

Abstract
The structural basis of simultaneous binding of two or more different drugs by any multidrug-binding protein is unknown and also how this can lead to a noncompetitive, uncompetitive or cooperative binding mechanism. Here, we describe the crystal structure of the Staphylococcus aureus multidrug-binding transcription repressor, QacR, bound simultaneously to ethidium (Et) and proflavin (Pf). The structure underscores the plasticity of the multidrug-binding pocket and reveals an alternative, Pf-induced binding mode for Et. To monitor the simultaneous binding of Pf and Et to QacR, as well as to determine the effects on the binding affinity of one drug when the other drug is prebound, a novel application of near-ultraviolet circular dichroism (UVCD) was developed. The UVCD equilibrium-binding studies revealed identical affinities of Pf for QacR in the presence or absence of Et, but significantly diminished affinity of Et for QacR when Pf is prebound, findings that are readily explicable by their structures. The principles for simultaneous binding of two different drugs discerned here are likely employed by the multidrug efflux transporters.
Keywords: multidrug binding, multidrug resistance, multidrug transporters, near-ultraviolet circular dichroism, QacR
Introduction
Multidrug resistance (MDR) has emerged as a major clinical problem and can arise through a number of mechanisms, including the action of efflux transporters that pump out a wide variety of structurally and chemically dissimilar drugs and cytotoxins (Bax et al, 2000; Walsh, 2003). The polyspecific drug binding by MDR efflux transporters presents not only challenges in terms of drug design but is also an enigma from a structural standpoint. How can one protein recognize such a large array of structurally and chemically dissimilar compounds? The recent structures of the Staphylococcus aureus multidrug-binding transcription repressor, QacR, bound to multiple cationic lipophilic drugs, have elucidated a fundamental mechanism for such binding by a single protein (Schumacher et al, 2001, 2002). In QacR, drug binding induces a coil to helix transition that results in the creation of an expansive drug-binding pocket of approximately 1100 Å3 that is rich in aromatic and acidic residues, the latter of which serve to neutralize the positively charged drugs. The presence of multiple aromatic residues in multidrug-binding pockets is likely universal and has been observed in the multidrug-binding proteins BmrR (Zheleznova et al, 1999) and PXR (Watkins et al, 2001), and recently in the MDR efflux transporter AcrB (Yu et al, 2003).
A notable feature of the QacR drug-binding pocket, also anticipated for other multidrug-binding proteins, is the presence of several ‘mini-pockets' within the larger drug-binding site. These mini-pockets specify different drug-binding loci and provided the first physical evidence, in the form of several snapshots, of the ‘multisite' model of multidrug binding (Schumacher et al, 2001). Additional support for a multisite model comes from the recent lower resolution crystal structures of the MDR transporter, AcrB, bound to four different drugs (Yu et al, 2003). These structures revealed that AcrB contains a huge, exposed drug-binding pocket in which each subunit of the trimer binds one drug (Murakami et al, 2002; Yu et al, 2003). Interestingly, the QacR and AcrB structures suggest that their multiple drug-binding sites could interact, as there are no obstructions or structural barriers between the continuous mini-pockets (Schumacher et al, 2001; Yu et al, 2003). This possibility could explain earlier work on MDR transporters that demonstrated both positive and negative cooperativity between certain drug classes, as well as noncooperative and uncooperative interactions between others (Tamai and Safa, 1991; Kolaczkowski et al, 1996; Dey et al, 1997; Shao et al, 1997; Pascaud et al, 1998; van Veen et al, 1998; Mitchell et al, 1999; Putman et al, 1999; Lewinson and Bibi, 2001; Loo et al, 2003a, 2003b). For example, extensive analysis on drug binding by the MDR transporter, LmrP, demonstrated a variety of double drug-binding interactions, including the competitive inhibition of Hoechst 33342 transport by quinine and verapamil, its noncompetitive inhibition by nicardipin and vinblastine, and its uncompetitive inhibition by TPP+ (Putman et al, 1999). Work on P-glycoprotein (P-gp) suggested that different dihydropyridines bind in sites that must overlap, as the presence of one drug decreased the affinity of a second drug (Pascaud et al, 1998). Recent photoaffinity labelling studies on a series of single cysteine P-gp mutants reveal that the binding site of MTS rhodamine overlaps those of cholchicine and calcein-AM but not Hoechst 33342 or MTS verapamil, and that the latter compound is proximal to the MTS rhodamine site in a common drug-binding pocket (Loo et al, 2003b). Finally, the Escherichia coli MdfA has been shown to bind both TPP+ and chloramphenicol simultaneously, whereby chloramphenicol stimulates TPP+ binding, again suggesting distinct but interacting sites (Lewinson and Bibi, 2001).
Understanding the structural basis of simultaneous binding of two different drugs by MDR proteins is critical for the full elucidation of the mechanism of multidrug recognition and transport, as well as in the development of MDR inhibitors. However, no structures exist for such a complex. Particularly intriguing are those studies suggesting that MDR proteins can bind different drugs simultaneously in overlapping sites (Kolaczkowski et al, 1996; Lewinson and Bibi, 2001; Loo et al, 2003b). Does a malleable drug-binding pocket adjust to permit the binding of the second drug, or do the drugs reorient themselves and make slightly different interactions to accommodate each other or a combination of both? To address this issue, we determined the structure of QacR bound simultaneously to two dissimilar drugs, proflavin (Pf) and ethidium (Et). In addition, we developed a novel application of near-ultraviolet circular dichroism (UVCD) to measure QacR–drug-binding constants under a variety of conditions and to monitor the simultaneous binding of Pf and Et to QacR in solution.
Results and discussion
Structure determination of QacR–Pf and a QacR–Pf–Et ternary complex
The structure of QacR bound to Pf (3,6-diaminoacridine), which is a bacteriostatic compound that is used as an antiseptic, was solved and refined to an Rwork of 23.2% and Rfree of 29.2% at 2.89 Å resolution. As observed in the structures of all other QacR–drug complexes, Pf binds with a stoichiometry of one drug per QacR dimer (Schumacher et al, 2001). As a critical component of binding, the Pf stacks with the side chains of residues Trp61 and Tyr93 (Figure 1A). These aromatic residues define the ‘Rhodamine 6G (R6G)' subsite of QacR (Schumacher et al, 2001). van der Waals contacts and hydrogen bonds to Pf are provided by the side chains of residues Leu54, Ile99 and Tyr103 and the side chains of residues Gln96 and Thr89, respectively. The delocalized positive charge of Pf is neutralized by QacR residues Glu57 and Glu58, which sandwich the Pf ring system.
Figure 1.
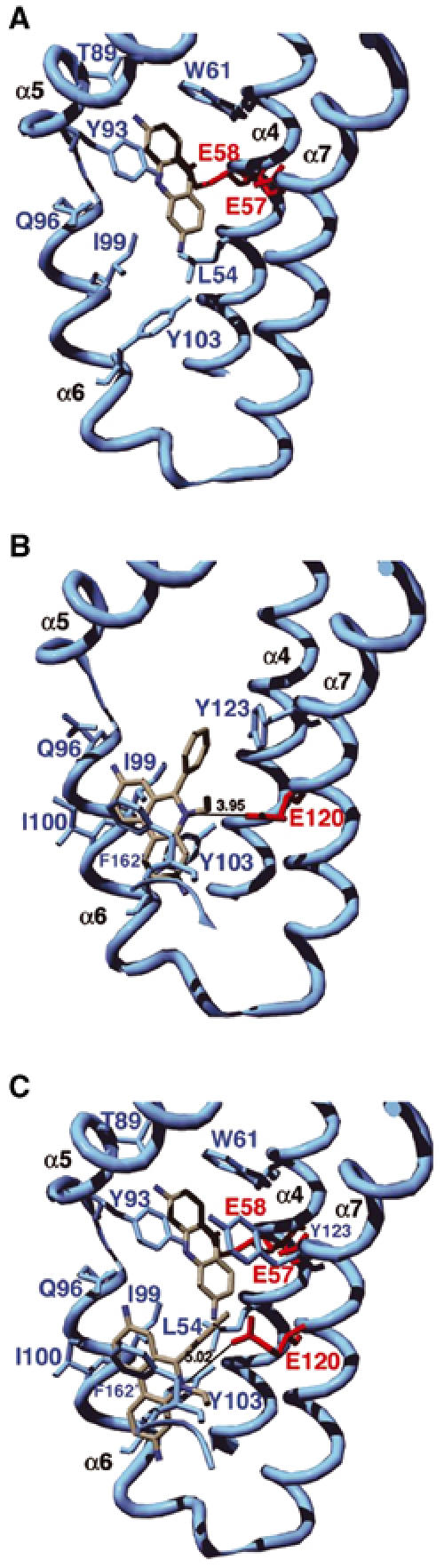
Ribbon diagrams of the drug-binding pocket of QacR. (A) QacR–Pf binary complex. (B) QacR–Et binary complex (Schumacher et al, 2001). (C) QacR–Pf–Et ternary complex. For clarity, only key drug-binding residues are shown. Acidic residues involved in drug neutralization are colored red and all other residues are colored light blue. Carbon, nitrogen and oxygen atoms of the drug molecules are colored white, blue and red, respectively. These figures and Figures 2A, C and 3A, B were made with SwissPdbViewer (Guex and Peitsch, 1997) and rendered with POVRAY (POV-Ray, Persistence of Vision Raytracer, version 3.1 (www.povray.org)).
The finding that Pf binds in the R6G subsite overcame a major obstacle in our efforts to crystallize a two-drug–QacR ternary complex. Although the structures of the QacR–R6G and QacR–Et binary complexes revealed that R6G and Et bind in distinct but partially overlapping mini-pockets of the multidrug-binding site (Schumacher et al, 2001), their superimposition showed that the two drugs would clash sterically (1.3 Å closest approach), indicating that these drugs would be unlikely to bind simultaneously (Figures 1B and 2A). By contrast, similar superimposition of the QacR–Pf and QacR–Et binary structures revealed that the closest approach of these drugs was 2.6 Å (Figure 2A), thus presenting a possible structural system for understanding simultaneous binding of two different drugs by a multidrug-binding protein.
Figure 2.
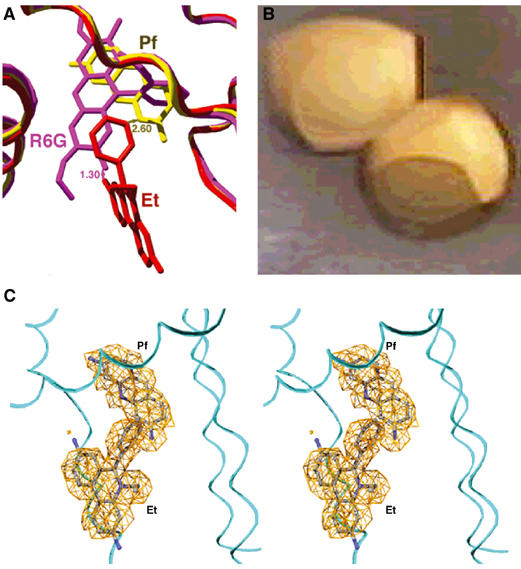
Simultaneous binding of Et and Pf by QacR. (A) Superimposition of QacR–R6G (magenta), QacR–Et (red) and QacR–Pf (yellow) structures showing the closest approaches of R6G–Et (1.3 Å) and Pf–Et (2.6 Å). (B) Crystals of the orange QacR–Pf–Et complex. (C) Stereoview of the simulated annealing composite omit map (orange mesh) in which Pf and Et were omitted (contoured at 3.0 σ). Carbon, nitrogen and oxygen atoms are colored gray, blue and red, respectively.
The QacR–Pf–Et complex was prepared and crystallized as described (Materials and methods; Schumacher et al, 2001). The presence of both drugs in the resulting crystals was suggested by their bright orange color, apparently resulting from QacR binding simultaneously to the red drug, Et, and the yellow drug, Pf (Figure 2B). Difference Fourier electron density maps confirmed the presence of both drugs in the multidrug-binding pocket, with one drug-binding pocket filled per dimer, again consistent with the established QacR–drug-binding stoichiometry (Schumacher et al, 2001). The QacR–Pf–Et structure was refined to Rwork and Rfree of 22.3% and 27.3%, respectively, to 2.96 Å resolution (Figures 1C, 2C and 3A, Table I). Superimposition of the QacR–Pf–Et ternary complex structure and the QacR–Et and QacR–Pf binary complex structures results in r.m.s.d.'s of less than 0.70 Å, demonstrating that no additional global structural changes are caused by dual drug binding to QacR. Moreover, the volume of the multidrug-binding pocket of the QacR–Pf–Et ternary complex does not need to expand to accommodate both drugs and remains ∼1100 Å3 (Brady and Stouten, 2000).
Figure 3.
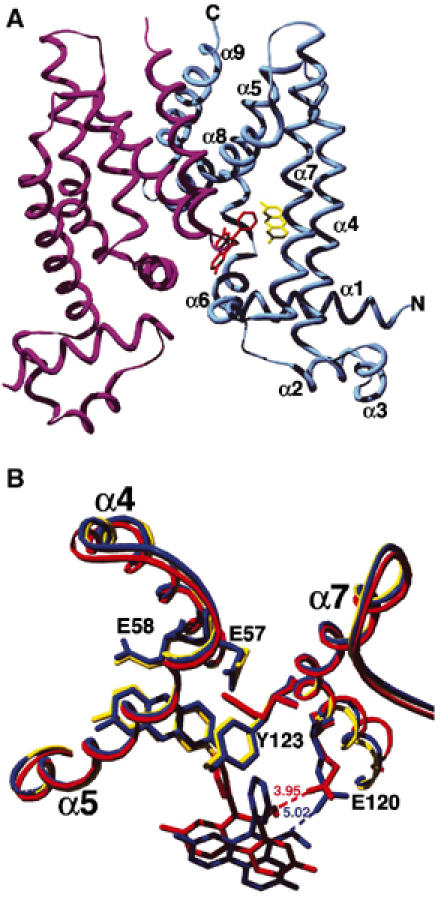
The structure of the ternary QacR–Pf–Et structure. (A) Ribbon diagram of the QacR–Pf–Et ternary complex. The nine α helices are labelled as are the N- and C-termini. The bound Et is shown as red sticks and the bound Pf as yellow sticks. (B) Superimposition of binary QacR–Et (red), binary QacR–Pf (yellow) and ternary QacR–Pf–Et (blue) complexes showing the reorientation of Et in the ternary complex.
Table 1.
Selected data collection and refinement statistics
| QacR–Pf | QacR–Pf–Et | |
|---|---|---|
| Unit cell constants (Å) | a=b=172.3, c=94.8 | a=b=171.8, c=94.6 |
| Resolution (Å) | 47.79–2.89 | 121.48–2.96 |
| Overall Rsym (%)a | 6.5 (44.0)b | 7.8 (38.9) |
| Overall I/σ (I) | 9.4 (2.0) | 8.0 (1.8) |
| Total reflections (#) | 285902 | 205502 |
| Unique reflections (#) | 32965 | 31274 |
| Completeness (%) | 97.6 | 98.7 |
| Rwork/Rfree (%)c | 23.2/29.2 | 22.3/27.3 |
| R.m.s.d. | ||
| Bond lengths (Å) | 0.007 | 0.013 |
| Bond angles (deg) | 1.23 | 1.47 |
| B-values (Å2) | 2.40 | 2.60 |
| Ramachandran analysis | ||
| Most favored (%/#) | 87.9/615 | 88.6/620 |
| Additional allowed (%/#) | 10.9/76 | 10.1/71 |
| Generously allowed (%/#) | 0.6/4 | 0.6/4 |
| Disallowed (%/#) |
0.7/5 |
0.7/5 |
| a Rsym=∑∑∣Ihkl−Ihkl(j)∣/∑NIhkl, where Ihkl(j) is the observed intensity and Ihkl is the final average value of intensity. | ||
| b Highest resolution shell data parameters are in parentheses. | ||
| c Rwork=∑∥Fobs∣−∣Fcalc∥/∑∣Fobs∣ and Rfree=∑∥Fobs∣−∣Fcalc∥/∑∣Fobs∣, where all reflections belong to a test set of 10% data randomly selected in CNS. | ||
Dual drug binding effected by creation of new binding pocket
In the QacR–Pf–Et ternary structure, Pf remains bound in the R6G pocket and protein–drug contacts are nearly identical to those observed in the QacR–Pf binary complex (Figure 1A and C, Table II). Again, the aromatic side chains of residues Trp61 and Tyr93 sandwich the drug and the side chains of Leu54, Ile99 and Tyr103 make van der Waals contacts. The hydrogen bonds provided by Gln96 and Thr89 are also retained and the side chains of residues Glu57 and Glu58 again neutralize the positive charge of the drug. The retention of the identical Pf-binding mode in the ternary and the binary complexes is substantiated by superimposition of the two structures, which shows that both Pf and the side chains that contact Pf are essentially identically disposed (Figure 3B, Table II). By sharp contrast, the Et molecule in the ternary structure has shifted considerably in the pocket compared to its location in the QacR–Et binary structure (Figures 1B, C and 3B). In its new location, Et does not clash with Pf but now engages in favorable van der Waals interactions with Pf, with a closest ring-to-ring approach of 4.0 Å. Thus, Pf appears to dominate binding and, once bound, does not adjust in the pocket upon the binding of Et. Rather, the Et molecule slides into a new pocket that is close to its preferred binding site (see below) but shifted.
Table 2.
QacR–drug interaction distancesa
| Et (binary) | Residue Et (Å) | Et (ternary) | Residue Et (Å) | Pf (binary) | Residue Pf (Å) | Pf (ternary) | Residue Pf (Å) |
|---|---|---|---|---|---|---|---|
| Q96 | 2.9 | Q96 | 3.0 | L54 | 3.8 | L54 | 3.9 |
| I99 | 4.3 | I99 | 5.1 | E57 | 3.9 | E57 | 4.1 |
| I100 | 3.4 | I100 | 3.6 | E58 | 3.6 | E58 | 3.2 |
| Y103 | 3.3 | Y103 | 3.1 | W61 | 3.4 | W61 | 3.6 |
| E120 | 4.0 | E120 | 5.0 | T89 | 2.8 | T89 | 2.8 |
| Y123 | 3.4 | Y123 | 3.5 | Y93 | 3.6 | Y93 | 3.8 |
| F162′ |
3.7 |
F162′ |
3.8 |
I99 |
4.1 |
I99 |
4.0 |
| a The distance indicates the shortest interatomic distance between each residue and the ligand. | |||||||
The new binding mode of Et in the ternary complex is optimized to maintain key hydrophobic contacts, particularly aromatic stacking interactions. Indeed, the distances from the side chains of Tyr103 and Phe162′ (where prime indicates the other subunit) to Et in the binary and ternary complexes remain the same, as do the positions of their side chains (Table II). Moreover, the aromatic moieties of Tyr103 and Phe162′ encase the positively charged nitrogen of Et, thus providing favorable cation–π interactions (Ma and Dougherty, 1997; Gallivan and Dougherty, 1999). The importance of such contacts in charge neutralization of this cationic drug is underscored by their repeated observation, most recently in the structure of the QacR–pentamidine complex (Murray et al, 2004). A significant change is seen in the Et binding site as a consequence of Pf binding, whereby the side chain of Tyr123 rotates to a position that is identical to that which it assumes in the QacR–Pf binary structure, again underscoring the dominance of Pf binding (Figure 3B). Despite this rotation, Tyr123 can still stack against the Et phenyl group (Figures 1B, C and 3B, Table II). Inspection of multiple QacR–cationic drug structures reveals that the Tyr123 side chain adopts primarily one of two conformations to maintain its drug-stacking interactions (Schumacher et al, 2001; Murray et al, 2004). The choice of conformer depends upon whether a drug is bound in the R6G or Et pocket and suggests that the role of this side chain is that of a drug subsite-locking mechanism. van der Waals contacts from residues Ile99 and Ile100 to Et are also maintained in the QacR–Et binary and QacR–Pf–Et ternary complexes. However, the Ile100 contact has been lengthened significantly in the ternary complex (Table II).
A notable finding to emerge from the QacR–Pf–Et ternary structure is the 1.0 Å increase in distance (4.0 Å in the binary complex to 5.0 Å in the ternary complex) from the side-chain carboxylate of the drug-neutralizing residue Glu120 to the positive charge of Et (Figure 3B, Table II). Thus, in the ternary complex, Et binding preserves primarily hydrophobic and aromatic contacts at the expense of charge–charge interactions. The primary utilization of aromatic residues in ligand binding is reminiscent of the complementarity-determining regions of antibodies (James et al, 2003). Moreover, the critical role that aromatic residues play in drug binding has been demonstrated for PXR, which binds to chemically diverse hormone and hormone-like compounds, as mutation of selected aromatic residues dramatically altered the ability of this protein to bind certain compounds (Watkins et al, 2001, 2003). Perhaps it is not surprising that aromatic residues are so critical in multidrug binding, as not only are they essential for the construction of the multidrug-binding pocket but are also required for drug-binding conformational transitions (Zheleznova et al, 1999; Schumacher et al, 2001, 2002). Moreover, the ‘flexibility' afforded by aromatic residues via rotation about their Cα–Cβ bonds also allows a drug to slide or shift in its pocket while still maintaining stacking interactions.
Use of near-UVCD to measure simultaneous drug-binding affinities
From the analysis of these and previous QacR–drug structures, several predictions can be made with regard to ligand-binding affinity and mode. First, the drugs R6G and Pf would exhibit competitive binding. Second, Pf binding, which is positioned identically in its binary and ternary complexes, would affect the binding affinity of Et. Given the increased distance between the Glu120 and Ile99 side chains and Et in the ternary complex, a decreased binding affinity of Et for QacR in the presence of Pf would be anticipated in comparison with the affinity of Et for QacR in the absence of Pf. On the other hand, Et should have little effect on Pf binding affinity. To address the energetic consequences of QacR–Pf binding on Et binding affinity, as well as QacR–Et binding on Pf binding affinity, we developed a novel ligand-binding assay that utilizes near-UVCD spectroscopy (Kelly and Price, 2000; Jordan et al, 2003). This assay exploited the saturable changes in the near-UVCD spectra, observed upon binding the three different drugs (R6G, Pf and Et) to QacR (Figure 4A), which were found to occur at different wavelengths for each drug. Using this technique, not only could equilibrium dissociation constants be quantified for QacR binding each drug individually, but also such constants could be obtained for simultaneous binding of more than one drug. It is worth noting that the induction of ellipticity in the near-UVCD spectrum is strictly an empirical phenomenon that cannot reliably be predicted a priori from structural knowledge of either the protein or the ligand and must, therefore, be measured directly for each specific case.
Figure 4.
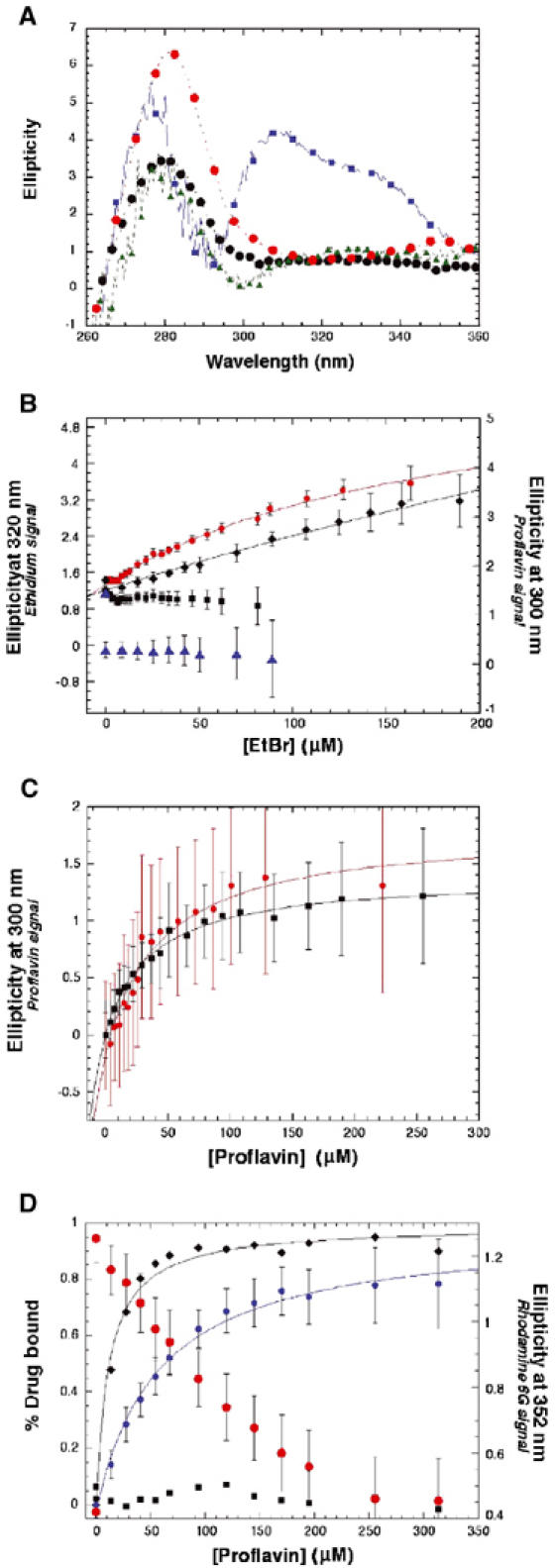
Near-UVCD analysis of single- and dual-drug titrations of QacR. All data were corrected for dilution and any necessary background contributions from the drugs used in the respective titration. Where shown, error bars represent one standard deviation of 600 measurements of the solution in the cuvette at each concentration of drug. Dissociation constants were obtained through a least-squares regression analysis (Kaleidagraph), which are shown as solid lines. (A) Near-UVCD spectra of drug-free and drug-bound QacR: drug-free QacR (black circles); with 25 μM R6G added (red circles); with 62 μM Et added (blue squares); with 86 μM Pf added (green triangles). Spectra were obtained at 0.5 nm resolution averaged over 2 s intervals with a 100 ms time constant. The drug-specific changes (285 and 352 nm for R6G, 300 nm for Pf and 320 nm for Et) were used to monitor drug binding in the single- and dual-drug-binding experiments in panels B–D and in Supplementary Figure 2. (B) QacR (2.5 μM dimer) binding Et in the absence of Pf (red circles, Kd=186±25 μM) and in the presence of 150 μM Pf (black diamonds, Kd ∼1 mM) plotted as the change in ellipticity (mdeg) at 320 nm (left y-axis) measured as in panel D. The change in ellipticity (mdeg) at 300 nm (right y-axis), indicating the presence of QacR-bound Pf, is shown for the Et titration with 0 μM Pf (black squares) and for the Et titration in the presence of 150 μM Pf (blue triangles). Note the presence of the Pf-bound signal throughout the course of the Et titration. Due to limits of solubility and diminishing signal-to-noise ratios at high concentrations, the Et titrations could not be carried out to saturation. (C) QacR (2.5 μM dimer) binding to Pf in the absence of Et (red circles, Kd=42±11 μM) and in the presence of 75 μM Et (black squares, Kd=35±4 μM) plotted as the change in ellipticity (mdeg) at 300 nm measured as in panel D. (D) QacR (7 μM dimer) binding to Pf in the absence of R6G (black diamonds) and in the presence of 17 μM R6G (blue circles) plotted as percent saturation (left y-axis) as determined relative to the asymptote of a least-squares regression fit (solid lines) of the raw data. Ellipticity (mdeg) at 352 nm (right y-axis), indicating the presence of QacR-bound R6G, is shown for the Pf titration in the absence of R6G (black squares) and for the Pf titration in the presence of 17 μM R6G (red circles). The decrease in R6G bound to QacR is proportional to the saturation of QacR with Pf crossing at ∼50%. The bound states of Pf and R6G were measured as the changes in ellipticity (mdeg) relative to drug-free QacR (7 μM) at 300 and 352 nm, respectively, averaged for 60 s with a 100 ms time constant at each increasing concentration of Pf. Note that the different affinities of QacR for Pf that are observed here are the result of the different experimental conditions employed. Using 2.5 μM QacR dimer, the affinity of QacR for Pf is Kd=42±11 μM. The Pf/R6G competition experiments necessitated higher protein concentrations (7 μM dimer) and the resulting Kd for the QacR–Pf complex is 11±1 μM. In the presence of 17 μM R6G, the Kd of Pf for QacR is increased five-fold (56±6 μM), a result expected for competitive binding with a higher affinity ligand.
Control experiments comparing the results of the near-UVCD assay with those obtained by other biochemical techniques were performed for QacR binding R6G. Titrations of QacR dimer at 2.5 μM with R6G, as measured by the change in ellipticity (mdeg) at wavelengths empirically determined to be specific for R6G binding, yielded a dissociation constant of 0.80±0.09 μM, and a similar titration of QacR dimer at 9.9 μM yielded a specific activity curve clearly indicating a 1:1 drug:dimer stoichiometry, consistent with previous data (Schumacher et al, 2001) (Supplementary Figure 1A and B). Isothermal titration calorimetry (ITC) analysis of QacR binding R6G under identical conditions yielded the same 1:1 drug:dimer stoichiometry and a dissociation constant of 0.79±0.09 μM (Supplementary Figure 2B). Titrations of R6G (1 nM) with QacR (0–3.4 μM dimer) under identical conditions, measured as the change in fluorescence polarization of R6G, resulted in a dissociation constant of 0.61±0.02 μM (Supplementary Figure 2A). Thus, the near-UVCD analysis of drug binding by QacR proved to be quite robust and reproduced the equilibrium dissociation constants and the drug:protein stoichiometry of QacR for R6G, which were determined by several different methods. Subsequent titrations of QacR individually with Et and Pf measured as above by near-UVCD at wavelengths specific for each drug resulted in dissociation constants of 186±25 and 42±11 μM, respectively (Figure 4B and C).
Dual drug-binding experiments were performed in which the drug-specific effects on the near-UVCD spectrum were exploited to monitor simultaneously the bound state of two different drugs. These experiments were performed by prebinding QacR with drug A and titrating with drug B while measuring the near-UVCD signal at the two drug-specific wavelengths such that the bound state of drug A could be monitored and the apparent dissociation constant of drug B in the presence of drug A could be measured. Titration of Pf into R6G-saturated QacR (17 μM R6G, 7 μM QacR dimer) demonstrated both the proportional displacement of bound R6G as Pf bound to QacR, and a five-fold decrease in the dissociation constant for Pf binding in the presence of saturating amounts of R6G: Kd=11±1 μM (in the absence of R6G); Kd=56±6 μM (in the presence of 17 μM R6G) (Figure 4D). The reverse experiment, that is, titration of R6G into Pf-saturated QacR (25 μM Pf, 7 μM QacR dimer), similarly demonstrated the displacement of Pf as R6G was bound (Supplementary Figure 1D). These experiments reveal that QacR binds Pf and R6G competitively in accord with our structural analysis. By contrast, performing the same dual drug experiments using Pf paired with Et revealed simultaneous binding by these compounds. Titration of Et into Pf-saturated QacR (150 μM Pf, 2.5 μM QacR dimer) demonstrated Et binding without any detectable displacement of bound Pf (Figure 4B). This result is consistent with the finding that the presence of 75 μM Et did not affect the dissociation constant of Pf: Kd=42±11 μM (in the absence of Et); Kd=35±4 μM (in the presence of Et) (Figure 4C). As anticipated from the structure of the ternary complex, the lengthening of the Glu120–Et contact when Pf is bound lowered the affinity of QacR for Et (Figure 4B). Thus, whereas the presence of Et does not affect the affinity of QacR for Pf, Pf strongly affects the affinity of QacR for Et. Moreover, these data show unequivocally that Pf and Et bind QacR simultaneously in solution.
The structure of the QacR–Pf–Et ternary complex reveals for the first time the structural mechanism for simultaneous binding of two different drugs by a multidrug-binding protein. Moreover, this structure, together with relevant binary QacR–drug complex structures and near-UVCD binding data, provides significant insight into the possible structural underpinnings of competitive, noncompetitive, uncompetitive and cooperative multidrug binding demonstrated by the MDR transporters, MdfA, LmrP, QacA and P-glycoprotein (Tamai and Safa, 1991; Kolaczkowski et al, 1996; Dey et al, 1997; Shao et al, 1997; Pascaud et al, 1998; van Veen et al, 1998; Putman et al, 1999; Mitchell et al, 1999; Lewinson and Bibi, 2001; Loo et al, 2003a, 2003b). The most easily understood category is competitive binding, as exemplified by R6G and Pf interacting with QacR, where the two drugs bind the identical mini-pocket and sterically interfere with the binding of the other. Noncompetitive binding is exhibited by Pf and Et, which display partial overlap of their binding pockets. Perhaps this case is described more accurately as uncompetitive because the exact Et-binding site found in the ternary complex is not used in the absence of Pf. Regardless of its exact characterization, the simultaneous binding of these two drugs is effected by the plasticity of the drug-binding pocket, aided primarily by the presence of a key aromatic residue, which by simple side-chain rotation permits Et to slide into a new site that is proximal to its binary complex-binding site. In this new location, Et still maintains key contacts to the protein, but suboptimally with a consequential reduction in binding affinity. However, the reverse case, in which binding of the first drug leads to the formation of a binding site of a second drug that is better than that taken in its binary complex, can be readily envisioned and would appear as cooperative binding. Thus, simple structural changes can explain seemingly complicated biochemical behavior. We anticipate that the multidrug transporters will utilize similar principles in their simultaneous binding of two or more different drugs.
Materials and methods
Protein preparation, crystallization, data collection and structure determination
QacR(C72A/C141S) protein was purified, subjected to reductive alkylation, mixed with drugs to give a final drug concentration of 100 μM, and crystallized as described (Grkovic et al, 1998; Schumacher et al, 2001). The QacR(C72A/C141S) protein displays wild-type DNA and drug-binding affinities, but, unlike wild-type QacR, does not aggregate. To crystallize the double drug–QacR ternary complex, reductively alkylated QacR was mixed with 100 μM Pf (final concentration) and allowed to incubate at 21°C for at least 3 h. After this time, 100 μM Et (final concentration) was added and the complex was crystallized by hanging drop vapor diffusion. All QacR–drug crystals are isomorphous and take the tetragonal space group P42212 (Table I). X-ray intensity data for the QacR–Pf and QacR–Pf–Et complex crystals were collected at the Stanford Synchrotron Radiation Laboratory beamline 9-1 and 11-1, respectively, at 100 K. All data were processed with MOSFLM. To determine the structures of the QacR–Pf and QacR–Pf–Et complexes, the QacR–dequalinium structure (Schumacher et al, 2001), which is the highest resolution QacR–drug complex structure (2.54 Å), was used as a starting model with the dequalinium and solvent removed. For each complex, the starting model was subjected to rigid body refinement followed by multiple cycles of simulated annealing (SA) and positional/thermal parameter refinement in CNS and rebuilding in O (Jones et al, 1991; Brünger et al, 1998). After initial refinement, the electron density of each drug was clearly visualized and Pf or Pf and Et were built into the model and subsequently subjected to refinement. The final refinement and model statistics are given in Table I.
Near-UVCD spectroscopy drug-binding experiments
QacR(C72A/C141S) was dialyzed 48 h against a UVCD buffer of 300 mM NaCl, 50 mM imidazole, 5% glycerol, 50 mM Tris (pH 7.65). Protein concentration was quantified using the Advanced Protein Assay Reagent (Cytoskeleton) referenced to a BSA standard curve. R6G, Et and Pf stock solutions were prepared at saturating concentrations in 10 ml aliquots of the remaining CD dialysate buffer and their final concentrations determined spectroscopically: 232 μM, 1.1 mM and 3.7 mM, respectively. Near-UVCD data were obtained with an AVIV 215 circular dichroism spectrometer with the bandwidth at 8.0 nm and with constant stirring of the sample in a 1 cm pathlength quartz cuvette at 25°C. Wavelength scans were performed from 360 to 260 nm with 0.5 nm resolution, averaged for 2 s/interval with a 100 ms time constant. Time-based measurements were collected at a single wavelength for 60 s with a 100 ms time constant and recorded as an average ellipticity±one standard deviation. Measurements were performed on 2.5 ml samples of 2.5–10.0 μM QacR dimer in 300 mM NaCl, 50 mM imidazole, 5% glycerol and 50 mM Tris (pH 7.65).
Single drug binding curves were obtained by collecting time-based measurements at the drug-specific wavelength(s): 285, 300 and 320 nm for R6G, Pf and Et, respectively, at increasing drug concentrations. Dual drug-binding experiments were performed by titrating drug ‘X' into a solution containing a known concentration of drug ‘Y' bound to QacR. At each increasing concentration of drug X, time-based measurements were performed at the wavelengths indicative of the binding of both drug X and drug Y: 352, 300 and 320 nm for R6G, Et and Pf, respectively. Standard curves were generated for each drug at each of the desired wavelengths to correct for background noise, which were found to be linear and varied depending on drug and wavelength from <1% for R6G at 295 nm to ∼40% for Et at 320 nm of the total measured change in ellipticity (mdeg) at the conclusion of the titration. All single- and double-drug-binding measurements were corrected for both drug-related background signal and sample dilution and plotted (±one standard deviation) as a function of ligand concentration.
Supplementary Material
Supplemental Figure 1.
Supplemental Figure 2.
Acknowledgments
MAS is a Burroughs Wellcome Career Development Awardee (992863). This work was supported by NIH grant AI 48593 (to RGB). Intensity data collection at the Stanford Synchrotron Radiation Laboratory (SSRL) was carried out under the auspices of the SSRL biotechnology program, which is supported by the National Institutes of Health, National Center for Research Resources, Biomedical Technology Program, and by the Department of Energy, Office of Biological and Environmental Research.
Data deposition The coordinates and structure factors for the QacR–Pf binary complex and QacR–Pf–Et ternary complex have been deposited with the Protein Data Bank under the accession codes 1QVT and 1QVU, respectively.
References
- Bax R, Mullan N, Verhoef J (2000) The millennium bugs—the need for and development of new antimicrobiols. Int J Antimicrob Agents 16: 51–59 [DOI] [PubMed] [Google Scholar]
- Brady GP Jr, Stouten PF (2000) Fast prediction and visualization of protein binding pockets with PASS. J Comput Aided Mol Des 14: 383–401 [DOI] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Crosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Dey S, Ramachandra M, Pastan I, Gottesman MM, Ambudkar SV (1997) Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc Natl Acad Sci USA 94: 10594–10599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallivan JP, Dougherty DA (1999) Cation–pi interactions in structural biology. Proc Natl Acad Sci USA 96: 9459–9464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grkovic S, Brown MH, Roberts J, Paulsen IT, Skurray RA (1998) QacR is a repressor protein that regulates expression of the Staphylococcus aureus multidrug efflux pump QacA. J Biol Chem 273: 17673–18665 [DOI] [PubMed] [Google Scholar]
- Guex N, Peitsch MC (1997) SWISS_MODEL and Swiss-Pdb viewer: an environment for comparative protein modelling. Electrophoresis 18: 2714–2723 [DOI] [PubMed] [Google Scholar]
- James LC, Roversi P, Tawfik DS (2003) Antibody multispecificity mediated by conformational diversity. Science 299: 1362–1367 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Jordan F, Nemeria NS, Zhang S, Yan Y, Arjunan P, Furey W (2003) Dual catalytic apparatus of the thiamin diphosphate coenzyme: acid-base via the 1′,4′-iminopyrimidine tautomer along its electrophile role. J Am Chem Soc 125: 12732–12738 [DOI] [PubMed] [Google Scholar]
- Kelly SM, Price NC (2000) The use of circular dichroism in the investigation of protein structure and function. Curr Prot Pept Sci 1: 349–384 [DOI] [PubMed] [Google Scholar]
- Kolaczkowski M, van der Rest M, Cybularz-Kolaczkowska A, Soumillion JP, Konings WN, Goffeau A (1996) Anticancer drugs, ionophoric peptides, and steroids as substrates of the yeast multidrug transporter Pdr5p. J Biol Chem 271: 31543–31548 [DOI] [PubMed] [Google Scholar]
- Lewinson O, Bibi E (2001) Evidence for simultaneous binding of dissimilar substrates by the Escherichia coli multidrug transporter MdfA. Biochemistry 40: 12612–12618 [DOI] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC, Clarke DM (2003a) Simultaneous binding of two different drugs in the pocket of the human multidrug resistance P-glycoprotein. J Biol Chem 278: 39706–39710 [DOI] [PubMed] [Google Scholar]
- Loo TW, Bartlett MC, Clarke DM (2003b) Methanethiosulfonate derivatives of rhodamine and verapamil activate human P-glycoprotein at different sites. J Biol Chem 278: 50136–50141 [DOI] [PubMed] [Google Scholar]
- Ma JC, Dougherty DA (1997) The Ca+ ion-pi interaction. Chem Rev 97: 1303–1324 [DOI] [PubMed] [Google Scholar]
- Mitchell BA, Paulsen IT, Brown MH, Skurray RA (1999) Bioenergetics of the staphylococcal multidrug export protein QacA. Identification of distinct drug binding sites for monovalent and bivalent cations. J Biol Chem 274: 3541–3548 [DOI] [PubMed] [Google Scholar]
- Murakami S, Nakashima R, Yamashita E, Yamaguchi A (2002) Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 419: 587–593 [DOI] [PubMed] [Google Scholar]
- Murray DS, Schumacher MA, Brennan RG (2004) Crystal structure of QacR–diamidine complexes reveal additional multidrug binding modes and a novel mechanism of drug charge neutralization. J Biol Chem 279: 14365–14371 [DOI] [PubMed] [Google Scholar]
- Pascaud C, Garrigos M, Orlowski S (1998) Multidrug resistance transporter P-glycoprotein has distinct but interacting binding sites for cytotoxic drugs and reversing agents. Biochem J 333: 351–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putman M, Koole LA, van Veen HW, Konings WN (1999) The secondary multidrug transporter LmrP contains multiple drug interaction sites. Biochemistry 38: 13900–13905 [DOI] [PubMed] [Google Scholar]
- Schumacher MA, Miller MC, Grkovic S, Brown MH, Skurray RA, Brennan RG (2001) Structural mechanism of QacR induction and multidrug recognition. Science 294: 2158–2163 [DOI] [PubMed] [Google Scholar]
- Schumacher MA, Miller MC, Grkovic S, Brown MH, Skurray RA, Brennan RG (2002) Structural basis for cooperative DNA binding by two dimers of the multidrug binding protein QacR. EMBO J 21: 1210–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y, Ayesh S, Stein WD (1997) Mutually co-operative interactions between modulators of P-glycoprotein. Biochim Biophys Acta 1360: 30–38 [DOI] [PubMed] [Google Scholar]
- Tamai L, Safa AR (1991) Azidopine noncompetitively interacts with vinblastine and cyclosporin A binding to P-glycoprotein in multidrug resistant cells. J Biol Chem 266: 16796–16800 [PubMed] [Google Scholar]
- van Veen HW, Callaghan R, Soceneantu L, Sardini A, Konings WN, Higgins CF (1998) A bacterial antibiotic-resistance gene that complements the human multidrug-resistance P-glycoprotein gene. Nature 391: 291–295 [DOI] [PubMed] [Google Scholar]
- Walsh C (2003) Antibiotics. Washington, DC: ASM Press [Google Scholar]
- Watkins RE, Maglich JM, Moore LB, Wisely GB, Noble SM, Davis-Searles PR, Lambert MH, Kliewer SA, Redinbo MR (2003) 2.1 Å crystal structure of human PXR in complex with the St. John's wort compound hyperforin. Biochemistry 42: 1430–1438 [DOI] [PubMed] [Google Scholar]
- Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR (2001) The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science 292: 2329–2333 [DOI] [PubMed] [Google Scholar]
- Yu EW, Aires JR, Nikaido H (2003) AcrB multidrug efflux pump of Escherichia coli: composite substrate-binding cavity of exceptional flexibility generates its extremely wide substrate specificity. J Bacteriol 185: 5637–5664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheleznova EE, Markham PN, Neyfakh AA, Brennan RG (1999) Structural basis of multidrug recognition by BmrR, a transcription activator of a multidrug transporter. Cell 96: 353–356 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1.
Supplemental Figure 2.