

Abstract
The transcription factor TFIIA is encoded by two genes, TFIIAαβ and TFIIAγ. In higher eukaryotes, the TFIIAαβ is translated as a precursor and undergoes proteolytic cleavage; the regulation and biological implications of the cleavage have remained elusive. We determined by Edman degradation that the TFIIAβ subunit starts at Asp 278. We found that a cleavage recognition site (CRS), a string of amino acids QVDG at positions −6 to −3 from Asp 278, is essential for cleavage. Mutations in the CRS that prevent cleavage significantly prolong the half-life of TFIIA. Consistently, the cleaved TFIIA is a substrate for the ubiquitin pathway and proteasome-mediated degradation. We show that mutations in the putative phosphorylation sites of TFIIAβ greatly affect degradation of the β-subunit. We propose that cleavage and subsequent degradation fine-tune the amount of TFIIA in the cell and consequently the level of transcription.
Keywords: ALF, CRS, degradation, TFIIA cleavage, ubiquitylation
Introduction
A critical step in transcription is the recruitment and assembly of the preinitiation complex on the promoter (Roeder, 1998; Lemon and Tjian, 2000). RNA polymerase II and the basal transcription factors are necessary and sufficient to support the basal transcription in vitro (Burley and Roeder, 1996; Orphanides et al, 1996). The basal transcription factor TFIIA has been shown to enhance transcription by interacting with TBP and stabilising its binding to DNA, thereby accelerating a rate-limiting step (Ranish et al, 1999). In addition, TFIIA possesses activator activity and can counteract negative cofactors like NC2/Dr1 and Dr2/PC3 (Ozer et al, 1994, 1998; Sun et al, 1994; Yokomori et al, 1994; DeJong et al, 1995). Basal transcription factors were originally defined as such because they were thought to be universally required for transcription. The identification of cell type- and gene-specific basal transcription factors such as the TBP paralogs, TLFs or the TFIIA paralog, ALF (Crowley et al, 1993; Rabenstein et al, 1999; Upadhyaya et al, 1999; Ozer et al, 2000; Veenstra et al, 2000; Martianov et al, 2002), implies a higher degree of complexity than previously assumed.
TFIIA is encoded by two genes: the small subunits are referred to as TOA2 in budding yeast and TFIIAγ in higher eukaryotes and show a high degree of overall homology, whereas the large subunits, called TOA1 in yeast and TFIIAαβ or TFIIA-L in humans, show extensive homology in both N- and C-terminus, and the central part is less conserved and structured (Ranish et al, 1992). A remarkable phenomenon in higher eukaryotes is the proteolytic cleavage of TFIIAαβ into TFIIAα and TFIIAβ subunits (DeJong and Roeder, 1993; Ma et al, 1993; Yokomori et al, 1993). The site and amino-acid requirements for cleavage have not been described until now, and it is also unclear whether cleavage is a specific and regulated process. Originally, only the cleavage products of native TFIIAαβ were detected in cell extracts in association with TFIIAγ. However, we recently identified a novel transcription complex, TAC, in embryonal carcinoma (EC) cells that contains TFIIAαβ along with TFIIAγ in a complex with TBP (Mitsiou and Stunnenberg, 2000, 2003). This observation and data presented in this study suggest that cleavage of TFIIAαβ precursor is a regulated process and that the cleaved (α+β+γ) and uncleaved (αβ+γ) TFIIA may have distinct cellular functions.
Key transcriptional regulatory mechanisms include post-translational modifications like acetylation, phosphorylation and ubiquitylation. Classically, ubiquitylation has been viewed as a protein disposal pathway, but cells also appear to use this system as a means of fine-tuning transcriptional regulation that involves nonproteolytic pathways (Muratani and Tansey, 2003). It has recently been demonstrated that ubiquitin–proteasome function is required for the transcriptional activity of ERα and VP16 activator (Lonard et al, 2000; Salghetti et al, 2001; Reid et al, 2003). Ubiquitylation of transcription factors can therefore affect their activity, accumulation and localisation.
In this study, we determine the N-terminal amino acid of TFIIAβ by Edman degradation and elucidate the mechanism and biological consequences of the cleavage process. Mutational analysis of the region surrounding the cleavage site revealed a cleavage recognition site (CRS), a string of four amino acids adjacent to the cleavage site that is essential for cleavage. We provide evidence that cleavage triggers degradation of TFIIA via the ubiquitin–proteasome pathway and determines its half-life. Our results further suggest that cleavage and degradation of TFIIA control the level of this basal transcription factor to meet the demands of the cell to adapt rapidly to transcription processes.
Results
Identification of the N-terminus of TFIIAβ
To facilitate rapid purification of TFIIA, we employed classical biochemical and immunoaffinity purification methods using a retrovirally transduced FM3A mouse cell line that stably expresses human TFIIAαβ with Myc and HA epitopes at the N- and C-termini, respectively. The transduced human TFIIAαβ protein is partially cleaved into TFIIAα and TFIIAβ subunits of the expected sizes (Figure 1A). Interestingly, overexpression of TFIIAαβ results in elevated levels of endogenous TFIIAγ (compare lane 1 with lane 2), probably resulting from stabilisation of endogenous TFIIAγ due to its interaction with transduced human TFIIAαβ. Thus, the transduced human TFIIAαβ is able to form a complex with mouse TFIIAγ.
Figure 1.
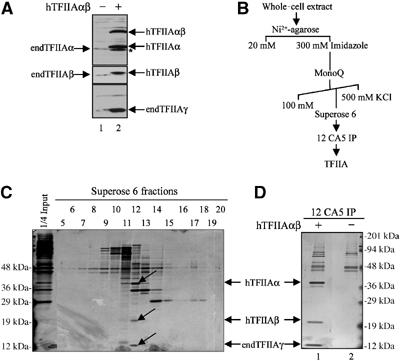
Expression and purification of TFIIA. (A) hTFIIAαβ stably expressed in transduced FM3A cells is cleaved to yield TFIIAα and TFIIAβ subunits. Extracts from nontransduced (lane 1) and stably transduced cells expressing Myc-HA-tagged hTFIIAαβ (lane 2) were analysed by SDS–PAGE and immunoblotting using antibodies against TFIIAα, TFIIAβ and TFIIAγ. The band marked with an asterisk is a minor TFIIAα product. (B) TFIIA purification scheme using whole-cell extracts from FM3A cells stably expressing hTFIIAαβ. (C) Superose 6 fractions were subjected to SDS–PAGE and silver staining. Fractions 12 and 13 contain TFIIA subunits (α, β and γ), as depicted by arrows. (D) TFIIA-containing fraction 12 from Superose 6 column (lane 1) or extract from nontransduced FM3A cells (lane 2) was subjected to immunoprecipitation using the 12 CA5 (HA) antibody against HA-tagged hTFIIAβ. The immunoprecipitates were eluted with an excess of the HA synthetic peptide and analysed by SDS–PAGE and silver staining. endTFIIA=endogenous TFIIA.
Whole-cell extracts prepared from a pool of transduced FM3A cells were used for purification of TFIIA as outlined in Figure 1B. Fractions from the Superose 6 column were analysed by SDS–PAGE and silver staining (Figure 1C) and by immunoblotting (data not shown). Fraction 12 containing the highest concentration of TFIIA was subjected to immunoprecipitation using the 12 CA5 monoclonal antibody against the HA epitope on the C-terminus of the β-subunit (Figure 1D). The immunoprecipitate contained, among others, polypeptides with molecular weights of about 40, 20 and 12 kDa, the expected sizes of the TFIIA subunits. Western blotting indeed identified these as the α-, β- and γ-subunits of TFIIA, respectively (data not shown). The 40-kDa protein and other slower migrating polypeptides (Figure 2D) were excised and analysed by mass spectrometry. A tryptic fragment of 11 amino acids conserved between mouse and human identified the 40-kDa polypeptide as the TFIIAα subunit. The identity of the slower migrating polypeptides indicated that they were most likely nonspecific contaminants (data not shown). The 20-kDa polypeptide corresponding to TFIIAβ was subjected to N-terminal sequence analysis (Edman degradation), which revealed Asp278 as the most N-terminal amino acid. The sequence of amino acids is presented in Figure 2A.
Figure 2.

Mutational analysis of the TFIIAαβ cleavage region. (A) Sequence alignment indicates conservation of the GTG/DT TFIIAαβ cleavage site (marked by arrow) among human, mouse, Xenopus, Drosophila and S. pombe. The 12 residues following the arrow were from cycles 1 to 12 in the Edman degradation. The amino acids of the CRS that are essential for cleavage are boxed. (B) Extracts from U2-OS cells transfected with plasmids expressing Myc-tagged hTFIIAαβ (wt or the Ala mutants of the indicated residues) and hTFIIAγ were analysed by SDS–PAGE and immunoblotting using antibodies against TFIIAα (upper panel) and TFIIAβ and TFIIAγ (lower panel). The asterisk marks the position of residue A267 that was not included in the analysis. (C) U2-OS cell extracts, from panel B were subjected to immunoprecipitation using an antibody against TFIIAγ. The immunoprecipitates were analysed by SDS–PAGE and immunoblotting as in (B). (D) Whole-cell extracts (WCE) from U2-OS cells transfected with plasmids expressing hTBP, Myc-tagged hTFIIAαβ (wt or the indicated mutants) and hTFIIAγ as indicated were used for EMSA with a synthetic oligonucleotide comprising the adenovirus 2 major late TATA box (lanes 4–13). Recombinant TBP and TFIIA were used giving rise to the DA (TBP–TFIIA–DNA) complex (lane 3).
Analysis of the cleavage site
To elucidate the molecular determinants of the cleavage, we performed a mutational analysis (alanine scan) of the region surrounding the cleavage site. Wild-type and mutant TFIIAαβ were cotransfected along with TFIIAγ into U2-OS cells and analysed by immunoblotting (Figure 2B). Interestingly, mutation of the amino acids at positions −6 to −3 with respect to the cleavage site (Q272A, V273A, D274A and G275A) either abolished cleavage or yielded only trace amounts of cleavage products. Mutation of the amino acid at +1, that is, the cleavage site (D278A), caused a significant reduction in the levels of the cleavage products TFIIAα and TFIIAβ as well as increased accumulation of TFIIAαβ. In contrast, mutation of the amino acids −2 and −1 (T276A and G277A, respectively) did not significantly affect cleavage. Mutations at −8, +3 and +5 (V270A, S280A and E282A) slightly affected cleavage and yielded elevated levels of the precursor. S280A affected the efficacy of the cleavage as well as yielded a doublet at the position of TFIIAβ. Whether this doublet is due to alternative cleavage or a post-translational modification is currently under investigation.
To test whether the different TFIIAαβ mutants and, in particular, the uncleaved mutant forms were assembled into functional complexes, coimmunoprecipitation experiments were performed under high-stringency conditions using an antibody against the TFIIAγ subunit (Figure 2C). Cleaved and the uncleaved TFIIAαβ were recovered with equal efficacy as judged from their relative abundance in the input and in the immunoprecipitates. Interestingly, both TFIIAβ polypeptides obtained with mutant S280A were efficiently coimmunoprecipitated with TFIIAγ, demonstrating that both peptides are part of a TFIIA complex.
Band shift assays performed with crude extracts from transfected U2-OS cells revealed that the mutated, uncleaved TFIIA facilitates a so-called DA mobility shift in the presence of TBP similar to that observed with wild-type TFIIA (Figure 2D, lanes 3, 5 and 8–13). Similarly, the uncleaved mutants of TFIIA raised activation of transcription from a tk promoter (Mitsiou and Stunnenberg, 2000) to levels similar to that observed with the wild-type protein (data not shown). Subcellular localisation experiments also showed that the uncleavable TFIIA was distributed similarly to the wild-type protein (data not shown).
Taken together, our data show that uncleavable and cleavable TFIIA equally participate in TFIIA complex formation and support TBP–TFIIA binding to DNA in vitro and transcriptional activation in vivo.
The cleavage site is highly conserved
The mutational analysis revealed a CRS, a string of four residues in the TFIIAα subunit essential for cleavage. Alignment of the region surrounding the cleavage site revealed a high degree of homology with TFIIAαβ from other organisms except budding yeast (Figure 2A). First, the Asp at position +1 is conserved in all species for which the sequence of TFIIAαβ is known. Second, the CRS is fully conserved in vertebrates and highly similar in Drosophila melanogaster, Caenorhabditis elegans and Schizosaccharomyces pombe. Furthermore, the spacing between the cleavage site and the CRS is conserved with the exception of C. elegans and S. pombe. The cleavage site is followed by several potential CKI and CKII phosphorylation sites (TSSEED) (Pinna, 2002). The high homology at and preceding the cleavage site is even more striking considering that the CRS is embedded in a part of TFIIAαβ that is of low sequence complexity and overall poorly conserved. Interestingly, a CRS is absent in S. cerevisiae and consistently TFIIA is not cleaved in this organism. Therefore, the high conservation of the CRS in addition to its critical role in the cleavage process strongly suggests that the CRS determines the position of the cleavage and furthermore predicts that cleavage of TFIIA also occurs in C. elegans and S. pombe.
Human ALF is cleaved
As the CRS is also highly conserved in the germ cell-specific TFIIA-like factor ALF, a TFIIA variant (Figure 3A), we examined whether human ALF is cleaved as well. Extracts from U2-OS cells transfected with Myc-tagged ALF alone or together with TFIIAγ were analysed by immunoblotting using the Myc antibody (Figure 3B). Western blotting identified a polypeptide that migrated at the position of full-length, recombinant ALF as well as a second ALF-derived polypeptide migrating with the relative mobility expected for the N-terminal moiety of ALF (left panels, lane 2). This is in accordance with a recent report that Xenopus laevis ALF is subject to cleavage in Xenopus oocytes (Han et al, 2003). Coimmunoprecipitation experiments further showed that both forms of ALF indeed form a complex with TFIIAγ (Figure 3B, right panels, lane 4). ALF could not be detected in the absence of TFIIAγ, suggesting that ALF, like TFIIAαβ, is unstable and formation of a complex between ALF and TFIIAγ is a prerequisite for its accumulation. Given the fact that a TFIIAγ paralogue has not been found in the genome, the recent study in Xenopus (Han et al, 2003) and our data in mammalian cells strongly suggest that TFIIAγ is the natural partner of TFIIAαβ as well as ALF.
Figure 3.
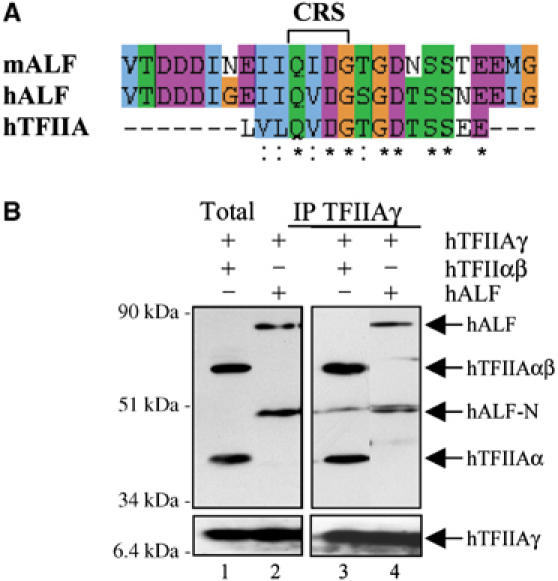
The TFIIA-like factor ALF is cleaved. (A) Sequence alignment among hTFIIAαβ and human and mouse ALF. The CRS is marked in the figure. (B) Extracts from U2-OS cells transfected with plasmids expressing hTFIIAγ together with Myc-tagged hTFIIAαβ (lanes 1 and 3) or Myc-tagged hALF (lanes 2 and 4) were subjected to immunoprecipitation using an antibody against TFIIAγ. The immunoprecipitates were analysed by SDS–PAGE and immunoblotting using the Myc antibody (upper panels) and the TFIIAγ-specific antibody (lower panels).
Cleavage of TFIIA affects its stability
Our results show that the uncleavable forms of TFIIAαβ such as G275A accumulated to higher levels than the wild-type protein (Figure 2B). Accordingly, TFIIAγ accumulated to higher levels when cotransfected with the uncleavable TFIIAαβ mutants than with wild-type TFIIAαβ. To investigate whether these mutations affected the stability of the different forms of TFIIA, we performed pulse-chase experiments to assess their half-life in vivo. Figure 4A shows that significant cleavage of TFIIAαβ occurred already during the pulse period, that is, within 1 h. Yet, cleavage took several hours to complete, suggesting that there is a limiting factor that is required for cleavage. Furthermore, the uncleavable mutant appeared to be more stable than wild-type TFIIA that undergoes cleavage (compare lanes 7–11 with lanes 2–6). Quantitation analysis revealed that equal amounts of wild-type products (TFIIAαβ+α+β) and uncleavable mutant TFIIA G275A were detected immediately after the pulse (Figure 4B), showing that both forms are produced at about equal rate. However, clear differences in accumulation could be observed after 8 and 24 h of chase, at which point uncleavable TFIIA G275A was roughly four times more abundant than wild-type protein. These data suggest that cleavage causes destabilisation of TFIIA. Experiments performed in the presence of cycloheximide (CHX) to prevent de novo protein synthesis demonstrated similar differences in accumulation of the wild-type and mutant forms of TFIIA (Figure 4C and data not shown). To extend our analysis, a number of mutants that displayed impaired cleavage were tested in identical settings (Figure 4D and E). Similarly, these results show that CRS mutants are stabilised up to four-fold compared to the wild-type protein.
Figure 4.

Uncleavable TFIIA is more stable than cleavable TFIIA. (A) U2-OS cells transfected with plasmids expressing hTFIIAγ together with Myc-tagged hTFIIAαβ wt (lanes 2–6) or mutant G275A (lanes 7–11) were labelled for 1 h with [35S]Trans followed by a chase for 1, 2, 4, 8 and 24 h. Extracts from these cells were subjected to immunoprecipitation using the Myc antibody followed by SDS–PAGE and fluorography. (B) Quantitation of labelled proteins from (A) was performed by Phosphoimager. The results represent the average of three independent experiments. (C) Extracts from U2-OS cells transfected with plasmids expressing hTFIIAγ and Myc-tagged hTFIIAαβ wild-type or mutant G275A and treated with CHX were analysed by SDS–PAGE and immunoblotting using antibodies against TFIIAα and GFP. Plasmid expressing GFP was cotransfected as the internal control. (D) Extracts from U2-OS cells transfected with plasmids expressing hTFIIAγ and Myc-tagged hTFIIAαβ wild type or mutants as indicated and treated with CHX were analysed by SDS–PAGE and immunoblotting using antibodies against TFIIAα. (E) Quantitation of (D) was performed by Phosphoimager. The result represents the average of three independent experiments.
Cleaved TFIIA is a substrate for the 26S proteasome
Having established that cleavage of TFIIA reduces its stability, we tested whether TFIIA is a substrate for the ubiquitin–proteasome pathway. Extracts from U2-OS cells cotransfected with TFIIAαβ and TFIIAγ and treated with the proteasome inhibitor MG132 were analysed by immunoblotting. Figure 5A shows that the TFIIAα and TFIIAβ subunits were stabilised upon treatment with MG132 (compare lane 2 with lane 1), indicating that these subunits are degraded by the 26S proteasome. In striking contrast, the uncleaved form of the protein was unchanged upon MG132 treatment, demonstrating that TFIIAαβ is not targeted for proteasome-mediated degradation. Stabilisation of endogenous p53 in the presence of MG132 served as an internal control (Woods and Vousden, 2001). Thus, the cleaved TFIIAα and TFIIAβ, but not TFIIAαβ, are degraded via the proteasome, which supports the notion that cleavage of TFIIA is a prerequisite for degradation and is in agreement with the half-life studies.
Figure 5.

Cleaved TFIIA is a substrate for proteasome-mediated degradation. (A) Inhibition of proteasome activity results in the stabilisation of hTFIIAα and hTFIIAβ subunits. Extracts from U2-OS cells transfected with plasmids expressing hTFIIAαβ and hTFIIAγ and treated (lane 2) or not (lane 1) with MG132 were analysed by SDS–PAGE and immunoblotting using antibodies against TFIIA (upper panel), TFIIAβ (middle panel) and p53 (lower panel). (B) Extracts from U2-OS cells transfected with plasmids expressing hTFIIAγ, Myc-tagged hTFIIAαβ and HA-tagged ubiquitin as indicated were subjected to immunoprecipitation under high-stringency conditions using an antibody against TFIIAβ. Extracts (lanes 1–4) and immunoprecipitates (lanes 5–8) were analysed by SDS–PAGE and immunblotting using antibodies against TFIIAα (lanes 1–4, upper panel), TFIIAβ and TFIIAγ (lanes 1–4, lower panel) and HA (lanes 5–8). (C) Extracts from U2-OS cells transfected with plasmids expressing hTFIIAγ and Myc-tagged hTFIIAαβ wild type or mutants as indicated were analysed by SDS–PAGE and immunoblotting using antibodies against TFIIAα, TFIIAβ and TFIIAγ. (D) Extracts from U2-OS cells transfected with plasmids expressing hTFIIAγ and Myc-tagged hTFIIAαβ wild type or mutants as indicated and treated with CHX were analysed by SDS–PAGE and immunoblotting using antibodies against TFIIAα and TFIIAβ. (E) Quantitation of (D) was performed by Phosphoimager. The result represents the average of three independent experiments. (F) Extracts from U2-OS cells transfected with plasmids expressing hTFIIAγ and Myc-tagged hTFIIAαβ and treated with CHX in the presence or the absence of α-amanitin or ActD as indicated were analysed by SDS–PAGE and immunoblotting using antibodies against TFIIAα and GFP (upper panel), TFIIAβ (middle panel) and TFIIA (lower panel). Plasmid expressing GFP was cotransfected as the internal control.
Given that most substrates for the proteasome are ubiquitylated, we next investigated whether cleaved TFIIA is ubiquitylated. Extracts from U2-OS cells cotransfected with an HA-tagged form of ubiquitin along with TFIIA were used for immunoprecipitation with an antibody against TFIIAβ under high-stringency conditions. Western blot analysis using the HA antibody revealed a characteristic laddering of ubiquitylated proteins only when TFIIA and HA–ubiquitin were coexpressed (Figure 5B, compare lanes 5–7 with lane 8). A weak laddering was observed when HA–ubiquitin was transfected alone, which may be due to ubiquitylation of endogenous TFIIA (lane 6). The smallest ubiquitylated polypeptide migrates at the position of ∼29 kDa (asterisk), consistent with the predicted size of monoubiquitylated TFIIAβ. Importantly, ubiquitylated TFIIA subunits could not be detected following immunoprecipitation using antibodies against TFIIAα and TFIIAγ (data not shown). Collectively, the above data suggest that ubiquitylation of TFIIAβ destabilises or even disrupts the TFIIA complex.
We have demonstrated that inhibiting TFIIA cleavage through mutations in the CRS increases the stability of the protein, linking processing of TFIIA to its degradation. Furthermore, we have shown that treating cells with the proteasome inhibitor stabilises the cleaved forms of TFIIA but not the uncleaved form, suggesting that TFIIAα and TFIIAβ are substrates for proteasomal degradation. Interestingly, cleavage of TFIIA creates an N-terminal aspartate, a secondary destabilising residue according to the N-end rule (Varshavsky 1996). To study the significance of the N-terminus of TFIIAβ for its stability, we generated two mutants: D278M, yielding a stabilising N-terminal residue, and D278R, yielding a primary destabilising N-terminus. Extracts from U2-OS cells transfected with TFIIAγ and wild-type or mutant TFIIAαβ (D278A, D278M and D278R, respectively) were analysed by Western blotting using antibodies against different TFIIA subunits (Figure 5C). In agreement with data presented in Figure 2, all D278 mutants showed reduced cleavage efficacy, indicating that residue D278 is important but not essential for cleavage, in contrast to residues within the CRS. Interestingly, while the steady-state levels of TFIIAγ remained the same, significant differences were observed with respect to TFIIAβ levels. TFIIAβ accumulated to a higher level when Asp was mutated to Met compared to Arg, supporting the hypothesis that TFIIAβ is a substrate for the N-end rule (Figure 5C, compare lane 4 with lane 5). D278A behaved similar to D278R, consistent with alanine being an unstable N-end residue. The effect was observed to a lesser extent for TFIIAα, suggesting that destabilisation of TFIIAβ also affects the stability of other subunits. Quantitation of the endogenous TFIIA subunits revealed that TFIIAβ is under-represented as compared to TFIIAα in extracts from U2-OS cells (data not shown), in agreement with the above observations.
To extend and corroborate these observations, we compared the turnover of TFIIAαβ, TFIIAα and TFIIAβ using extracts from U2-OS cells transfected with wild-type or mutant TFIIAαβ (D278M and D278R) in the presence of CHX. Figure 5D and E show that the stability of TFIIA is dependent on the identity of residue 278 and that both D278M and D278R mutants of TFIIAαβ exhibit reduced cleavage as compared to wild-type protein. Interestingly, mutant D278M stabilises the TFIIAβ subunit, whereas D278R increases its degradation (Figure 5D and E). The above results support our notion that cleavage of TFIIAαβ is linked to the turnover of the protein and that TFIIAβ is a substrate for N-end rule degradation.
Our observation that TFIIAβ is likely degraded via the proteasome combined with recent publications showing that transcription is required for the proteasome-mediated degradation of liganded hERα and SREBP (Reid et al, 2003; Sundqvist and Ericsson, 2003) led us to assess whether degradation of TFIIA subunits is linked to transcription. Therefore, TFIIA was expressed in U2-OS cells and the effect of transcription inhibitors was tested in the presence of CHX. Figure 5F shows that treatment with α-amanitin or actinomycin D (ActD) neither affects cleavage nor degradation of TFIIA subunits. Similar results were also obtained with pulse-chase experiments in the presence of transcription inhibitors (data not shown). Taken together, our data indicate that degradation of TFIIAβ via the proteasome, probably via the N-end rule, occurs independent of its transcriptional activity.
TFIIAβ stability is regulated by residues C-terminal of the cleavage site
Our results demonstrate that the cleaved TFIIA is degraded by the proteasome and indicate that TFIIAβ is ubiquitylated. As mentioned previously, the TFIIAβ subunit contains three conserved, potential phosphorylation sites directly C-terminal of the cleavage site (TSS). The single mutations T279A and S280A displayed no apparent effect on cleavage or stability, whereas S281A reduced the steady-state levels of TFIIAαβ (Figures 2 and 6A, lanes 2–4), suggesting that S281A is unstable or cleaved at a higher rate than wild-type TFIIAαβ. To remove all phosphorylation sites simultaneously, we mutated T279/S280/S281 to alanines (TSS-A). This had a dramatic effect on the stability of TFIIAα and particularly the TFIIAβ, which was only detectable after long exposure (Figure 6A, compare lanes 1 and 5). Similarly, mutating the recognition sites of casein kinases I and II by the triple mutation E282A/E283A/D284A dramatically destabilised the β-subunit (data not shown). The β-specific antibody is raised against the most C-terminal 76 amino acids of the precursor, and recognition by the antibody of the respective TFIIAαβ mutants is unaffected, arguing against reduced epitope recognition. In addition, identical results were obtained using antibodies against the C-terminal HA tag of the β-subunit.
Figure 6.
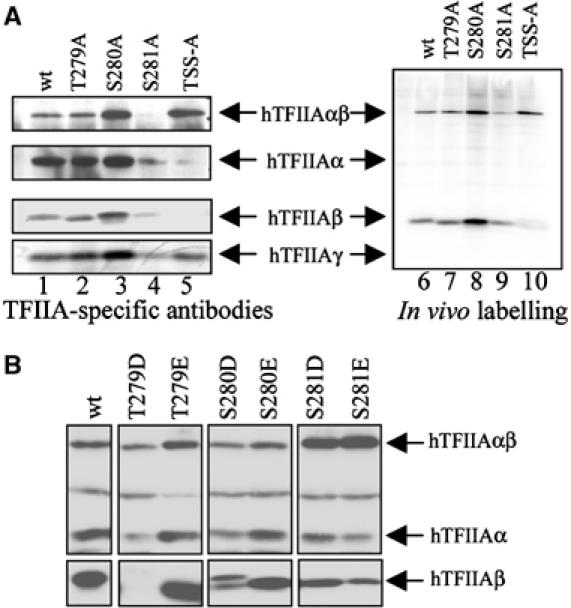
Residues adjacent to the cleavage site are important for the stability of TFIIAβ. (A) Extracts from U2-OS cells transfected with plasmids expressing hTFIIAγ and Myc-tagged hTFIIAαβ wild type or mutants as indicated and labelled with [32P]PO4 were subjected to Ni-NTA purification and immunoprecipitation with the Myc antibody followed by analysis with SDS–PAGE and immunoblotting using antibodies against TFIIAα, TFIIAβ and TFIIAγ or fluorography. (B) Extracts from U2-OS cells transfected with plasmids expressing hTFIIAγ and hTFIIAαβ wild type or mutants as indicated were analysed by SDS–PAGE and immunoblotting using antibodies against TFIIAα, TFIIAβ and TFIIAγ.
To assess directly whether residues 279–281 are phosphorylated, we performed in vivo labelling experiments using TFIIA wild type, the mutants T279A, S280A and S281A, and the triple mutant TSS-A. Figure 6A shows that TFIIAαβ and TFIIAβ, but not TFIIAα and TFIIAγ, are phosphorylated (lane 6). None of the mutations significantly affected the overall 32P incorporation into TFIIA and the phosphorylation level roughly corresponded to the protein level in all cases (Figure 6A, compare lanes 1–5 with lanes 6–10). Mimicking constitutive phosphorylation by mutating T279/S280/S281 to Asp or Glu yielded ambiguous results. Whereas T279D and S280D destabilised the β-subunit, T279E, S280E, S281E and S281D behaved similar to wild type (Figure 6B).
In conclusion, we have found that the residues immediately C-terminal to the newly created N-terminus are critical for the stability of TFIIAβ and consequently to TFIIA.
Discussion
It has been known for quite some time that TFIIA undergoes co- or post-translational cleavage (DeJong and Roeder, 1993; Ma et al, 1993; Yokomori et al, 1993), but neither the cleavage site nor the biological implications of the cleavage have been described. In the present study, we characterised the cleavage site of TFIIAαβ and shed light on the function of the cleavage. We identified the CRS, a string of four residues N-terminal of the cleavage site that is essential for the cleavage process and showed that the identity of the amino acid at the cleavage site is less critical. The CRS partially overlaps with a region in TFIIAαβ that has the propensity to form a β-sheet and is directly followed by a highly acidic, probably unstructured part. Given the high conservation of the charged region following the conserved CRS, it is likely that the cleavage site and the CRS are surface exposed.
The amino-acid sequence of the cleavage site and CRS do not match the recognition site of a known protease. The only protease that is known to cleave N-terminal to an Asp is the endopeptidase N-Asp from Pseudomonas fragi, which does not appear to have a homologue in eukaryotes. Recent studies addressing the processing of MLL, a human homologue of the Drosophila trithorax protein, revealed a cleavage recognition site, QV/LDG, which is virtually identical to the CRS in TFIIAαβ (Yokoyama et al, 2002; Hsieh et al, 2003). Furthermore, in MLL, the CRS also precedes a highly acidic stretch containing multiple potential phosphorylation sites. MLL is cleaved in the CRS, between D and G, while we identified the cleavage site of TFIIA more C-terminal of the CRS. Currently, we cannot exclude the possibility that cleavage of TFIIA occurs within the CRS and that the N-terminal D278 is generated by a secondary cleavage or exopeptidase activity. Identification and characterisation of the CRS-specific TFIIAαβ protease will shed more light on the regulatory role of the cleavage and the possible link between TFIIA and MLL processing.
In our experiments, the uncleaved and cleaved forms of TFIIA behave similarly with respect to TBP–DNA binding and transcriptional activity. While these assays are generally used to assess the ‘classical' characteristics of TFIIA like stabilising TBP–DNA interactions, their in vivo value is rather limited, as they do not provide insight into potential promoter-specific differences. Although it seems likely that cleaved and uncleaved TFIIA may have distinct roles as could be inferred from the presence of the uncleaved TFIIA in the TAC complex, appropriate assays are currently under investigation. However, the present study provides evidence that cleavage has an important consequence for the half-life of TFIIA. Inhibition of cleavage as obtained with the mutation in the CRS impairs or prevents protein degradation, leading to accumulation of the protein, which is consistent with the observed increased half-life of the uncleaved TFIIA.
Our results demonstrate that the cleaved but not the uncleaved TFIIA complex is a substrate of the 26S proteasome. Several pieces of evidence suggest that ubiquitylation occurs on TFIIAβ followed by its degradation, and subsequently the TFIIAα and TFIIAγ subunits are degraded. First, the stabilising effect of the proteasome inhibitor MG132 is seen with the α- and β-subunits but not with the TFIIAαβ precursor. Second, the smallest ubiquitylated polypeptide is approximately 29 kDa, consistent with monoubiquitylated TFIIAβ. Third, under high-stringency conditions, the polyubiquitylated forms of TFIIA can only be immunoprecipitated with antibodies raised against TFIIAβ but not TFIIAα or TFIIAγ. Lastly, the instability of the β-subunit is markedly enhanced by mutating sites adjacent to the cleavage site, indicating an important role for these residues in TFIIA degradation and suggesting that degradation of different subunits can occur partly independent of each other. The precise role of the putative phosphorylation sites can only be speculated upon at present. It is likely that phosphorylation of residues T279-S281 protects the β-subunit against proteasomal degradation, and that dephosphorylation accelerates degradation of TFIIAβ as well as the other subunits of TFIIA, similar to what has been reported for c-fos and NF-κB (Coronella-Wood et al, 2004; Yeh et al, 2004).
Cleavage of TFIIA creates an N-terminal aspartate, which is a secondary destabilising residue according to the N-end rule pathway (Varshavsky, 1996). This N-terminal aspartate, although highly conserved in TFIIA and ALF of all higher eukaryotes, is important but not essential for cleavage. It is therefore likely that the aspartate is conserved mostly to render the cleavage product unstable. Recent studies have shown that caspase-mediated cleavage of DIAP1 (Drosophila IAP1) converts the more stable full-length protein into a highly unstable Asn-bearing N-degron and that its subsequent degradation by the N-end rule pathway is essential for the regulation of apoptosis (Ditzel et al, 2003; Varshavsky, 2003). It is therefore conceivable that TFIIAαβ represents a stable pro-N-degron and that its cleavage results in the production of the unstable Asp-bearing N-degron (TFIIAβ). Whether TFIIA indeed represents another physiological N-end rule metazoan substrate remains to be investigated.
We hypothesise that the biological significance and implications of cleavage is to generate a destabilising N-terminus that triggers destruction of the protein to fine-tune the level of this basal transcription factor and consequently the transcriptional activity of the cell. The level of expression of the TFIIA protease and its activity are likely to have consequences for transcriptional initiation and reinitiation. The regulation of cleavage of TFIIA and subsequent degradation of the protein may provide directionality to the transcription process and prevent recycling of the basal factor.
Materials and methods
Plasmids, mutagenesis and antibodies
Expression plasmids encoding hTBP (pSG5-hTBP), Myc-tagged hTFIIAαβ (pSG5-Myc-hTFIIAαβ) and hTFIIAγ (pSG5-hTFIIAγ) have been described earlier (Mitsiou and Stunnenberg, 2000). Expression plasmid encoding Myc-HA-tagged hTFIIAαβ (pSG5-Myc-HA-hTFIIAαβ) was constructed by insertion of a double-stranded oligonucleotide encoding for the HA epitope at the 3′-end of hTFIIAαβ in pSG5-Myc-hTFIIAαβ. pSRα-tk-neo-Myc-HA-hTFIIAαβ was constructed by subcloning the appropriate fragment from pSG5-Myc-HA-hTFIIAαβ into the EcoRI site of pSRα-tk-neo. Plasmid expressing Myc-tagged hALF (pSG5-Myc-ALF) was constructed by subcloning the appropriate fragment from pRSET-ALF (provided by J DeJong) into pSG5-Myc plasmid (Mitsiou and Stunnenberg, 2000). Plasmid pSG5-Myc-hTFIIAαβ was used for mutagenesis according to the manufacturer's instructions (Quick Site-directed Mutagenesis, Stratagene). Plasmid expressing enhanced green fluorescence protein (pEGFP-N1) was from Clontech.
The monoclonal antibodies Myc, HA and SL39 have been described previously (Mitsiou and Stunnenberg, 2000). Polyclonal antibodies against hTFIIA were as follows: αN specific (against the N-terminus, amino acids 1–63), β specific (against amino acids 301–376) and γ specific against the TFIIAγ subunit. Polyclonal antibody against GFP was purchased from Clontech.
Cell culture, retroviral transduction, transient transfections, treatment with CHX, α-amanitin, ActD and the proteasome inhibitor MG132, pulse-chase labelling and in vivo phosphate labelling
U2-OS, HeLa, COS7 and Bosc23 cells were maintained in DMEM supplemented with 10% FCS. P19 EC cells were maintained as described (Berkenstam et al, 1992). FM3A cells were maintained in RPMI medium supplemented with 5% FCS. FM3A cells stably expressing hTFIIAαβ were generated by retroviral transduction. The supernatant from Bosc23 packaging cells transfected with plasmid pSRα-tk-neo-TFIIAαβ was used to transduce FM3A cells at a density of 100 000 cells/ml, followed by selection with G418 (800 μg/ml) (Takebe et al, 1988; Pear et al, 1993).
Transient transfections were performed as described previously (Mitsiou and Stunnenberg, 2000). Transfected cells were washed with PBS, incubated with fresh medium for 24 h and then treated or not with CHX (20 μg/ml) alone or together with α-amanitin (2.5 μg/ml) or ActD (50 ng/ml), for the indicated periods of time. For treatment with the proteasome inhibitor MG132, transfected cells were incubated with 20 μM MG132 for 5 h at 37°C.
For pulse-chase labelling, 48 h after transfection, cells were incubated in methionine- and cysteine-free DMEM supplemented with 10% dialysed FBS (against 0.15 M NaCl, cutoff 10 kDa), for 15 min at 37°C. After starvation, cells were labelled in DMEM supplemented with 10% FBS and 20 μCi/ml Tran35S label (ICN) for 1 h, followed by different chase periods in the fresh medium.
For in vivo phosphate labelling, 36 h after transfection, cells were incubated in phosphate-free DMEM supplemented with 0.5 mCi/ml [32P]PO4 (ICN) for 4 h.
Ni2+-agarose affinity chromatography, ion exchange and gel filtration chromatography, protein extracts, SDS–PAGE, immunoblotting immunoprecipitation, fluorography and electrophoretic mobility shift assay (EMSA)
Purification of TFIIA from FM3A extracts by Ni2+-NTA-agarose affinity chromatography and MonoQ column was carried out as described (DeJong and Roeder, 1993). Fractions containing TFIIA were loaded on a Superose 6 column in 20 mM Tris (pH 7.3), 100 mM KCl, 20% glycerol, 0.2 mM EDTA, 5 mM DTT and 0.5 mM PMSF. Fractions were collected and assayed by immunoblotting. Preparation of cell extracts, SDS–PAGE and immunoblotting have been described (Berkenstam et al, 1992). For quantitative immunoblotting, proteins were detected by ECL plus (Amersham) and analysed by PhosphorImager (Molecular Dynamics). EMSA, immunoprecipitation and fluorography were as described previously (Mitsiou and Stunnenberg, 2000).
Chemical sequencing and Edman degradation analysis
Chemical sequencing was carried out using a Procise 494 instrument from Applied Biosystems (AB) as described (Tempst et al, 1994). Step-wise liberated PTH amino acids were identified using an ‘on-line' HPLC system (AB) equipped with a PTH C18 (2.1 × 220 mm2; 5 μm particle size) column (AB).
Gel-resolved proteins were digested with trypsin, partially fractionated and the resulting peptide mixtures analysed by matrix-assisted laser-desorption/ionisation reflectron time-of-flight (MALDI-reTOF) mass spectrometry (MS) (Reflex III; BRUKER Daltonics, Bremen, Germany), as described (Erdjument-Bromage et al, 1998), and also using an electrospray ionisation triple quadrupole MS/MS instrument (API300; ABI/MDS SCIEX, Thornhill, Canada) modified with an ultrafine ionisation source (Geromanos et al, 2000). Selected precursor or fragment ion masses from the MALDI-TOF MS or NanoES-MS/MS spectra were taken to search the human segment of a protein nonredundant database, as described (Winkler et al, 2002). MS/MS spectra were also inspected for y″ ion series to compare with the computer-generated fragment ion series of the predicted tryptic peptides.
Acknowledgments
We thank members of the Stunnenberg laboratory and Susanne Mandrup for discussions and critical reading of the manuscript. We thank D Reinberg, RG Roeder, J DeJong, O Witte and PM Lieberman for providing plasmids and antibodies. TH was partly financed by the Norwegian Research Council, project number 137372/300. PT was supported by NCI Cancer Center Support Grant P30 CA08748.
References
- Berkenstam A, Ruiz MM, Barettino D, Horikoshi M, Stunnenberg HG (1992) Cooperativity in transactivation between retinoic acid receptor and TFIID requires an activity analogous to E1A. Cell 69: 401–412 [DOI] [PubMed] [Google Scholar]
- Burley SK, Roeder RG (1996) Biochemistry and structural biology of transcription factor IID (TFIID). Annu Rev Biochem 65: 769–799 [DOI] [PubMed] [Google Scholar]
- Coronella-Wood J, Terrand J, Sun H, Chen QM (2004) c-Fos phosphorylation induced by H2O2 prevents proteasomal degradation of c-Fos in cardiomyocytes. J Biol Chem (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Crowley TE, Hoey T, Liu JK, Jan YN, Jan LY, Tjian R (1993) A new factor related to TATA-binding protein has highly restricted expression patterns in Drosophila. Nature 361: 557–561 [DOI] [PubMed] [Google Scholar]
- DeJong J, Bernstein R, Roeder RG (1995) Human general transcription factor TFIIA: characterization of a cDNA encoding the small subunit and requirement for basal and activated transcription. Proc Natl Acad Sci USA 92: 3313–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJong J, Roeder RG (1993) A single cDNA, hTFIIA/α, encodes both the p35 and p19 subunits of human TFIIA. Genes Dev 7: 2220–2234 [DOI] [PubMed] [Google Scholar]
- Ditzel M, Wilson R, Tenev T, Zachariou A, Paul A, Deas E, Meier P (2003) Degradation of DIAP1 by the N-end rule pathway is essential for regulating apoptosis. Nat Cell Biol 5: 467–473 [DOI] [PubMed] [Google Scholar]
- Erdjument-Bromage H, Lui M, Lacomis L, Grewal A, Annan RS, McNulty DE, Carr SA, Tempst P (1998) Examination of micro-tip reversed-phase liquid chromatographic extraction of peptide pools for mass spectrometric analysis. J Chromatogr A 826: 167–181 [DOI] [PubMed] [Google Scholar]
- Geromanos S, Freckleton G, Tempst P (2000) Tuning of an electrospray ionization source for maximum peptide-ion transmission into a mass spectrometer. Anal Chem 72: 777–790 [DOI] [PubMed] [Google Scholar]
- Han SY, Xie W, Hammes SR, DeJong J (2003) Expression of the germ cell-specific transcription factor ALF in Xenopus oocytes compensates for translational inactivation of the somatic factor TFIIA. J Biol Chem 278: 45586–45593 [DOI] [PubMed] [Google Scholar]
- Hsieh JJ, Ernst P, Erdjument-Bromage H, Tempst P, Korsmeyer SJ (2003) Proteolytic cleavage of MLL generates a complex of N- and C-terminal fragments that confers protein stability and subnuclear localization. Mol Cell Biol 23: 186–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemon B, Tjian R (2000) Orchestrated response: a symphony of transcription factors for gene control. Genes Dev 14: 2551–2569 [DOI] [PubMed] [Google Scholar]
- Lonard DM, Nawaz Z, Smith CL, O'Malley BW (2000) The 26S proteasome is required for estrogen receptor-alpha and coactivator turnover and for efficient estrogen receptor-alpha transactivation. Mol Cell 5: 939–948 [DOI] [PubMed] [Google Scholar]
- Ma D, Watanabe H, Mermelstein F, Admon A, Oguri K, Sun X, Wada T, Imai T, Shiroya T, Reinberg D, Handa H (1993) Isolation of a cDNA encoding the largest subunit of TFIIA reveals functions important for activated transcription. Genes Dev 7: 2246–2257 [DOI] [PubMed] [Google Scholar]
- Martianov I, Brancorsini S, Gansmuller A, Parvinen M, Davidson I, Sassone-Corsi P (2002) Distinct functions of TBP and TLF/TRF2 during spermatogenesis: requirement of TLF for heterochromatic chromocenter formation in haploid round spermatids. Development 129: 945–955 [DOI] [PubMed] [Google Scholar]
- Mitsiou DJ, Stunnenberg HG (2000) TAC, a TBP–sans–TAFs complex containing the unprocessed TFIIAαβ precursor and the TFIIAγ subunit. Mol Cell 6: 527–537 [DOI] [PubMed] [Google Scholar]
- Mitsiou DJ, Stunnenberg HG (2003) p300 is involved in formation of the TBP–TFIIA-containing basal transcription complex, TAC. EMBO J 22: 4501–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muratani M, Tansey WP (2003) How the ubiquitin–proteasome system controls transcription. Nat Rev Mol Cell Biol 4: 192–201 [DOI] [PubMed] [Google Scholar]
- Orphanides G, Lagrange T, Reinberg D (1996) The general transcription factors of RNA polymerase II. Genes Dev 10: 2657–2683 [DOI] [PubMed] [Google Scholar]
- Ozer J, Lezina LE, Ewing J, Audi S, Lieberman PM (1998) Association of transcription factor IIA with TATA binding protein is required for transcriptional activation of a subset of promoters and cell cycle progression in Saccharomyces cerevisiae. Mol Cell Biol 18: 2559–2570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozer J, Moore PA, Bolden AH, Lee A, Rosen CA, Lieberman PM (1994) Molecular cloning of the small (gamma) subunit of human TFIIA reveals functions critical for activated transcription. Genes Dev 8: 2324–2335 [DOI] [PubMed] [Google Scholar]
- Ozer J, Moore PA, Lieberman PM (2000) A testis-specific transcription factor IIA (TFIIAtau) stimulates TATA-binding protein-DNA binding and transcription activation. J Biol Chem 275: 122–128 [DOI] [PubMed] [Google Scholar]
- Pear WS, Nolan GP, Scott ML, Baltimore D (1993) Production of high-titer helper-free retroviruses by transient transfection. Proc Natl Acad Sci USA 90: 8392–8396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinna LA (2002) Protein kinase CK2: a challenge to canons. J Cell Sci 115: 3873–3878 [DOI] [PubMed] [Google Scholar]
- Rabenstein MD, Zhou S, Lis JT, Tjian R (1999) TATA box-binding protein (TBP)-related factor 2 (TRF2), a third member of the TBP family. Proc Natl Acad Sci USA 96: 4791–4796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranish JA, Lane WS, Hahn S (1992) Isolation of two genes that encode subunits of the yeast transcription factor IIA. Science 255: 1127–1129 [DOI] [PubMed] [Google Scholar]
- Ranish JA, Yudkovsky N, Hahn S (1999) Intermediates in formation and activity of the RNA polymerase II preinitiation complex: holoenzyme recruitment and a postrecruitment role for the TATA box and TFIIB. Genes Dev 13: 49–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid G, Hubner MR, Metivier R, Brand H, Denger S, Manu D, Beaudouin J, Ellenberg J, Gannon F (2003) Cyclic, proteasome-mediated turnover of unliganded and liganded ERalpha on responsive promoters is an integral feature of estrogen signaling. Mol Cell 11: 695–707 [DOI] [PubMed] [Google Scholar]
- Roeder RG (1998) Role of general and gene-specific cofactors in the regulation of eukaryotic transcription. Cold Spring Harb Symp Quant Biol 63: 201–218 [DOI] [PubMed] [Google Scholar]
- Salghetti SE, Caudy AA, Chenoweth JG, Tansey WP (2001) Regulation of transcriptional activation domain function by ubiquitin. Science 293: 1651–1653 [DOI] [PubMed] [Google Scholar]
- Sun X, Ma D, Sheldon M, Yeung K, Reinberg D (1994) Reconstitution of human TFIIA activity from recombinant polypeptides: a role in TFIID-mediated transcription. Genes Dev 8: 2336–2348 [DOI] [PubMed] [Google Scholar]
- Sundqvist A, Ericsson J (2003) Transcription-dependent degradation controls the stability of the SREBP family of transcription factors. Proc Natl Acad Sci USA 100: 13833–13838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebe Y, Seiki M, Fujisawa J, Hoy P, Yokota K, Arai K, Yoshida M, Arai N (1988) SR alpha promoter: an efficient and versatile mammalian cDNA expression system composed of the simian virus 40 early promoter and the R-U5 segment of human T-cell leukemia virus type 1 long terminal repeat. Mol Cell Biol 8: 466–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempst P, Geromanos S, Elicone C, Erdjument-Bromage H (1994) Improvements in microsequencer performance for low picomole sequence analysis. Methods 6: 248–261 [Google Scholar]
- Upadhyaya AB, Lee SH, DeJong J (1999) Identification of a general transcription factor TFIIAalpha/beta homolog selectively expressed in testis. J Biol Chem 274: 18040–18048 [DOI] [PubMed] [Google Scholar]
- Varshavsky A (1996) The N-end rule: functions, mysteries, uses. Proc Natl Acad Sci USA 93: 12142–12149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshavsky A (2003) The N-end rule and regulation of apoptosis. Nat Cell Biol 5: 373–376 [DOI] [PubMed] [Google Scholar]
- Veenstra GJ, Weeks DL, Wolffe AP (2000) Distinct roles for TBP and TBP-like factor in early embryonic gene transcription in Xenopus. Science 290: 2312–2315 [DOI] [PubMed] [Google Scholar]
- Winkler GS, Lacomis L, Philip J, Erdjument-Bromage H, Svejstrup JQ, Tempst P (2002) Isolation and mass spectrometry of transcription factor complexes. Methods 26: 260–269 [DOI] [PubMed] [Google Scholar]
- Woods DB, Vousden KH (2001) Regulation of p53 function. Exp Cell Res 264: 56–66 [DOI] [PubMed] [Google Scholar]
- Yeh PY, Yeh KH, Chuang SE, Song YC, Cheng AL (2004) Suppression of MEK/ERK signaling pathway enhances cisplatin-induced NF-κB activation by protein phosphatase 4-mediated NF-κB p65 Thr-dephosphorylation. J Biol Chem 279: 26143–26148 [DOI] [PubMed] [Google Scholar]
- Yokomori K, Admon A, Goodrich JA, Chen JL, Tjian R (1993) Drosophila TFIIA-L is processed into two subunits that are associated with the TBP/TAF complex. Genes Dev 7: 2235–2245 [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Kitabayashi I, Ayton PM, Cleary ML, Ohki M (2002) Leukemia proto-oncoprotein MLL is proteolytically processed into 2 fragments with opposite transcriptional properties. Blood 100: 3710–3718 [DOI] [PubMed] [Google Scholar]
- Yokomori K, Zeidler MP, Chen JL, Verrijzer CP, Mlodzik M, Tjian R (1994) Drosophila TFIIA directs cooperative DNA binding with TBP and mediates transcriptional activation. Genes Dev 8: 2313–2323 [DOI] [PubMed] [Google Scholar]