

Abstract
The ataxia-telangiectasia mutated and rad3-related (ATR) kinase orchestrates cellular responses to DNA damage and replication stress. Complete loss of ATR function leads to chromosomal instability and cell death. However, heterozygous ATR mutations are found in human cancers with microsatellite instability, suggesting that ATR haploinsufficiency contributes to tumorigenesis. To test this possibility, we generated human cell line and mouse model systems in which a single ATR allele was inactivated on a mismatch repair (MMR)-deficient background. Monoallelic ATR gene targeting in MLH1-deficient HCT 116 colon carcinoma cells resulted in hypersensitivity to genotoxic stress accompanied by dramatic increases in fragile site instability, and chromosomal amplifications and rearrangements. The ATR+/− HCT 116 cells also displayed compromised activation of Chk1, an important downstream target for ATR. In complementary studies, we demonstrated that mice bearing the same Atr+/−/Mlh1−/− genotype were highly prone to both embryonic lethality and early tumor development. These results demonstrate that MMR proteins and ATR functionally interact during the cellular response to genotoxic stress, and that ATR serves as a haploinsufficient tumor suppressor in MMR-deficient cells.
Keywords: ATR, mismatch repair, MLH1, tumor suppressor
Introduction
To maintain the integrity of their genomes, eukaryotic organisms have evolved a complex network of signaling and effector proteins that allow these cells to recognize damaged or abnormally structured DNA, and to slow the rate of cell-cycle progression until the initiating lesion is repaired. These genome surveillance mechanisms are termed cell-cycle checkpoints (Elledge, 1996; Melo and Toczyski, 2002). Defects in the checkpoint signaling machinery are associated with increased rates of cell death and aging, and may promote the development and/or progression of cancer in humans (Hartwell, 1992; Teixeira da Costa and Lengauer, 2002).
Two members of the phosphoinositide 3-kinase-related kinase family have been identified as proximal components of the checkpoint signaling apparatus assembled in response to DNA strand breaks and DNA replication errors. The ataxia-telangiectasia mutated (ATM) and ataxia-telangiectasia rad3-related (ATR) kinases are rapidly recruited to DNA-damage sites where they relay signals to the damage response network through the phosphorylation of specific sets of target proteins on serine or threonine residues (Abraham, 2001; Shiloh, 2003). The current paradigm suggests that ATM and ATR play only partially overlapping roles in checkpoint activation by different forms of DNA damage. ATM functions primarily as a first responder to a particularly lethal form of DNA damage, the DNA double-strand break (DSB). In contrast, ATR is centrally and indispensably involved in the maintenance of DNA replication fidelity. Through mechanisms that are only partially understood, ATR monitors the progress of active DNA replication forks, and triggers checkpoint activation when fork progression is stalled by abnormally structured or damaged DNA, or by exposure to agents such as hydroxyurea (HU) and aphidicolin (APH) (Casper et al, 2002; Cha and Kleckner, 2002; Cimprich, 2003; Dart et al, 2004; Ward et al, 2004). In the absence of ATR, mammalian cells rapidly accumulate chromosomal abnormalities and lose viability in the absence of any extrinsic genotoxic agents (Brown and Baltimore, 2000; de Klein et al, 2000). This proliferation-associated cytotoxicity is likely due to a profound breakdown in DNA replication fidelity as the cells traverse S phase.
The mismatch repair (MMR) system also plays a central role in genome maintenance during S phase, through the correction of base misincorporation errors arising during normal DNA replication. The core MMR machinery in mammals consists of two heterodimeric protein complexes, MutS and MutL (Harfe and Jinks-Robertson, 2000). In somatic cells, the best characterized of the MutS complexes contains MSH2 partnered with either MSH3 or MSH6, while the major MutL heterodimer in somatic cells contains MLH1 and PMS2. Notably, loss of function mutations in MSH2 and MLH1 is common in hereditary nonpolyposis colon carcinoma and other cancers bearing the microsatellite mutator phenotype (Peltomaki, 2003). In addition to its role in postreplicative DNA repair, the MMR machinery has been linked to genome surveillance pathways. During recombinational DNA repair, the MMR proteins detect base mispairing in recombination intermediates, and block illegitimate recombination events between divergent sequences in the host genome (Harfe and Jinks-Robertson, 2000). Several lines of evidence indicate that the MMR proteins also communicate with the ATM- and ATR-dependent checkpoint signaling pathways. Treatment of mammalian cells with ionizing radiation (IR) during S phase triggers an ATM-dependent checkpoint response that causes an acute suppression of replicative DNA synthesis (Painter and Young, 1980). This response requires the MutS and MutL complexes, and may involve direct interactions between MLH1 and ATM on the one hand, and MSH2 and the ATM target protein, Chk2, on the other (Brown et al, 2003). Other results implicate MSH2 and MLH1 as damage sensors for the ATR-dependent checkpoint signaling cascade in cells exposed to DNA-methylating agents (Cejka et al, 2003; Wang and Qin, 2003). In this case, the MMR proteins may communicate with ATR through MSH2 (Wang and Qin, 2003).
In this study, we demonstrate that disruption of a single ATR allele in an MLH1-deficient human colon cancer cell line causes a dramatic increase in chromosomal instability, together with hypersensitivity to killing by multiple forms of genotoxic stress. To determine whether these genome surveillance and repair defects translate into an increased cancer predisposition, we generated mice bearing the same ATR/MLH1 phenotype (i.e., Atr+/− Mlh1−/− mice), and found that these animals were prone to tumor development at an earlier age than their Atr+/− or Mlh1−/− counterparts. These results indicate that loss of a single ATR allele leads to high-level genetic instability in MMR-deficient cells, and suggest that ATR functions as a haploinsufficient tumor suppressor on an MMR-deficient background.
Results
To generate a cell-based system for studies of potential interactions between ATR and the MMR machinery, we performed somatic cell gene targeting in the human colon carcinoma cell line HCT 116. This cell line is MMR defective due to the expression of markedly reduced levels of MLH1 (Papadopoulos et al, 1994), and has served as the host for previous gene-targeting efforts (Waldman et al, 1995; Brown et al, 1997; Bunz et al, 1998; Cahill et al, 1998; Lengauer et al, 1998). Using a promoterless targeting vector (Cao et al, 2002) that contains a selectable marker (Neor) flanked by 3.1 and 4.5 kb homologous sequences of ATR, we deleted most of exons 4 and 5 of the ATR gene through homologous recombination. After screening by PCR with the primer pair A1–C1 (Figure 1A), we identified eight out of 158 clones that contained one wild-type and one targeted ATR allele. Homologous integration of the targeting cassette was further confirmed by PCR with a second primer pair (A2–C2), and results of the PCR analyses from three independent ATR+/− clones are presented in Figure 1B. Immunoblot analysis indicated that the selected clones expressed ATR at 40–60% of the level found in the parental HCT 116 cell line (Figure 1C).
Figure 1.
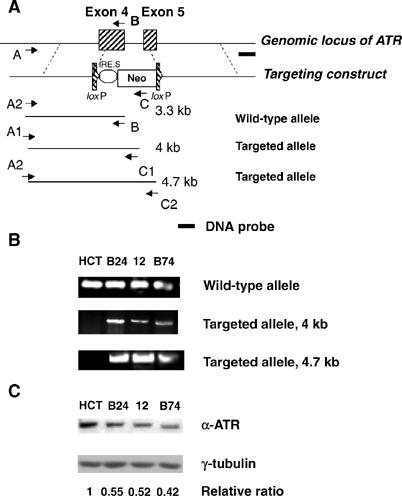
Impaired survival of ATR+/− cells after genotoxic stress. (A) Schematic diagram of ATR-targeting construct. The neomycin resistance (Neo) gene-targeting construct is flanked by two loxP sites to allow Cre recombinase-mediated excision of the drug resistance cassette for targeting of the second ATR allele (not performed in the present study). Primers A, B, and C were used in the PCR screen to identify targeted clones. ‘A' primers (A1 and A2) recognize regions of the ATR gene located outside of the 5′-flanking region used in the targeting cassette. ‘C' primers (C1 and C2) hybridize to different sequences in the neomycin resistance marker (Neo). The dark line labeled ‘Probe' shows the source of ATR genomic probe used or the Southern blot analysis in Figure 6. (B) Identification of targeted clones by PCR analysis with primer pairs indicated in panel A. (C) Expression of ATR protein in HCT 116-derived clones. Sample lanes are identified in panel B legend. The relative ratio indicates the level of ATR protein in each sample lane after normalization of each sample to the tubulin loading control. The resulting ratios were then normalized to that obtained with the HCT 116 cell extract.
Owing to the type of promoterless targeting vector used in this gene disruption strategy, it is formally possible that an amino-terminal ATR fragment (∼100 amino acids, maximum length) encoded by exons 1–3 and the 5′-end of exon 4 could be expressed from the targeted locus. Unfortunately, the existing ATR antibodies do not allow us to test whether such a fragment exists; hence, we cannot rule out the possibility that this fragment is expressed and contributes to the phenotype of the ATR+/− HCT 116 cells.
A previous report showed that HCT 116 cells were sensitized to killing by ultraviolet (UV) light, and the heightened UV sensitivity was reversed by reconstituting the cells with a human chromosome 3 fragment containing the MLH1 gene (HCT 116.chr3 cells) (Koi et al, 1994; Mellon et al, 1996). Cell survival after UV irradiation is also partially dependent on ATR function (Cliby et al, 1998; Abraham, 2001). To determine whether disruption of one ATR allele further sensitized HCT 116 cells to genotoxic stress, we performed clonogenic survival assays with IR-, UV light-, or HU-treated ATR−/+ HCT 116 cells. Monoallelic disruption of the ATR gene led to a further increase in the sensitivity of HCT 116 cells to all three forms of genotoxic stress, whereas two drug-resistant clones that had integrated the targeting vector outside of the ATR locus did not display heightened sensitivity to these DNA-damaging agents (Supplementary Figure 1 and data not shown). To insure that the decreases in clonogenic survival were related to the reduced level of ATR expression, ATR+/− clone B24 cells were transfected with an ATR expression plasmid (Figure 2A). Reconstitution of ATR expression to the wild-type level completely reversed UV sensitivity in clone B24 cells (Figure 2B). We conclude that ATR+/− HCT 116 cells are haploinsufficient for ATR function with respect to recovery from UV light-induced genotoxic stress.
Figure 2.
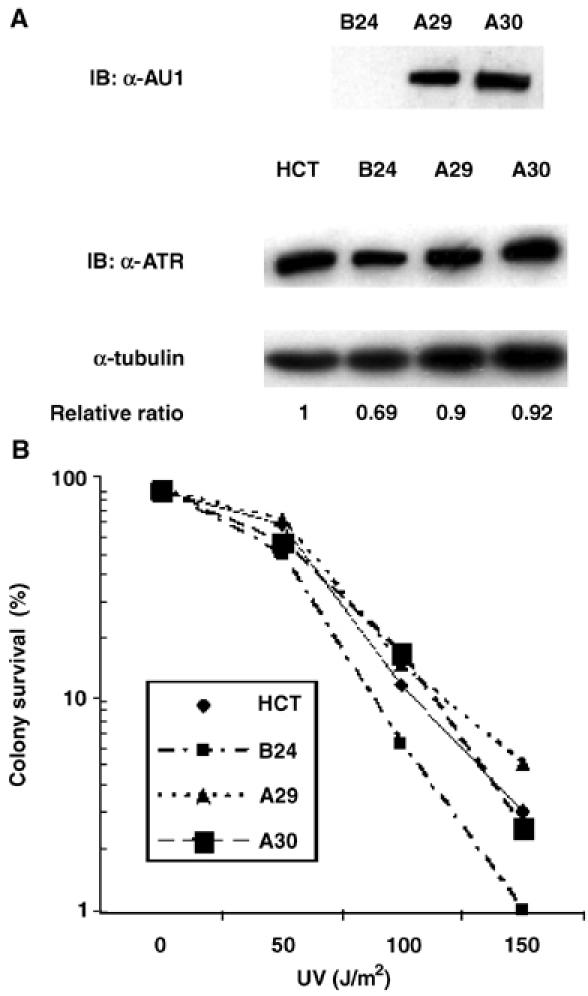
Reconstitution of ATR function in ATR+/− clones. (A) ATR+/− B24 cells were stably transfected with an AU1-tagged ATR expression plasmid. Transfected clones A29 and A30 were selected for clonogenic survival assays. (B) Clonogenic survival of HCT 116, ATR+/− B24, and clone A29 and A30 cells was determined after exposure to the indicated doses of UV.
In humans, a splicing abnormality that leads to decreased ATR expression causes Seckel syndrome, a disorder associated with chromosomal instability (O'Driscoll et al, 2003). To determine whether ATR+/− HCT 116 cells exhibit increased chromosomal instability, we examined metaphase spreads from the parental cell line and three ATR+/− clones. HCT 116 cells exhibited a relatively stable chromosomal content, with the presence of signature alterations in several chromosomes (marked by open arrows in Figure 3B; summarized in Figure 3A), which have been observed previously in this cell line (Masramon et al, 2000). A more detailed inspection revealed that only one out of 11 metaphases of these cells showed additional, minor chromosomal alterations (data not shown). These observations are consistent with the notion that gross chromosomal alterations are rare in cells with microsatellite instability (MSI) (Papadopoulos et al, 1994; Peltomaki, 2003). Similar findings were obtained in analyses of metaphase spreads from MLH-1-reconstituted HCT 116.chr3 cells (results not shown). In contrast, the ATR+/− clones contained numerous chromosomal abnormalities, including translocations, deletions, rearrangements, and random chromosome losses and gains (Figure 3A). More importantly, the chromosomal instability was an ongoing process, since individual cells from the same passage and same cell population of each ATR+/− clone displayed different patterns of chromosomal aberrations (Figure 3C and D). In addition to chromosome gains, losses, and translocations, gene amplification events were also observed. Homogenously staining regions (HSRs), which usually mark gene amplifications, appeared in two of the three ATR+/− clones (see below). These results reinforce the notion that partial loss of ATR function is sufficient to cause a dramatic increase in chromosomal instability in HCT 116 cells, even in the absence of exogenous genotoxic agents.
Figure 3.
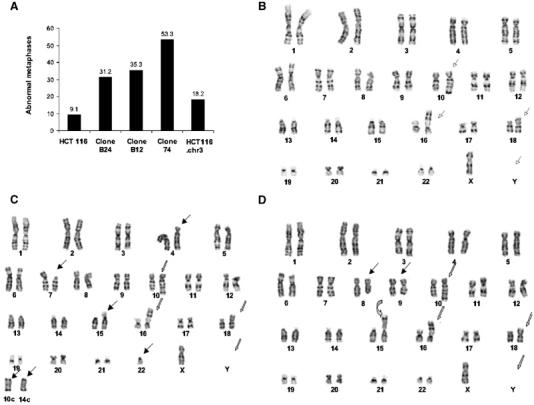
Spontaneous chromosomal instability in ATR+/− cells. (A) Karyotypic analysis. The plot shows the percentage of metaphases with abnormalities for each cell line. The total number of metaphase spreads examined for each cell line were as follows: HCT 116, 11; clone B24, 16; clone 12, 15; clone B74, 17; HCT116.chr3: 11. (B) Representative metaphase spread from HCT 116 cells. Karyotype was stably maintained as 45, X, −Y, add 10q, add 16p, add 18p. The four signature alterations in these chromosomes are marked with open arrows. (C, D) Karyotype of two representative cells from the same ATR+/− clone B74 population. In addition to the four signature chromosomal alterations detected in the parental HCT 116 cells (open arrows), random gross chromosomal aberrations (solid arrows) and an HSR (curved arrow) located between the centromere and the dark band were observed in this metaphase spread.
Complete loss of ATR function in mammalian cells interferes with the DNA-damage-induced G2/M checkpoint (Cliby et al, 1998; Brown and Baltimore, 2003). To assess the integrity of the G2/M checkpoint in ATR+/− HCT 116 cells, asynchronous cultures were treated with IR, and then incubated for 24–96 h in the presence of nocodazole to capture cycling cells in mitosis. IR exposure caused a persistent block to M-phase entry in HCT 116 cells, as expected for cells with an intact G2 checkpoint (Bunz et al, 1998) (Figure 4A). In contrast, the ATR+/− HCT 116 cell populations failed to hold the mitotic block beyond 24–48 h postirradiation (Figure 4A). Both the parental HCT 116 and ATR+/− clones accumulated in mitosis at comparable rates in the presence of nocodazole alone (Figure 4B), indicating that partial loss of ATR function had no intrinsic effect on the rate of G2- to M-phase progression. Thus, inactivation of one ATR allele interferes with the maintenance but not the initiation of the DNA-damage induced G2 checkpoint in HCT 116 cells.
Figure 4.
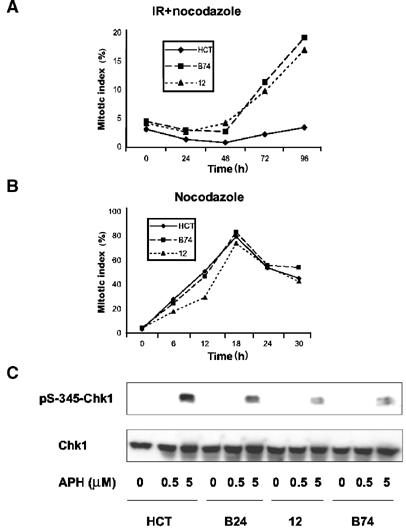
S and G2 checkpoint defects in ATR+/− cells. (A) Parental HCT 116 cells and ATR+/− clone B74 and 12 cells were treated with nocodazole at 30 min after IR exposure. Approximately 200 cells were counted to determine mitotic index. Results shown are representative of two to three experiments for each sample. (B) The same cells were treated with nocodazole only to monitor normal G2-to-M-phase progression. (C) Cells were treated for 1 h with the indicated concentrations of APH, and detergent-soluble proteins were separated by SDS–PAGE and immunoblotted with the indicated antibodies.
A key downstream mediator of ATR-dependent checkpoint signaling is the protein kinase Chk1 (Bartek and Lukas, 2003). To determine whether partial loss of ATR function led to impaired signal relay to Chk1, we examined the phosphorylation of this protein at Ser-345, a known target site for modification by ATR (Zhao and Piwnica-Worms, 2001). HCT 116 cells and three ATR+/− heterozygous clones were treated with APH to induce DNA replication stress, and Chk1 phosphorylation was examined by immunoblotting. Relative to the parental HCT 116 cells, all three of the ATR+/− lines showed impaired phosphorylation of Chk1 after APH exposure (Figure 4C). Thus, a reduction in ATR gene dosage caused a partial defect in Chk1 phosphorylation and presumably activation in response to stalled DNA replication forks.
A recent report demonstrated that severe depletion of ATR protein promoted fragile site instability in MMR-competent human cells (Casper et al, 2002). To determine whether ATR+/− HCT116 cells were prone to fragile site expression, we treated both the parental and ATR+/− HCT 116 cells with a low concentration (0.1 μM) of APH to induce DNA replicative stress. Cells were harvested after 18 h in culture, and mitotic spreads were scored for chromosomal breaks and gaps. Both of the ATR+/− HCT 116 clones showed a >2-fold increase in the numbers of gaps and breaks per cell than did the parental HCT 116 cells (Figure 5A and B). These results indicate that ATR is a haploinsufficient suppressor of fragile site expression in HCT 116 cells.
Figure 5.
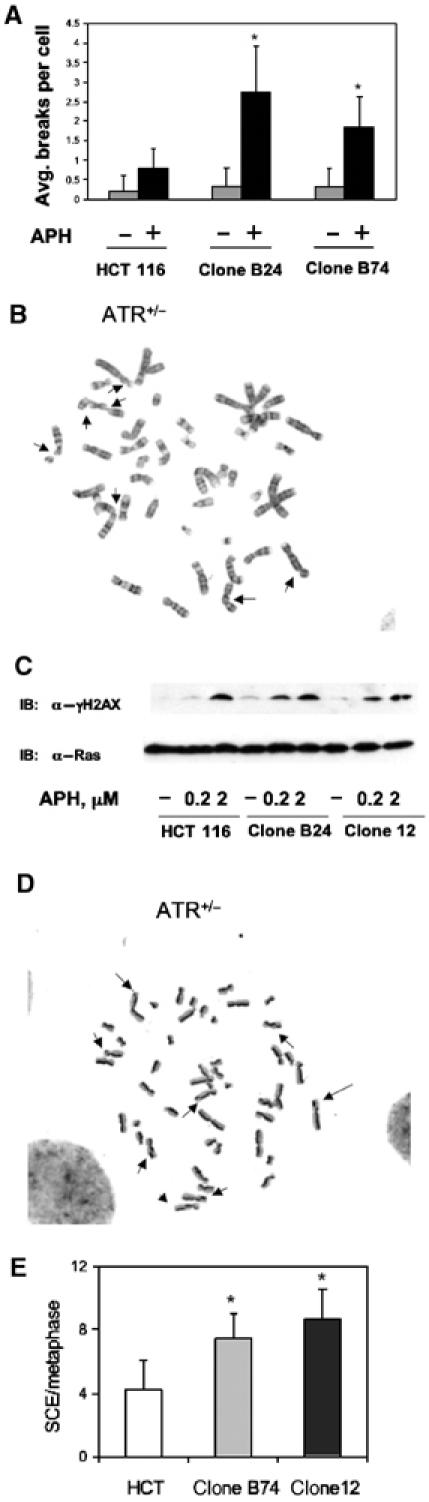
Fragile site expression and SCE in ATR+/− cells. (A) Average numbers of chromosome breaks and gaps per metaphase observed in 25 metaphase spreads from each cell line. Fragile sites were induced by cellular exposure to 0.1 μM APH. *P<0.05. (B) Representative metaphase spread from APH-treated ATR+/− cells. The solid arrows point to fragile sites. (C) Enhanced phosphorylation of histone H2AX in ATR+/− cells after APH treatment. Cells from the parental line and two ATR+/− clones were treated as indicated, and cell extracts were analyzed by immunoblotting with anti-γH2AX antibody. (D) Representative metaphase from ATR+/− cells showing SCE. Sister chromatids were stained differentially and imaged as light or dark strands. The solid arrows point to SCE sites. (E) Average SCE frequencies in HCT 116 cells (n=20 metaphases) and two ATR+/− clones (n=25 metaphases per clone). *P<0.005.
Expression of fragile sites is accompanied by DNA DSB, which in turn triggers the ATM/ATR-dependent phosphorylation of histone H2AX (Brown and Baltimore, 2003). To determine whether APH-induced replicative stress provoked an exaggerated DNA-damage response in ATR+/− HCT 116 cells, we treated these cells with a low (0.2 μM) or high (2.0 μM) concentration of APH. Relative to the parental HCT 116 cells, both of the ATR+/− clones (clones 12 and B74) consistently expressed higher levels of phosphorylated H2AX (γH2AX) in the absence of any extrinsic genotoxic stress, consistent with the idea that these cells accrued elevated amounts of spontaneously damaged DNA during the normal cell cycle (Figure 5C). The difference between the parental HCT 116 cells and the ATR+/− clones was considerably enhanced after the cells were subjected to low-level replicative stress with 0.2 μM APH. In contrast, both the ATR+/+ and ATR+/− HCT 116 cells displayed strong γH2AX responses after DNA replication was blocked with a 10-fold higher concentration of APH. Thus, partial loss of ATR function in HCT 116 cells renders these cells more prone to accumulate DNA damage in the presence of low-level replicative stress.
Common fragile sites are considered ‘hot spots' for a variety of genomic rearrangements, including sister chromatid exchanges (SCEs) and chromosomal translocations and deletions. In accordance with the increase in fragile site instability in ATR+/− HCT 116 cells, an examination of metaphase spreads containing differentially stained sister chromatids revealed an approximate two-fold increase in SCE frequency in these cells (Figure 5D and E).
Homologous integration of the ATR gene-targeting vector was initially determined by PCR with two different primer pairs (Figure 1A and B). During efforts to confirm these results by Southern blot analysis of genomic DNA, we observed that six of seven ATR+/− HCT 116 clones selected for further analysis displayed a highly complex pattern of hybridizing bands, which were suggestive of amplification and/or rearrangement events at the ATR gene loci (Figure 6A). Fluorescence in situ hybridization (FISH), with a probe generated from the ATR cDNA sequence or from a 6 kb genomic fragment, yielded no detectable signal in either nontransfected HCT 116 cells or in a drug-resistant clone that arose from a nonhomologous integration event (unpublished observation). Thus, the germline ATR alleles were not visible in this assay, likely due to the small size of the probe and/or the sensitivity of the detection method. In contrast, extensive regions of hybridization were detected in interphase and metaphase spreads from two of the ATR+/− clones (Figure 6B). In clone B74, the amplification event produced HSRs on a chromosome other than chromosome 3, which normally harbors the ATR locus (Figure 6C). A trivial explanation for these findings is that the particular gene-targeting vector used in these studies sensitized the ATR locus to opportunistic amplification and rearrangement events. Consequently, we generated a distinct ATR-targeting vector designed to integrate into a different region of the ATR gene. Transfection of HCT 116 cells with this targeting vector also yielded homologous integrants that had incurred amplification events at the ATR gene locus, as detected by FISH analysis (results not shown). These results suggest that, over the course of the clonal expansion of the ATR gene-targeted cells, instability at the targeted locus itself led to the accumulation of multiple chromosomal rearrangements involving the ATR gene.
Figure 6.
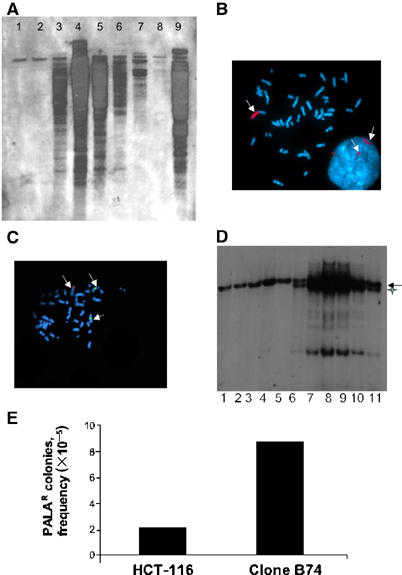
Gene amplification and rearrangement events in ATR+/− cells. (A) Southern blot analysis with the DNA probe shown in Figure 1A. The ATR+/− clones used in this experiment were used at passages 3–5 after recovery from primary frozen stocks, and approximately 35 to 45 population doublings had occurred before the cells were harvested for these experiments. Lane 1, HCT 116; lane 2, a nonhomologously targeted clone; lane 4, clone 12; lane 8, clone B24; lane 9, clone B74. Lanes 3 and 5–7 show results from additional ATR+/− clones coisolated with the three named clones selected for further analyses. (B) Amplification of a chromosomal region containing the ATR locus (marked with red fluorophore) in metaphase and interphase nuclei from ATR+/− cells. (C) Appearance of amplified ATR locus in other chromosomes. The green fluorophore marks chromosome 3 alpha satellite (centromeric) DNA. Regions stained with red and green fluorophores are marked by arrows. (D) Amplification and rearrangement of p21/WAF1 locus during gene targeting. Lane 1, HCT 116 cells; lanes 2–5, hygromycin-resistant clones derived from HCT 116 cells transfected with the p21/WAF1-targeting vector; lane 6, ATR+/− cells (clone 12); lanes 7–11, hygromycin-resistant clones derived from ATR+/− cells transfected with the p21/WAF1-targeting vector. Southern blot was performed as described (Waldman et al, 1995). Restriction fragment from the wild-type p21/WAF1 allele is indicated with an arrow and the additional p21/WAF1-derived fragment found in ATR+/− cells (lane 6) is indicated with a cross. (E) PALA-induced CAD gene amplification. PALA-resistant cell colonies were scored as described in the text. Results shown are representative of two independent trials.
The dramatic destabilization of the ATR gene induced by homologous integration of the targeting vector could reflect either a locus-specific event or a more global destabilizing effect of ATR deficiency on genes that have undergone homologous recombination. To further examine these possibilities, we determined whether targeting of a distinct gene locus, p21/WAF1, led to similar locus-specific amplification and/or rearrangement events in the ATR+/− HCT 116 cells. HCT 116 cells and ATR+/− clone 12 cells were transfected with a p21/WAF1-targeting vector (Waldman et al, 1995), and the p21/WAF1 locus was subsequently analyzed by Southern blot analysis. Interestingly, the nontransfected ATR+/− clone contained an additional genomic DNA restriction fragment (indicated with a cross) that hybridized with the p21/WAF1 probe, suggesting that one of the p21/WAF1 alleles was already altered in these cells. Integration of the p21/WAF1-targeting construct into its cognate gene locus triggered further alterations in the chromosomal region containing this gene (Figure 6D). Thus, these results suggest that ATR+/− HCT 116 cells are generally predisposed to chromosomal amplification and rearrangement events, particularly at gene loci involved in homologous recombination events.
To examine more specifically the impact of ATR gene dosage on gene amplification frequencies in HCT 116 cells, we performed an N-phosphonacetyl-L-aspartate (PALA) resistance study. Chronic exposure to PALA selects for the outgrowth of cells that have amplified the CAD (carbamyl-P synthetase/aspartate transcarbamylase/dihydro-orotase) gene loci (Chen et al, 2001). The ATR+/− cells gave rise to PALA-resistant colonies at a four-fold higher frequency than did the parental HCT 116 cells (Figure 6E), confirming that loss of one ATR allele increases significantly the frequency of gene amplification events in these cells.
The results obtained with HCT 116 cells indicate that ATR functions as a haploinsufficient genome maintenance protein on an MLH1-deficient background. To validate this conclusion, we crossed Atr+/− mice (Brown and Baltimore, 2000) with Mlh1+/− mice (Baker et al, 1996), and then intercrossed the Atr+/− Mlh1+/− progeny to obtain Atr+/− Mlh1−/− animals. The strain background for the Mlh1+/− animals was C57BL/6, and either C57BL/6 or 129/Sv for Atr+/− mice. Analysis of the progeny of these crosses revealed that the Atr+/−Mlh1−/− genotype was significantly under-represented in weaned pups from both strain backgrounds (Table I). This phenomenon was particularly evident with respect to the C57BL/6 × C57BL/6 intercrosses, in which the Atr+/−Mlh1−/− animals were present at only 11% of the frequency predicted by Mendelian genetics (Table I). To determine whether the low numbers of Atr+/−Mlh1−/− pups stemmed from postnatal death of animals before genotyping, we genotyped an additional 126 newborn pups from 21 independent litters. The results revealed a similar reduction in the ratio of newborn Atr+/−Mlh1−/− mice, indicating that the loss of these animals was caused by developmental failure in utero, rather than early postnatal death. The increased embryonic lethality associated with Atr+/− Mlh1−/− presumably reflects a catastrophic loss of genome integrity associated with cell proliferation in the developing embryo.
Table 1.
Genotypic analysis of 10-day-old pups derived from intercrosses of Atr+/− Mlh1+/− mice on two different breeding backgrounds
| Genotype |
|||||||
|---|---|---|---|---|---|---|---|
| WT | Atr+/− | Mlh1+/− | Atr+/− Mlh1+/− | Mlh1−/− | Atr+/− Mlh1−/− | Total | |
| BL/6 × BL/6 | |||||||
| No. of pups | 33 | 81 | 91 | 106 | 80 | 7 | 398 |
| Expected proportion | 1/12 | 2/12 | 2/12 | 4/12 | 1/12 | 2/12 | |
| a/e ratio | 0.997 | 1.22* | 1.37** | 0.8** | 2.42*** | 0.11*** | |
| 129/SV × BL/6 | |||||||
| No. of pups | 28 | 37 | 38 | 109 | 56 | 19 | 287 |
| Expected ratio | 1/12 | 1/6 | 1/6 | 1/3 | 1/12 | 1/6 | |
| a/e ratio | 1.18 | 0.772 | 0.79 | 1.15 | 2.35*** | 0.4*** | |
| Expected proportion of viable pups of each genotype (per 12 total pups) is shown for each strain intercross, together with actual/expected (a/e) ratio of animals bearing each genotype.*P<0.05; **P<0.005; ***P<0.001. | |||||||
We subsequently treated Atr+/−Mlh1−/− mouse embryonic fibroblasts (MEFs) with 0.1 μM APH and examined the integrity of mitotic chromosomes. As reported previously (Baker et al, 1996; Brown and Baltimore, 2000), the numbers of chromosomal breaks and gaps per metaphase (mean±s.d. values) in MEFs from the Atr+/− (0.8±0.7, P<0.05) or the Mlh1−/− mice (0.94±0.64, P<0.005) were significantly increased in comparison to wild-type MEFs (0.4±0.5) after cellular exposure to APH-induced replicative stress. However, the Atr+/−Mlh1−/− MEFs were clearly sensitized to APH-induced chromosome breakage and gap formation (2.17±1.34, P<0.005). Furthermore, the frequency of spontaneous SCEs in mouse dermal fibroblasts derived from Atr+/−Mlh1−/− mice was also increased by 50% relative to wild-type and Atr+/− dermal fibroblasts. Thus, these cytogenetic data support the notion that monoallelic ATR gene inactivation substantially increases genetic instability in cells that lack MLH1 expression.
Genetic instability is frequently associated with cancer development and progression (Lengauer et al, 1998; Teixeira da Costa and Lengauer, 2002). Consequently, we comparatively examined the live F1 progeny of intercrosses between Atr+/−Mlh1+/− mice for the development of histologically detectable malignancies. In all, 19 litters of F1 mice were genotyped and monitored for cancer development between 4 and 9 months after birth (Table II). Littermates were killed for examination when one of the animals in the group manifested symptoms of disease. Over this time frame, we found that 71% of the Atr+/−Mlh1−/− mice developed cancer, particularly lymphomas and intestinal adenocarcinomas (see Supplementary Table I for a comprehensive data summary), whereas their Atr+/− and Atr+/−Mlh1+/− littermates were tumor free, and only 2/12 (17%) of Mlh1−/− littermates exhibited a pathologically detectable tumor. These results indicate that tumor development is markedly accelerated by monoallelic Atr gene inactivation on an MLH1-null genetic background.
Table 2.
Summary of tumor incidence results from Atr+/− and Mlh−/− mice and their littermates
| Age |
||||||
|---|---|---|---|---|---|---|
| 4–6 weeks |
6–9 weeks |
|||||
| Tumor type |
Tumor type |
|||||
| Genotype | Tumor incidence | Lymphocytic | Epitheliala | Tumor incidence | Lymphocytic | Epitheliala |
| Atr+/− | 0/2 | — | — | 0/8 | — | — |
| Mlh−/− | 0/5 | — | — | 2/7 | 1 | 1 |
| Atr+/− Mlh+/− | 0/0 | — | — | 0/3 | — | — |
| Atr+/− Mlh−/− | 5/8 | 5 | 0 | 10/13 | 5 | 5 |
| Nineteen litters (indicated as groups) of mice were observed for 4–9 months after birth. The animals were killed when tumor growth was evident or the mice became moribund. Control littermates were killed at the same ages for comparative pathological analysis. See Supplementary Table 1 for a more detailed presentation of these data. | ||||||
| aAll epithelial tumors were intestinal adenocarcinomas. | ||||||
Discussion
The results of this study indicate that a defect in the MMR machinery unmasks a strong gene dosage effect for ATR in the regulation of cell-cycle checkpoint functions, chromosome dynamics, and tumor suppression. As ATR expression is required for viability is cycling cells (Brown and Baltimore, 2000; de Klein et al, 2000), biallelic inactivation of ATR may not be tolerated, even in late-stage malignant cells. However, the present findings demonstrate that ATR haploinsufficiency leads to high-level genetic instability and accelerated tumorigenesis in MMR-deficient hosts. This mechanism may explain the appearance of heterozygous mutations in ATR in gastric and endometrial cancers bearing the MSI phenotype (Menoyo et al, 2001; Vassileva et al, 2002). Our results suggest that such cancers are haploinsufficient for ATR function, and that monoallelic ATR gene inactivation is positively selected during tumor evolution, since this event fuels genetic instability and tumor progression.
The present study complements a recent report that identified the MutS subunit, MSH2, as an ATR-associated protein that was required for transmission of checkpoint signals through ATR in cells treated with the DNA-methylating agent N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) (Wang and Qin, 2003). In co-immunoprecipitation assays, we have also observed a constitutive association between the MutL subunit, MLH1, and the ATRIP–ATR complex (unpublished results). These results suggest that MutS/MutL complexes serve as DNA-damage sensors for ATR–ATRIP or, alternatively, that the MMR proteins process mismatch-containing DNA heteroduplexes into repair intermediates that can be recognized by ATR–ATRIP. The DNA-damage-processing model is supported by the observation that binding of replication protein A to single-stranded DNA is required for ATR-dependent hChk1 activation in MNNG-treated cells (Wang and Qin, 2003).
While our results are compatible with the existence of an ‘MSH2–ATR signaling module' (Wang and Qin, 2003), the present findings add further support to the idea that the MMR proteins and ATRIP–ATR also function in separate checkpoint signaling cascades. An intact MMR system appears to be required for full activation of an ATM-dependent, S-phase checkpoint triggered by the induction of DSBs in replicating DNA (Brown et al, 2003). Loss of MLH1 or MSH2 would disrupt this ATM-dependent S-phase checkpoint, rendering the host cells wholly reliant on ATR for the suppression of DNA synthesis in the face of DNA DSBs encountered during S phase. A second major point of convergence between the MMR system and ATR may relate to the maintenance of recombination fidelity. We propose that the predisposition of MMR-defective cells to perform illegitimate recombination events (Harfe and Jinks-Robertson, 2000) is exacerbated by partial loss of ATR function, opening the gate to promiscuous recombination, and consequent gene amplifications, chromosome breaks and translocations. From the cancer perspective, our results suggest that an intact ATR–hChk1 pathway presents a major barrier to the progression of MMR-deficient tumors from the microsatellite mutator phenotype to gross chromosomal instability.
One prominent manifestation of the rampant genome destabilization induced by reduced expression of ATR was apparent amplification of the ATR gene itself in the ATR+/− HCT 116 cells. We were not able to determine definitively whether the targeted ATR locus, the nondisrupted allele, or both gene loci were altered in these cells. Moreover, the apparent amplification of ATR gene sequences was never associated with an increase in ATR protein expression (unpublished results), suggesting that the amplifications involved segments of the ATR gene, rather than the entire gene locus. However, the increased propensity of the ATR+/− cells to incur gene amplification events was clearly not restricted to the targeted ATR allele, in that both the p21/WAF1 gene and the CAD gene complex were rearranged and/or amplified at increased frequencies after monoallelic ATR gene disruption. The increased rate of gene amplification in the ATR+/− clones is consistent with the hyper-recombinogenic phenotype discussed above, although other mechanisms, such as re-replication of DNA due to inappropriate firing of origins of replication, cannot be ruled out.
A striking outcome of the present study was that cells from mice bearing the same ATR–MLH1 genotype as our ATR+/− HCT 116 cells also demonstrated fragile site instability and a hyper-recombinogenic phenotype. The increased level of genetic instability in Atr+/−Mlh−/− mice presumably underlies the dramatic decrease in the number of live pups born with this phenotype, relative to their Atr+/− littermates expressing at least one functional Mlh1 allele. The increased embryonic lethality associated with the Atr+/−Mlh−/− genotype supports the major conclusion from our human HCT 116 cell studies, that is, loss of MLH1 sensitizes cells to partial loss of ATR function. Previous reports indicated that the Atr+/− mice used in the present study develop tumors at a modest frequency by 18 months of age (Brown and Baltimore, 2000), while the Mlh1−/− animals were considerably more tumor prone (mean age of onset, 8–9 months) (Prolla et al, 1998). Using the same mouse strains studied in these earlier reports, we recorded no tumors in the Atr+/− animals, and found that only 17% of the Mlh−/− animals were tumor positive over the 4–9 months observation period. The reason for the apparent lower frequency of tumor development in our cohort of Mlh1−/− mice, relative to that observed by Prolla et al (1998), remains unclear, but it should be emphasized that nearly all of our Mlh1−/− mice were killed before 9 months of age, in parallel with their respective, tumor-bearing Atr+/−Mlh1−/− littermates. Therefore, our data do not allow us to draw any firm conclusions with regard to the mean age of tumor appearance in the Mlh1−/− versus Atr+/−Mlh1−/− mice. Nonetheless, we clearly observed a much higher incidence (71%) of pathologically detectable tumors, mainly lymphomas, intestinal adenomas, and adenocarcinomas, in the Atr+/−Mlh1−/− mice at 4–9 months of age. Interestingly, the tissue locations of the tumors in the Atr+/−Mlh1−/− mice were identical to those reported previously in Mlh1−/− mice bearing two wild-type Atr alleles (Prolla et al, 1998), suggesting that partial loss of ATR function leads to increased and/or accelerated tumor formation in the same cell populations that were most susceptible to neoplastic transformation in the absence of MLH1 alone.
In summary, the results of this study indicate that ATR functions as a haploinsufficient genome maintenance protein and tumor suppressor in MMR-deficient cells. While ATR and MMR proteins form a DNA-damage-responsive signaling module (Wang et al, 2003), an intact MMR system is not essential for ATR function, at least when genotoxic stress is induced by agents (e.g., IR, UV) other than alkylating drugs. The MMR–ATR signaling module may be most relevant to the overall checkpoint functions of ATR when the initiating DNA lesion (e.g., postreplication DNA base mispairing) is of a type that is normally corrected by MMR. The results of this study focus attention on a much less direct interdependence between ATR and the MMR system, wherein normal ATR expression is crucial for the prevention of gross chromosomal instability in MMR-deficient cells. Whether such chromosomal instability actually drives tumorigenesis, particularly during the early stages of malignant progression, is the subject of an ongoing debate (Cahill et al, 1999; Nowak et al, 2002; Teixeira da Costa and Lengauer, 2002). Nonetheless, these findings should provoke more detailed investigations of tumors that progress from the MSI phenotype to gross chromosomal instability. In these tumors, genetic or epigenetic alterations that lead to reduced function of ATR or its downstream targets (e.g., hChk1) may provide a facile route to high-level genome destabilization, accelerated tumor progression, and resistance to therapeutic agents. From the therapeutic perspective, tumors that have crossed the threshold from MSI to chromosomal instability through this avenue might prove hypersensitive to agents that place further stress on the ATR–hChk1 pathway (e.g., camptothecins) or to drugs targeted directly against these two critical checkpoint kinases.
Materials and methods
Construction of targeting vectors and screening of targeted clones by PCR
The promoterless targeting vector (Cao et al, 2002) was kindly provided by Dr Youjia Cao. Details with regard to construction of the ATR-targeting construct are available on request. The targeting plasmid was linearized with KasI and transfected into HCT 116 cells with Fugene 6 reagent (Roche). At 48 h posttransfection, cells were transferred into 96-well plates and grown in selection medium containing geneticin (0.4 mg/ml) (Gibco). Genomic DNA was extracted from each sample well with the DNeasy tissue kit (Qiagen), and was subjected to PCR with the primers depicted schematically in Figure 1A. PCR products were resolved by agarose-gel electrophoresis. The sequences of the oligonucleotide primers used to distinguish the endogenous and targeted ATR alleles were as follows: ATR-A1, CCGTGATGTTGCTTGATTTCATC; ATR-B, CTTCTATGGAGGTAAACCAAGTC; ATR-C1, CCAGTCATAGCCGAATAGCCTC; ATR-A2, TTCATCCAGCATATCATGAAATCC; and ATR-C2, TACCGTAAAGCACGAGGAAGC.
Cell culture
HCT 116 cells were cultured in McCoy's 5A medium (Gibco) with 10% fetal bovine serum (FBS). Primary murine fibroblasts were cultured from day 14.5 embryos and primary dermal fibroblasts were isolated from 2-day-old mice as described (Datto et al, 1999).
Immunoprecipitation and immunoblot analysis
Antibodies used for immunoprecipitation and immunoblotting were as follows (sources within parentheses): α–γH2AX (Ward and Chen, 2001) (gift from Dr Junjie Chen, Division of Oncology Research, Mayo Clinic), α-Ras (Oncogene Research Products), α-ATR (Affinity Bioreagents), α-AU1 (Covance), α-tubulin (Sigma), α-phospho-Chk1(Ser-345) (Cell Signaling Technology), α-Chk1 (Santa Cruz), and α-MLH1 (BD Biosciences).
Cells were grown to 70% confluency and were harvested by scraping into 0.4 ml lysis buffer A (50 mM HEPES pH 7.4, 300 mM sodium chloride, 0.5% NP-40, 1 mM magnesium chloride, 1.5 mM EGTA, 1 mM dithiothreitol, 0.1 mM sodium orthovanadate, 10 ng/ml microcystin, 5 μg/ml aprotinin, and 5 μg/ml leupeptin). Total cellular extracts were prepared, resolved by SDS–PAGE, and transferred to PVDF membranes. Protein blots were probed with the indicated antibodies, and detection was performed with the Supersignal West Pico Chemiluminescent Substrate kit (Pierce, Rockford, IL).
For γH2AX immunoblotting, cells were lysed in 2 × SDS gel sample buffer, boiled for 5 min, and vortexed. Total cellular extracts were prepared and resolved by 4–20% SDS–PAGE.
Clonogenic survival assay
Cells were harvested from log-phase growing cultures, and were plated in duplicate into 60 mm dishes. After 18 h, cells were treated with IR, UV light, or HU as indicated in the figure legends. Culture medium was replaced 24 h after HU treatment. Cells were then cultured for 12 days before fixation and staining with 0.1% Coomassie blue. The plates were photographed with a digital camera, and staining intensities were quantified with the Image-Pro Plus software program (Media Cybernetics, Silver Spring, MD).
Nocodazole capture assay
Cells were treated with 12 Gy IR, and at 30 min postirradiation, nocodazole (500 ng/ml) (Sigma) was added to each sample well. The cells were harvested after the indicated times, and suspended cells were fixed in methanol:acetic acid (3:1). The fixed cells were stained with Giemsa, and the mitotic index was determined from a minimum of 200 cells per sample.
Chromosome preparation
Cells were treated for 30 min with colcemid (50 ng/ml) (Irvine Scientific, Santa Ana, CA). Metaphase chromosome spreads were prepared according to standard cytogenetic procedures. Cells were dropped onto slides and baked for 45 min at 95°C before G banding, FISH, or SCE. Cells were imaged, and karyotypic analyses were carried out with a Cytovision version 2.7 computer-assisted karyotyping system (Applied Imaging, Santa Clara, CA). Abnormalities were described according to the International System for Human Cytogenetic Nomenclature (ISCN, 1995).
Analysis of SCE
Cells were cultured for 44 h in 10 μM bromodeoxyuridine prior to harvest. Slides made from cell suspensions were stained with Hoechst 33258, exposed to UV light, and then stained with Giemsa as described (German and Alhadeff, 1994). Metaphase images were captured with the Cytovision version 2.7 karyotyping system. Numbers of SCEs per metaphase were counted for 20 or 25 metaphase cells in each sample preparation.
Fragile site analysis
Cells were treated with 0.1 μM APH, and chromosome spreads were prepared as described above. Metaphase images were captured as described above. The numbers of breaks and gaps per metaphase were counted for 20 or 25 metaphase cells in each case.
PALA resistance assay
PALA was kindly provided by Dr Robert Shultz (Developmental Therapeutics Program, Division of Cancer Treatment, National Cancer Institute). The concentration of PALA that inhibited cell growth by 50% (IC50) was determined as described (Perry et al, 1992). Cellular resistance to PALA was examined at a drug concentration of six times the IC50 in deoxynucleotide- and ribonucleotide-deficient McCoy's 5A (Gibco) supplemented with 10% dialyzed FBS. The medium was changed every 5 days, and colonies were stained with 0.1% Coomassie blue after 15 days of continuous drug selection. Colonies containing more than 100 cells were scored as positive. Frequencies were corrected for differences in the plating efficiency of each test cell population.
FISH analysis
Slides were prepared as described above, but not baked. The fluorescently labeled chromosome 3 alpha satellite (centromeric) probe was purchased from Cytocell (Rainbow Scientific, Windsor, CT). Fluorescent probes derived from the ATR cDNA and ATR genomic sequences were prepared with a nick translation kit (Gibco). Hybridization and probe detection were performed according to the manufacturer's instructions and standard procedures. Hybridization was detected with a kit for biotin-labeled probes with the Texas red fluorophore (Ventana Medical Systems Inc.). Chromosomes were counterstained with 4′,6-diamidino-2-phenylindole. Cells were analyzed with a Zeiss Axiophot microscope equipped with the Chroma 8300 filter set and Quips Pathvision Software (Applied Imaging, Santa Clara, CA). Selected images were captured with a CCD camera (Sensys, Photometrics).
Targeting of the p21/WAF1 gene and Southern blot analysis
The p21/WAF1-targeting vector (Waldman et al, 1995) was provided by Dr Bert Vogelstein (Johns Hopkins University). The p21/WAF1-targeting vector was digested with SalI and transfected into HCT 116 cells and ATR+/− cells. At 48 h after transfection, cells were trypsinized and plated into medium containing 100 μg/ml hygromycin. After expansion of drug-resistant clones, genomic DNA (5 μg) was extracted and digested with BglII. After electrophoresis through a 0.8% agarose gel, samples were transferred to a nitrocellulose membrane, and hybridized with a radiolabeled p21/WAF1 cDNA probe (Waldman et al, 1995).
Genotyping by PCR, necropsy, and pathology
Tail DNA was prepared from the progeny of Atr+/−Mlh1+/− intercrosses, and genotypes were determined by PCR as described (Baker et al, 1996; Brown and Baltimore, 2000). Littermates were analyzed as individual groups, with Atr+/−, Mlh1−/−, and Atr+/− Mlh1+/− siblings serving as controls. Mice were killed at 4–9 months of age. Blood samples were collected for complete blood counts, and were analyzed for white blood cell differentiation. Animals were autopsied and tissues fixed in 10% buffered formalin and embedded in paraffin. Sections were stained with hematoxylin and eosin by a standard protocol. Selected lymphomas were phenotyped for lymphocytic lineage with anti-CD3 or anti-CD20 antibody.
Statistical analysis
The Student's t-test for unpaired samples was used for comparisons of average numbers of chromosome breaks and gaps, and for comparisons of SCE frequencies. The actual ratios of live pups of different genotypes born from Atr+/−Mlh+/− intercrosses (see Table I) were compared to the predicted ratios by χ2 analysis.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 1 Legend
Supplementary Table I
Acknowledgments
We thank Dr Bert Vogelstein for the p21-hygro-targeting vector, Drs David Baltimore and Eric Brown for the Atr+/− mice, Dr R Michael Liskay for the MLH1+/− mice, Dr Robert Schultz for the PALA reagent, Dr Junjie Chen for the anti-γH2AX antibody, Dr Youjia Cao for the promoter-less vector, Dr Scott Trasti for assistance with pathological analyses, and Diane Otterness and Yong Yu for expert technical assistance. This work was supported by grants from the National Institutes of Health, Johnson & Johnson, and the Ataxia-Telangiectasia Children's Project.
References
- Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15: 2177–2196 [DOI] [PubMed] [Google Scholar]
- Baker SM, Plug AW, Prolla TA, Bronner CE, Harris AC, Yao X, Christie DM, Monell C, Arnheim N, Bradley A, Ashley T, Liskay RM (1996) Involvement of mouse Mlh1 in DNA mismatch repair and meiotic crossing over. Nat Genet 13: 336–342 [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas J (2003) Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3: 421–429 [DOI] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D (2000) ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 14: 397–402 [PMC free article] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D (2003) Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev 17: 615–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JP, Wei W, Sedivy JM (1997) Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 277: 831–834 [DOI] [PubMed] [Google Scholar]
- Brown KD, Rathi A, Kamath R, Beardsley DI, Zhan Q, Mannino JL, Baskaran R (2003) The mismatch repair system is required for S-phase checkpoint activation. Nat Genet 33: 80–84 [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B (1998) Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282: 1497–1501 [DOI] [PubMed] [Google Scholar]
- Cahill DP, Kinzler KW, Vogelstein B, Lengauer C (1999) Genetic instability and Darwinian selection in tumours. Trends Biochem Sci 24: M57–M60 [PubMed] [Google Scholar]
- Cahill DP, Lengauer C, Yu J, Riggins GJ, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B (1998) Mutations of mitotic checkpoint genes in human cancers. Nature 392: 300–303 [DOI] [PubMed] [Google Scholar]
- Cao Y, Janssen EM, Duncan AW, Altman A, Billadeau DD, Abraham RT (2002) Pleiotropic defects in TCR signaling in a Vav-1-null Jurkat T-cell line. EMBO J 21: 4809–4819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casper AM, Nghiem P, Arlt MF, Glover TW (2002) ATR regulates fragile site stability. Cell 111: 779–789 [DOI] [PubMed] [Google Scholar]
- Cejka P, Stojic L, Mojas N, Russell AM, Heinimann K, Cannavo E, di Pietro M, Marra G, Jiricny J (2003) Methylation-induced G(2)/M arrest requires a full complement of the mismatch repair protein hMLH1. EMBO J 22: 2245–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha RS, Kleckner N (2002) ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297: 602–606 [DOI] [PubMed] [Google Scholar]
- Chen S, Bigner SH, Modrich P (2001) High rate of CAD gene amplification in human cells deficient in MLH1 or MSH6. Proc Natl Acad Sci USA 98: 13802–13807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA (2003) Fragile sites: breaking up over a slowdown. Curr Biol 13: R231–233 [DOI] [PubMed] [Google Scholar]
- Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH (1998) Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J 17: 159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dart DA, Adams KE, Akerman I, Lakin ND (2004) Recruitment of the cell cycle checkpoint kinase ATR to chromatin during S-phase. J Biol Chem 279: 16433–16440 [DOI] [PubMed] [Google Scholar]
- Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF (1999) Targeted disruption of Smad3 reveals an essential role in transforming growth factor beta-mediated signal transduction. Mol Cell Biol 19: 2495–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smi B, Carr AM, Lehmann AR, Hoeijmakers JH (2000) Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol 10: 479–482 [DOI] [PubMed] [Google Scholar]
- Elledge SJ (1996) Cell cycle checkpoints: preventing an identity crisis. Science 274: 1664–1672 [DOI] [PubMed] [Google Scholar]
- German J, Alhadeff B (1994) Sister chromatid exchange analysis. In Current Protocols in Human Genetics, Dracopoli NCea (ed), Vol. 8, pp 1–10. New York: John Wiley & Sons [DOI] [PubMed] [Google Scholar]
- Harfe BD, Jinks-Robertson S (2000) DNA mismatch repair and genetic instability. Annu Rev Genet 34: 359–399 [DOI] [PubMed] [Google Scholar]
- Hartwell L (1992) Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell 71: 543–546 [DOI] [PubMed] [Google Scholar]
- ISCN (1995), In An International System for Cytogenetic Nomenclature, Mittleman F (ed) Basel: S Karger [Google Scholar]
- Koi M, Umar A, Chauhan DP, Cherian SP, Carethers JM, Kunkel TA, Boland CR (1994) Human chromosome 3 corrects mismatch repair deficiency and microsatellite instability and reduces N-methyl-N′-nitro-N-nitrosoguanidine tolerance in colon tumor cells with homozygous hMLH1 mutation. Cancer Res 54: 4308–4312 [PubMed] [Google Scholar]
- Lengauer C, Kinzler KW, Vogelstein B (1998) Genetic instabilities in human cancers. Nature 396: 643–649 [DOI] [PubMed] [Google Scholar]
- Masramon L, Ribas M, Cifuentes P, Arribas R, Garcia F, Egozcue J, Peinado MA, Miro R (2000) Cytogenetic characterization of two colon cell lines by using conventional G-banding, comparative genomic hybridization, and whole chromosome painting. Cancer Genet Cytogenet 121: 17–21 [DOI] [PubMed] [Google Scholar]
- Mellon I, Rajpal DK, Koi M, Boland CR, Champe GN (1996) Transcription-coupled repair deficiency and mutations in human mismatch repair genes. Science 272: 557–560 [DOI] [PubMed] [Google Scholar]
- Melo J, Toczyski D (2002) A unified view of the DNA-damage checkpoint. Curr Opin Cell Biol 14: 237–245 [DOI] [PubMed] [Google Scholar]
- Menoyo A, Alazzouzi H, Espin E, Armengol M, Yamamoto H, Schwartz S Jr (2001) Somatic mutations in the DNA damage-response genes ATR and CHK1 in sporadic stomach tumors with microsatellite instability. Cancer Res 61: 7727–7730 [PubMed] [Google Scholar]
- Nowak MA, Komarova NL, Sengupta A, Jallepalli PV, Shih Ie M, Vogelstein B, Lengauer C (2002) The role of chromosomal instability in tumor initiation. Proc Natl Acad Sci USA 99: 16226–16231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA (2003) A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 33: 497–501 [DOI] [PubMed] [Google Scholar]
- Painter RB, Young BR (1980) Radiosensitivity in ataxia-telangiectasia: a new explanation. Proc Natl Acad Sci USA 77: 7315–7317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM, Adams MD, Venter JC, Hamilton SR, Petersen GM, Watson P, Lynch HT, Peltomäki P, Mecklin J-P, de la Chapelle A, Kinzler KW, Vogelstein B (1994) Mutation of a mutL homolog in hereditary colon cancer. Science 263: 1625–1629 [DOI] [PubMed] [Google Scholar]
- Peltomaki P (2003) Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol 21: 1174–1179 [DOI] [PubMed] [Google Scholar]
- Perry ME, Rolfe M, McIntyre P, Commane M, Stark GR (1992) Induction of gene amplification by 5-aza-2′-deoxycytidine. Mutat Res 276: 189–197 [DOI] [PubMed] [Google Scholar]
- Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, Bronner CE, Zheng B, Gordon M, Reneker J, Arnheim N, Shibata D, Bradley A, Liskay RM (1998) Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat Genet 18: 276–279 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155–168 [DOI] [PubMed] [Google Scholar]
- Teixeira da Costa L, Lengauer C (2002) Exploring and exploiting instability. Cancer Biol Ther 1: 212–225 [DOI] [PubMed] [Google Scholar]
- Vassileva V, Millar A, Briollais L, Chapman W, Bapat B (2002) Genes involved in DNA repair are mutational targets in endometrial cancers with microsatellite instability. Cancer Res 62: 4095–4099 [PubMed] [Google Scholar]
- Waldman T, Kinzler KW, Vogelstein B (1995) p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 55: 5187–5190 [PubMed] [Google Scholar]
- Wang X, Zou L, Zheng HY, Wei QY, Elledge SJ, Li L (2003) Genomic instability and endoreduplication triggered by RAD17 deletion. Genes Dev 17: 965–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Qin J (2003) MSH2 and ATR form a signaling module and regulate two branches of the damage response to DNA methylation. Proc Natl Acad Sci USA 100: 15387–15392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward IM, Chen J (2001) Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem 276: 47759–47762 [DOI] [PubMed] [Google Scholar]
- Ward IM, Minn K, Chen J (2004) UV-induced ataxia-telangiectasia-mutated and Rad3-related (ATR) activation requires replication stress. J Biol Chem 279: 9677–9680 [DOI] [PubMed] [Google Scholar]
- Zhao H, Piwnica-Worms H (2001) ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 21: 4129–4139 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 1 Legend
Supplementary Table I