 7.14 – 7.07 (m, 1H), 6.99 (dd, J = 8.7, 1.9 Hz, 1H), 4.08 (t, J = 5.8 Hz, 2H), 3.54 (bs, 4H), 3.44 – 3.33 (m, 1H), 2.94 (bs, 4H), 2.73 (s, 3H), 2.65 (bs, 2H), 1.89 (bs, 4H), 1.47 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ 180.01, 158.26, 156.85, 154.54, 148.44, 142.02, 136.76, 127.82, 126.54, 125.89, 121.88, 121.68, 116.06, 115.25, 106.10, 77.48, 77.16, 76.84, 68.18, 58.44, 53.39, 51.60, 34.25, 29.84, 27.34, 26.00, 23.33, 23.03; HPLC 99%, RT 4.746 min; MS (ESI) m/z 475.1 [M + H]+. HRMS m/z [M + H]+ calcd for C28H34N4OS 475.2532, found 475.2510.
8-(4-(4-((2-Ethylbenzo[d]thiazol-5-yl)oxy)butyl)piperazin-1-yl)-3,4-dihydro-2H-benzo[b][1,4]oxazine (50)
Compound 50 (52 mg) was prepared as white solid via the same procedure as preparing 1 starting from 2-ethylbenzo[d]thiazol-5-ol (16) and 1,4-dibromobutane, yield 56%. 1H NMR (400 MHz, CDCl3): δ 7.66 (d, J = 8.8 Hz, 1H), 7.46 (d, J = 2.4 Hz, 1H), 6.98 (dd, J = 8.8, 2.4 Hz, 1H), 6.69 (t, J = 8.0 Hz, 1H), 6.39 (dd, J = 8.0, 1.3 Hz, 1H), 6.32 (dd, J = 7.9, 1.3 Hz, 1H), 4.35 – 4.29 (m, 2H), 4.06 (t, J = 6.3 Hz, 2H), 3.68 (bs, 1H), 3.45 – 3.38 (m, 2H), 3.12 (dd, J = 15.2, 7.6 Hz, 6H), 2.66 (bs, 4H), 2.52 – 2.45 (m, 2H), 1.92 – 1.82 (m, 2H), 1.73 (dt, J = 9.6, 7.2 Hz, 2H), 1.46 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ 174.89, 158.36, 154.66, 141.60, 136.74, 134.14, 126.80, 121.80, 120.93, 115.24, 110.80, 108.65, 106.08, 68.30, 65.35, 58.51, 53.60, 50.89, 40.94, 27.96, 27.42, 23.68, 13.93; HPLC 99%, RT 4.275 min; MS (ESI) m/z 453.1 [M + H]+.
8-(4-(4-((2-Isopropylbenzo[d]thiazol-5-yl)oxy)butyl)piperazin-1-yl)-3,4-dihydro-2H-benzo[b][1,4]oxazine (51)
Compound 51 (69 mg) was prepared as white solid via the same procedure as preparing 1 starting from 2-isopropylbenzo[d]thiazol-5-ol (17) and 1,4-dibromobutane, yield 69%. 1H NMR (400 MHz, CDCl3): δ 7.67 (d, J = 8.8 Hz, 1H), 7.46 (d, J = 2.4 Hz, 1H), 6.98 (dd, J = 8.7, 2.5 Hz, 1H), 6.69 (t, J = 8.0 Hz, 1H), 6.38 (dd, J = 8.0, 1.4 Hz, 1H), 6.33 (dd, J = 7.9, 1.4 Hz, 1H), 4.34 – 4.27 (m, 2H), 4.06 (t, J = 6.1 Hz, 2H), 3.70 (bs, 1H), 3.44 – 3.33 (m, 3H), 3.16 (bs, 4H), 2.77 (bs, 4H), 2.62 – 2.54 (m, 2H), 1.93 – 1.75 (m, 4H), 1.47 (d, J = 6.9 Hz, 6H); 13C NMR (101 MHz, CDCl3): δ 180.01, 158.23, 154.53, 141.18, 136.67, 134.19, 126.53, 121.87, 120.99, 115.22, 110.97, 108.67, 106.09, 68.13, 65.36, 58.32, 53.40, 50.33, 40.91, 34.24, 27.29, 23.26, 23.02; HPLC 99%, RT 4.529 min; MS (ESI) m/z 467.1 [M + H]+.
5-(4-(4-(2,3-Dihydrobenzo[b][1,4]dioxin-5-yl)piperazin-1-yl)butoxy)-2-ethylbenzo[d]thiazole (52)
Compound 52 (58 mg) was prepared as white solid via the same procedure as preparing 1 starting from 2-ethylbenzo[d]thiazol-5-ol (16) and 1,4-dibromobutane, yield 63%. 1H NMR (400 MHz, CDCl3): δ 7.66 (d, J = 8.8 Hz, 1H), 7.46 (d, J = 2.4 Hz, 1H), 6.98 (dd, J = 8.8, 2.4 Hz, 1H), 6.77 (t, J = 8.1 Hz, 1H), 6.58 (dd, J = 8.2, 1.1 Hz, 1H), 6.54 (dd, J = 8.0, 1.2 Hz, 1H), 4.36 – 4.28 (m, 2H), 4.28 – 4.21 (m, 2H), 4.06 (t, J = 6.3 Hz, 2H), 3.12 (dd, J = 15.2, 7.6 Hz, 6H), 2.66 (bs, 4H), 2.53 – 2.44 (m, 2H), 1.93 – 1.81 (m, 2H), 1.80 – 1.67 (m, 2H), 1.46 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, CDCl3): δ 174.90, 158.34, 154.66, 144.20, 141.98, 136.59, 126.80, 121.79, 120.76, 115.22, 111.98, 110.82, 106.07, 68.27, 64.47, 64.12, 58.46, 53.53, 50.91, 27.95, 27.38, 23.65, 13.92; HPLC 99%, RT 4.502 min; MS (ESI) m/z 454.1 [M + H]+.
5-(4-(4-(2,3-Dihydrobenzo[b][1,4]dioxin-5-yl)piperazin-1-yl)butoxy)-2-isopropylbenzo[d]thiazole (53)
Compound 53 (53 mg) was prepared as white solid via the same procedure as preparing 1 starting from 2-isopropylbenzo[d]thiazol-5-ol (17) and 1,4-dibromobutane, yield 67%. 1H NMR (400 MHz, CDCl3): δ 7.67 (d, J = 8.7 Hz, 1H), 7.47 (d, J = 2.4 Hz, 1H), 6.98 (dd, J = 8.8, 2.2 Hz, 1H), 6.70 (t, J = 7.9 Hz, 1H), 6.41 – 6.36 (m, 1H), 6.35 – 6.30 (m, 1H), 4.35 – 4.28 (m, 2H), 4.06 (t, J = 6.1 Hz, 2H), 3.45 – 3.34 (m, 3H), 3.15 (bs, 4H), 2.75 (bs, 4H), 2.61 – 2.52 (m, 2H), 1.93 – 1.74 (m, 4H), 1.51 – 1.43 (m, 6H); 13C NMR (101 MHz, CDCl3): δ 180.01, 158.26, 154.54, 141.26, 136.69, 134.18, 126.52, 121.87, 120.99, 115.24, 110.94, 108.68, 106.09, 68.17, 65.37, 58.36, 53.44, 50.44, 40.92, 34.25, 27.32, 23.35, 23.03; HPLC 99%, RT 4.333 min; MS (ESI) m/z 468.1 [M + H]+. HRMS m/z [M + H]+ calcd for C26H34N3O3S 468.2322, found 468.2317.
5-(4-(4-(2,3-Dichlorophenyl)piperidin-1-yl)butoxy)-N-methylbenzo[d]thiazol-2-amine (54)
Compound 54 (51 mg) was prepared as white solid via the same procedure as preparing 1 starting from 2-(methylamino)benzo[d]thiazol-5-ol (22) and 1,4-dibromobutane, yield 53%. 1H NMR (400 MHz, CDCl3): δ 7.42 (d, J = 8.6 Hz, 1H), 7.34 (dd, J = 7.8, 1.5 Hz, 1H), 7.29 – 7.23 (m, 1H), 7.19 (t, J = 7.9 Hz, 1H), 7.07 (d, J = 2.4 Hz, 1H), 6.68 (dd, J = 8.6, 2.4 Hz, 1H), 4.02 (t, J = 5.9 Hz, 2H), 3.50 (d, J = 11.8 Hz, 2H), 3.36 – 3.26 (m, 1H), 3.08 (s, 3H), 2.93 – 2.85 (m, 2H), 2.72 – 2.57 (m, 3H), 2.39 – 3.25 (m, 2H), 2.08 – 1.93 (m, 4H), 1.92 – 1.82 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 169.76, 158.25, 153.89, 133.30, 131.72, 128.72, 127.85, 125.57, 125.48, 122.27, 121.20, 110.42, 104.26, 67.76, 58.16, 55.86, 53.85, 38.77, 31.81, 30.47, 27.16, 22.62; HPLC 99%, RT 4.607 min; MS (ESI) m/z 464.0 [M + H]+. HRMS m/z [M + H]+ calcd for C23H28Cl2N3OS 464.1330, found 464.1319.
N-Methyl-5-(4-(4-(2-methylquinolin-8-yl)piperazin-1-yl)butoxy)benzo[d]thiazol-2-amine (55)
Compound 55 (57 mg) was prepared as white solid via the same procedure as preparing 1 starting from 2-(methylamino)benzo[d]thiazol-5-ol (22) and 1,4-dibromobutane, yield 55%. 1H NMR (400 MHz, CDCl3): δ 8.00 (d, J = 8.4 Hz, 1H), 7.43 (d, J = 8.5 Hz, 2H), 7.37 (t, J = 7.7 Hz, 1H), 7.26 (d, J = 8.4 Hz, 1H), 7.14 – 7.07 (m, 2H), 6.71 (dd, J = 8.6, 2.3 Hz, 1H), 5.47 (s, 1H), 4.05 (t, J = 6.0 Hz, 2H), 3.71 (bs, 4H), 3.20 (bs, 4H), 3.08 (s, 3H), 2.94 – 2.80 (m, 2H), 2.73 (s, 3H), 2.09 – 1.97 (m, 2H), 1.97 – 1.83 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 169.72, 158.25, 157.17, 153.91, 147.44, 141.81, 136.91, 127.84, 125.89, 122.32, 122.28, 121.86, 121.19, 116.47, 110.45, 104.24, 67.77, 58.06, 52.93, 50.24, 31.80, 27.12, 26.02, 22.40; HPLC 99%, RT 4.086 min; MS (ESI) m/z 462.1 [M + H]+.
5-(4-(4-(3,4-Dihydro-2H-benzo[b][1,4]oxazin-8-yl)piperazin-1-yl)butoxy)-N-methylbenzo[d]thiazol-2-amine (56)
Compound 56 (74 mg) was prepared as white solid via the same procedure as preparing 1 starting from 2-(methylamino)benzo[d]thiazol-5-ol (22) and 1,4-dibromobutane, yield 59%. 1H NMR (400 MHz, CDCl3): δ 7.42 (d, J = 8.6 Hz, 1H), 7.06 (d, J = 2.4 Hz, 1H), 6.69 (dd, J = 13.0, 5.1 Hz, 2H), 6.39 – 6.31 (m, 2H), 4.31 – 4.24 (m, 2H), 4.02 (t, J = 5.9 Hz, 2H), 3.804 (bs, 1H), 3.46 (t, J = 4.5 Hz, 4H), 3.43 – 3.37 (m, 3H), 3.35 – 3.12 (m, 4H), 3.08 (s, 3H), 3.04 – 2.97 (m, 2H), 2.11 (dt, J = 15.5, 7.7 Hz, 2H), 1.93 – 1.82 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 169.81, 158.06, 153.84, 139.38, 136.41, 134.41, 122.39, 121.24, 111.72, 110.27, 108.82, 104.23, 67.45, 65.45, 57.55, 52.55, 47.95, 40.82, 31.80, 26.80, 21.54; HPLC 99%, RT 3.652 min; MS (ESI) m/z 454.1 [M + H]+.
5-(4-(4-(2,3-Dihydrobenzo[b][1,4]dioxin-5-yl)piperazin-1-yl)butoxy)-N-methylbenzo[d]thiazol-2-amine (57)
Compound 57 (86 mg) was prepared as white solid via the same procedure as preparing 1 starting from 2-(methylamino)benzo[d]thiazol-5-ol (22) and 1,4-dibromobutane, yield 61%. 1H NMR (400 MHz, CDCl3): δ 7.42 (d, J = 8.6 Hz, 1H), 7.09 (d, J = 2.4 Hz, 1H), 6.77 (t, J = 8.1 Hz, 1H), 6.70 (dd, J = 8.6, 2.4 Hz, 1H), 6.62 – 6.57 (m, 1H), 6.53 (dd, J = 8.0, 1.2 Hz, 1H), 5.55 (s, 1H), 4.35 – 4.28 (m, 2H), 4.27 – 4.21 (m, 2H), 4.02 (t, J = 5.7 Hz, 2H), 3.19 (bs, 4H), 3.08 (s, 3H), 2.80 (bs, 4H), 2.60 (t, J = 6.9 Hz, 2H), 1.90 – 1.75 (m, 4H); 13C NMR (101 MHz, CDCl3): δ 169.74, 158.40, 153.88, 144.22, 141.44, 136.55, 122.07, 121.13, 120.83, 112.24, 110.94, 110.50, 104.21, 68.01, 64.48, 64.10, 58.25, 53.27, 50.21, 31.80, 27.29, 23.15; HPLC 99%, RT 3.968 min; MS (ESI) m/z 455.0 [M + H]+.
6-(4-(4-(2,3-Dichlorophenyl)piperidin-1-yl)butoxy)-2-methylbenzo[d]thiazole (58)
Compound 58 (52 mg) was prepared as white solid via the same procedure as preparing 1 starting from 2-methylbenzo[d]thiazol-6-ol and 1,4-dibromobutane, yield 58%. 1H NMR (400 MHz, CDCl3): δ 7.81 (d, J = 8.9 Hz, 1H), 7.36 (d, J = 7.9 Hz, 1H), 7.31 (d, J = 7.7 Hz, 1H), 7.28 (d, J = 2.5 Hz, 1H), 7.21 (t, J = 7.9 Hz, 1H), 7.00 (dd, J = 8.9, 2.5 Hz, 1H), 4.07 (t, J = 5.8 Hz, 2H), 3.73 (d, J = 11.5 Hz, 2H), 3.42 (ddd, J = 12.3, 9.0, 3.3 Hz, 1H), 3.18 – 3.08 (m, 2H), 2.92 (t, J = 11.4 Hz, 2H), 2.78 (s, 3H), 2.69 – 2.51 (m, 2H), 2.26 – 2.15 (m, 2H), 2.07 (d, J = 13.8 Hz, 2H), 1.99 – 1.89 (m, 2H); 13C NMR (101 MHz, CDCl3): δ 164.82, 156.32, 148.22, 142.24, 137.07, 133.48, 131.53, 129.34, 128.20, 125.68, 122.99, 115.36, 105.25, 67.63, 57.66, 53.44, 37.50, 39.82, 28.70, 26.82, 21.39, 20.12; HPLC 99%, RT 4.712 min; MS (ESI) m/z 449.1 [M + H]+.
5-Chloro-7-((4-(pyridin-2-yl)piperazin-1-yl)methyl)quinolin-8-ol (59)
Compound 59 was purchased from www.molport.com from the supplier Vitas-M laboratory Ltd. (catalog number STK973418) and was also obtained from David Sibley and Benjamin Free at NIH. The identity and purity of compound 59 was determined by LC-MS.
Experimental procedures for in vitro pharmacology assays
General procedures
Experimental procedures for the secondary radioligand binding for the GPCRs listed in Table 4 are available online through the Psychoactive Drug Screening Program (PDSP) website: http://pdsp.med.unc.edu/. The PDSP Assay Protocol book is freely available at http://pdsp.med.unc.edu/UNC-CH%20Protocol%20Book.pdf. Protocols of the cAMP biosensor, β-arrestin recruitment Tango, D2R radioligand binding, and bioluminescent resonance energy transfer (BRET) assays are detailed below. Human D2L receptors were used for these assays except the BRET assay, whereas mouse D2L receptors were used.
D2R Gi/o-mediated cAMP inhibition Assay
D2R Gi/o-mediated cAMP inhibition assays were performed in parallel with D2R β-Arrestin recruitment Tango assays. HEK293T cells co-expressing the cAMP biosensor GloSensor-22F (Promega) and hD2L receptors were seeded (15,000 cells/40 µL/well) into white 384 clear-bottom, tissue culture plates in DMEM containing 1% dialyzed FBS. Next day, drug dilutions were made in diluted in HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4. The same drug dilutions used for the D2R Gi/o-mediated cAMP inhibition assays were also used for D2R β-Arrestin recruitment Tango assay, which was performed in parallel. Next, media was removed on 384 well plates and 20 µL of drug buffer (HBSS, 20 mM HEPES, pH 7.4) was added per well and allowed to equilibrate for at least 15 minutes room temperarture. To start the assay, cells were treated with 10 µL per well of 3× drug diluted in HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4 using a FLIPR (Molecular Devices). After 15 min, cAMP accumulation was initiated by addition of 10 µL/well of 0.3 µM isoproterenol (final concentration) in GloSensor reagent. Luminescence per well per second was read on a Wallac TriLux microbeta plate counter. Data were normalized to maximum cAMP inhibition by quinpirole (100%) and basal cAMP accumulation by isoproterenol (0%). Data were analyzed using the sigmoidal dose-response function built into GraphPad Prism 5.0. Notably, HEK293T cells expressing the GloSensor-22F alone (no hD2R) were assayed in parallel and displayed no inhibition of isoproterenol-stimulated cAMP, either by quinpirole or by the test compounds, suggesting that the effect observed in hD2L-expressing cells was due to compound acting via the recombinant receptor (Figure S1).
D2R β-Arrestin Recruitment Tango Assay
Recruitment of β-arrestin to agonist-stimulated D2L receptors was performed using a previously described “Tango”-type assay.41,52 Briefly, HTLA cells stably expressing β-arrestin-TEV protease and a tetracycline transactivator-driven luciferase were plated in 15-cm dishes in DMEM containing 10% FBS and transfected (via calcium phosphate) with 10 µg of a D2V2-TCS-tTA construct. The next day, cells were plated in white, clear-bottom, 384-well plates (Greiner; 15,000 cells/well, 40 µL/well) in DMEM containing 1% dialyzed FBS. The following day, media was decanted and exchanged for fresh DMEM media containing 1% dialyzed FBS. The same drug dilutions used for the D2R Gi/o-mediated cAMP inhibition assay were used for the Tango assay. Cells were treated with 20 µL/well of drug using a FLIPR and after at least 20 h, the medium was removed and replaced with 1× BriteGlo reagent (Promega), and luminescence per well was read using a TriLux plate reader (1 s/well). Data were normalized to vehicle (0%) and quinpirole (100%) controls and analyzed using the sigmoidal dose-response function built into GraphPad Prism 5.0.
HEK-D2 Membrane Preparation and Radioligand Binding Assay
HEK-D2 membrane preparation. HEK293T cells were transfected in 15-cm dishes (in DMEM containing 10% FBS) with human D2L similarly described for D2 Gi/o-mediated cAMP inhibition assays. After 48 hour transfection, cells were washed with PBS, pH 7.4, and lysed with cold hypotonic lysis buffer (1 mM HEPES, 2 mM EDTA, pH 7.4), resuspensed, and centrifuged at 30,000 × g for 30 min. The supernatant was removed and membrane pellets were resuspended in standard binding buffer (50 mM Tris-HCl, 10 mM MgCl2, 0.5 mM EDTA, pH 7.4), mechanically homogenized, aliquoted, and stored at −80 °C until used for radioligand binding assays.
Membranes prepared as above were resuspended and 50 µL was added to each well of a polypropylene 96-well plate containing (per well) 50 µL of 0.1 nM [3H]N-methylspiperone (final concentration), and reference or test ligands at various concentrations ranging from 10 pM to 32 µM (final concentrations). After a 2.5-h incubation in the dark at room temperature, the reactions were harvested onto 0.3% PEI-soaked Filtermax GF/A filters (Wallac) and washed three times with ice-cold 50 mM Tris, pH 7.4, using a Perkin-Elmer Filtermate 96-well harvester. Filters were dried and placed on a hot plate (100 °C), and Melitilex-A (Wallac) scintillant was applied. Filters were allowed to cool and counted on a Wallac TriLux microbeta counter (1 min/well). Bound [3H]N-methylspiperone binding to filtered membranes was plotted as a function of log [ligand] and the data were analyzed using One-site-Fit Ki model built into Prism 5.0 (GraphPad software).
Bioluminescence Resonance Energy Transfer (BRET) Arrestin Assay
31 To measure D2R-mediated β-Arrestin recruitment, HEK293T cells were co-transfected in a 1:1:15 ratio with mouse D2Long containing a C-terminal renilla luciferase (RLuc8), GRK2, and Venus-tagged N-terminal β-arrestin2. Next day, transfected cells were plated in poly-lysine coated 96-well white clear bottom cell culture plates in plating media (DMEM + 1% dialyzed FBS) at a density of 40–50,000 cells in 200 µl per well and incubated overnight. Next day, media was decanted and cells were washed twice with 60 uL of drug buffer (1× HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4). Then 60 uL of drug buffer was added per well and drug stimulation was initiated with addition of 30 uL of drug (3×) per well. Plates were incubated at room temperature for at least 5 minutes before addition of 10 uL per well of RLuc substrate, coelenterazine h (Promega, 5 µM final concentration). Plates were read for both luminescence at 485 nm and fluorescent eYFP emission at 530 nm for 1 second per well using a Mithras LB940. The ratio of eYFP/RLuc was calculated per well and the net BRET ratio was calculated by subtracting the eYFP/RLuc per well from the eYFP/RLuc ratio in wells without Venus-β-Arrestin present. The net BRET ratio was plotted as a function of drug concentration using Graphpad Prism 5 (Graphpad Software Inc., San Diego, CA). Data were normalized to % quinpirole or dopamine stimulation and analyzed using nonlinear regression log(agonist) vs. response.
DiscoverX D2R β-arrestin-2 Recruitment Assay
β-arrestin-2 recruitment was measured by using the PathHunter enzyme complementation assay (DiscoverX, 93-0579E2CPM2M), which uses CHO-K1 cells stably expressing the human D2Rlong and β-arrestin-2 fused to a split β-galactosidase enzyme. Assays were performed exactly according to the manufacturer’s instructions, except drugs were diluted in drug buffer used for all other assays (1× HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4). Cells were incubated with drug for 90 minutes at 37°C for 90 minutes as per the manufacturer’s instructions. Luminescence was read one second per well on a Wallac TriLux microbeta plate counter. Data were normalized to maximum β-arrestin-2 recruitment by quinpirole (100%) and analyzed using nonlinear regression log(agonist) vs. response.
BRET Gαi1-γ2 Dissociation Assay
To measure D2R-mediated Gαi1-γ2 dissociation, HEK293T cells were co-transfected in a 1:5:5:5 ratio of Gαi1-RLuc, Gβ1, GFP2-Gγ2, and human D2Rlong, respectively. Gαi1-RLuc, Gβ1 and GFP2-Gγ2 constructs were generously provided by Dr. Michel Bouvier. Cells were plated and assays were performed exactly similar to the BRET arrestin assay, except the substrate used was Coelenterazine 400a (Nanolight, 5 µM final concentration). Plates were read after 5 minute drug incubation measuring luminescence at 400 nm and fluorescent GFP2 emission at 515 nm for 1 second per well using a Mithras LB940. The ratio of GFP2/RLuc was calculated per well and plotted as a function of drug concentration using Graphpad Prism 5 (Graphpad Software Inc., San Diego, CA). Data were normalized to % quinpirole stimulation and analyzed using nonlinear regression log(agonist) vs. response.
Supplementary Material
Acknowledgments
The research described here was supported by the grant U19MH082441 (to J.J. and B.L.R.) from the National Institutes of Health.
ABBREVIATIONS USED
- GPCR
G protein-coupled receptor
- SFSR
structure-functional selectivity relationships
- D2R
dopamine D2 receptor
- cAMP
cyclic adenosine monophosphate
- LHS
left-hand side
- RHS
right-hand side
- BRET
bioluminescence resonance energy transfer
- GRK2
G protein–coupled receptor kinase 2
- D1R
dopamine D1 receptor
- D3R
dopamine D3 receptor
- D4R
dopamine D4 receptor
- D5R
dopamine D5 receptor
- 5-HT
5-hydroxytryptamine
- DAT
dopamine transporter
- SERT
serotonin transporter
- H1
histamine receptor 1
- H2
histamine receptor 2
- TFA
trifluoroacetic acid
- DCM
dichloromethane
Footnotes
ASSOCIATED CONTENT
Supporting Information. 1H and 13C NMR spectra of compounds 1, 24, 46, 49, 53 and 54. Kd and BMAX estimates comparing D2R GloSensor and D2R Tango cell types. GloSensor cAMP inhibition with no D2R expressed. D2R G protein antagonism by compound 1. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Shin S, Hisahara S. Dopamine receptors and Parkinson's Disease. Int. J. Med. Chem. 2011;2011:1–16. doi: 10.1155/2011/403039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gray JA, Roth BL. The pipeline and future of drug development in schizophrenia. Mol. Psychiatry. 2007;12:904–922. doi: 10.1038/sj.mp.4002062. [DOI] [PubMed] [Google Scholar]
- 3.Beaulieu JM, Espinoza S, Gainetdinov RR. Dopamine receptors - IUPHAR Review 13. Br. J. Pharmacol. 2015;172:1–23. doi: 10.1111/bph.12906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 5.Noble EP. D2 dopamine receptor gene in psychiatric and neurologic disorders and its phenotypes. Am. J. Med. Genet., Part B. 2003;116B:103–125. doi: 10.1002/ajmg.b.10005. [DOI] [PubMed] [Google Scholar]
- 6.Fan XL, Xu M, Hess EJ. D2 dopamine receptor subtype-mediated hyperactivity and amphetamine responses in a model of ADHD. Neurobiol. Dis. 2010;37:228–236. doi: 10.1016/j.nbd.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roth BL, Sheffler DJ, Kroeze WK. Magic shotguns versus magic bullets: selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discovery. 2004;3:353–359. doi: 10.1038/nrd1346. [DOI] [PubMed] [Google Scholar]
- 8.Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am. J. Psychiatry. 1991;148:1474–1486. doi: 10.1176/ajp.148.11.1474. [DOI] [PubMed] [Google Scholar]
- 9.Leucht S, Corves C, Arbter D, Engel RR, Li CB, Davis JM. Second-generation versus first-generation antipsychotic drugs for schizophrenia: a meta-analysis. Lancet. 2009;373:31–41. doi: 10.1016/S0140-6736(08)61764-X. [DOI] [PubMed] [Google Scholar]
- 10.Caroff SN, Hurford I, Lybrand J, Campbell EC. Movement disorders induced by antipsychotic drugs: implications of the CATIE Schizophrenia trial. Neurologic Clinics. 2011;29:127–148. doi: 10.1016/j.ncl.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koster LS, Carbon M, Correll CU. Emerging drugs for schizophrenia: an update. Expert Opin. Emerging Drugs. 2014;19:511–531. doi: 10.1517/14728214.2014.958148. [DOI] [PubMed] [Google Scholar]
- 12.Leo RJ, Regno PD. A typical antipsychotic use in the treatment of psychosis in primary care. Prim. Care Companion J. Clin. Psychiatry. 2000;2:194–204. doi: 10.4088/pcc.v02n0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swainston Harrison T, Perry CM. Aripiprazole: a review of its use in schizophrenia and schizoaffective disorder. Drugs. 2004;64:1715–1736. doi: 10.2165/00003495-200464150-00010. [DOI] [PubMed] [Google Scholar]
- 14.Shapiro DA, Renock S, Arrington E, Chiodo LA, Liu LX, Sibley DR, Roth BL, Mailman R. Aripiprazole, a novel atypical antipsychotic drug with a unique and robust pharmacology. Neuropsychopharmacology. 2003;28:1400–1411. doi: 10.1038/sj.npp.1300203. [DOI] [PubMed] [Google Scholar]
- 15.Gopalakrishna G, Aggarwal A, Lauriello J. Long-acting injectable aripiprazole: how might it fit in our tool box? Clin. Schizophr. Relat. Psychoses. 2013;7:87–92. [PubMed] [Google Scholar]
- 16.Allen JA, Yost JM, Setola V, Chen X, Sassano MF, Chen M, Peterson S, Yadav PN, Huang XP, Feng B, Jensen NH, Che X, Bai X, Frye SV, Wetsel WC, Caron MG, Javitch JA, Roth BL, Jin J. Discovery of beta-arrestin-biased dopamine D-2 ligands for probing signal transduction pathways essential for antipsychotic efficacy. Proc. Natl. Acad. Sci. U. S. A. 2011;108:18488–18493. doi: 10.1073/pnas.1104807108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H, Javitch JA, Roth BL, Christopoulos A, Sexton PM, Miller KJ, Spedding M, Mailman RB. Functional selectivity and classical concepts of quantitative pharmacology. J. Pharmacol. Exp. Ther. 2007;320:1–13. doi: 10.1124/jpet.106.104463. [DOI] [PubMed] [Google Scholar]
- 18.Mailman RB. GPCR functional selectivity has therapeutic impact. Trends Pharmacol. Sci. 2007;28:390–396. doi: 10.1016/j.tips.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Beaulieu JM, Gainetdinov RR, Caron MG. The Akt-GSK-3 signaling cascade in the actions of doparnine. Trends Pharmacol. Sci. 2007;28:166–172. doi: 10.1016/j.tips.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 20.Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discovery. 2013;12:205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- 21.Chen X, Sassano MF, Zheng LY, Setola V, Chen M, Bai X, Frye SV, Wetsel WC, Roth BL, Jin J. Structure-functional selectivity relationship studies of beta-arrestin-biased dopamine D-2 receptor agonists. J. Med. Chem. 2012;55:7141–7153. doi: 10.1021/jm300603y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park SM, Chen M, Schmerberg CM, Dulman RS, Rodriguiz RM, Caron MG, Jin J, Wetsel WC. Effects of beta-arrestin-biased dopamine D2 receptor ligands on Schizophrenia-like behavior in hypoglutamatergic mice. Neuropsychopharmacology. 2016;41:704–715. doi: 10.1038/npp.2015.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shonberg J, Herenbrink CK, Lopez L, Christopoulos A, Scammells PJ, Capuano B, Lane JR. A Structure-activity analysis of biased agonism at the dopamine D2 receptor. J. Med. Chem. 2013;56:9199–9221. doi: 10.1021/jm401318w. [DOI] [PubMed] [Google Scholar]
- 24.Hiller C, Kling RC, Heinemann FW, Meyer K, Hubner H, Gmeiner P. Functionally selective dopamine D-2/D-3 receptor agonists comprising an enyne moiety. J. Med. Chem. 2013;56:5130–5141. doi: 10.1021/jm400520c. [DOI] [PubMed] [Google Scholar]
- 25.Szabo M, Herenbrink CK, Christopoulos A, Lane JR, Capuano B. Structure-activity relationships of privileged structures lead to the discovery of novel biased ligands at the dopamine D-2 receptor. J. Med. Chem. 2014;57:4924–4939. doi: 10.1021/jm500457x. [DOI] [PubMed] [Google Scholar]
- 26.Moller D, Kling RC, Skultety M, Leuner K, Hubner H, Gmeiner P. Functionally selective dopamine D-2, D-3 receptor partial agonists. J. Med. Chem. 2014;57:4861–4875. doi: 10.1021/jm5004039. [DOI] [PubMed] [Google Scholar]
- 27.Free RB, Chun LS, Moritz AE, Miller BN, Doyle TB, Conroy JL, Padron A, Meade JA, Xiao JB, Hu X, Dulcey AE, Han Y, Duan LH, Titus S, Bryant-Genevier M, Barnaeva E, Ferrer M, Javitch JA, Beuming T, Shi L, Southall NT, Marugan JJ, Sibley DR. Discovery and characterization of a G protein-biased agonist that inhibits beta-arrestin recruitment to the D2 dopamine receptor. Mol. Pharmacol. 2014;86:96–105. doi: 10.1124/mol.113.090563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Houslay MD. Arrestin times for developing antipsychotics and beta-blockers. Sci. Signaling. 2009;2:1–5. doi: 10.1126/scisignal.266pe22. [DOI] [PubMed] [Google Scholar]
- 29.Mottola DM, Kilts JD, Lewis MM, Connery HS, Walker QD, Jones SR, Booth RG, Hyslop DK, Piercey M, Wightman RM, Lawler CP, Nichols DE, Mailman RB. Functional selectivity of dopamine receptor agonists. I. Selective activation of postsynaptic dopamine D2 receptors linked to adenylate cyclase. J. Pharmacol. Exp. Ther. 2002;301:1166–1178. doi: 10.1124/jpet.301.3.1166. [DOI] [PubMed] [Google Scholar]
- 30.Kilts JD, Connery HS, Arrington EG, Lewis MM, Lawler CP, Oxford GS, O'Malley KL, Todd RD, Blake BL, Nichols DE, Mailman RB. Functional selectivity of dopamine receptor agonists. II. Actions of dihydrexidine in D2L receptor-transfected MN9D cells and pituitary lactotrophs. J. Pharmacol. Exp. Ther. 2002;301:1179–1189. doi: 10.1124/jpet.301.3.1179. [DOI] [PubMed] [Google Scholar]
- 31.Masri B, Salahpour A, Didriksen M, Ghisi V, Beaulieu JM, Gainetdinov RR, Caron MG. Antagonism of dopamine D2 receptor/beta-arrestin 2 interaction is a common property of clinically effective antipsychotics. Proc. Natl. Acad. Sci. U. S. A. 2008;105:13656–13661. doi: 10.1073/pnas.0803522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 33.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 34.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 35.Oshiro Y, Sato S, Kurahashi N, Tanaka T, Kikuchi T, Tottori K, Uwahodo Y, Nishi T. Novel antipsychotic agents with dopamine autoreceptor agonist properties: Synthesis and pharmacology of 7-[4-(4-phenyl-1-piperazinyl)butoxy]-3,4-dihydro-2(1H)-quinolinone derivatives. J. Med. Chem. 1998;41:658–667. doi: 10.1021/jm940608g. [DOI] [PubMed] [Google Scholar]
- 36.Zhu JT, Chen ZX, Xie HB, Li S, Wu YM. A general and straightforward method for the synthesis of 2-trifluoromethylbenzothiazoles. Org. Lett. 2010;12:2434–2436. doi: 10.1021/ol1006899. [DOI] [PubMed] [Google Scholar]
- 37.Weissman SA, Zewge D. Recent advances in ether dealkylation. Tetrahedron. 2005;61:7833–7863. [Google Scholar]
- 38.Shi DF, Bradshaw TD, Wrigley S, McCall CJ, Lelieveld P, Fichtner I, Stevens MFG. Antitumor benzothiazoles .3. Synthesis of 2-(4-aminophenyl)benzothiazoles and evaluation of their activities against breast cancer cell lines in vitro and in vivo. J. Med. Chem. 1996;39:3375–3384. doi: 10.1021/jm9600959. [DOI] [PubMed] [Google Scholar]
- 39.Jordan AD, Luo C, Reitz AB. Efficient conversion of substituted aryl thioureas to 2-aminobenzothiazoles using benzyltrimethylammonium tribromide. J. Org. Chem. 2003;68:8693–8696. doi: 10.1021/jo0349431. [DOI] [PubMed] [Google Scholar]
- 40.Kimple AJ, Soundararajan M, Hutsell SQ, Roos AK, Urban DJ, Setola V, Temple BRS, Roth BL, Knapp S, Willard FS, Siderovski DP. Structural determinants of G-protein alpha subunit selectivity by regulator of G-protein signaling 2 (RGS2) J. Biol. Chem. 2009;284:19402–19411. doi: 10.1074/jbc.M109.024711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kroeze WK, Sassano MF, Huang XP, Lansu K, McCorvy JD, Giguere PM, Sciaky N, Roth BL. PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 2015;22:362–369. doi: 10.1038/nsmb.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsuruta K, Frey EA, Grewe CW, Cote TE, Eskay RL, Kebabian JW. Evidence that LY-141865 specifically stimulates the D-2 dopamine receptor. Nature. 1981;292:463–465. doi: 10.1038/292463a0. [DOI] [PubMed] [Google Scholar]
- 43.Yale HL. The trifluoromethyl group in medicinal chemistry. J. Med. Pharm. Chem. 1959;1:121–133. doi: 10.1021/jm50003a001. [DOI] [PubMed] [Google Scholar]
- 44.Feenstra RW, de Moes J, Hofma JJ, Kling H, Kuipers W, Long SK, Tulp MTM, van der Heyden JAM, Kruse CG. New 1-aryl-4-(biarylmethylene)piperazines as potential atypical antipsychotics sharing dopamine D-2-receptor and serotonin 5-HT1A-receptor affinities. Bioorg. Med. Chem. Lett. 2001;11:2345–2349. doi: 10.1016/s0960-894x(01)00425-5. [DOI] [PubMed] [Google Scholar]
- 45.Wagaw S, Rennels RA, Buchwald SL. Palladium-catalyzed coupling of optically active amines with aryl bromides. J. Am. Chem. Soc. 1997;119:8451–8458. [Google Scholar]
- 46.Kenakin T, Christopoulos A. OPINION Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discovery. 2013;12:205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- 47.Herenbrink CK, Sykes DA, Donthamsetti P, Canals M, Coudrat T, Shonberg J, Scammells PJ, Capuano B, Sexton PM, Charlton SJ, Javitch JA, Christopoulos A, Lane JR. The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun. 2016;7 doi: 10.1038/ncomms10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Klewe IV, Nielsen SM, Tarpo L, Urizar E, Dipace C, Javitch JA, Gether U, Egebjerg J, Christensen KV. Recruitment of beta-arrestin2 to the dopamine D2 receptor: Insights into anti-psychotic and anti-parkinsonian drug receptor signaling. Neuropharmacology. 2008;54:1215–1222. doi: 10.1016/j.neuropharm.2008.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burris KD, Molski TF, Xu C, Ryan E, Tottori K, Kikuchi T, Yocca FD, Molinoff PB. Aripiprazole, a novel antipsychotic, is a high-affinity partial agonist at human dopamine D2 receptors. J. Pharmacol. Exp. Ther. 2002;302:381–389. doi: 10.1124/jpet.102.033175. [DOI] [PubMed] [Google Scholar]
- 50.Winpenny D, Clark M, Cawkill D. Biased ligand quantification in drug discovery: from theory to high throughput screening to identify new biased opioid receptor agonists. Br. J. Pharmacol. 2016;173:1393–1403. doi: 10.1111/bph.13441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gottlieb HE, Kotlyar V, Nudelman A. NMR chemical shifts of common laboratory solvents as trace impurities. J. Org. Chem. 1997;62:7512–7515. doi: 10.1021/jo971176v. [DOI] [PubMed] [Google Scholar]
- 52.Barnea G, Strapps W, Herrada G, Berman Y, Ong J, Kloss B, Axel R, Lee KJ. The genetic design of signaling cascades to record receptor activation. Proc. Natl. Acad. Sci. U. S. A. 2008;105:64–69. doi: 10.1073/pnas.0710487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.