

Abstract
Drug resistance compromises control of malaria. Here, we show that resistance to a commonly used antimalarial medication, atovaquone, is apparently unable to spread. Atovaquone pressure selects parasites with mutations in cytochrome b, a respiratory protein with low but essential activity in the mammalian blood phase of the parasite life cycle. Resistance mutations rescue parasites from the drug but later prove lethal in the mosquito phase, where parasites require full respiration. Unable to respire efficiently, resistant parasites fail to complete mosquito development, arresting their life cycle. Because cytochrome b is encoded by the maternally inherited parasite mitochondrion, even outcrossing with wild-type strains cannot facilitate spread of resistance. Lack of transmission suggests that resistance will be unable to spread in the field, greatly enhancing the utility of atovaquone in malaria control.
A tovaquone, a component of the safe and effective antimalarial medication Malarone, kills both the blood and liver stages of malaria (1). The rollout of cheap generics should see increased atovaquone usage, and atovaquone derivatives are in development (1). Atovaquone is prone to resistance (1), and it has been assumed that this resistance will spread (2, 3), as it has for other antimalarials (4, 5). However, the target of atovaquone, cytochrome b (cytB) (6–9), has unique genetics (10–12) and experiences differential selection across the malaria parasite life cycle (13), which prompted us to investigate whether atovaquone resistance can spread via the mosquito vector.
We tested three atovaquone-resistant strains of the rodent malaria parasite Plasmodium berghei, each with different mutations in their mitochondrial DNA-encoded cytB gene (14, 15), for transmissibility from mouse to mosquito and back to mouse (Table 1). Anopheles stephensi mosquitoes were fed on mice infected with either the parental PbANKA strain or one of the three atovaquone-resistant mutants, and sexual development of parasites in mosquitoes was assayed (Table 1). All three atovaquone-resistant parasite lines produced wild-type numbers of active male gametes (exflagellation) (Table 1). Parasites carrying the PbM133I and PbY268C mutations in their cytB gene were able to self-fertilize, generate ookinetes, and successfully produce oocysts, but the oocysts produced had developmental defects (Fig. 1, A and B, and Table 1). Parasites with the PbY268N mutation were defective in the ability to self-fertilize and infect the mosquito host (Table 1) due to severely impaired female gamete activation (Fig. 1C). From 17 attempted mosquito infections, no parasite carrying an atovaquone-resistant cytB mutation was able to generate the sporozoite stages in the mosquito salivary glands or was able to infect a naïve mouse (Table 1). We conclude that the rodent malaria atovaquone-resistant cytB mutants tested—which represent a good cross section of the clinical atovaquone-resistant genotypes, including the common Y268 locus (16)—are unable to transmit from mouse to mouse via A. stephensi mosquitoes when self-fertilizing.
Table 1. Atovaquone-resistant mutants in rodent and human malaria parasites fail to produce sporozoites in mosquitoes, and bite-back experiments with mice yielded no resistance transmission.
All values are ±SEM. IC50, median inhibitory concentration; wt, wild type; nd, no data available; na, not applicable.
| Parasite genotype nuclear/mitochondrial | Atovaquone IC50 (nM) | Number of infections | Exflagellations per 104 red blood cells | Ookinetes per mosquito (n = no. of mosquitoes) | Midgut infection % infected oocytes per mosquito (n = no. of mosquitoes) | Sporozoites per mosquito (n = no. of mosquitoes) | Transmission to naïve mice | Time to patency (days) |
|---|---|---|---|---|---|---|---|---|
| Pb wt/wt | 9.1 ± 0.4 | 6 | 5.3 ± 1.6 | 2327.6 ± 810.7 (58) |
100 109.8 ± 21.7 (85) |
10,348 ± 3279 | 3/3 | 4.3 ±0.3 |
| Pb wt/M133I | 250 ± 41 | 4 | 5.5 ± 1.9 | 1488 ±549 (38) |
59 ± 16 23.2 ± 9.8 (69) |
0 | 0/4 | na |
| Pb wt/Y268C | 23,695 ± 915 | 3 | 4.5 ± 1.3 | 725 ± 52.0 (30) |
50 ± 10 7.0 ± 2.9 (47) |
0 | 0/5 | na |
| Pb wt/Y268C P0 | 19,080 ± 1119 | 4 | 6.7 ± 1.4 | 347 ± 97.0 (29) |
52 ± 17 17.1 ± 10.2 (85) |
0 | 0/6 | na |
| Pb wt/Y268N | 11,625 ± 1225 | 6 | 7.3 ± 1.9 | 41.7 ± 41.7* (81) |
17± 11 2.7 ± 1.7 (80) |
0 | 0/5 | na |
| Pf NF54e/wt | 2.25 ± 1.13 | 4 | 20.0 ± 9.3 | 1918 ± 225 (30) |
97.3 ± 1.7 98.5 ± 38.5 (83) |
nd | na | na |
| Pf NF54e/M133I | 16.2 ± 3.9 | 4 | 9.3 ± 3.8 | 0 | 1.5 ± 1.5 0.02 ± 0.02* (144) |
nd | na | na |
| Pf NF54e/V259L | 35.3 ± 2.5 | 4 | 7.0 ± 4.3 | 0 | 3 ± 1 0.03 ± 0.01 (154) |
nd | na | na |
Parasites were detected from only a single experiment.
Fig. 1. Atovaquone-resistant parasites generate small, malformed oocysts in the mosquito that fail to form infectious sporozoites.
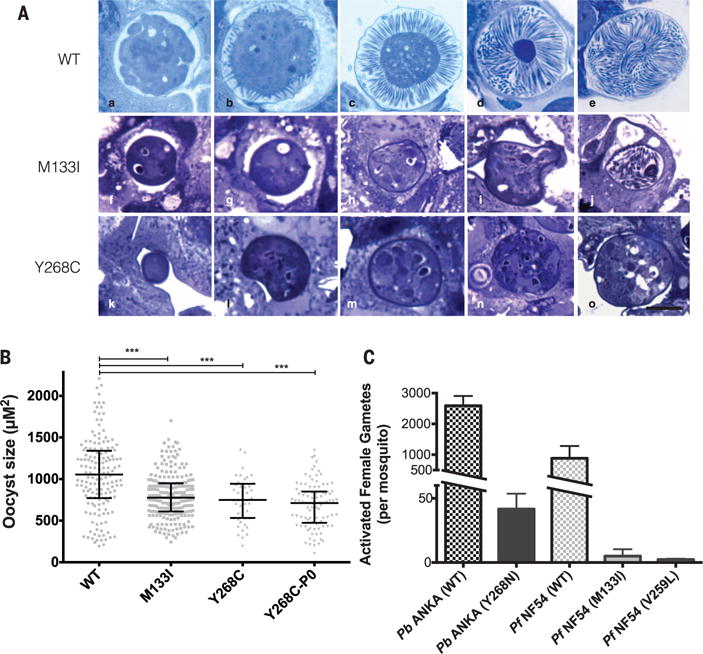
(A) (a to e) Developmental series of oocyst sporogony in PbANKA (atovaquone sensitive and wild type) over ~18 days. The sporoblast buds off hundreds of long, thin sporozoites within the cyst wall. (f to j) Atovaquone-resistant mutant (PbM133I) has smaller oocysts with dense cytoplasm, and sporozoite budding is minimal. Rare oocysts (j) form short, thick sporozoites that do not emerge. (k to o) Atovaquone-resistant mutant (PbY268C) also has small, dense, misshapen oocysts that fail to bud off any sporozoites. Scale bar, 25 μm. (B) Quantification of oocyst area at 12 days after feeding, showing reduced size of atovaquone-resistant parasites. Bars represent median with interquartile range. ***P < 0.001); difference between three atovaquone-resistant lines is not significant (P > 0.05); Dunn’s multiple comparison test. (C) Number of activated females in P. berghei (all activated forms 24 hours after feeding) and P. falciparum (females and zygotes present 20 hours after feeding).
To determine whether outcrossing with atovaquone-sensitive parasites could help transmit the atovaquone resistance genes, we generated crosses of our atovaquone-resistant P. berghei lines with atovaquone-sensitive parasites. These experiments simulate what might happen if a mosquito bit an individual (or separate individuals) infected with both atovaquone-resistant and atovaquone-sensitive parasites, which can then mate in the mosquito gut. They allow us to assess whether the presence of wild-type copies of the cytB genes from one parent can complement a mutation in the other, as observed with deletions of electron transport components encoded in the nuclear genome (17). We first crossed PbY268C with an atovaquone-sensitive line (wild-type cytB) carrying a mutation (15) in the nucleus-encoded dihydrofolate reductase (dhfr) gene conferring pyrimethamine resistance (PbdhfrS110N) by pooling blood from separate infected mice and then membrane-feeding mosquitoes. Sporozoites were produced, and all 14 naïve mice bitten by these mosquitoes (three trials) developed blood-stage infections. Genotyping of these progeny [passage zero (P0)] showed that outcrossing had occurred because 2 out of 14 mice carried parasites with both the wild-type and pyrimethamine-resistant (S110N) alleles of Pbdhfr. However, all 14 mice carried parasites with only wild-type, atovaquone-sensitive cytB alleles; outcrossing did not facilitate transmission of atovaquone resistance.
To explore this further, and quantify the level of outcrossing, we fed A. stephensi mosquitoes from mice infected with equal numbers of an atovaquone-sensitive line that constitutively expresses green fluorescent protein (GFP) from the nucleus (18) and one of our three atovaquone-resistant lines (PbM133I, PbY268C, or PbY268N), which lack GFP. All crosses between sensitive (PbGFP) parasites and our atovaquone-resistant parasites successfully infected the mosquitoes, and these mosquitoes were able to infect naïve mice by biting (Table 2). Presence in the progeny of both the GFP and the wild-type (no GFP) nuclear markers confirmed outcrossing (Table 2). However, all the progeny had wild-type cytB genotype and were sensitive to atovaquone (Table 2 and fig. S1), reaffirming that the resistance mutations cannot be complemented and transmitted, even when one parent carries a wild-type version of the gene. We conclude that outcrossing cannot assist transmission of the commonly occurring cytB mutations conferring atovaquone resistance.
Table 2. Outcrossing atovaquone-resistant rodent malaria lines to sensitive lines does not facilitate resistance transmission because resistance is maternally inherited.
All values are ±SEM; wt, wild type; nd, no data available; na, not applicable.
| Cross nuclear genotype/mitochondrial genotype | Number of infections | Exflagellations per104 red blood cells | Midgut infection % infected oocytes per mosquito (n = no. of mosquitoes) | Sporozoites per mosquito (n = no. of mosquitoes) | Transmission to naïve mice | Time to patency (days) | Nuclear genotype P0 | Mitochondrial genotype P0 |
|---|---|---|---|---|---|---|---|---|
| GFP/wt × GFP/wt | 3 | 6.07 ± 1.88 | 93 ± 6 211.2 ± 40.4 (30) |
12,433 ± 1822 (29) |
4/4 | 4 ± 0 | 100% GFP | wt |
| wt/M133I × GFP/wt | 3 | 6.77 ± 2.16 | 91 ± 6 140.7 ± 76.3 (31) |
10,600 ± 3139 (55) |
4/4 | 4.25 ±0.3 | 60 ± 4% GFP | wt |
| wt/Y268C × GFP/wt | 3 | 1.83 ± 0.73 | 69 ± 6 27.0 ± 15.0 (42) |
3133 ± 2533 (57) |
3/4 | 4 ± 0 | 50 ± 8% GFP | wt |
| wt/Y268N × GFP/wt | 3 | 6.73 ± 2.13 | 77 ± 12 16.8 ± 8.1 (29) |
6076 ± 2899 (45) |
4/4 | 4.5 ± 0.3 | 72 ± 17% GFP | wt |
| s48|45ko/wt × s48|45ko/wt | 1 | 1.7 | 0 | 0 | 0/1 | na | na | na |
| nek-4ko/wt × nek-4ko/wt | 2 | 9.0 ± 6.2 | 0 (27) | na | na | na | na | na |
| s48|45ko/wt × nek-4ko/wt | 1 | 10.2 | 44 9.9 (15) |
750 (17) |
1/1 | 5 | s48|45ko and nek-4ko | nd |
| GFP/wt × nek-4ko/wt | 2 | 11 ± 3.8 | 85 ± 15 31.8 ± 26.8 (22) |
4575 ± 4425 (25) |
2/2 | 4 ± 0 | 57 ± 5% GFP | wt |
| wt/M133I × s48|45ko/wt | 1 | 2.2 | 67 7.3 (12) |
18,900 (10) |
1/1 | 4 | wt and s48|45ko | wt |
| wt/Y268C × s48|45ko/wt | 1 | 6.4 | 100 16 (5) |
8500 (10) |
1/1 | 4 | wt and s48|45ko | wt |
| wt/Y268N × s48|45ko/wt | 1 | 8.1 | 90 8.9 (9) |
2125 (10) |
1/1 | 4 | wt and s48|45ko | wt |
| wt/M133I × nek-4ko/wt | 4 | 5.5 ± 1.5 | 48 ± 14 8.9 ± 3.6 (73) |
0 (80) |
0/5 | na | na | na |
| wt/Y268C × nek-4ko/wt | 5 | 7.3 ± 4.3 | 24 ± 15 5.6 ± 3.6 (99) |
500* (108) |
1/7 | 8 | wt | Y268C |
| wt/Y268CP0 × nek-4ko/wt | 3 | 9.3 ± 4.5 | 41 ± 10 6.2 ± 2.6 (73) |
0 (76) |
0/5 | na | na | na |
| wt/Y268N × nek-4ko/wt | 3 | 12.6 ± 2.1 | 43 ± 21 18.8 ± 16.7 (4) |
0 (31) |
0/3 | na | na | na |
Sporozoites detected in only one infection trial.
The malaria parasite cytB gene is encoded on maternally inherited mitochondrial DNA (10–12), which implies that known forms of atovaquone resistance must be maternally inherited. We reasoned that the block in mosquito-stage development when atovaquone-resistant parasites attempt to self-fertilize (Table 1) is due to the mutation in the cytochrome b protein in the mitochondrion, which is only carried by the female gamete and effectively renders cytB mutant females sterile. To confirm this, we crossed our three P. berghei atovaquone-resistant lines with parasites genetically modified to be either male sterile (genotype Pbs48|45ko) or female sterile (genotype Pbnek-4ko) (19, 20). If atovaquone resistance is indeed linked to mitochondrial inheritance, we expect normal genetic recombination from crosses to male-deficient parasites but no progeny from attempted crosses to a second female-deficient line. After confirming the phenotypes of the tester parasite lines (18–20), we crossed each of them with our three P. berghei cytB mutants. Crosses to the male-deficient line resulted in recombinant progeny (Table 2), confirming previous results from outcrossing to wild type. Again, though, all progeny from these crosses had wild-type, atovaquone-sensitive cytB genotype (Table 2), so they must have acquired their mitochondria from Pbs48|45ko female gametes (Fig. 2C).
Fig. 2. The genetics of inheritance of mitochondrial DNA–encoded atovaquone resistance mutations in cytB prevent transmission.
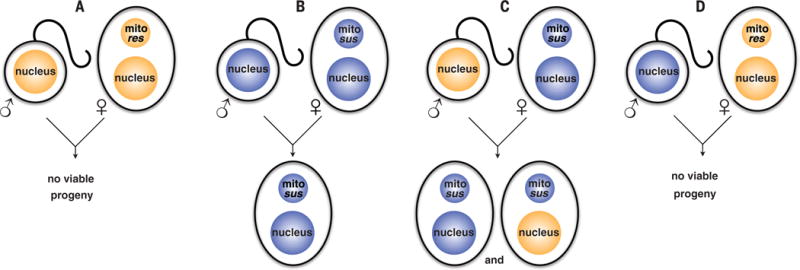
(A) Parasites with atovaquone-resistant cytB genes in their mitochondrion (mito res) cannot generate viable progeny by self-fertilization and cannot transmit. (B) Susceptible (mito sus) parasites produce susceptible progeny when self-fertilizing. (C) Sperm from resistant lines are able to fertilize eggs from susceptible lines and recombinant progeny ensue, but all inherit a susceptible mitochondrion from the female parent. (D) Sperm from susceptible lines fertilizing eggs carrying a mitochondrion-encoded resistance allele. The progeny develop poorly in mosquitoes, are not viable, and cannot transmit.
Crossing atovaquone-resistant lines with the female infertile Pbnek-4ko (20) resulted in no progeny in 16 of 17 attempts to transmit to a naïve mouse (Table 2), largely confirming our hypothesis that the atovaquone-resistant mutants are effectively female sterile. In a single instance, parasites carrying the Y268C mutation were transmitted but with a markedly reduced efficiency (8 days to patency) (Table 2). Three independent cloned lines of the parasites recovered from this sole transmission event (named PbY268C P0) were unable to retransmit when either self-fertilized or backcrossed to Pbnek-4ko parasites (Tables 1 and 2); passage had not improved their transmissibility. In sum, from 44 separate transmission attempts involving 750 mosquito bites, atovaquone resistance transmission was only observed once, and this mutant was unable to transmit further despite seven attempts. We conclude that the cytB mutations in the mitochondrial DNA of atovaquone-resistant rodent malaria parasites render them effectively female sterile and hence largely unable to pass on the resistance gene, which must be inherited through the mitochondrion (Fig. 2).
To determine whether the impact of cytB mutations conferring atovaquone resistance on transmission is similar in the human malaria parasite (P. falciparum), we selected atovaquone-resistant lines by repeated exposure to sublethal concentrations of a drug during in vitro culture (16). Two clones, with different mutations in cytB (M133I and V259L), were established (Table 1). In vitro cultured gametocytes were fed to A. gambiae mosquitoes, and oocyst numbers were counted 7 days after infection (Table 1). The parental line (NF54e) retained normal mosquito infectivity (Table 1). However, the two atovaquone-resistant mutants were severely impaired in their mosquito infectivity and in the number of oocysts produced when infection did occur (Table 1). The severe defect in activation of female gametes phenocopies the reduced number of activated females in the rodent malaria PbY268N (Fig. 1C). We conclude that human P. falciparum malaria parasites carrying atovaquone resistance mutations in cytB are unable to successfully infect A. gambiae mosquitoes, which strongly suggests that human malaria parasites will also be unable to transmit atovaquone-resistant mutations efficiently.
Our findings show that common, clinically relevant atovaquone resistance mutations block transmission of malaria by the mosquito vector and that this phenotype is a consequence of maternal inheritance of the mitochondrion. Why are cytB mutations “genetic time bombs” that affect the mosquito stages of the parasite so severely? All clinically recovered atovaquone resistance mutations are in the quinol oxidase (Qo) site of the mitochondrion-encoded cytochrome b protein and prevent atovaquone from displacing ubiquinone from complex III in the mitochondrial electron transport chain (1, 6–9, 16). Importantly, ubiquinone → cytochrome b electron transport operates at only minimal levels during the malaria parasite blood phase, which relies solely on aerobic glycolysis (21). Nevertheless, nominal transport is essential, primarily as an electron sink for pyrimidine biosynthesis (22). We hypothesize that the modest levels of mitochondrial electron transport during blood phase offer relaxed selection on cytB—which is multicopy and easily mutable (3, 23)—allowing respiration-deficient mutants (8, 24) with reduced atovaquone binding (9) to be readily selected by drug pressure. However, when these mutants switch to the mosquito phase—which relies on full aerobic respiration with an active tricarboxylic acid cycle (25), robust electron transport (17, 26, 27), and mitochondrial adenosine triphosphatase activity (28)—the respiration deficits of the cytB mutants (8, 24) prevent them from completing their development and generating infectious sporozoites. This results in a block of transmission of atovaquone resistance genotypes to new hosts—a block that cannot be overcome by outcrossing because cytB is maternally inherited.
Cytochrome b is thus a rather unique malaria drug target. Its genetics are constrained by maternal inheritance (10–12, 29), there is no recombination of mitochondrial DNA (23), and markedly different selection regimes in the mammalian versus the mosquito hosts (17, 25, 26, 28) all combine to restrict the parasite’s options to disseminate mutations conferring resistance to atovaquone, even though they can arise relatively quickly in patients (2, 3). These constraints likely apply to other cytochrome b targeting drugs currently under development (30–32) and perhaps to drugs targeting the maternally inherited apicoplast (10–12, 29), an endosymbiotic organelle drug target that also has differential activity across the life cycle.
Supplementary Material
Acknowledgments
This work was supported by grants from the National Health and Medical Research Council (Australia); the Australian Research Council; the Indonesian Ministry of Research, Technology, and Higher Education; the U.S. National Institutes of Health (grants AI031478 and RR00052); the Japanese Society for Promotion of Science (JSPS) KAKENHI (grant 23117004 to M. M. and 26253025 to K. K.); and Japanese Science and Technology Agency/Japan International Cooperation Agency Science and Technology Research Partnership for Sustainable Development (SATREPS; no. 10000284 to K. K.). We acknowledge support of the Program for Promotion of Basic and Applied Researches for Innovations in Bio-oriented Industry (BRAIN) and the Science and Technology Research Promotion Program for Agriculture, Forestry, Fisheries and Food Industry. J.E.S. was a JSPS Ph.D. fellow (RONPAKU program) and an Endeavor Fellow from Scope Global, Australia. A.S. is affiliated with TropIQ Health Sciences (http://tropiq.nl). We thank the Johns Hopkins Malaria Research Institute mosquito and parasite core facilities for help with mosquito rearing and P. falciparum cultures, S. Narulitha of the Eijkman Institute for Molecular Biology for the novel P. berghei PbM133I atovaquone resistance mutant, and Walter Reed Army Institute of Research for the P. falciparum NF54e parasites. Data are archived at figshare (https://figshare.com) under doi 10.4225/49/56DE29B278684.
Footnotes
SUPPLEMENTARY MATERIALS
www.sciencemag.org/content/352/6283/349/suppl/DC1
Materials and Methods
References (33–39)
REFERENCES AND NOTES
- 1.Nixon GL, et al. J Antimicrob Chemother. 2013;68:977–985. doi: 10.1093/jac/dks504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maude RJ, Nguon C, Dondorp AM, White LJ, White NJ. Malar J. 2014;13:380. doi: 10.1186/1475-2875-13-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cottrell G, Musset L, Hubert V, Le Bras J, Clain J. Antimicrob Agents Chemother. 2014;58:4504–4514. doi: 10.1128/AAC.02550-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klein EY. Int J Antimicrob Agents. 2013;41:311–317. doi: 10.1016/j.ijantimicag.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ashley EA, et al. N Engl J Med. 2014;371:411–423. doi: 10.1056/NEJMoa1314981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fry M, Pudney M. Biochem Pharmacol. 1992;43:1545–1553. doi: 10.1016/0006-2952(92)90213-3. [DOI] [PubMed] [Google Scholar]
- 7.Srivastava IK, Morrisey JM, Darrouzet E, Daldal F, Vaidya AB. Mol Microbiol. 1999;33:704–711. doi: 10.1046/j.1365-2958.1999.01515.x. [DOI] [PubMed] [Google Scholar]
- 8.Siregar JE, et al. Parasitol Int. 2015;64:295–300. doi: 10.1016/j.parint.2014.09.011. [DOI] [PubMed] [Google Scholar]
- 9.Birth D, Kao WC, Hunte C. Nat Commun. 2014;5:4029. doi: 10.1038/ncomms5029. [DOI] [PubMed] [Google Scholar]
- 10.Creasey A, et al. Mol Biochem Parasitol. 1994;65:95–98. doi: 10.1016/0166-6851(94)90118-x. [DOI] [PubMed] [Google Scholar]
- 11.Creasey AM, et al. Curr Genet. 1993;23:360–364. doi: 10.1007/BF00310900. [DOI] [PubMed] [Google Scholar]
- 12.Vaidya AB, Morrisey J, Plowe CV, Kaslow DC, Wellems TE. Mol Cell Biol. 1993;13:7349–7357. doi: 10.1128/mcb.13.12.7349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jacot D, Waller RF, Soldati-Favre D, MacPherson DA, MacRae JI. Trends Parasitol. 2016;32:56–70. doi: 10.1016/j.pt.2015.09.001. [DOI] [PubMed] [Google Scholar]
- 14.Syafruddin D, Siregar JE, Marzuki S. Mol Biochem Parasitol. 1999;104:185–194. doi: 10.1016/s0166-6851(99)00148-6. [DOI] [PubMed] [Google Scholar]
- 15.Siregar JE, Syafruddin D, Matsuoka H, Kita K, Marzuki S. Parasitol Int. 2008;57:229–232. doi: 10.1016/j.parint.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 16.Korsinczky M, et al. Antimicrob Agents Chemother. 2000;44:2100–2108. doi: 10.1128/aac.44.8.2100-2108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boysen KE, Matuschewski K. J Biol Chem. 2011;286:32661–32671. doi: 10.1074/jbc.M111.269399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Franke-Fayard B, et al. Mol Biochem Parasitol. 2004;137:23–33. doi: 10.1016/j.molbiopara.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 19.van Dijk MR, et al. Cell. 2001;104:153–164. doi: 10.1016/s0092-8674(01)00199-4. [DOI] [PubMed] [Google Scholar]
- 20.Reininger L, et al. J Biol Chem. 2005;280:31957–31964. doi: 10.1074/jbc.M504523200. [DOI] [PubMed] [Google Scholar]
- 21.MacRae JI, et al. BMC Biol. 2013;11:67. doi: 10.1186/1741-7007-11-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Painter HJ, Morrisey JM, Mather MW, Vaidya AB. Nature. 2007;446:88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 23.Preston MD, et al. Nat Commun. 2014;5:4052. doi: 10.1038/ncomms5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fisher N, et al. J Biol Chem. 2012;287:9731–9741. doi: 10.1074/jbc.M111.324319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ke H, et al. Cell Reports. 2015;11:164–174. doi: 10.1016/j.celrep.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hino A, et al. J Biochem. 2012;152:259–268. doi: 10.1093/jb/mvs058. [DOI] [PubMed] [Google Scholar]
- 27.Tanaka TQ, Hirai M, Watanabe Y, Kita K. Parasitol Int. 2012;61:726–728. doi: 10.1016/j.parint.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 28.Sturm A, Mollard V, Cozijnsen A, Goodman CD, McFadden GI. Proc Natl Acad Sci USA. 2015;112:10216–10223. doi: 10.1073/pnas.1423959112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Okamoto N, Spurck TP, Goodman CD, McFadden GI. Eukaryot Cell. 2009;8:128–132. doi: 10.1128/EC.00267-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nam TG, et al. ACS Chem Biol. 2011;6:1214–1222. doi: 10.1021/cb200105d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stickles AM, et al. Am J Trop Med Hyg. 2015;92:1195–1201. doi: 10.4269/ajtmh.14-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong CK, et al. Chem Biol. 2011;18:1602–1610. doi: 10.1016/j.chembiol.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.