

Abstract
Background and Aims
In hereditary hemochromatosis, iron deposition in the liver parenchyma may lead to fibrosis, cirrhosis and hepatocellular carcinoma. Most cases are ascribed to a common mutation in the HFE gene, but the extent of clinical expression is greatly influenced by the combined action of yet unidentified genetic and/or environmental modifying factors. In mice, transcription factor NRF2 is a critical determinant of hepatocyte viability during exposure to acute dietary iron overload. We evaluated if the genetic disruption of Nrf2 would prompt the development of liver damage in Hfe-/- mice (an established model of human HFE-hemochromatosis).
Methods
Wild-type, Nrf2-/-, Hfe-/- and double knockout (Hfe/Nrf2-/-) female mice on C57BL/6 genetic background were sacrificed at the age of 6 (young), 12–18 (middle-aged) or 24 months (old) for evaluation of liver pathology.
Results
Despite the parenchymal iron accumulation, Hfe-/- mice presented no liver injury. The combination of iron overload (Hfe-/-) and defective antioxidant defences (Nrf2-/-) increased the number of iron-related necroinflammatory lesions (sideronecrosis), possibly due to the accumulation of toxic oxidation products such as 4-hydroxy-2-nonenal-protein adducts. The engulfment of dead hepatocytes led to a gradual accumulation of iron within macrophages, featuring large aggregates. Myofibroblasts recruited towards the injury areas produced substantial amounts of collagen fibers involving the liver parenchyma of double-knockout animals with increased hepatic fibrosis in an age-dependent manner.
Conclusions
The genetic disruption of Nrf2 promotes the transition from iron accumulation (siderosis) to liver injury in Hfe-/- mice, representing the first demonstration of spontaneous hepatic fibrosis in the long term in a mouse model of hereditary hemochromatosis displaying mildly elevated liver iron.
Keywords: Iron, Hepatocyte, Macrophage, Sideronecrosis, Oxidative stress
Graphical abstract
Highlights
-
•
Despite the parenchymal iron overload, single Hfe-/- mice present no liver injury.
-
•
Hfe and Nrf2 double knockout mice develop liver fibrosis with aging.
-
•
Fibrosis is triggered by iron-related hepatocellular death (sideronecrosis).
-
•
Nrf2 genetic disruption increases susceptibility to oxidative/electrophilic stress.
-
•
NRF2 status is a potential determinant of liver injury in hemochromatosis.
1. Introduction
HFE-associated hereditary hemochromatosis (HH) is a common autosomal disease characterized by excessive intestinal absorption of dietary iron and its accumulation in tissues, with secondary organ damage ascribed to oxidative stress [1]. The liver, being the main body iron reservoir, is particularly exposed to iron toxicity [2]. Thus, HH patients have increased risk of hepatic fibrosis, cirrhosis and hepatocellular carcinoma, with symptoms often starting in the fourth decade of life [1]. While most HH patients are homozygous for the p.C282Y variant of the HFE gene, there is great phenotypic variability among C282Y homozygotes concerning both the degree of iron loading and the risk of liver disease [3], [4]. Organ disease develops only in a minority of C282Y homozygotes, most commonly as a result of alcohol toxicity, obesity-related steatosis, diabetes but also due to other genetic/environmental modifying factors that are still unidentified [1], [3], [4], [5], [6], [7]. The Hfe-/- mouse develops spontaneous parenchymal iron overload similar to human HH [8]. Despite the increasing hepatic iron overload, Hfe-/- mice present no hepatic fibrosis or cirrhosis [9], [10], which suggests the existence of efficient adaptive mechanisms that protect mice from the noxious effect of iron overload.
The transcription factor NRF2 (Nfe2l2/Nrf2) plays a key role in the adaptation to oxidative/electrophilic stress by regulating the transcriptional induction of a series of cytoprotective genes including antioxidant and xenobiotic conjugating enzymes, ubiquitin/proteasomes, chaperones and heat-shock proteins [11]. Several studies have demonstrated that livers of Nrf2-/- mice are more susceptible to oxidative/electrophilic stress than those of wild-type mice [12], [13], and NRF2 is increasingly regarded as an important modifier of chronic liver diseases [14]. Recently, we have reported that iron activates NRF2 in mouse hepatocytes and that NRF2 is important for the viability of hepatocytes during exposure to acute dietary iron overload [15]. To test the hypothesis that NRF2-mediated resistance to oxidative/electrophilic stress may be a modifier of disease progression in HFE-HH, we aimed to evaluate if the genetic suppression of Nrf2 would predispose the Hfe-/- mouse to spontaneous liver damage. For that purpose, a double knockout (Hfe/Nrf2-/-) was generated by crossing Hfe-/- and Nrf2-/- mice on C57BL/6 genetic background. Since the disease has a late onset in humans, we evaluated the effect of each genotype on liver pathology and antioxidant defenses in mice at the ages of 6 (young), 12–18 (middle-aged) and 24 months (old).
2. Materials and methods
2.1. Reagents
All chemicals and reagents were purchased from Sigma-Aldrich (Munich, Germany), unless otherwise stated.
2.2. Animals
To generate Hfe and Nrf2 compound homozygous mutant animals, Hfe-null mice [8] were mated with Nrf2-null mice [16] to generate double heterozygous mutant mice (Hfe+/- and Nrf2+/-) on C57BL/6 genetic background. These mice were then intercrossed to generate either compound homozygous mutant mice (Hfe/Nrf2-/-) or ones with various other combinations of mutant and wild-type alleles. Polymerase chain reaction (PCR) analysis was performed to determine the mouse genotype. Primers are listed in Supplementary Table 1.
Animals were housed at the Instituto de Biologia Molecular e Celular animal facility in a temperature and light-controlled environment, with free access to standard rodent chow (4RF21 containing 480 mg/kg iron sulfate heptahydrate, Mucedola, Milan, Italy) and water. Young (6 months old), middle-aged (12–18 months old) and old (24 months old) female mice were sacrificed for tissue collection. In each genotype, the middle-aged group comprised an equal number of individuals at 12 and 18 months of age. Blood was obtained by retro-orbital bleeding under terminal anesthesia with isoflurane. Liver fragments were snap-frozen in liquid nitrogen and stored at −80 °C for subsequent RNA and antioxidant enzyme activity analysis. All animals received humane care according to the criteria outlined by the Federation of European Laboratory Animal Science Associations for the care and handling of laboratory animals (EU Directive 2010/63/EU); experimental procedures were approved by the Instituto de Biologia Molecular e Celular Animal Ethics Committee.
2.3. Histology and immunohistochemistry
Tissue samples were fixed in neutral formalin 10% and embedded in paraffin. Following deparaffinization with xylene and hydration by a passage through a grade of alcohols, 3 µm-thick sections were stained with hematoxylin-eosin, Perls’ Prussian blue reaction for ferric iron and Sirius red using standard procedures. Histological iron grading was performed using the grading system of Deugnier et al. [17], which takes into account the size of iron deposits, as well as their cellular and lobular location, leading to three scores: hepatocytic (ranging 0–36), sinusoidal (ranging 0–12) and portal (ranging 0–12). All slides (stained with Perls' method) were examined by the same observer, in a blind manner (i.e. with no previous knowledge of the animals' genotypes). Evaluation of necroinflammatory lesions (sideronecrosis) was performed according to Deugnier et al. [17]. Staging of liver fibrosis was performed by an anatomic pathologist (JML) according to Kleiner et al. [18], as follows: 0) none; 1) mild perisinusoidal or periportal; 2) perisinusoidal and portal/periportal; 3) bridging fibrosis; and 4) cirrhosis. The quantification of collagen type I (yellow-orange birefringence) in liver sections stained with Sirius red was performed under polarized light and expressed as the percentage of fibrotic area. Digital images of 3 different fields at ×100 magnification were analyzed using ImageJ software (available at http://rsbweb.nih.gov/ij/index.html) to assess the percentage of stained area compared with the total evaluated liver area.
Fragmented DNA was detected through TdT-mediated dUTP nick-end labeling (TUNEL) staining as described by Silva-Gomes et al.[15]. For the detection of F4/80, samples were blocked with 5% bovine serum albumin and 0.05% Tween 20 in phosphate buffer and incubated with rat anti-F4/80 (Serotec, Oxford, UK), followed by goat anti-rat IgG AlexaFluor 647 (Life Technologies, Carlsblad, CA, USA). For the detection of alpha-smooth muscle actin (α-SMA), samples were permeabilized in 0.2% Triton X-100 in phosphate buffer, then subjected to antigen retrieval in microwave with citrate buffer, blocked with mouse-on-mouse blocking solution (Vector, Burlingame, CA, USA) and incubated with mouse anti-α-SMA, followed by goat anti-mouse IgG AlexaFluor488 (Life Technologies, Carlsblad, CA, USA). Cell nuclei were stained with 4′,6-diamidino-2-phenylindole. Samples were analyzed in a wide field fluorescence microscope (Zeiss AxioImager Z1, Carl Zeiss, Jena, Germany).
2.4. Transmission electron microscopy
Liver tissue samples were fixed and embedded in Epon as described by Silva-Gomes et al. [15]. Semithin sections (approximately 0.5 µm thick), stained with toluidine blue and methylene blue, were used to search for areas of interest. Ultrathin sections, either unstained or stained with uranyl acetate and lead citrate, were evaluated by electron microscopy as described by Silva-Gomes et al. [15]. Elemental analysis was performed on a JEM 1400 electron microscope (Jeol, Tokyo, Japan) fit with a Schottky field emission gun, a high-angle annular dark field detector and an X-MAX 80 mm2 ultrathin window energy-dispersive X-ray spectroscopy detector (Oxford Instruments, Oxford, UK). Spectra were collected from 3 distinct electron-dense and 3 distinct electron-lucent areas at a 20.2° tilt angle with a stationary electron probe in scanning transmission electron microscope mode (at an accelerating voltage of 80 kV).
2.5. Tissue iron quantification
Non-heme iron content in tissue samples was measured as described by Silva-Gomes et al. [15].
2.6. Hydroxyproline content
Liver samples (10 mg) were homogenized in ultrapure water and hydroxyproline was quantified using the Hydroxyproline assay kit (Sigma-Aldrich).
2.7. Assessment of total glutathione content and glutathione-S-transferase activity
Liver samples were collected, homogenized in cold phosphate buffer (25 mM KH2PO4, 30 mM Na2HPO4·2H2O and 0.1% Triton X-100, pH 7.0) and protein content was quantified using the Bio-Rad DC protein assay kit (Hercules, CA, USA). Homogenates were centrifuged (16,000g, 10 min, 4 °C) and the supernatants were aliquoted and stored at −80 °C. Glutathione-S-transferase activity and the total glutathione content were determined as described by Assunção et al. [19].
2.8. Western blotting
Liver samples were homogenized in lysis buffer (300 mM sodium chloride, 50 mM Tris-HCl pH 7.4, 1% Triton X-100) containing protease inhibitors. Total protein content of lysates was measured using the RC/DC Protein Assay (Bio-Rad, Hercules, CA) and 30 µg resolved by electrophoresis on 12% SDS-polyacrylamide gels. Proteins were then transferred to 0.45 µm nitrocellulose Hybond-C Extra membranes (GE Healthcare, Portugal). After blocking with 5% dry milk in Tris-buffered saline containing 0.05% Tween 20 (TBS-T) for 1 h, membranes were incubated overnight with anti 4- hydroxynonenal rabbit antiserum clone HNE11-S (1:500; Alpha Diagnostic International, Santo Antonio, Texas, USA) or for 1 h with rabbit polyclonal antibody anti-p62 (SQSTM1) from MBL (1:1000; Nagoya, Japan), followed by a horseradish peroxidase-conjugated sheep anti-rabbit IgG (Life Technologies, Carlsblad, CA, USA) for 1 h at room temperature. After washing with TBS-T, signal was developed with a chemiluminescence substrate (Millipore, Billerica, MA, USA) and the blots exposed to CL-X Posure films (Pierce, Rockford, IL). For normalization of protein loading, blots were stripped and reprobed with a rabbit antibody against β-actin (Abcam, Cambridge, UK).
2.9. Real-time reverse-transcription-PCR
RNA isolation and analysis of gene expression were performed as described by Silva-Gomes et al. [15]. The quantity of genes involved in iron homeostasis (hepcidin antimicrobial peptide, Hamp), antioxidant response [NAD(P)H dehydrogenase, quinone 1, Nqo1; glutathione S-transferase, alpha 1, Gsta1] and fibrogenesis (collagen, type I, alpha 1, Col1a1; tissue inhibitor of metalloproteinase 1, Timp1) was normalized against the quantity of the endogenous control gene (hypoxanthine guanine phosphoribosyl transferase, Hprt). Primer sequences are listed in Supplementary Table 1.
2.10. Serum parameters
Serum biochemical markers of iron metabolism and liver toxicity were analyzed as described by Silva-Gomes et al. [15].
2.11. Statistical analysis
All data were statistically analyzed by using the GraphPad Prism 6.07 software (GraphPad Software, CA, USA) and the averaged values are presented as mean and standard deviation. Differences among multiple group means were compared by one-way or two-way analysis of variance (when testing for mouse genotype alone or mouse genotype vs. age, respectively) with Dunnet's multiple comparison test. The association between variables was assessed using Pearson correlation. Statistical significance was assumed at p<0.05.
3. Results
Hfe/Nrf2+/- mice gave birth to viable double knock-outs (Hfe/Nrf2-/-) according to Mendelian frequencies (data not shown). Young Hfe/Nrf2-/- adults were apparently healthy and fertile. At 6 months of age, their average body weight was not different from age-matched wild-types or single knockouts. From middle-age on, however, they weighed significantly less than age-matched wild-types. This was in contrast with Hfe-/- animals, which sustained gain weight until sacrifice, compared to wild-type counterparts (Table 1).
Table 1.
Body weight, relative liver weight and serum ALT.
| Body weight | Relative liver weight | Serum ALT | ||
|---|---|---|---|---|
| Wild-type | N | (g) | (%) | (U/L) |
| Young | 8 | 24.2±1.9 | 4.5±0.3 | 26.4±3.0 |
| Middle-aged | 15 | 30.6±3.6 | 4.7±0.7 | 33.7±9.7 |
| Old | 9 | 33.3±3.6 | 4.4±0.5 | 27.0±4.0 |
| Nrf2-/- | ||||
| Young | 8 | 22.9±3.1 | 3.5±0.7a | 25.8±5.2 |
| Middle-aged | 15 | 30.5±3.5 | 4.1±0.4a | 34.7±6.9 |
| Old | 8 | 29.4±5.1a | 4.0±0.5 | 32.8±6.4 |
| Hfe-/- | ||||
| Young | 10 | 24.5±1.6 | 3.9±0.4 | 25.2±3.1 |
| Middle-aged | 14 | 30.8±3.5 | 4.5±0.4 | 29.8±7.4 |
| Old | 8 | 38.9±4.5a | 3.9±0.6 | 31.5±4.3 |
| Hfe/Nrf2-/- | ||||
| Young | 6 | 24.1±1.6 | 4.0±0.8 | 27.7±3.8 |
| Middle-aged | 11 | 26.8±2.4a | 3.8±0.8a | 35.6±10.3 |
| Old | 9 | 26.5±1.6a | 3.9±0.6 | 45.3±24.5a |
p<0.05 vs. age-matched wild-type mice. ALT, alanine aminotransferase.
Concerning the liver, Hfe/Nrf2-/- animals presented a trend for lower relative liver weight, which reached statistical significance only in the middle-aged group (Table 1). Serum alanine aminotransferase activity was mildly increased in old Hfe/Nrf2-/- mice (Table 1). Nonetheless, livers of old Hfe/Nrf2-/- mice were macroscopically distinct upon dissection (Fig. 1A). Whereas wild-type and single knock-out livers displayed smooth surface with perceptible individual lobes, old Hfe/Nrf2-/- livers presented an irregular surface and a contracted, distorted and nodular appearance, consistent with fibrosis. Since the activation of fibrogenesis is a major consequence of parenchymal iron overload in the livers of HFE-HH patients [4], we evaluated the presence of fibrosis by quantifying the amount of hydroxyproline and the expression of pro-fibrotic genes in the liver of old mice. In line with the macroscopic features, Hfe/Nrf2-/- livers contained significantly higher hydroxyproline content (Fig. 1B) and mRNA expression of Col1a1 and Timp1 compared with age-matched wild-types (Figs. 1C and D). Fibrosis was further assessed in paraffin-embedded liver sections of mice of all age groups using Sirius red stain. In wild-type and single knock-out livers, collagen fibers were usually restricted to the blood vessels encircling connective tissue (Fig. 2A). In Hfe/Nrf2-/- livers, collagen fibers were more abundant, encompassing both periportal and pericellular fibrosis (Fig. 2A). Quantification of fibrotic area revealed that fibrosis was present in young Hfe/Nrf2-/- mice, preceding the loss of body weight (Fig. 2B). The staging of fibrosis revealed its progression with ageing; the occurrence of bridging fibrosis was observed in Hfe/Nrf2-/- animals at old age (Fig. 2C). Cirrhosis was not detected.
Fig. 1.

Liver macroscopy and fibrosis markers in old mice. (A) Macroscopic appearance of livers of representative old mice demonstrating the irregular surface and the contracted, distorted and nodular appearance of the Hfe/Nrf2-/- livers (Bar=1 cm). (B) Hydroxyproline content of mouse livers, N=4. (C) and (D) Expression of Col1a1 and Timp1 mRNA in mouse livers, N=7–9. *, p<0.05 vs. age-matched wild-type mice; n.s., p>0.05 vs. age-matched wild-type mice.
Fig. 2.

Histological evaluation of liver fibrosis. (A) Sirius red staining for collagen in paraffin-embedded liver sections. Images are representative of N=6–10. Bar=500 µm. (B) Quantification of fibrotic area in mouse livers of all ages and genotypes, N=6–10. (C) Staging of liver fibrosis in wt (circles), Nrf2-/-(squares), Hfe-/- (triangles) and Hfe/Nrf2-/- (inverted triangles) mice of all age groups. *, p<0.05 vs. age-matched wild-type mice; n.s., p>0.05 vs. age-matched wild-type mice.
Liver fibrosis can be triggered by different stimuli and is important in several pathologies that lead to liver failure [20]. To investigate whether iron was implicated in the etiopathogenesis of liver fibrosis in the present experimental model, we assessed systemic and hepatic iron overload in mice of different genotype and age groups. We found no significant differences when comparing the levels of serum iron, transferrin saturation or liver non-heme iron of Nrf2-/- mice with those of age-matched wild-types (Figs. 3A–C). As observed in untreated HH patients, serum iron (Fig. 3A) and transferrin saturation (Fig. 3B) were significantly raised in Hfe-/- mice of all ages. Liver iron stores were also significantly higher, increasing along animal ageing (Fig. 3C). In HH, increased iron absorption is believed to be caused by insufficient hepcidin expression [4]. In line with a previous report from Ahmad et al. [21], the lack of HFE associated with low steady-state levels of hepatic Hamp mRNA expression, as expected for the amount of iron accrued in the liver (Fig. 3D). Serum iron (Fig. 3A) and transferrin saturation (Fig. 3B) were also significantly increased in young and middle-aged Hfe/Nrf2-/- mice, but not in old animals. Nonetheless, Hfe/Nrf2-/- mice revealed raised liver iron stores throughout life, like their Hfe-/- siblings (Fig. 3C), possibly related to inadequate hepatic Hamp expression (Fig. 3D). Perls method was performed to evaluate the amount of iron in liver tissue (Fig. 4A). Histological grading revealed that in wild-type and in Nrf2-/- mice iron was faint or absent (Fig. 4B). In Hfe-/- mice, iron was almost restricted to the liver parenchyma (Fig. 4B), with a decreasing gradient from the periportal to the centrilobular zones (Fig. 4A). In Hfe/Nrf2-/- mice, iron was detected firstly in periportal hepatocytes, but with age there was a steady iron accumulation in sinusoidal cells (Fig. 4B). From middle-age on, Hfe/Nrf2-/- livers displayed mixed (parenchymal and mesenchymal) liver iron overload pattern, as observed in secondary hemosiderosis (Fig. 4A). Electron microscopy disclosed accumulation of electron-dense material, consistent with iron, within pericanalicular hepatocyte lysosomes (Supplementary Figure 1A), as reported in human HH [17], but also the presence of ferritin and hemosiderin within non-hepatocyte cells (Supplementary Figure 1B), as described by others [22]. Large aggregates of non- parenchymal cells rich in a granular yellow-brown pigment strongly positive with Perls method were observed (Fig. 4A). Energy-dispersive X-ray spectroscopy elemental analysis confirmed that the electron-dense granules of non-parenchymal cells were iron- and oxygen-rich, consistent with ferric oxide components of hemosiderin (Supplementary Figure 1C). By immunofluorescence, we observed that these non- parenchymal cells contained also autofluorescent material (lipofuscin-type) and displayed macrophage immunophenotype (F4/80+) (Fig. 4C). Hence, Hfe/Nrf2-/- livers contained a significantly higher number of iron-rich macrophages than wild-types (Fig. 4D).
Fig. 3.

Systemic and hepatic iron parameters. (A) Serum iron and (B) Transferrin saturation, N=6–14. (C) Hepatic non-heme iron concentration, N=6–14. (D) Ratio of hepcidin mRNA expression to hepatic iron concentration, N=4–12. *, p<0.05 vs. age-matched wild-type mice; n.s., p>0.05 vs. age-matched wild-type mice.
Fig. 4.

Hepatic tissue iron distribution. (A) Perls’ prussian blue stain displaying distinct hepatic iron overload in Hfe-/- and Hfe/Nrf2-/- mice of all age groups. Insets show iron overload in periportal hepatocytes. Bar=500 µm or 50 µm (insets). (B) Histological iron grading, N=6–12. (C) Immunofluorescence micrograph of a liver section from a middle-aged Hfe/Nrf2-/- mouse stained for F4/80 depicting a large aggregate of macrophages (F4/80+ cells) containing autofluorescent material. Bar=20 µm. (D) Quantification of iron-rich macrophages in liver sections, N=5–11. *, p<0.05 vs. age-matched wild-type mice; n.s., p>0.05 vs. age-matched wild-type mice.
Liver reticuloendothelial system cells may become iron-laden through phagocytosis of erythrocytes and iron-overloaded apoptotic hepatocytes. Since we have recently reported that Nrf2-/- hepatocytes are more susceptible to the injury of dietary iron [15], we searched for evidence of hepatocellular injury in Hfe/Nrf2-/- mice. Notably, we found bonafide necroinflammatory foci comprising histiocytes, siderophages, hepatocyte necrosis and apoptotic bodies (Fig. 5A). Discrete features of hepatocyte apoptotic cell death included cytoplasmic shrinkage and nuclear chromatin condensation and fragmentation (Fig. 5A). The frequency of the necroinflammatory foci increased with age (Fig. 5B), in parallel with the increase of TUNEL-positive hepatocytes (Fig. 5C). Remarkably, the progression of these two features with age matched the increase in the number of iron-rich macrophages (Fig. 4D) and in the percentage of fibrotic area (Fig. 2B), indicating that reticuloendothelial iron loading and fibrosis were triggered by iron-related necroinflammation (sideronecrosis). This assumption is supported, at least in part, by the analysis of liver histology. Indeed, Hfe/Nrf2-/- mice displayed iron overload of non-hepatocyte cells located in the sideronecrosis foci, consistent with phagocytosis of dead hepatocytes by reticuloendothelial cells (Figs. 6A and 7A). Additionally, the resolution of the necroinflammatory foci paralleled the gradual increase of collagen fibers (Fig. 6A), and collagen accumulation arose in areas of reticuloendothelial iron loading (Fig. 6B), as highlighted by the Sirius red and Perls method features. Hepatic stellate cells (HSCs) are considered the main fibrogenic cell type in the liver, and are responsible for the production of collagen and other types of extracellular matrix [23]. By transmission electron microscopy, we observed the presence of activated-type HSC nearby the sideronecrosis foci (Fig. 7A), and in the proximity of aggregates of phagocytes that had engulfed leftovers of all forms of necrosis/apoptosis, including lipofuscin-containing vesicles (residual bodies), electron-dense granules consistent with hemosiderin, and cell debris (Fig. 7B). By immunofluorescence, we established the presence of well differentiated myofibroblasts (expressing alpha-smooth muscle actin, α-SMA) contiguous with collagen bundles close to the large aggregates of lipofuscin/hemosiderin-rich macrophages (Fig. 7C). To analyse quantitatively the association between sideronecrosis and fibrosis, we pooled middle-aged and old animals of all genotypes, and for each individual we plotted the percentage of fibrotic area against the hepatic non-heme iron concentration or the number of necroinflammatory foci. Fig. 8A shows that fibrosis correlated with the non-heme hepatic iron content in the pooled population (r=0.5001, p<0.0001), but the strength of the correlation was lowered probably by the fact that Hfe-/- mice are somehow protected from fibrosis. On the other hand, a stronger correlation was found between liver fibrosis and the number of necroinflammatory foci (r=0.8472, p<0.0001) (Fig. 8B).
Fig. 5.

Histological evaluation of necroinflammatory lesions. (A) Representative necroinflammatory foci in Hfe/Nrf2-/- mouse livers containing apoptotic bodies and iron pigment in histiocytes. Center left and far left, HE stain, bar=50 µm; center right, semithin section stained with methylene blue/toluidine blue, bar=20 µm; far right, electron micrograph stained with uranyl acetate/lead citrate. Bar=5 µm. Open arrows point to hepatocyte apoptotic bodies (note contracted cytoplasm and condensed and fragmented nuclear chromatin). (B) Quantification of necroinflammtory foci in liver sections, N=6–11. (C) Quantification of TUNEL-positive cells in liver sections, N=4–8. *, p<0.05 vs. age-matched wild-type mice; n.s., p>0.05 vs. age-matched wild-type mice.
Fig. 6.

Histological evidence of concurrent necroinflammation, reticuloendothelial iron loading and fibrosis. (A) HE (left), Perls (center) and Sirius red (right) stains illustrating concurrent hepatocyte sideronecrosis, iron accumulation in histiocytes and initial pericellular fibrosis, respectively, in young Hfe/Nrf2-/- mice (top images). Arrows point to hepatocyte apoptotic bodies. Bottom images disclose aggregates of hemosiderin-rich non-hepatocyte cells surrounded by collagen fibers in a middle-aged Hfe/Nrf2-/- mouse. Bars=50 µm. (B) Sirius red stain for fibrosis and Perls stain for siderosis in the same liver from old Hfe-/- and Hfe/Nrf2-/- mice, representative of N=6–9. Bar=200 µm.
Fig. 7.

Detection of collagen-producing cells at necroinflammatory foci. (A) Electron micrograph of a sideronecrosis focus in a middle-aged Hfe/Nrf2-/- mouse liver displaying the accumulation of products of cell degeneration within hepatic phagocytes (arrows) and the active-type hepatic stellate cells next to collagen bundles (arrowheads and inset). Bar=5 µm (Inset, 2 µm). (B) Electron micrograph showing hemosiderin (long arrow)- and lipofuscin-containing residual bodies (small arrow and inset)-rich nodule. An activated-type stellate cell (star) containing numerous small lipid droplets, expanded endoplasmic reticulum and extensive extracellular collagen bundles (arrow head) is depicted nearby a hemosiderin-rich macrophage. Bar=5 µm (Inset, 0.5 µm). (C) Micrograph depicting the presence of myofibroblasts (α-SMA+) surrounding a cluster of hepatic macrophages containing autofluorescent material in the liver of a middle-aged Hfe/Nrf2-/- mouse. Bar=10 µm.
Fig. 8.

Correlation between necroinflammation and liver fibrosis. (A) and (B) Correlations between the percentage of hepatic fibrosis and the liver iron stores or the number of necroinflammatory foci in pooled middle-aged and old mice of all genotypes.
The data presented so far show that, unlike Hfe-/- counterparts, Hfe/Nrf2-/- livers displayed sideronecrosis-associated fibrosis, which cannot be attributed only to the iron loading. Hepatic iron toxicity is generally considered to be due to increased oxidative stress leading to hepatocellular death (sideronecrosis) [2]. We hypothesized that the genetic disruption of Nrf2 rendered Hfe/Nrf2-/- hepatocytes more prone to iron-induced oxidative/electrophilic stress. Thus, a series of parameters associated with oxidative/electrophilic stress was evaluated in middle-aged livers. In line with our hypothesis, the mRNA expression of the prototypical NRF2 target genes Nqo1 (Fig. 9A) and Gsta1 (Fig. 9B), and the enzymatic activity of glutathione-S-transferase (Fig. 9C) were significantly lower in middle-aged Nrf2-/- and Hfe/Nrf2-/- mouse livers compared with age-matched wild-type animals. Levels of total glutathione, the major hepatocellular antioxidant, were also significantly lower in Nrf2-/- and Hfe/Nrf2-/- livers than in wild-type (Fig. 9D). The alterations of these antioxidant markers were expected to cause increased oxidative/electrophilic stress and cell damage. We estimated the amount of 4-hydroxy-2-nonenal (4-HNE), a cytotoxic aldehyde product of the peroxidation of polyunsaturated fatty acids that is able to diffuse across membranes and bind covalently to functional proteins [24]. According to our hypothesis, higher levels of 4-HNE-protein adducts were present in liver homogenates of Hfe/Nrf2-/- mice compared with wild-type counterparts (Fig. 9E). Likewise, the expression of p62/SQSTM1 protein, which is known to be increased in chronic liver diseases in response to oxidative stress and to the accumulation of toxic protein aggregates [25], was significantly greater in Hfe/Nrf2-/- livers (Fig. 9F). Taken together, the data presented above indicate that the accumulation of toxic oxidation products, such as 4-HNE, likely contributed to hepatocellular injury and concomitantly to liver fibrosis in the present study.
Fig. 9.

Assessment of oxidative/electrophilic stress parameters in middle-aged mice. (A) Steady-state mRNA expression of Nqo1 and (B) Gsta1 in mouse livers, N=8–9. (C) Glutathione S-transferase enzymatic activity in liver homogenates, N=9–12. (D) Total glutathione in liver homogenates, N=3–4. (E) Detection of 4-HNE protein adducts in mouse livers, N=6–8. (F) Quantification of p62 protein in mouse livers, N=3. *, p<0.05 vs. age-matched wild-type mice; n.s., p>0.05 vs. age-matched wild-type mice; °, p<0.05 vs. age-matched Nrf2-/- and Hfe-/- mice.
4. Discussion
In human HH, the long-term deposition of iron in the liver can lead to organ damage, including hepatic fibrosis and cirrhosis. The development of hepatic injury in HH is highly variable and cannot be ascribed solely to the severity of iron overload. Clinical cofactors influence disease progression and genetic factors may also play a role [1], [3]. In rodents, the ability of iron to stimulate the progression of experimentally-induced hepatic fibrosis has been reported; alcoholic liver fibrosis is potentiated by dietary iron [26] and carbon tetrachloride-induced liver fibrogenesis is exacerbated in mice fed carbonyl iron [27] and in hemojuvelin knockout mice [28]. Conversely, an iron-deficient diet attenuates the development of liver fibrosis after treatment of rats with thioacetamide or bile-duct ligation [29]. Yet, hepatic fibrosis and cirrhosis have not been observed in the mouse models of HFE-HH [9], [10] and hemojuvelin-associated HH [30]; this evokes the existence of efficient adaptive mechanisms that render the mouse liver able to bear the increased iron toxicity. Such unidentified mechanisms may also be implicated in the low penetrance of the HFE mutation in humans.
Transcription factor NRF2 is a master regulator of cellular detoxification responses and redox status [13] that protects mice against hepatotoxin-induced fibrosis [12]. We have previously reported the development of extensive hepatocyte necrosis in Nrf2-null mice fed an excessive amount of dietary iron [15]; this occurred in the absence of fibrosis, probably because the iron overload (equivalent to the hepatic iron stores of middle-aged Hfe-/- mice) was induced over a short period of time (2 weeks). In the present study, we used the Hfe-/- mouse, a model of human HFE-HH that displays several features of human C282Y homozygotes: mild parenchymal iron overload with decreasing hepatocytic gradient from the periportal to centrilobular zones, consistent with increased intestinal absorption of iron [17]; insufficient hepcidin production in spite of excessive iron deposition in the hepatic parenchyma [31]; elevated serum iron and transferrin saturation [32]; and normal alanine aminotransferase [33]. We established, so far for the first time, that the genetic disruption of Nrf2 in the Hfe-/- mouse increased hepatocyte apoptosis/sideronecrosis, in association with foci of inflammation, and a gradual shift of iron from hepatocytes to the sinusoidal macrophages (F4/80+). Serum alanine aminotransferase activity was only mildly increased in Hfe/Nrf2-/- mice, which fits with the possibility that mild iron overload is insufficient to cause extensive hepatocellular necrosis/apoptosis. Conversely, necroinflammatory foci of Hfe/Nrf2-/- mice were set locally associated to the presence of hepatic histiocytes, which may have originated from reticuloendothelial macrophages (Kupffer cells) and/or from circulating monocytes.
Remarkably, these are well-characterized iron-related histological lesions reported in the livers of human HH patients [17], where mesenchymal iron deposits are usually not observed until the liver cell iron content is high enough to cause cell necrosis [1,2]. In middle-aged and old Hfe/Nrf2-/- animals, the iron-rich macrophages aggregated to form large siderotic nodules, which were composed of hemosiderin, lipofuscin and other products of cell degeneration. Erythrocytes flooding areas of parenchymal collapse (as seen in Fig. 5A) may have further contributed to pigment deposition within hepatic histiocytes upon phagocytosis. In this etiopathogenic process, HSCs seem to evolve into myofibroblast-like cells, presumably by the release of factors such as reactive oxygen species, lipid peroxidation products and transforming growth factor beta( TGF-β) [23]. α-SMA+ cells are known to be responsible for collagen overproduction in injured liver areas. In Hfe/Nrf2-/- animals, collagen bundles were detected in the injured areas and eventually led to the establishment of hepatic fibrosis in an age-dependent manner.
The present study describes, for the first time, a mouse model of fibrosis that conveys several features reported in the pathology of human HH: etiology (chronic iron accumulation), development over months/years, hepatocellular necrosis/apoptosis, and reticuloendothelial system iron deposits [17], [34]. Delima et al. [35] reported increased collagen deposition in the portal tracts of mice after the simultaneous disruption of Hfe and another major regulator of iron homeostasis, the transferrin receptor 2. Also, a recent study reported the development of hepatic fibrosis in Hamp-/- mice fed an iron-rich diet [36]. In both studies, however, the effect was attributed to the more severe hepatic iron loading when compared with single mutant animals or with animals fed standard diet, respectively. In our model, Hfe/Nrf2-/- mice appeared to accumulate more liver iron than Hfe-/- single knockouts (Figs. 3C and 4B). Although the effect did not reach statistical significance, the data suggest a possible role for NRF2 in the regulation of iron homeostasis that so far has not been reported. On the other hand, serum iron and transferrin saturation were not significantly elevated in old Hfe/Nrf2-/- mice, which may have possibly resulted from a number of factors related with old age, including malnutrition, reduced efficiency of iron absorption, and/or chronic inflammation [37]. Nevertheless, fibrosis developed in Hfe/Nrf2-/- mice with concentrations of hepatic iron that are ~16-fold below the reported threshold for the development of hepatic fibrosis in HH patients [38], and was associated with focal necroinflammation. Therefore, it seems reasonable to assume, based in our study that iron accumulation (siderosis) per se does not predict the development of liver injury in our model of human HFE-hemochromatosis, which may also occur in HH patients; among other factors, oxidative/electrophilic stress seems to be required.
Excessive iron within hepatocytes catalyzes lipid peroxidation of cellular organelle membranes to produce reactive aldehydes such as 4- HNE, which may form aldehyde-protein adducts. Higher amounts of 4-HNE-protein adducts were previously found in the liver of rats fed an iron-rich diet [39] and of HH patients [40]. The accumulation of toxic oxidation products, such as HNE, may cause lysosomal, mitochondrial and microsomal dysfunction, causing hepatocyte injury or death [2,41]. The livers of Hfe/Nrf2-/- mice disclosed lower basal levels of Nqo1 and Gsta1 mRNA expression and of glutathione-S-transferase activity. NQO1 [42], [43] and glutathione-S-transferases [44] are involved in the detoxification of reactive molecules, including reactive oxygen species and a range of harmful electrophiles produced during membrane lipid peroxidation (e.g. 4-HNE), in the cytosol, mitochondria and endoplasmic reticulum. One such mechanism is the conjugation of 4-HNE with glutathione, which can occur spontaneously or can be catalysed by glutathione-S-transferases [45], [46]. In Nrf2-/- and Hfe/Nrf2-/- livers, we have observed lower levels of both glutathione and glutathione-S-transferase activity, which may have contributed to the increased levels of 4-HNE-protein adducts and of p62/SQSTM1 protein (a surrogate for the intracellular accumulation of toxic protein aggregates) of the double-knockouts. These alterations seem to account for the increased death of Hfe/Nrf2-/- hepatocytes observed in the present study and for the accumulation of lipofuscin-like autofluorescent storage material (ceroid) within hepatic phagocytes. Engulfment of dead bodies by Kupffer cells and HSCs increases the expression of profibrogenic factors, and persistent activation of these cells promotes the development of fibrosis [47]. HNE was also reported to up-regulate the expression of pro-fibrogenic genes (e.g. Col1a1 and Timp1) by hepatic stellate cell cultures [48]. In this study we established that an increased susceptibility to oxidative/electrophilic stress boosts the progression from iron accumulation (siderosis) to liver fibrosis in a model of HH, and identified a genetic modifier – the transcription factor NRF2 – which is a novel factor that seems to play a role (so far never reported) in the iron load liver pathogenesis of HH. Our results raise the possibility that NRF2 status might be a modifier of disease progression towards iron related liver injury, and that C282Y homozygotes with compromised NRF2 signaling may be at an increased risk of developing iron-related hepatic fibrosis or cirrhosis, even with mild increase of hepatic iron.
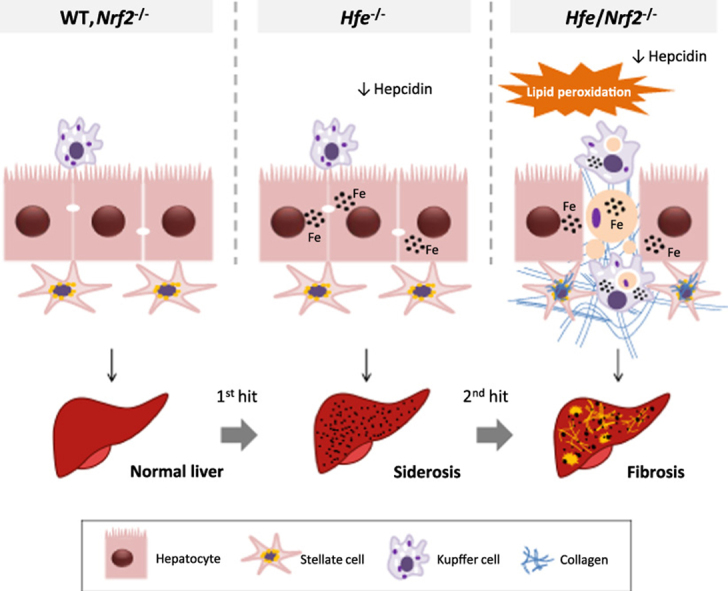
In conclusion, we established that the genetic suppression of Nrf2 increases the frequency of iron-related necro-inflammatory events (sideronecrosis) and liver fibrosis in a mouse model of HFE-HH, which may be due to an increased susceptibility to oxidative/electrophilic stress. A schematic diagram of the proposed role for NRF2 in limiting oxidative damage and liver injury in HH, based in our experimental model, is presented in Fig. 10.
Fig. 10.

Schematic diagram of the working model proposed for NRF2 in limiting oxidative damage and liver injury in HH. In Hfe-/- mice, similar to human HFE-HH, low hepcidin expression resulted in increased iron absorption and its pericanalicular accumulation within hepatocytes. Macrophages remain iron-depleted. While the genetic disruption of Nrf2 per se had no significant impact on iron homeostasis, the combination of iron overload (Hfe-/-) and compromised antioxidant defences (Nrf2-/-) resulted in lipid peroxidation and increased number of iron-related necroinflammatory lesions (sideronecrosis). The engulfment of dead hepatocytes led to a gradual accumulation of iron within macrophages. Myofibroblasts recruited towards the injury areas synthesized substantial amounts of collagen fibers involving the liver parenchyma with increased hepatic fibrosis in an age-dependent manner. The diagram denotes the progressive nature of HFE-HH, where iron accumulation (siderosis) (1st hit) and oxidative stress (2nd hit) are both required for the development of liver injury.
Conflict of interest
The authors have no conflicts of interest to declare.
Financial support
This work was supported by National funds through Fundação para a Ciência e a Tecnologia/Ministério da Educação e Ciência (PTDC/SAU-FCF/101177/2008, PTDC/BIM-MET/0739/2012 and SFRH/BPD/108207/2015), by FEDER funds through the COMPETE – Operational Competitiveness Programme (FCOMP-01-0124-FEDER-011062 and FCOMP-01-0124-FEDER-028447) and Project Norte-01-0145-FEDER-000012, supported by Norte Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (FEDER), and by Reitoria da Universidade do Porto/Santander through PP-IJUP2011-122.
Author's contributions
Study concept and design: TLD.
Acquisition of data: CC, AGS, ASG, MJM, TLD.
Analysis and interpretation of data: SSG, MJM, GP, JML, TLD.
Drafting of the manuscript: TLD.
Critical revision of the manuscript for important intellectual content: all authors.
All the authors approved the final version of the manuscript.
Acknowledgements
The authors are grateful to Rui Fernandes (Histology and Electron Microscopy service, Instituto de Biologia Molecular e Celular) for technical assistance in transmission electron microscopy, to João Neves for help in animal dissection and to Corelab staff at Centro Hospitalar do Porto for serum biochemistry analyses.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2016.11.013.
Appendix A. Supplementary material
Supplementary material
.
References
- 1.Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139:393–408. doi: 10.1053/j.gastro.2010.06.013. e2. [DOI] [PubMed] [Google Scholar]
- 2.Pietrangelo A. Mechanisms of iron hepatotoxicity. J. Hepatol. 2016;65:226–227. doi: 10.1016/j.jhep.2016.01.037. [DOI] [PubMed] [Google Scholar]
- 3.Deugnier Y., Mosser J. Modifying factors of the HFE hemochromatosis phenotype. Expert Rev. Gastroenterol. Hepatol. 2008;2:531–540. doi: 10.1586/17474124.2.4.531. [DOI] [PubMed] [Google Scholar]
- 4.Powell L.W., Seckington R.C., Deugnier Y. Haemochromatosis. Lancet. 2016;388:706–716. doi: 10.1016/S0140-6736(15)01315-X. [DOI] [PubMed] [Google Scholar]
- 5.Powell E.E., Ali A., Clouston A.D., Dixon J.L., Lincoln D.J., Purdie D.M. Steatosis is a cofactor in liver injury in hemochromatosis. Gastroenterology. 2005;129:1937–1943. doi: 10.1053/j.gastro.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 6.Wood M.J., Powell L.W., Ramm G.A. Environmental and genetic modifiers of the progression to fibrosis and cirrhosis in hemochromatosis. Blood. 2008;111:4456–4462. doi: 10.1182/blood-2007-11-122374. [DOI] [PubMed] [Google Scholar]
- 7.Wood M.J., Powell L.W., Dixon J.L., Ramm G.A. Clinical cofactors and hepatic fibrosis in hereditary hemochromatosis: the role of diabetes mellitus. Hepatology. 2012;56:904–911. doi: 10.1002/hep.25720. [DOI] [PubMed] [Google Scholar]
- 8.Bahram S., Gilfillan S., Kuhn L.C., Moret R., Schulze J.B., Lebeau A. Experimental hemochromatosis due to MHC class I HFE deficiency: immune status and iron metabolism. Proc. Natl. Acad. Sci. USA. 1999;96:13312–13317. doi: 10.1073/pnas.96.23.13312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lebeau A., Frank J., Biesalski H.K., Weiss G., Srai S.K., Simpson R.J. Long-term sequelae of HFE deletion in C57BL/6×129/O1a mice, an animal model for hereditary haemochromatosis. Eur. J. Clin. Investig. 2002;32:603–612. doi: 10.1046/j.1365-2362.2002.01026.x. [DOI] [PubMed] [Google Scholar]
- 10.Rodrigues P., Lopes C., Mascarenhas C., Arosio P., Porto G., De Sousa M. Comparative study between Hfe-/- and beta2m-/- mice: progression with age of iron status and liver pathology. Int. J. Exp. Pathol. 2006;87:317–324. doi: 10.1111/j.1365-2613.2006.00491.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharm. Toxicol. 2013;53:401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu W., Hellerbrand C., Kohler U.A., Bugnon P., Kan Y.W., Werner S. The Nrf2 transcription factor protects from toxin-induced liver injury and fibrosis. Lab Investig. 2008;88:1068–1078. doi: 10.1038/labinvest.2008.75. [DOI] [PubMed] [Google Scholar]
- 13.Klaassen C.D., Reisman S.A. Nrf2 the rescue: effects of the antioxidative/electrophilic response on the liver. Toxicol. Appl. Pharm. 2010;244:57–65. doi: 10.1016/j.taap.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang W., Jiang Y.F., Ponnusamy M., Diallo M. Role of Nrf2 in chronic liver disease. World J. Gastroenterol. 2014;20:13079–13087. doi: 10.3748/wjg.v20.i36.13079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Silva-Gomes S., Santos A.G., Caldas C., Silva C.M., Neves J.V., Lopes J. Transcription factor NRF2 protects mice against dietary iron-induced liver injury by preventing hepatocytic cell death. J. Hepatol. 2014;60:354–361. doi: 10.1016/j.jhep.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Itoh K., Chiba T., Takahashi S., Ishii T., Igarashi K., Katoh Y. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 17.Deugnier Y.M., Loreal O., Turlin B., Guyader D., Jouanolle H., Moirand R. Liver pathology in genetic hemochromatosis: a review of 135 homozygous cases and their bioclinical correlations. Gastroenterology. 1992;102:2050–2059. doi: 10.1016/0016-5085(92)90331-r. [DOI] [PubMed] [Google Scholar]
- 18.Kleiner D.E., Brunt E.M., Van Natta M., Behling C., Contos M.J., Cummings O.W. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 19.Assuncao M., Santos-Marques M.J., Monteiro R., Azevedo I., Andrade J.P., Carvalho F. Red wine protects against ethanol-induced oxidative stress in rat liver. J. Agric. Food Chem. 2009;57:6066–6073. doi: 10.1021/jf900576h. [DOI] [PubMed] [Google Scholar]
- 20.Elpek G.Ö. Cellular and molecular mechanisms in the pathogenesis of liver fibrosis: an update. World J. Gastroenterol. 2014;20:7260–7276. doi: 10.3748/wjg.v20.i23.7260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahmad K.A., Ahmann J.R., Migas M.C., Waheed A., Britton R.S., Bacon B.R. Decreased liver hepcidin expression in the Hfe knockout mouse. Blood Cells Mol. Dis. 2002;29:361–366. doi: 10.1006/bcmd.2002.0575. [DOI] [PubMed] [Google Scholar]
- 22.Fairweather-Tait S.J., Wawer A.A., Gillings R., Jennings A., Myint P.K. Iron status in the elderly. Mech. Ageing Dev. 2014;136–137:22–28. doi: 10.1016/j.mad.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iancu T.C. Ultrastructural aspects of iron storage, transport and metabolism. J. Neural Transm. 2011;118:329–335. doi: 10.1007/s00702-011-0588-7. [DOI] [PubMed] [Google Scholar]
- 24.Shimizu I., Shimamoto N., Saiki K., Furujo M., Osawa K. In: Lipid Peroxidation in Hepatic Fibrosis. Catala A., editor. Lipid Peroxidation: InTech; 2012. [Google Scholar]
- 25.Barrera G., Pizzimenti S., Ciamporcero E., Daga M., Ullio C., Arcaro A. Role of 4-hydroxynonenal-protein adducts in human diseases. Antioxid. Redox Signal. 2015;22:1681–1702. doi: 10.1089/ars.2014.6166. [DOI] [PubMed] [Google Scholar]
- 26.Khan M.F., Wu X., Tipnis U.R., Ansari G.A., Boor P.J. Protein adducts of malondialdehyde and 4-hydroxynonenal in livers of iron loaded rats: quantitation and localization. Toxicology. 2002;173:193–201. [PubMed] [Google Scholar]
- 27.Niemelä O., Parkkila S., Britton R.S., Brunt E., Janney C., Bacon B. Hepatic lipid peroxidation in hereditary hemochromatosis and alcoholic liver injury. J. Lab Clin. Med. 1999;133:451–460. doi: 10.1016/s0022-2143(99)90022-7. [DOI] [PubMed] [Google Scholar]
- 28.Taniguchi K., Yamachika S., He F., Karin M. p62/SQSTM1-Dr. Jekyll and Mr. Hyde that prevents oxidative stress but promotes liver cancer. FEBS Lett. 2016;590:2375–2397. doi: 10.1002/1873-3468.12301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tsukamoto H., Horne W., Kamimura S., Niemela O., Parkkila S., Yla-Herttuala S. Experimental liver cirrhosis induced by alcohol and iron. J. Clin. Investig. 1995;96:620–630. doi: 10.1172/JCI118077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arezzini B., Lunghi B., Lungarella G., Gardi C. Iron overload enhances the development of experimental liver cirrhosis in mice. Int. J. Biochem Cell Biol. 2003;35:486–495. doi: 10.1016/s1357-2725(02)00298-4. [DOI] [PubMed] [Google Scholar]
- 31.Sebastiani G., Gkouvatsos K., Maffettone C., Busatto G., Guido M., Pantopoulos K. Accelerated CCl4-induced liver fibrosis in Hjv-/- mice, associated with an oxidative burst and precocious profibrogenic gene expression. PLoS One. 2011;6:e25138. doi: 10.1371/journal.pone.0025138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otogawa K., Ogawa T., Shiga R., Nakatani K., Ikeda K., Nakajima Y. Attenuation of acute and chronic liver injury in rats by iron-deficient diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008;294:R311–R320. doi: 10.1152/ajpregu.00735.2007. [DOI] [PubMed] [Google Scholar]
- 33.Huang F.W., Pinkus J.L., Pinkus G.S., Fleming M.D., Andrews N.C. A mouse model of juvenile hemochromatosis. J. Clin. Investig. 2005;115:2187–2191. doi: 10.1172/JCI25049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bridle K.R., Frazer D.M., Wilkins S.J., Dixon J.L., Purdie D.M., Crawford D.H. Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis. Lancet. 2003;361:669–673. doi: 10.1016/S0140-6736(03)12602-5. [DOI] [PubMed] [Google Scholar]
- 35.Batey R.G., Lai Chung Fong P., Shamir S., Sherlock S. A non-transferrin-bound serum iron in idiopathic hemochromatosis. Dig. Dis. Sci. 1980;25:340–346. doi: 10.1007/BF01308057. [DOI] [PubMed] [Google Scholar]
- 36.Adams P.C., Speechley M., Barton J.C., McLaren C.E., McLaren G.D., Eckfeldt J.H. Probability of C282Y homozygosity decreases as liver transaminase activities increase in participants with hyperferritinemia in the hemochromatosis and iron overload screening study. Hepatology. 2012;55:1722–1726. doi: 10.1002/hep.25538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stal P., Broome U., Scheynius A., Befrits R., Hultcrantz R. Kupffer cell iron overload induces intercellular adhesion molecule-1 expression on hepatocytes in genetic hemochromatosis. Hepatology. 1995;21:1308–1316. doi: 10.1002/hep.1840210514. [DOI] [PubMed] [Google Scholar]
- 38.Delima R.D., Chua A.C., Tirnitz-Parker J.E., Gan E.K., Croft K.D., Graham R.M. Disruption of hemochromatosis protein and transferrin receptor 2 causes iron-induced liver injury in mice. Hepatology. 2012;56:585–593. doi: 10.1002/hep.25689. [DOI] [PubMed] [Google Scholar]
- 39.Lunova M., Goehring C., Kuscuoglu D., Mueller K., Chen Y., Walther P. Hepcidin knockout mice fed with iron-rich diet develop chronic liver injury and liver fibrosis due to lysosomal iron overload. J. Hepatol. 2014;61:633–641. doi: 10.1016/j.jhep.2014.04.034. [DOI] [PubMed] [Google Scholar]
- 40.Bassett M.L., Halliday J.W., Powell L.W. Value of hepatic iron measurements in early hemochromatosis and determination of the critical iron level associated with fibrosis. Hepatology. 1986;6:24–29. doi: 10.1002/hep.1840060106. [DOI] [PubMed] [Google Scholar]
- 41.Bacon B.R., Britton R.S. The pathology of hepatic iron overload: a free radical – mediated process? Hepatology. 1990;11:127–137. doi: 10.1002/hep.1840110122. [DOI] [PubMed] [Google Scholar]
- 42.Kwon J., Han E., Bui C.B., Shin W., Lee J., Lee S. Assurance of mitochondrial integrity and mammalian longevity by the p62-Keap1-Nrf2-Nqo1 cascade. EMBO Rep. 2012;13:150–156. doi: 10.1038/embor.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong H., Shertzer H.G., Genter M.B., Gonzalez F.J., Vasiliou V., Jefcoate C. Mitochondrial targeting of mouse NQO1 and CYP1B1 proteins. Biochem. Biophys. Res. Commun. 2013;435:727–732. doi: 10.1016/j.bbrc.2013.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hayes J.D., Flanagan J.U., Jowsey I.R. Glutathione transferases. Annu. Rev. Pharm. Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 45.Khan M.F., Srivastava S.K., Singhal S.S., Chaubey M., Awasthi S., Petersen D.R. Iron-induced lipid peroxidation in rat liver is accompanied by preferential induction of glutathione S-transferase 8-8 isozyme. Toxicol. Appl. Pharm. 1995;131:63–72. doi: 10.1006/taap.1995.1047. [DOI] [PubMed] [Google Scholar]
- 46.Völkel W., Alvarez-Sánchez R., Weick I., Mally A., Dekant W., Pähler A. Glutathione conjugates of 4-hydroxy-2(E)-nonenal as biomarkers of hepatic oxidative stress-induced lipid peroxidation in rats. Free Radic. Biol. Med. 2005;38:1526–1536. doi: 10.1016/j.freeradbiomed.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 47.Guicciardi M.E., Gores G.J. Apoptosis: a mechanism of acute and chronic liver injury. Gut. 2005;54:1024–1033. doi: 10.1136/gut.2004.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zamara E., Novo E., Marra F., Gentilini A., Romanelli R.G., Caligiuri A. 4-Hydroxynonenal as a selective pro-fibrogenic stimulus for activated human hepatic stellate cells. J. Hepatol. 2004;40:60–68. doi: 10.1016/s0168-8278(03)00480-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material