

Abstract
Retinoids and interferons are signaling molecules with pronounced anticancer activity. We show that in both acute promyelocytic leukemia and breast cancer cells the retinoic acid (RA) and interferon signaling pathways converge on the promoter of the tumoricidal death ligand TRAIL. Promoter mapping, chromatin immunoprecipitation and RNA interference reveal that retinoid-induced interferon regulatory factor-1 (IRF-1), a tumor suppressor, is critically required for TRAIL induction by both RA and IFNγ. Exposure of breast cancer cells to both antitumor agents results in enhanced TRAIL promoter occupancy by IRF-1 and coactivator recruitment, leading to strong histone acetylation and synergistic induction of TRAIL expression. In coculture experiments, pre-exposure of breast cancer cells to RA and IFNγ induced a dramatic TRAIL-dependent apoptosis in heterologous cancer cells in a paracrine mode of action, while normal cells were not affected. Our results identify a novel TRAIL-mediated tumor suppressor activity of IRF-1 and suggest a mechanistic basis for the synergistic antitumor activities of certain retinoids and interferons. These data argue for combination therapies that activate the TRAIL pathway to eradicate tumor cells.
Keywords: apoptosis, interferon, IRF, retinoic acid, tumor suppressor
Introduction
The cancer therapeutic and preventive action of retinoids is very well established in a large number of in vitro and animal systems, and addition of retinoic acid (RA) to the therapeutic protocol of acute promyelocytic leukemia (APL) has led to a dramatically increased cure rate of APL patients (Melnick and Licht, 1999; Minucci and Pelicci, 1999; Altucci and Gronemeyer, 2001; Piazza et al, 2001; Altucci and Gronemeyer, 2004). APL originates from multiple signaling aberrations caused by the PML-RARα oncofusion. These comprise in addition to a block of differentiation due to the aberrant formation of heterochromatin over RA target genes (Minucci and Pelicci, 1999), increased blast survival due to the inhibition of p53 by deacetylation and increased self-renewal of stem cells (Pearson et al, 2000; Alcalay et al, 2003, PG Pelicci, personal communication). While RA therapy of APL is the prototypic cancer ‘differentiation therapy', it must not be overlooked that retinoids have cancer therapeutic activities beyond the induction of differentiation (Altucci and Gronemeyer, 2001; Sun and Lotan, 2002; Altucci and Gronemeyer, 2004). For example, RA not only induces blast differentiation but also triggers blast eradication through apoptosis by inducing tumor necrosis factor-related apoptosis-inducing ligand (TRAIL, also referred to as Apo2L or TNFSF10) (Altucci et al, 2001). TRAIL is a fascinating member of the TNF family because it has a dual role in tumor defense: (i) in a cell autonomous manner TRAIL induces apoptosis selectively in the cancer cells, while normal cells are largely insensitive to TRAIL-mediated killing (Ashkenazi and Dixit, 1999; Walczak et al, 1999; Wang and El-Deiry, 2003b); and (ii) TRAIL signaling is critically involved in immune surveillance against tumor development (Takeda et al, 2001, 2002, 2004) and is also required for optimal graft-versus-tumor activity of T cells (Schmaltz et al, 2002). The therapeutic value and possible toxicity of recombinant soluble TRAIL are intensively discussed and require further assessment in suitable in vivo systems (for further information and references, see Supplementary Material). Irrespective of these discussions, it has been convincingly demonstrated that TRAIL−/− mice display no overt phenotype but an increased susceptibility to tumor initiation and metastasis (Cretney et al, 2002; Sedger et al, 2002). While these results support a central role of TRAIL signaling in tumor defense, the mechanism by which TRAIL and its receptors induce cancer-selective death has remained elusive despite the identification of a plethora of modulators of TRAIL-mediated apoptosis (Burns and El-Deiry, 2001; Aza-Blanc et al, 2003).
Retinoids signal through multiple receptor isotypes (α, β, γ) of the RAR and RXR families; it is generally believed that RAR–RXR heterodimers are the species transmitting the signal in vivo (for details and references, see Laudet and Gronemeyer, 2002). In addition to the unknown mechanistic link between apoptosis induction and RA action described above, several questions linked to the anticancer action of retinoids are unresolved; one concerns the nature and specific action of the RAR isotype that exerts a growth regulatory action. For example, in myeloid cells, it is generally RARα that mediates the differentiative and apoptogenic response even though RARγ is equally expressed (Chen et al, 1996). Conversely, in keratinocytes, RARγ is the principal receptor contributing to all-trans-RA-mediated growth arrest (Goyette et al, 2000). Finally, primitive endodermal differentiation of F9 embryo carcinoma cells requires RARγ2, while parietal differentiation requires RARγ2 and RARα1 (Taneja et al, 1997). The second unresolved question concerns the tumor suppressive function of RARβ2, which is frequently deleted or epigenetically silenced during cancer progression (reviewed by Altucci and Gronemeyer, 2001) and may be related to particularities of RARβ (Germain et al, 2002). The third issue concerns the role and mechanistic basis of AP1 transrepression. It has been shown that RA blocks tumor promotion in models of chemical skin carcinogenesis (reviewed by Altucci and Gronemeyer, 2001). The corresponding mechanistic basis remains to be established.
IFNs, antiviral and immunomodulatory proteins, are also important negative growth factors that inhibit cell proliferation and induce apoptosis (Belardelli et al, 2002; Ikeda et al, 2002). Genetic studies have confirmed their role in cancer immunosurveillance (Dunn et al, 2002). Notably, the TRAIL signaling pathway is critically involved in natural killer cell-mediated and IFNγ-dependent tumor surveillance (Takeda et al, 2001, 2002, 2004; Smyth et al, 2003).
Interestingly, there is evidence that IFNs and RA can synergize in their antiproliferative activity in vitro and mouse xenograft models, and that combination therapies may be effective in some solid tumors, such as squamous cell carcinomas (Lippman et al, 1997; Altucci and Gronemeyer, 2001). Indeed, several clinical trials are ongoing (Ortiz et al, 2002).
In the present study, we reveal the molecular mechanism by which RA induces TRAIL expression in a cell autonomous manner. We observe that in both NB4 promyelocytic and breast cancer cells RA-induced interferon regulatory factor-1 (IRF-1) causes TRAIL promoter activation and that IFNs synergize with RA. Synergistically induced TRAIL strongly increases the killing of heterologous tumor cells in a paracrine mode of action, while normal cells are not affected. Our results indicate the existence of cancer surveillance by the TRAIL signaling pathway that can be activated by known cancer therapeutics to act in a cell autonomous manner. Apparently, TRAIL acts as a central executor of the tumor-selective apoptogenic component of a diverse set of anticancer drugs and their corresponding signaling pathways.
Results
Treatment with 9-cis-RA of NB4 APL or SK-BR-3 breast cancer cells leads to the ligand-dependent appearance of a DNase I hypersensitive site (DHS site I*) in the promoter (schematically illustrated in Figure 1A and B) of the tumor necrosis factor-related apoptosis-inducing ligand TRAIL/Apo2L (Bodmer et al, 2002), while two more distal DHS sites (II and III in Figure 1B) were ligand independent and were seen only in breast cancer cells. Transient transfection into SK-BR-3 cells of a chimeric luciferase reporter gene (pTRL1) driven by the TRAIL promoter confirmed that the 2 kb sequence upstream of the transcription initiation site (Gong and Almasan, 2000; Wang et al, 2000) contains RA-inducible elements (Figure 1C). Promoter mapping narrowed these elements down to 165 bp and revealed similar 6.5-fold RA inducibility of pTRL1, 2 and 3. Notably, pTRL4, which contains only 35 bp upstream of the transcription initiation, retained RA inducibility but displayed severely impaired basal activity. Removal of sequences between –1905 and –165 increased basal but not RA-induced transcriptional activity of the reporter (Figure 1C; compare pTRL1 and pTRL3). These results indicated that (i) the 35 bp upstream sequence contains at least one element that can mediate RA inducibility, (ii) additional constitutively or RA-inducible elements between –35 and –165 cooperate with this element(s) to increase basal and RA-induced activities and (iii) elements upstream of −165 silence TRAIL promoter activity.
Figure 1.
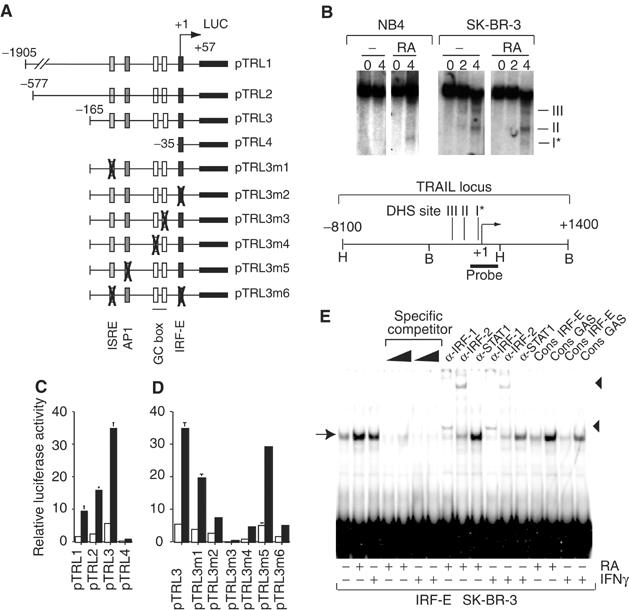
Mapping of RA-responsive sites in the TRAIL promoter region. (A) Illustration of pTRL reporter genes containing the TRAIL upstream regulatory region and site-specific mutants derived from pTRL3. (B) DNase I hypersensitive mapping of the TRAIL locus schematically depicted below. Nuclei of vehicle- (‘−') or RA-treated NB4 and SK-BR-3 cells were treated with increasing amounts of DNase I (0, 2 and 4 U; indicated at the top). Hypersensitive regions are shown as DHS I, II and III. The asterisk indicates that DHS I is the only RA-induced site in both NB4 and SK-BR-3. (C) Luciferase activity in SK-BR-3 transiently transfected with the indicated pTRL reporters in the presence (black bars) or absence (white bars) of RA. (D) Transfection experiment as in (C) but using pTRL3 derivatives with mutated response elements. (E) EMSA with the TRAIL IRF-E and nuclear extracts from SK-BR-3 cells treated with RA or IFNγ, as indicated below. Complexes (arrow) were competed with cold probe or supershifted with antibodies (triangles) directed against IRF-1, IRF-2 and STAT1, as depicted at the top.
Because no obvious RA response element could be detected in the 165 bp RA-responsive sequence, we systematically mutated each putative cis-acting element in the background of pTRL3 to test if heterologous response elements (Figure 1A) would mediate this response (Figure 1D). Mutation of either of the two IFN-responsive elements, ISRE (Levy et al, 1988) and IRF-E (Tanaka et al, 1993), strongly decreased and their simultaneous mutation abrogated, albeit not completely, RA induction. No significant effect was seen upon mutating the AP1 site. Mutating the promoter-proximal GC box knocked out basal activity completely and RA did not generate a measurable signal. The corresponding mutation of the promoter-distal GC box severely decreased basal, but still allowed significant RA-induced activity. To assess if individual elements could autonomously mediate RA inducibility, we studied the corresponding reporter genes driven by a heterologous promoter. Interestingly, none of these elements mounted a significant RA response when placed into a tk promoter-driven reporter (data not shown). Together, these results demonstrate a complex orchestration of the RA responsiveness of the TRAIL promoter in that (i) the integrity of multiple enhancer elements is required for full RA induction, (ii) the upstream enhancer elements function only in the context of the homologous promoter and (iii) its basal activity is affected by mutations/deletions of the upstream enhancer and silencer elements. These features are reminiscent of the complexity of the IFNβ promoter (Wathelet et al, 1998), whose viral response is similarly mediated by factors precisely arranged on a short promoter sequence. Further studies are required to assess if the factors assembled on the TRAIL promoter constitute an enhanceosome.
Since mutations of either IFN response element resulted in a major decrease of RA inducibility, we tested these sites in electrophoretic mobility shift assays (EMSAs). EMSAs with extracts of SK-BR-3 cells demonstrated that RA induced the expression of a protein(s) that bind to the IRF-E (Figure 1E) and, more weakly, the ISRE (data not shown). A complex of identical mobility was formed when cells were exposed to IFNγ, which is known to induce factor(s) binding to IRF-Es (Boehm et al, 1997). Both the RA- and IFNγ-induced complexes could be competed off the specific probe by TRAIL and consensus IRF-E (Tanaka et al, 1993) oligonucleotides; no competition was seen with a consensus GAS (Boehm et al, 1997) site. Antibody supershifts revealed the presence of IRF-1 and IRF-2, but not STAT1, in these complexes. We conclude that RA, like IFNγ, induces the formation of IRF-1- and IRF-2-containing complexes at the IRF-E and ISRE sites of the TRAIL promoter. Apparently, these complexes cooperate with GC box-binding factors to generate a high level of RA-induced TRAIL transcription.
We reasoned that the induction of IRFs could account for RA-induced TRAIL transcription. Indeed, chromatin immunoprecipitation (ChIP) assays revealed that RA induced acetylation of histones H3 and H4 at the IRF-1 promoter (Figure 2A) and increased IRF-1 mRNA (data not shown) and protein levels in NB4 and SK-BR-3 cells (Figure 2B), albeit less efficiently than IFNα or γ. While RA was previously reported to induce STAT1 (Kolla et al, 1996; Matikainen et al, 1997), we saw no increase of STAT1 protein levels or phosphorylation (data not shown) in MCF7, H3396 or SK-BR-3 breast cancer cells. We also saw no significant increase of two other IRF family members, IRF-2 and p48 (Figure 2B and data not shown). Given that STAT1 was not detected by EMSA supershifts and interacts with ISREs (Levy et al, 1988) as a complex with p48, we ruled out a significant implication of these two factors in RA-dependent induction of TRAIL and concentrated on analyzing the role of RA-induced IRF-1. In transient transfections, increased levels of exogenous IRF-1 induced strong expression of endogenous TRAIL in both MCF7 and SK-BR-3 cells (Figure 2C). This was apparently due to IRF-1-responsive elements in the promoter-proximal region, as IRF-1 was a potent enhancer of pTRL3 transcription (Figure 2D). Notably, IRF-1 action required the integrity of both the IRF-E and ISRE, as mutation of either site (pTRL3m1, pTRL3m2) severely impaired activation of this reporter gene (Figure 2D). No induction was seen when both sites were mutated (pTRL3m6) indicating the absence of further IRF-1 response elements in this TRAIL promoter region.
Figure 2.
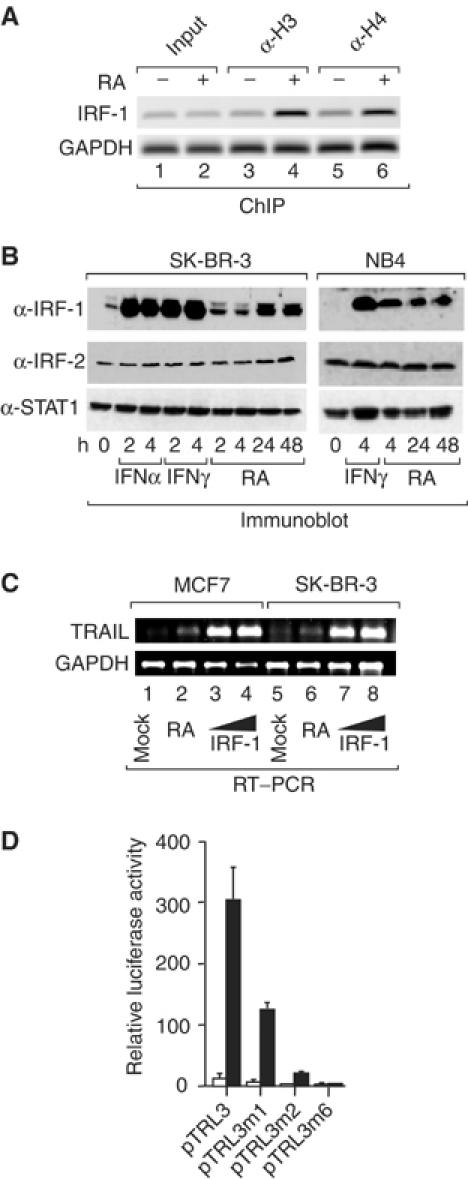
RA-induced IRF-1 is sufficient to induce TRAIL promoter activity. (A) SK-BR-3 cells were treated with RA for 3 h and ChIPs performed using antibodies directed against acetylated histones H3 (lanes 3 and 4) or H4 (lanes 5 and 6). Immunoprecipitated chromatin was analyzed by PCR with primers specific for promoter-proximal regions of the IRF-1 or GAPDH genes. (B) SK-BR-3 and NB4 cells were treated with IFNα, IFNγ or RA as indicated. Shown are immunoblots generated with anti-IRF-1, anti-IRF-2 and anti-STAT1 antibodies. (C) Semiquantitative RT–PCR performed with primers for TRAIL or GAPDH mRNA using total RNA of MCF7 (lanes 1–4) or SK-BR-3 (lanes 5–8) either treated with RA for 48 h (lanes 2 and 6) or transiently transfected with empty (pCDNA3; lanes 1 and 5) or IRF-1 expression vector (2 μg, lanes 3 and 7; or 5 μg, lanes 4 and 8). (D) SK-BR-3 cells were transiently cotransfected with the indicated pTRL reporters and either pCDNA3 (white bars) or the pCDNA3-based IRF-1 expression vector (black bars).
The above data support a mechanism by which RA-induced IRF-1 stimulates TRAIL expression through the IRF-E and ISRE sites. Indeed, ChIP assays confirmed that RA exposure of SK-BR-3 cells results in de novo recruitment of IRF-1 to the TRAIL promoter whereas the constitutive association of IRF-2 remains unchanged (Figure 3A). Similar results were obtained using NB4 cells with the only difference that IRF-1 displayed some promoter occupancy in the absence of RA (Figure 3B). To investigate whether IRF-1 is critically required for RA-induced TRAIL expression, we knocked down IRF-1 in H3396 cells by RNA interference. Western blotting confirmed that the siRNA reduced RA-induced IRF-1 protein levels by more than 90% (Figure 3C). Importantly, induction of TRAIL mRNA levels by RA was completely abolished when IRF-1 was knocked down (Figure 3D). Thus, IRF-1 is critically involved in mediating RA-induced TRAIL expression in breast cancer cells, and is likely to have the same role in APL cells.
Figure 3.
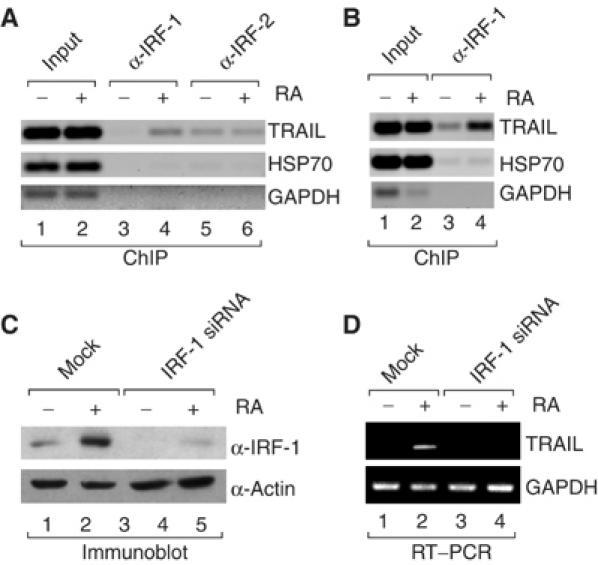
IRF-1 is recruited to the TRAIL promoter in vivo and required for RA-induced TRAIL expression. (A, B) ChIP assays using antibodies to IRF-1 and IRF-2 (indicated at the top) and SK-BR-3 (A) or NB4 (B) cells treated with RA for 36 h. Immunoprecipitated chromatin was analyzed by PCR using primers specific for the TRAIL, HSP70 or GAPDH promoters. (C) siRNA to IRF-1 specifically knocks down expression of IRF-1 protein. H3396 breast cancer cells were mock transfected or transfected with siRNA to IRF-1 at 200 nM and treated with RA for 36 h. Shown is the corresponding immunoblot using antibody to IRF-1 or actin. (D) H3396 cells were transfected and treated as in (C) and total RNA was isolated. Semiquantitative RT–PCR was performed with primers specific for TRAIL or GAPDH mRNAs.
That an IFN-inducible factor mediates the apoptogenic action of RA suggested that the two signaling pathways could synergistically converge on the TRAIL promoter. Transactivation experiments with pTRL3 and mutants thereof (Figure 1A) demonstrated that (i) the RA–IFNγ synergy is mediated by elements within the 165 bp promoter-proximal sequence, (ii) the IRF-E is crucial for this synergy (compare pTRL3 and pTRL3m2) and (iii) the ISRE does not contribute to the RA–IFNγ synergy but rather to the magnitude of the individual and combined responses (Figure 4A). Similar results were obtained with H3396 cells. Synergy was not restricted to type II IFNs, as synergistic induction of TRAIL expression was seen also with RA and IFNβ (Supplementary Figure 1). To gain a mechanistic insight into this phenomenon, we investigated transcription factor recruitment to the TRAIL promoter by ChIP assays and real-time PCR. IRF-1 recruitment is apparently a key component of the synergistic response, as a strong and more than additive occupancy of the TRAIL promoter by IRF-1 is seen after simultaneous exposure of H3396 cells to RA and IFNγ (Figure 4B). This leads to increased CBP recruitment (Figure 4C) and results in increased histone H3 acetylation (Figure 4D). On its own IFNγ does not recruit CBP more efficiently than RA, even though it recruits IRF-1 more efficiently (Figure 4B), suggesting that additional coactivators contribute to the transcriptional response of IFNγ-induced IRF-1 on the TRAIL promoter. These additional coactivators may not exert HAT activity, as H3 acetylation in response to RA and IFNγ is additive rather than synergistic (Figure 4D). Irrespective of which additional coregulators may mediate the IRF-1 action on the TRAIL promoter, its synergistic recruitment leads to a strong increase of TRAIL mRNA (Figure 4E) and protein (Figure 4F).
Figure 4.

RA and IFNγ synergize to induce TRAIL. (A) Luciferase activities of SK-BR-3 cells transfected with pTRL reporters and treated with RA and/or INFγ as depicted. (B) H3396 cells were treated with RA, IFNγ or both as indicated and ChIP assays were performed using anti-IRF-1 antibodies. Immunoprecipitated chromatin was analyzed by PCR using primers specific for the TRAIL or GAPDH promoters. A representative ChIP experiment analyzed on an ethidium bromide gel and data from three independent ChIP experiments analyzed by real-time PCR are shown. (C, D) H3396 cells were treated as in (B) and ChIP assays were performed using no antibody (‘No Ab') or anti-CBP or anti-acetylhistone H3 antibodies. The graphs depict real-time PCR data as percent input immunoprecipitated. (E, F) SK-BR-3 cells were treated as in (A) and semiquantitative RT–PCR (E) with primers for TRAIL and GAPDH or Western blot analyses (F) with anti-TRAIL or anti-actin antibodies were performed as indicated.
A comparison of the kinetics of induction of IRF-1 expression with its kinetics of recruitment to the TRAIL promoter revealed that the combination of RA and IFNγ alters the IRF-1 protein expression profile compared to the induction seen with the individual compounds. Moreover, this altered profile correlates precisely with the kinetics of IRF-1 recruitment assuming that this recruitment is cooperative as previously established. Indeed, while RA is a late and weak inducer of IRF-1, and while IFNγ produces a transient peak of expression, the combination of both results in a robust sustained induction of IRF-1 expression above that seen with either compound alone (Figure 5A). These expression profiles correlate with the IRF-1 recruitment kinetics to the TRAIL promoter (Figure 5B). Importantly, RA–IFNγ cotreatment leads to a clearly synergistic and sustained occupancy of IRF-1 at the TRAIL promoter, which is followed by the recruitment of RNA polymerase II (Figure 5C). Note the steep increase in the IRF-1 recruitment kinetics by the cotreatment (Figure 5B), which is highly indicative of a cooperative binding of IRF-1 (Fujii et al, 1999). Ultimately, the steady-state level of TRAIL mRNA is dramatically increased by more than 70-fold (Figure 5D).
Figure 5.
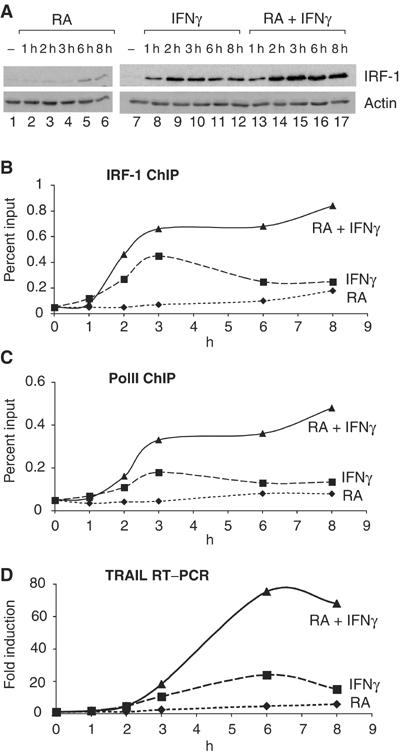
RA and IFNγ cooperatively recruit IRF-1 and PolII to the TRAIL promoter. (A) Western blot of IRF-1 expression in H3396 cells treated with RA, IFNγ or both as indicated. (B) H3396 cells were treated as indicated and ChIP assays were performed using anti-IRF-1 antibodies. Immunoprecipitated chromatin was analyzed by real-time PCR using primers specific for the TRAIL promoter. Data from two independent ChIP experiments (expressed as % input) are shown. (C) Similar to (B) but anti-PolII antibodies were used to perform the ChIP. (D) H3396 cells were treated as depicted, RNA was isolated and quantitative PCR was performed with cDNA (diluted 1/40) from a reverse transcription reaction using 3 μg RNA (representative of three experiments).
The observed RA–IFNγ synergy for TRAIL induction may translate not only into homologous cell death but, more importantly, may also elicit apoptosis in a paracrine and tumor-selective fashion. To test this option, we exposed cocultured SK-BR-3 breast cancer and CMFDA cell tracker-labeled Jurkat T cells to RA and/or IFNγ (Figure 6A). In contrast to SK-BR-3 cells, Jurkat cells are virtually insensitive to RA and/or IFNγ (apoptosis <2%; data not shown). Death was measured as propidium iodide (PI) incorporation by the CMFDA-positive (Jurkat) and -negative (SK-BR-3) populations using FACS analysis. RA or IFNγ alone induced a low level of cell death in cocultured Jurkat T cells (3–5%; top right and bottom left). Importantly however, concomitant exposure to both drugs dramatically induced death not only in the SK-BR-3 (bottom right, top left quadrant) but also in the cocultured Jurkat cells (40%; top right quadrant). To investigate if paracrine apoptosis was due to induced TRAIL, we used neutralizing TRAIL receptor–Fc chimeras, which are known to neutralize TRAIL action. Indeed, heterologous cell kill in SK-BR-3/Jurkat cocultures exposed to RA and IFNγ could be significantly inhibited through the addition of TRAIL receptor 2–Fc chimeras; under otherwise identical conditions, the addition of the corresponding FAS or TNF receptor–Fc chimeras had no effect (Figure 6B). Because TRAIL is believed to act in a tumor-selective manner, we tested if normal cord blood CD4+ naive T cells would be sensitive to the paracrine action of TRAIL produced in SK-BR-3 cells upon exposure to RA and IFNγ. However, whereas there was an induction of 18% death in Jurkat cells in this experimental setting (1 day coculture; see illustration and top panels in Figure 6C), normal T cells (bottom) were virtually resistant to paracrine death induction.
Figure 6.
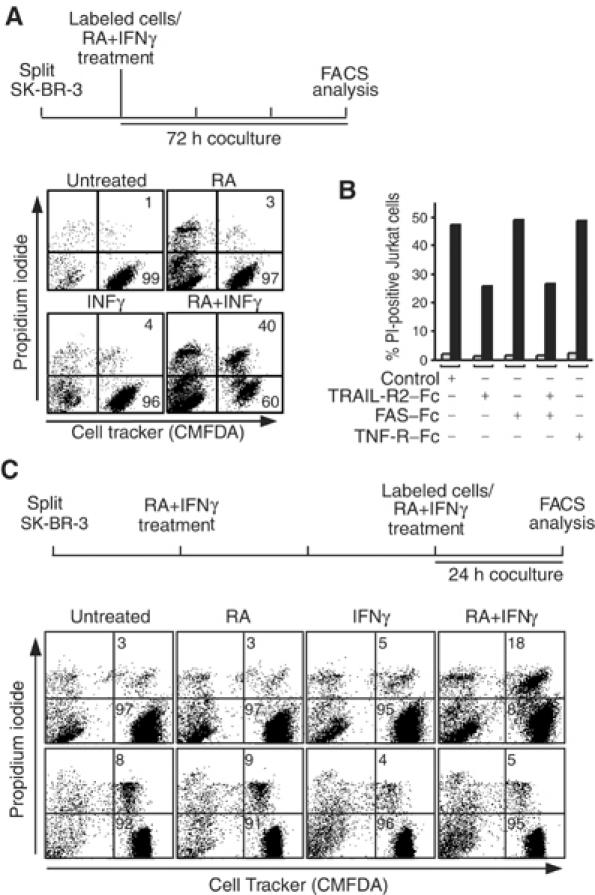
RA and IFNγ synergize to kill leukemic, but not normal, T cells in a paracrine mode of action involving TRAIL. (A) Coculture experiment for paracrine death induction performed as outlined at the top. SK-BR-3 effector cells were cocultured for 72 h with Cell Tracker-labeled Jurkat target cells at a 10:1 ratio in the absence or presence of RA and/or IFNγ. Death induced in target cells was analyzed by PI incorporation and FACS analysis; paracrine killed target cells are gated to the right top quadrants, and the left quadrants gate the Cell Tracker-negative effector cells. Numbers indicate the percentage of PI-positive and -negative target cells. (B) Bar graph showing the percentage of dead Jurkat cells in a representative experiment performed as in (A) with SK-BR-3 effector cells in the absence (gray bars) or presence of RA and INFγ (black bars) and presence of neutralizing receptor–Fc chimeras as shown; similar results were obtained in three independent experiments. Note that only the TRAIL receptor chimera inhibits paracrine death. (C) Leukemia cell-selective paracrine death induction analyzed by coculturing SK-BR-3 effector and either Jurkat leukemia (upper panels) or normal CD4+ T (lower panels) target cells as outlined at the top. The effector cells were treated with RA and INFγ for 48 h before labeled target cells were cocultured for a further 24 h at a two-fold excess of effector cells. FACS data are presented as in (A); only the percentage of paracrine death is given.
Discussion
It is well established that retinoids have cancer chemotherapeutic and preventive activities beyond the mere induction of differentiation in leukemic cell models in vitro and APL patients in vivo (Altucci and Gronemeyer, 2001; Sporn and Suh, 2002; also see Introduction). Indeed, we have recently provided evidence that the induction of TRAIL, a death ligand that has been shown to kill selectively cancer in a variety of in vitro and in vivo settings (Almasan and Ashkenazi, 2003), contributes to the antileukemic action of RA (Altucci et al, 2001). In this study, we set out to identify the molecular pathway responsible for the induction of TRAIL. Importantly, we observed that RA induces TRAIL expression not only in hematopoietic but also in breast cancer cells. Our study identifies IRF-1 as the factor critically involved in mediating the retinoid response to TRAIL. Using promoter mapping, RNA interference and ChIP, we show that in both breast cancer and myeloid cells RA-induced IRF-1 binds to bona fide IRF-E and ISRE elements in the proximal region of the TRAIL promoter, thereby causing induction of TRAIL expression. In addition to IRF-1, IRF-2 is also bound to the TRAIL promoter; however, we did not observe any modulation of its TRAIL promoter association in response to RA and/or IFN. Thus, as it has been reported for the cell cycle regulation in NIH 3T3 cells (Tanaka and Taniguchi, 2000), it is apparently the modulation of the IRF-1:IRF-2 ratio that triggers TRAIL expression.
The apparent convergence of the RA and IFN signaling pathways on TRAIL was reminiscent of observations demonstrating that combination of RA and IFN results in synergistic inhibition of cell proliferation in many cancer cell systems. Indeed, RA has shown promise for cancer therapy outside of the hematopoietic system, often in combination with other cancer therapeutic compounds, for example, IFNs or chemotherapy (for review, see Altucci and Gronemeyer, 2001). The basis of this synergism is not well understood, but it had been observed that RA induces IRF-1 expression in several different types of tumor cells in culture (Matikainen et al, 1996; Pelicano et al, 1997; Percario et al, 1999; Um et al, 2000). We rationalized that synergistic induction of IRF-1 by RA and IFN may lead to synergistic induction of TRAIL, which would then cause the antiproliferative effect. Therefore, we explored the effect of using both signaling molecules on TRAIL expression in breast cancer cells, as a model for solid cancers. At the molecular level, the combination of both agents indeed resulted in synergistic recruitment of IRF-1 to the TRAIL promoter, most likely due to the cooperative binding of IRF-1 to multiple GAAA repeats (Escalante et al, 1998; Fujii et al, 1999), and concomitant increase in histone acetylation. Surprisingly, we observed that whereas the individual signals resulted in recruitment of IRF-1, we found only small increases in CBP recruitment, suggesting that other coactivators may contribute to IRF-1-mediated transactivation. In other systems, such as the IFNβ enhanceosome, CBP has been shown to bind to IRF family members, including IRF-1, and to be involved in IFNβ transactivation; however, in this case, IRF-1 acts in concert with a plethora of other transcription and architectural factors (Thanos and Maniatis, 1995). Other IRF family members, in particular IRF-3/IRF-7, are known to recruit CBP for activation only under conditions where they have formed hetero- and/or homodimers or are bound to DNA as multimers (Wathelet et al, 1998; Morin et al, 2002). Such a scenario could also explain the strong recruitment of CBP under the conditions of cotreatment where much higher levels of IRF-1 are bound at the TRAIL promoter. Moreover, our results show that other factors binding to nearby cis-acting elements (such as the GC boxes) contribute to the magnitude of the response, as well as to the basal level of TRAIL expression. In this respect, it is worth noting that IRF-1 has been observed to crosstalk to SP1 on the human CDK2 promoter (Xie et al, 2003) but the corresponding molecular mechanism has remained elusive. Collectively, these results suggest that (a) complex interactive structure(s) resembling the IFNβ enhanceosome (Thanos and Maniatis, 1995) may form at the TRAIL promoter. Thus, it is likely that, as in the case of IFNβ where only the stereospecific assembly of several factors allows the efficient recruitment of CBP (Merika et al, 1998), the synergistic activation of TRAIL expression requires the establishment of a similar structure to warrant efficient CBP recruitment.
The link between IRF-1 and TRAIL provides a novel insight into molecular mechanisms by which IRF-1 exerts its tumor suppressor activity in various systems (Tanaka and Taniguchi, 2000). Indeed, while initial gene deletion experiments suggested that IRF-1 acted as a ‘tumor susceptibility gene' (Nozawa et al, 1999), recent data obtained with older IRF-1−/− mice (Eason et al, 2003) and the frequent loss or epigenetic modification of the IRF-1 locus in hematopoietic and solid cancers indicate that IRF-1 is a bona fide tumor suppressor. The molecular basis underlying the tumor suppressive activity of IRF-1 is still incompletely understood, albeit some features have been worked out. These include (i) the deficiency of IRF-1−/− mouse embryo fibroblasts (MEFs) in their ability to undergo DNA damage-induced cell cycle arrest, most probably due to the presence of both IRF-1-and p53-binding sites in the p21 promoter (Tanaka et al, 1996), (ii) the p53-independent DNA damage-induced apoptosis in activated mature T lymphocytes (Tamura et al, 1995), (iii) the reversion of oncogene-transformed cells by ectopic expression of IRF-1 (Tanaka et al, 1994b) and (iv) the elimination of activated ras-transformed MEFs by apoptosis upon DNA damage induction, which was not seen in IRF-1−/− cells under otherwise identical conditions (Tanaka et al, 1994a). Our finding that TRAIL is activated by IRF-1 may contribute to these tumor suppressor activities and explain the p53-independent IRF-1-dependent apoptosis of oncogene-transformed cells. Moreover, the synergy between IRF-1 and p53 should be reconsidered in view of the p53-induced expression of the TRAIL receptor DR5 (Wu et al, 1997; Kim et al, 2001; Wang and El-Deiry, 2003a) and the observation that IFNs induce p53 at the transcriptional level (Takaoka et al, 2003).
We show that synergy between RA and IFN results in a potent paracrine cell death of heterologous cancer cells. This paracrine killing has important implications, because (i) tumor cells resistant against one or both of the inducers can be nevertheless eliminated by neighboring cells that express TRAIL, (ii) normal cells are apparently resistant to TRAIL (also see Supplementary Material) but may express it (it will be interesting to study in this context if there are any differences in the TRAIL induction between cancer and normal cells) and (iii) combining two synergizing drugs has the advantage that the concentrations of the individual drugs can be reduced to limit side effects.
In parallel to these studies, we and others have observed that histone deacetylase inhibitors (HDACi) induce TRAIL expression in acute myeloid leukemia (AML) cells in vitro, AML patients blasts ex vivo and in AML leukemia mouse models (Insinga et al, 2004; Nebbioso et al, 2004). Together with the data presented here, these studies identify TRAIL as a central executor of the cancer cell-selective apoptogenic action of several known anticancer drugs and their corresponding signaling pathways, which form an intricate tumor suppressive signaling network (Figure 7). It is important to point out that these actions occur in a cell autonomous manner and represent a drug-modulable tumor defense system that acts independently of, and adds to the immunosurveillance through NK cells, which also involves TRAIL. The recognition of TRAIL and its receptor(s) as key mediators of the antitumor activities of certain retinoids, IFNs or HDAC inhibitors, and its link to two established tumor suppressors, IRF-1 and p53, supports the central role of this death signaling pathway in tumor defense and may pave the way toward novel apoptogenic anticancer therapies.
Figure 7.
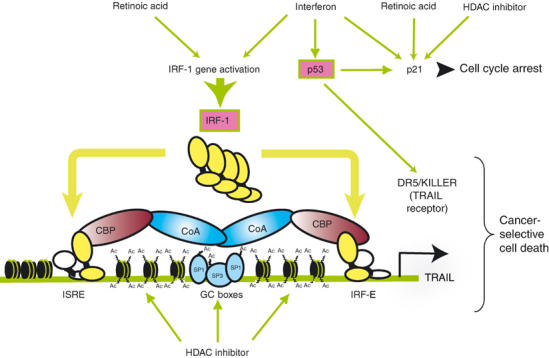
Sketch illustrating the convergence of three different anticancer signals on the TRAIL promoter. TRAIL is integrated in a tumor suppressive signaling network that is triggered by RA, IFN and HDAC inhibitors. RA and IFN synergistically induce expression of IRF-1, which activates the promoter of TRAIL through the ISRE and IRF-E sites. HDAC inhibitors lead to acetylation of histone and SP1 family members, thereby inducing TRAIL promoter activation. IFN also induces p53 expression, which stimulates the TRAIL receptor DR5 and the cell cycle inhibitor p21 whose expression is also upregulated by RA and HDAC inhibitors, thus promoting cell cycle arrest. Pink boxes indicate tumor suppressors; CoA, coactivator.
Materials and methods
Cell lines and cell culture
SK-BR-3 and MCF7 cells were maintained in DMEM supplemented with 10% FCS, 100 U/ml penicillin and 100 μg/ml streptomycin. Jurkat, H3396 and NB4 cells were maintained in RPMI medium supplemented with 2 mM L-glutamine (Life Technologies), 10% FCS and antibiotics. Cryopreserved cord blood CD4+ T cells (Cambrex) were thawed, washed and placed in culture at 106 cells/ml in RPMI with 10% human serum, 100 U/ml penicillin and 100 μg/ml streptomycin. Cells were kept in culture for no more than 6 days.
Plasmids, reagents and antibodies
pTRL reporters were constructed as follows: the −3907 to +56 region of the human TRAIL promoter was amplified from human genomic DNA by PCR and inserted into pGEM-T Easy (Promega) to generate pGEM-TR. All pTRL reporters are derivatives of this sequence inserted into pGL3 basic (Promega). Sequences of primers used for cloning or mutagenesis and details of plasmid constructions are available upon request. The following reagents were used: RA (9-cis-RA) (Sigma) at 1 μM, human recombinant IFNγ (15 ng/ml) and IFNα (1000 U/ml; Sigma). Rabbit polyclonal antibodies against acetylhistone H4 and H3 (Upstate), IRF-1, IRF-2, actin, CBP, PolII (Santa Cruz), Stat1 (Santa Cruz, Cell Signalling) and TRAIL (R&D) were used for ChIPs, immunoblots and EMSAs.
Reporter assays
Briefly, 1 day after seeding, SK-BR-3 or H3396 cells were transfected using Fugene (Roche). At 16 h after transfection, cells were treated with ligands (see figure legends) for 24 h. Cells were lysed and luciferase assays were performed (Promega). A CMV-driven β-galactosidase expression vector was used to monitor transfection efficiencies. Normalized values are reported as the mean±s.d.; each value originates from at least three transfections performed in duplicate.
DNase I hypersensitive mapping
DNase I hypersensitive mapping of the TRAIL locus was performed on SK-BR-3 and NB4 cells treated with RA for 48 and 72 h, respectively. Nuclei were isolated and aliquots were treated with increasing amounts of DNase I. Genomic DNA was digested with BglI, followed by overnight digestion with proteinase K, phenol/chloroform extraction and ethanol precipitation. In all, 50 μg of digested sample was run on a 0.8% agarose gel and transferred to Hybond-N+ paper. The blot was incubated with 107 cpm 32P-labeled probe (Figure 1B), washed extensively with SSC and exposed to Kodak Biomax for 3–5 days.
Electrophoretic mobility assay and Western blot analysis
EMSAs were performed by incubating 6 μg of nuclear extract with or without anti-IRF-1, anti-IRF-2 or anti-STAT1 antibodies (Santa Cruz Biotechnology), cold TRAIL-IRF-E (100 × or 200 ×), cold consensus IRF-E (200 ×) or consensus GAS (200 ×) probes for 20 min on ice. In all, 50 000 cpm of 32P-labeled TRAIL IRF-E (5′-ACAACTCATTCGCTTTCATTTCCTCACTGA-3′) was added and reactions were incubated for 15 min at room temperature. The total volume of the reaction was 20 μl. Protein–DNA complexes were resolved on nondenaturing 5% polyacrylamide gels in 0.5 × TBE. For immunoblot analysis, 30 μg aliquots of whole-cell extracts in Laemmli sample buffer were separated by 10% SDS–PAGE and immunoblotted according to standard procedures.
Chromatin immunoprecipitation
ChIP assays for the IRF-1 and GAPDH promoters were performed with the anti-acetylhistone H3 and H4 assay kit as recommended (Upstate). ChIP assays for TRAIL and HSP70 genes were carried out as described (Nissen and Yamamoto, 2000). Amplified products were run on a 1.2% agarose gel and visualized on a Typhoon Scanner. Quantitative real-time PCR was performed on a Roche ‘Lightcycler'. All primer sequences will be provided on request.
Reverse transcriptase polymerase chain reaction
Total RNA was isolated (TRIZOL) from treated cells and 5 μg of RNA was used in a reverse transcription reaction recommended (Invitrogen). PCR was performed using equal amounts of cDNA with primers for TRAIL or GAPDH mRNAs. PCR products were run on a 1.2% agarose gel and visualized on a Typhoon Scanner.
Paracrine death induction assays
Jurkat and cord blood CD4+ T cells were labeled with Cell Tracker Green CMFDA (Molecular Probes) according to the supplier's instructions. Cell Tracker-labeled cells were incubated with SK-BR-3 cells in the absence or presence of 1 μM RA and/or 15 ng/ml IFNγ as illustrated in Figure 5A and C. Floating cells (T cells and detached SK-BR-3) were collected, pelleted, resuspended in PBS containing 1 μg/ml PI and analyzed by FACS. CMFDA-positive cells were regarded as target (Jurkat or CD4+ T) cells, and CMFDA-negative cells as effector (SK-BR-3) cells. PI-negative cells were considered as living, and PI-positive cells as dead. For neutralization experiments, 1 μg/ml of either TRAIL-R2:Fc, FAS:Fc or TNF:Fc chimeras (R&D) was added twice to the coculture, 48 and 24 h before FACS analysis.
RNA interference
H3396 cells were seeded 24 h before transfecting (Oligofectamine, Invitrogen) them with 200 nM siRNA for IRF-1 (Dharmacon) according to the manufacturer's instructions and retransfecting them after 24 h. After 16 h, cells were treated with RA for 36 h and lysed for RNA or protein extraction.
Supplementary Material
Supplementary Material
Acknowledgments
We are grateful to Lucia Altucci for multiple discussions and suggestions throughout these studies, and for critical reading of the manuscript. We thank Cathie Erb, Michele Lieb and Astrid Pornon for technical assistance and Keiko Ozato for providing the IRF-1 cDNA. NC was supported by the Association pour la Recherche sur le Cancer, AJ-L by a Marie Curie and EV by a MENRT fellowship. This work was supported by the INSERM, the CNRS, the Hôpital Universitaire de Strasbourg, the AICR, the ARC, the Fondation de France, the European Community (QLG1-CT2001-01935 and QLK3-CT2002-02029) and Bristol-Myers Squibb.
References
- Alcalay M, Meani N, Gelmetti V, Fantozzi A, Fagioli M, Orleth A, Riganelli D, Sebastiani C, Cappelli E, Casciari C, Sciurpi MT, Mariano AR, Minardi SP, Luzi L, Muller H, Di Fiore PP, Frosina G, Pelicci PG (2003) Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J Clin Invest 112: 1751–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasan A, Ashkenazi A (2003) Apo2L/TRAIL: apoptosis signaling, biology, and potential for cancer therapy. Cytokine Growth Factor Rev 14: 337–348 [DOI] [PubMed] [Google Scholar]
- Altucci L, Gronemeyer H (2001) The promise of retinoids to fight against cancer. Nat Rev Cancer 1: 181–193 [DOI] [PubMed] [Google Scholar]
- Altucci L, Gronemeyer H (2004) Retinoids and TRAIL: two cooperating actors to fight against cancer. In Vitamins and Hormones, Litwack G (ed) Vol. 67, pp 319–345. Academic Press: Elsevier [DOI] [PubMed] [Google Scholar]
- Altucci L, Rossin A, Raffelsberger W, Reitmair A, Chomienne C, Gronemeyer H (2001) Retinoic acid-induced apoptosis in leukemia cells is mediated by paracrine action of tumor-selective death ligand TRAIL. Nat Med 7: 680–686 [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Dixit VM (1999) Apoptosis control by death and decoy receptors. Curr Opin Cell Biol 11: 255–260 [DOI] [PubMed] [Google Scholar]
- Aza-Blanc P, Cooper CL, Wagner K, Batalov S, Deveraux QL, Cooke MP (2003) Identification of modulators of TRAIL-induced apoptosis via RNAi-based phenotypic screening. Mol Cell 12: 627–637 [DOI] [PubMed] [Google Scholar]
- Belardelli F, Ferrantini M, Proietti E, Kirkwood JM (2002) Interferon-alpha in tumor immunity and immunotherapy. Cytokine Growth Factor Rev 13: 119–134 [DOI] [PubMed] [Google Scholar]
- Bodmer JL, Schneider P, Tschopp J (2002) The molecular architecture of the TNF superfamily. Trends Biochem Sci 27: 19–26 [DOI] [PubMed] [Google Scholar]
- Boehm U, Klamp T, Groot M, Howard JC (1997) Cellular responses to interferon-gamma. Annu Rev Immunol 15: 749–795 [DOI] [PubMed] [Google Scholar]
- Burns TF, El-Deiry WS (2001) Identification of inhibitors of TRAIL-induced death (ITIDs) in the TRAIL-sensitive colon carcinoma cell line SW480 using a genetic approach. J Biol Chem 276: 37879–37886 [DOI] [PubMed] [Google Scholar]
- Chen JY, Clifford J, Zusi C, Starrett J, Tortolani D, Ostrowski J, Reczek PR, Chambon P, Gronemeyer H (1996) Two distinct actions of retinoid-receptor ligands. Nature 382: 819–822 [DOI] [PubMed] [Google Scholar]
- Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ (2002) Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol 168: 1356–1361 [DOI] [PubMed] [Google Scholar]
- Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3: 991–998 [DOI] [PubMed] [Google Scholar]
- Eason DD, LeBron C, Coppola D, Moscinski LC, Livingston S, Sutton ET, Blanck G (2003) Development of CD30+ lymphoproliferative disease in mice lacking interferon regulatory factor-1. Oncogene 22: 6166–6176 [DOI] [PubMed] [Google Scholar]
- Escalante CR, Yie J, Thanos D, Aggarwal AK (1998) Structure of IRF-1 with bound DNA reveals determinants of interferon regulation. Nature 391: 103–106 [DOI] [PubMed] [Google Scholar]
- Fujii Y, Shimizu T, Kusumoto M, Kyogoku Y, Taniguchi T, Hakoshima T (1999) Crystal structure of an IRF–DNA complex reveals novel DNA recognition and cooperative binding to a tandem repeat of core sequences. EMBO J 18: 5028–5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain P, Iyer J, Zechel C, Gronemeyer H (2002) Coregulator recruitment and the mechanism of retinoic acid receptor synergy. Nature 415: 187–192 [DOI] [PubMed] [Google Scholar]
- Gong B, Almasan A (2000) Genomic organization and transcriptional regulation of human Apo2/TRAIL gene. Biochem Biophys Res Commun 278: 747–752 [DOI] [PubMed] [Google Scholar]
- Goyette P, Feng Chen C, Wang W, Seguin F, Lohnes D (2000) Characterization of retinoic acid receptor-deficient keratinocytes. J Biol Chem 275: 16497–16505 [DOI] [PubMed] [Google Scholar]
- Ikeda H, Old LJ, Schreiber RD (2002) The roles of IFN gamma in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev 13: 95–109 [DOI] [PubMed] [Google Scholar]
- Insinga A, Monestiroli S, Ronzoni S, Gelmetti V, Marchesi F, Altucci L, Gronemeyer H, Nervi C, Minucci S, Pelicci PG (2004) Inhibitors of histone deacetylases induce apoptosis selectively in leukemia cells through activation of the death receptor pathway. (submitted) [DOI] [PubMed]
- Kim K, Takimoto R, Dicker DT, Chen Y, Gazitt Y, El-Deiry WS (2001) Enhanced TRAIL sensitivity by p53 overexpression in human cancer but not normal cell lines. Int J Oncol 18: 241–247 [DOI] [PubMed] [Google Scholar]
- Kolla V, Lindner DJ, Xiao W, Borden EC, Kalvakolanu DV (1996) Modulation of interferon (IFN)-inducible gene expression by retinoic acid. Up-regulation of STAT1 protein in IFN-unresponsive cells. J Biol Chem 271: 10508–10514 [DOI] [PubMed] [Google Scholar]
- Laudet V, Gronemeyer H (2002) The Nuclear Receptor Facts Book. San Diego: Academic Press [Google Scholar]
- Levy D, Reich N, Kessler D, Pine R, Darnell JE Jr (1988) Transcriptional regulation of interferon-stimulated genes: a DNA response element and induced proteins that recognize it. Cold Spring Harb Symp Quant Biol 53 (Part 2): 799–802 [DOI] [PubMed] [Google Scholar]
- Lippman SM, Lotan R, Schleuniger U (1997) Retinoid-interferon therapy of solid tumors. Int J Cancer 70: 481–483 [DOI] [PubMed] [Google Scholar]
- Matikainen S, Ronni T, Hurme M, Pine R, Julkunen I (1996) Retinoic acid activates interferon regulatory factor-1 gene expression in myeloid cells. Blood 88: 114–123 [PubMed] [Google Scholar]
- Matikainen S, Ronni T, Lehtonen A, Sareneva T, Melen K, Nordling S, Levy DE, Julkunen I (1997) Retinoic acid induces signal transducer and activator of transcription (STAT) 1, STAT2, and p48 expression in myeloid leukemia cells and enhances their responsiveness to interferons. Cell Growth Differ 8: 687–698 [PubMed] [Google Scholar]
- Melnick A, Licht JD (1999) Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood 93: 3167–3215 [PubMed] [Google Scholar]
- Merika M, Williams AJ, Chen G, Collins T, Thanos D (1998) Recruitment of CBP/p300 by the IFN beta enhanceosome is required for synergistic activation of transcription. Mol Cell 1: 277–287 [DOI] [PubMed] [Google Scholar]
- Minucci S, Pelicci PG (1999) Retinoid receptors in health and disease: co-regulators and the chromatin connection. Semin Cell Dev Biol 10: 215–225 [DOI] [PubMed] [Google Scholar]
- Morin P, Genin P, Doly J, Civas A (2002) The virus-induced factor VIF differentially recognizes the virus-responsive modules of the mouse IFNA4 gene promoter. J Interferon Cytokine Res 22: 77–86 [DOI] [PubMed] [Google Scholar]
- Nebbioso A, Clarke N, Bontempo P, Volz E, Alvarez R, Schiavone EM, Weisz A, Bresciani F, de Lera AR, Gronemeyer H, Altucci L (2004) Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. (submitted) [DOI] [PubMed]
- Nissen RM, Yamamoto KR (2000) The glucocorticoid receptor inhibits NFkappaB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev 14: 2314–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozawa H, Oda E, Nakao K, Ishihara M, Ueda S, Yokochi T, Ogasawara K, Nakatsuru Y, Shimizu S, Ohira Y, Hioki K, Aizawa S, Ishikawa T, Katsuki M, Muto T, Taniguchi T, Tanaka N (1999) Loss of transcription factor IRF-1 affects tumor susceptibility in mice carrying the Ha-ras transgene or nullizygosity for p53. Genes Dev 13: 1240–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz MA, Bayon Y, Lopez-Hernandez FJ, Piedrafita FJ (2002) Retinoids in combination therapies for the treatment of cancer: mechanisms and perspectives. Drug Resist Update 5: 162–175 [DOI] [PubMed] [Google Scholar]
- Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, Pelicci PG (2000) PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 406: 207–210 [DOI] [PubMed] [Google Scholar]
- Pelicano L, Li F, Schindler C, Chelbi-Alix MK (1997) Retinoic acid enhances the expression of interferon-induced proteins: evidence for multiple mechanisms of action. Oncogene 15: 2349–2359 [DOI] [PubMed] [Google Scholar]
- Percario ZA, Giandomenico V, Fiorucci G, Chiantore MV, Vannucchi S, Hiscott J, Affabris E, Romeo G (1999) Retinoic acid is able to induce interferon regulatory factor 1 in squamous carcinoma cells via a STAT-1 independent signalling pathway. Cell Growth Differ 10: 263–270 [PubMed] [Google Scholar]
- Piazza F, Gurrieri C, Pandolfi PP (2001) The theory of APL. Oncogene 20: 7216–7222 [DOI] [PubMed] [Google Scholar]
- Schmaltz C, Alpdogan O, Kappel BJ, Muriglan SJ, Rotolo JA, Ongchin J, Willis LM, Greenberg AS, Eng JM, Crawford JM, Murphy GF, Yagita H, Walczak H, Peschon JJ, van den Brink MR (2002) T cells require TRAIL for optimal graft-versus-tumor activity. Nat Med 8: 1433–1437 [DOI] [PubMed] [Google Scholar]
- Sedger LM, Glaccum MB, Schuh JC, Kanaly ST, Williamson E, Kayagaki N, Yun T, Smolak P, Le T, Goodwin R, Gliniak B (2002) Characterization of the in vivo function of TNF-alpha-related apoptosis-inducing ligand, TRAIL/Apo2L, using TRAIL/Apo2L gene-deficient mice. Eur J Immunol 32: 2246–2254 [DOI] [PubMed] [Google Scholar]
- Smyth MJ, Takeda K, Hayakawa Y, Peschon JJ, van den Brink MR, Yagita H (2003) Nature's TRAIL—on a path to cancer immunotherapy. Immunity 18: 1–6 [DOI] [PubMed] [Google Scholar]
- Sporn MB, Suh N (2002) Opinion: chemoprevention: an essential approach to controlling cancer. Nat Rev Cancer 2: 537–543 [DOI] [PubMed] [Google Scholar]
- Sun SY, Lotan R (2002) Retinoids and their receptors in cancer development and chemoprevention. Crit Rev Oncol Hematol 41: 41–55 [DOI] [PubMed] [Google Scholar]
- Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, Sasaki S, Imai K, Shibue T, Honda K, Taniguchi T (2003) Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 424: 516–523 [DOI] [PubMed] [Google Scholar]
- Takeda K, Smyth MJ, Cretney E, Hayakawa Y, Kayagaki N, Yagita H, Okumura K (2002) Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J Exp Med 195: 161–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Smyth MJ, Cretney E, Hayakawa Y, Yamaguchi N, Yagita H, Okumura K (2001) Involvement of tumor necrosis factor-related apoptosis-inducing ligand in NK cell-mediated and IFN-gamma-dependent suppression of subcutaneous tumor growth. Cell Immunol 214: 194–200 [DOI] [PubMed] [Google Scholar]
- Takeda K, Yamaguchi N, Akiba H, Kojima Y, Hayakawa Y, Tanner JE, Sayers TJ, Seki N, Okumura K, Yagita H, Smyth MJ (2004) Induction of tumor-specific T Cell immunity by anti-DR5 antibody therapy. J Exp Med 199: 437–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura T, Ishihara M, Lamphier MS, Tanaka N, Oishi I, Aizawa S, Matsuyama T, Mak TW, Taki S, Taniguchi T (1995) An IRF-1-dependent pathway of DNA damage-induced apoptosis in mitogen- activated T lymphocytes. Nature 376: 596–599 [DOI] [PubMed] [Google Scholar]
- Tanaka N, Ishihara M, Kitagawa M, Harada H, Kimura T, Matsuyama T, Lamphier MS, Aizawa S, Mak TW, Taniguchi T (1994a) Cellular commitment to oncogene-induced transformation or apoptosis is dependent on the transcription factor IRF-1. Cell 77: 829–839 [DOI] [PubMed] [Google Scholar]
- Tanaka N, Ishihara M, Lamphier MS, Nozawa H, Matsuyama T, Mak TW, Aizawa S, Tokino T, Oren M, Taniguchi T (1996) Cooperation of the tumour suppressors IRF-1 and p53 in response to DNA damage. Nature 382: 816–818 [DOI] [PubMed] [Google Scholar]
- Tanaka N, Ishihara M, Taniguchi T (1994b) Suppression of c-myc or fosB-induced cell transformation by the transcription factor IRF-1. Cancer Lett 83: 191–196 [DOI] [PubMed] [Google Scholar]
- Tanaka N, Kawakami T, Taniguchi T (1993) Recognition DNA sequences of interferon regulatory factor 1 (IRF-1) and IRF-2, regulators of cell growth and the interferon system. Mol Cell Biol 13: 4531–4538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Taniguchi T (2000) The interferon regulatory factors and oncogenesis. Semin Cancer Biol 10: 73–81 [DOI] [PubMed] [Google Scholar]
- Taneja R, Rochette-Egly C, Plassat JL, Penna L, Gaub MP, Chambon P (1997) Phosphorylation of activation functions AF-1 and AF-2 of RAR alpha and RAR gamma is indispensable for differentiation of F9 cells upon retinoic acid and cAMP treatment. EMBO J 16: 6452–6465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanos D, Maniatis T (1995) Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 83: 1091–1100 [DOI] [PubMed] [Google Scholar]
- Um SJ, Kim EJ, Hwang ES, Kim SJ, Namkoong SE, Park JS (2000) Antiproliferative effects of retinoic acid/interferon in cervical carcinoma cell lines: cooperative growth suppression of IRF-1 and p53. Int J Cancer 85: 416–423 [PubMed] [Google Scholar]
- Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, Smith C, Smolak P, Goodwin RG, Rauch CT, Schuh JC, Lynch DH (1999) Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med 5: 157–163 [DOI] [PubMed] [Google Scholar]
- Wang Q, Ji Y, Wang X, Evers BM (2000) Isolation and molecular characterization of the 5′-upstream region of the human TRAIL gene. Biochem Biophys Res Commun 276: 466–471 [DOI] [PubMed] [Google Scholar]
- Wang S, El-Deiry WS (2003a) Requirement of p53 targets in chemosensitization of colonic carcinoma to death ligand therapy. Proc Natl Acad Sci USA 100: 15095–15100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, El-Deiry WS (2003b) TRAIL and apoptosis induction by TNF-family death receptors. Oncogene 22: 8628–8633 [DOI] [PubMed] [Google Scholar]
- Wathelet MG, Lin CH, Parekh BS, Ronco LV, Howley PM, Maniatis T (1998) Virus infection induces the assembly of coordinately activated transcription factors on the IFN-beta enhancer in vivo. Mol Cell 1: 507–518 [DOI] [PubMed] [Google Scholar]
- Wu GS, Burns TF, McDonald ER III, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, Wu G, el-Deiry WS (1997) KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet 17: 141–143 [DOI] [PubMed] [Google Scholar]
- Xie RL, Gupta S, Miele A, Shiffman D, Stein JL, Stein GS, van Wijnen AJ (2003) The tumor suppressor interferon regulatory factor 1 interferes with SP1 activation to repress the human CDK2 promoter. J Biol Chem 278: 26589–26596 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material