

Abstract
The ability of enzymes to distinguish between fatty acyl groups can involve molecular measuring devices termed hydrocarbon rulers, but the molecular basis for acyl-chain recognition in any membrane-bound enzyme remains to be defined. PagP is an outer membrane acyltransferase that helps pathogenic bacteria to evade the host immune response by transferring a palmitate chain from a phospholipid to lipid A (endotoxin). PagP can distinguish lipid acyl chains that differ by a single methylene unit, indicating that the enzyme possesses a remarkably precise hydrocarbon ruler. We present the 1.9 Å crystal structure of PagP, an eight-stranded β-barrel with an unexpected interior hydrophobic pocket that is occupied by a single detergent molecule. The buried detergent is oriented normal to the presumed plane of the membrane, whereas the PagP β-barrel axis is tilted by approximately 25°. Acyl group specificity is modulated by mutation of Gly88 lining the bottom of the hydrophobic pocket, thus confirming the hydrocarbon ruler mechanism for palmitate recognition. A striking structural similarity between PagP and the lipocalins suggests an evolutionary link between these proteins.
Keywords: crystal structure, endotoxin, lipid A, phospholipids, signal transduction
Introduction
The assembly and maintenance of biological membranes depend critically on the specificity of lipid acyl group transfer reactions (Vance and Vance, 2002). As lipids are key components of biological membranes, many enzymes of lipid metabolism are integral membrane proteins. While significant advances have been made in the structural study for some classes of membrane proteins, only two structures are known for integral membrane enzymes of lipid metabolism. These are OMPLA and PagP, and both are β-barrel proteins from the outer membrane of Escherichia coli. The outer membrane of Gram-negative bacteria is an asymmetric bilayer, with a periplasmic phospholipid leaflet and an outer leaflet that is normally restricted to the lipid A (endotoxin) component of lipopolysaccharide (Kamio and Nikaido, 1976). The phospholipase OMPLA is a 12-stranded antiparallel β-barrel with its active site located at a Ca2+-stabilized dimeric interface (Snijder et al, 1999). OMPLA is thought to function in the removal of phospholipids that aberrantly migrate into the outer membrane outer leaflet (Dekker, 2000), and has little specificity for the length of the acyl chain of the lipid substrate.
In contrast, PagP is an outer membrane acyltransferase that catalyzes the transfer of a palmitate group from the sn-1 position of a phospholipid to the N-linked R-3-hydroxymyristate chain on the proximal glucosamine unit of lipid A (Figure 1). The simplest lipopolysaccharide structure encountered by PagP in outer membranes is lipid A, a phosphorylated and acylated disaccharide of glucosamine, bearing two 3-deoxy-D-manno-2-octulosonic acid (Kdo) sugars (Raetz and Whitfield, 2002). Despite the presence of 18-carbon acyl groups in bacterial phospholipids, only 16-carbon palmitoyl groups are transferred by PagP to lipid A in vivo (Guo et al, 1998), and PagP has the unusual ability to distinguish between phospholipid acyl groups that differ by only a single carbon atom (Brozek et al, 1987; Bishop et al, 2000). The active site residues of PagP map to the extracellular surface of the outer membrane, indicating that phospholipids must first migrate into the outer leaflet before the reaction can proceed (Hwang et al, 2002).
Figure 1.
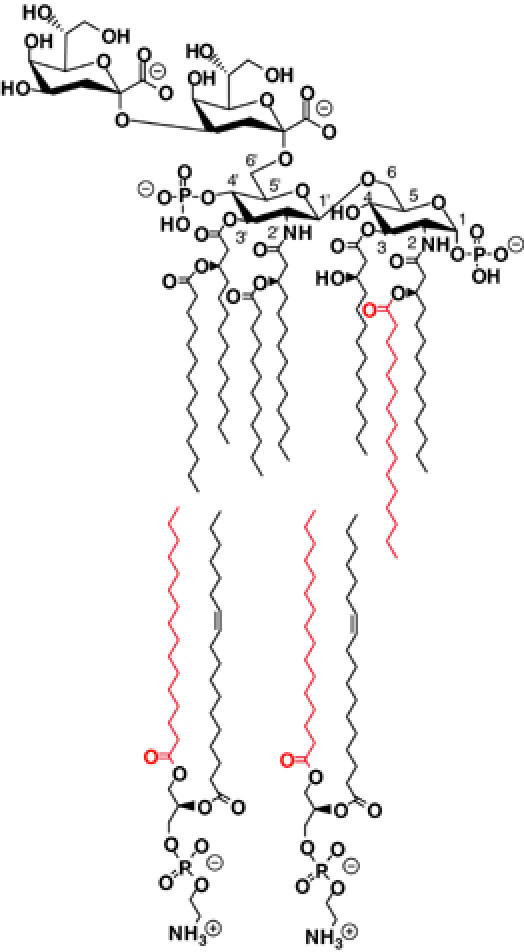
The asymmetric outer membrane of Gram-negative bacteria consists of an inner phospholipid leaflet (indicated by the two molecules of phosphatidylethanolamine in the lower part of the panel) and an outward-facing leaflet of lipid A bearing minimally two 3-deoxy-D-manno-2-octulosonic acid (Kdo) sugars (indicated by a Kdo2-lipid A in the upper part of the panel, shown as the palmitoylated product of the PagP reaction). PagP catalyzes the transfer of a palmitate chain (shown in red) from the sn-1 position of a phospholipid to the lipid A acceptor. This transfer requires the migration of the phospholipid donor to the outer leaflet of the outer membrane, presumably by mechanisms that do not directly involve PagP.
The product of the PagP reaction is palmitoylated lipid A, which provides resistance to vertebrate antimicrobial peptides (Guo et al, 1998) and antagonizes endotoxin signaling through the human toll-like receptor 4 pathway (Tanamoto and Azumi, 2000; Muroi et al, 2002; Kawasaki et al, 2004). PagP has been shown to promote infections of the mammalian respiratory tract by providing bacterial resistance to host immune defenses (Robey et al, 2001; Preston et al, 2003), including antibody-mediated complement lysis (Pilione et al, 2004). The pagP gene is regulated by virulence-associated signal transduction pathways including Salmonella PhoP/PhoQ (Guo et al, 1998) and Bordetella BvgA/BvgS (Preston et al, 2003), and lipid A palmitoylation appears to be a regulated process in other pathogens of humans (Rebeil et al, 2004), insects (Derzelle et al, 2004), and plants (Fukuoka et al, 2001).
The ability of enzymes to distinguish between fatty acyl groups with varying numbers of carbon atoms may involve molecular measuring devices termed ‘hydrocarbon rulers' (Wyckoff et al, 1998). Hydrocarbon rulers have been described in the structures of certain soluble enzymes (Tarshis et al, 1996; Long et al, 1998; Wyckoff et al, 1998); however, the molecular basis for acyl-chain recognition in any membrane-bound enzyme remains to be defined. The global fold and dynamics of E. coli PagP have been determined by NMR spectroscopy, and revealed an eight-stranded antiparallel β-barrel preceded by an N-terminal amphipathic α-helix (Hwang et al, 2002). Key residues for catalysis mapped to a highly dynamic cell surface loop, but neither the position of the amino-acid side chains nor the presence of any bound ligands or detergents was determined in the NMR structure. Here we describe the crystal structure of PagP at 1.9 Å resolution, which unexpectedly reveals a bound detergent molecule in the β-barrel interior. Based on this observation, we determined the detergent inhibition properties of the enzyme, and used site-directed mutagenesis experiments to verify the role of the interior detergent-binding site in acyl-chain recognition. These studies provide the first molecular description of a hydrocarbon ruler in an integral membrane enzyme.
Results and discussion
Expression and purification of PagP
We were able to crystallize the purified protein either solubilized from native E. coli membranes or refolded from inclusion bodies. Natively folded protein was purified from membranes via selective solubilization with the detergent lauryldimethylamine oxide (LDAO), followed by NiNTA chromatography, as previously described (Bishop et al, 2000). For the refolded protein, PagP was targeted to insoluble aggregates by expressing the protein without its native leader sequence (Hwang et al, 2002). Inclusion bodies were isolated and solubilized in GuHCl, and the protein was refolded by fast dilution into solutions of 0.5% LDAO and further purified by ion-exchange chromatography. The yield of refolded protein was typically greater than 50%. Both the natively folded and refolded forms of the protein exhibited similar temperature-dependent shifts in electrophoretic mobility by SDS–PAGE, displayed indistinguishable specific activities, and crystallized under similar conditions in the same space group with comparable unit cells. We continued the structural studies with the refolded protein because this material produced larger and more reproducible crystals.
Structure and organization of PagP in outer membranes
We determined the crystal structure of E. coli PagP to 1.9 Å resolution. As expected, PagP is an eight-stranded antiparallel β-barrel with a periplasmic N-terminal amphipathic α-helix (Table I and Figure 2A). The average Cα r.m.s.d. with the average NMR backbone structure (Hwang et al, 2002) is 1.8 Å, excluding the leading α-helix and all connecting loops (Figure 3A). The enzyme has a barrel shear number of 10 and is thus topologically similar to OmpA (Pautsch and Schulz, 2000) and NspA (Vandeputte-Rutten et al, 2003), although the PagP barrel is considerably shorter (Figure 2). The barrel cross-section is approximately circular. Consistent with previous structures of β-barrel outer membrane proteins, short loops are found facing the periplasmic surface, while larger, more flexible loops face the cell exterior (Koebnik et al, 2000). In all, 10 residues from the first outward-facing loop L1 (Tyr38–Asn47) are not visible in the electron density maps, and this loop displayed highly variable conformations in the NMR ensemble of structures (Hwang et al, 2002; Figure 3A). None of the intermolecular contacts in the crystal lattice suggest that PagP is a dimer or higher order oligomer, and we assume that the protein is monomeric in its native state.
Table 1.
Crystallographic statistics
| Diffraction data | Native | NaBr | CsCl | SeMet |
|---|---|---|---|---|
| Resolution (Å) | 1.9 | 2.7 | 2.6 | 2.6 |
| Unit cell (Å) | ||||
| a,b | 50.67 | 51.02 | 51.12 | 50.60 |
| c | 158.44 | 158.31 | 158.23 | 158.06 |
| Wavelength (Å) | 0.9505 | 0.9050 | 0.9505 | 1.54 |
| Reflections collected | 227 607 | 48 970 | 71 093 | 49 340 |
| Unique reflections | 17 041 | 5967 | 6925 | 6217 |
| Completeness (%) | 99.5 (99.9) | 93.9 (94.5) | 99.0 (100.0) | 89.8 (93.4) |
| 〈I〉/〈σI〉 | 38.1 (9.0) | 17.9 (5.6) | 21.2 (7.7) | 29.6 (4.2) |
| Rsym (%) | 5.0 (33.7) | 8.6 (45.2) | 7.8 (38.5) | 6.2 (39.8) |
| |
|
|
|
|
| Refinement |
Native |
|
|
|
| Resolution (Å) | 20.0–1.90 | |||
| Data cutoff F/σ(F) | 0 | |||
| R/Rfree, 5% (%) | 21.65/25.55 | |||
| R.m.s.d. bond lengths (Å) | 0.015 | |||
| R.m.s.d. bond angles (deg) | 1.44 | |||
| Number of atoms/residues | ||||
| PagP | 1225/157 | |||
| LDAO | 66/3 complete, 2 partial | |||
| Acetate | 20/3 | |||
| MPD | 16/2 | |||
| Waters | 38 | |||
| Numbers in parentheses refer to the highest resolution shell. | ||||
Figure 2.
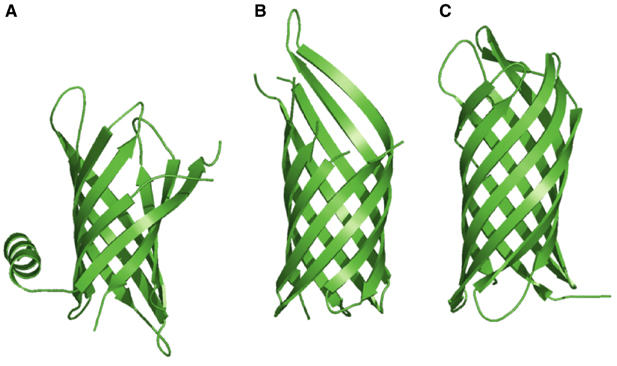
Ribbon diagrams of (A) PagP, (B) OmpA (Protein Data Bank code 1QJP; Pautsch and Schulz, 2000), and (C) NspA (Protein Data Bank code 1P4T; Vandeputte-Rutten et al, 2003).
Figure 3.
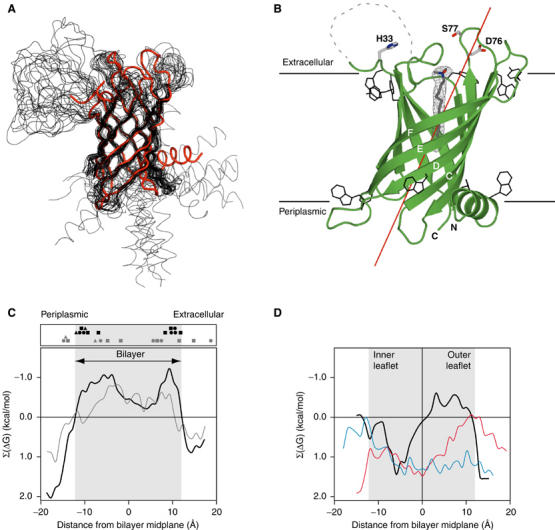
(A) Superposition of the PagP crystal structure (red) with the 20 lowest energy NMR structures in DPC micelles (black) (Hwang et al, 2002; Protein Data Bank code 1MM4). The leading α-helix is at the lower right, and loop L1 is at the upper left. (B) Ribbon diagram of PagP. 2∣∣Fo∣−∣Fc∣∣ electron density for the internal LDAO ligand is shown in gray mesh, and the aromatic residues located at the presumed membrane/water interfaces are shown in black. Residues 38–47 of extracellular loop L1 are not visible in the electron density maps and are indicated by the dashed segment. The barrel axis is tilted approximately 25° from the membrane normal and is shown as a red line. The horizontal lines represent the presumed position of the outer membrane bilayer. (C) Hydrophobicity profiles (Wimley, 2002) for the outward-facing PagP residues as a function of membrane position are shown as solid lines. Negative Σ(ΔG) values indicate regions that are more hydrophobic. The gray line and symbols present results for the protein positioned with the β-barrel axis aligned along the membrane normal, while the black line and symbols are for the protein tilted as in panel B. The solid symbols represent the Cγ positions of the Trp (squares), Tyr (circles), and Phe (triangles) residues that form the inner (Trp17, Tyr23, Trp89, Trp93, Phe101, Tyr133, Phe161) and outer (Trp32, Trp51, Trp81, Tyr119, Tyr142, Tyr153) aromatic belts. (D) The hydrophobicity profiles for the inward-facing residues of PagP (black), OmpA (blue), and NspA (red) indicate an unusually hydrophobic interior in the upper half of the PagP β-barrel.
While all previously described outer membrane proteins are oriented in the membrane with their β-barrel axis normal to the plane of the bilayer (Wimley, 2002), a representation of the protein colored according to side-chain hydrophobicity clearly revealed a nonpolar band of appropriate width to span the membrane, but this band was not aligned with the barrel axis. A quantitative analysis of the distribution of the exposed hydrophobic/hydrophilic surfaces of the protein indicates a 25° tilt of the PagP barrel axis with respect to the membrane normal (Figure 3). The hydrophobicity profile of the untilted protein is not consistent with that seen in other membrane proteins (Wimley, 2002), since the middle hydrophobic region is too shallow and there is no clear and sharp difference between the central membrane-embedded and the flanking solvent-exposed regions. In the tilted position, however, the protein surface shows the expected hydrophobic zone of appropriate width (roughly 24 Å), with sharp transitions on either side to strongly hydrophilic regions (Figure 3C). A tilted barrel is also supported by the positions of aromatic belt residues (Schulz, 2002), which cluster strongly at the membrane interface regions only when the barrel is in this position (Figures 3 and 4). The protein in this orientation presents a relatively flat surface on either side of the membrane, without prominent protruberances. The spaces caused by the tilting of the main barrel are filled with the N-terminal amphipathic α-helix on the periplasmic side, and a β-bulge at Pro28 near the end of strand A on the opposite extracellular side (lower right and upper left of Figure 3B, respectively). The amphipathic helix lies along the lipid/water interface, with a polar face exposed to the periplasmic space, and the opposite face interacting with the barrel and the inner phospholipid leaflet of the outer membrane. This helix contacts the periplasmic ends of strands B and C, which do not have aromatic belt residues, and contributes the conserved W17 to the belt. Although the helix was not positioned relative to the rest of the protein in the NMR structure (Figure 3A), we expect that the orientation of the helix relative to the barrel in the crystal structure represents a true contact that occurs in bilayers.
Figure 4.
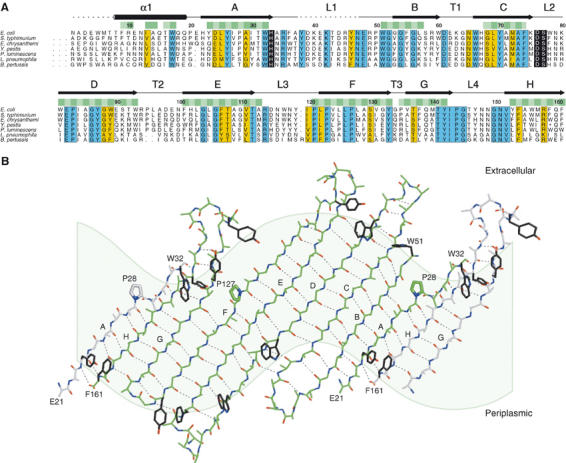
(A) Multiple sequence alignment of PagP homologues from E. coli, Salmonella typhimurium, Erwinia chrysanthemi, Yersinia pestis, Photorhabdus luminescens, Legionella pneumophila, and Bordetella pertussis. To date, the enzyme has been found primarily in pathogenic Gram-negative bacteria, and a representative species from each genus is included in the alignment. Residues highlighted in blue boxes are identical in all known homologues, and conserved residues are shown in yellow boxes. The putative catalytic residues are included in black boxes. Residues that are located within the bilayer are indicated with horizontal green bars, with the lipid-exposed positions shown in darker green. The residue numbering is based on the E. coli PagP sequence of the mature protein without the leading signal sequence. Disordered regions in the crystal structure are indicated by the dashed lines. The N-terminal amino acid of the mature E. coli protein was determined by Edman degradation (Bishop et al, 2000). The N-terminal amino acids of the other homologues following the removal of the signal sequences have not yet been experimentally determined. (B) An ‘unrolled' barrel was generated by projecting atomic positions onto a cylinder aligned with the barrel axis and laid flat. The light green shading indicates the position of the membrane, which follows a sinusoidal path because of the tilting of the barrel in the bilayer. One central PagP molecule is drawn with green carbon atoms, while flanking strands are shown with white carbon atoms. Residues 8–20 of the leading α-helix and residues 33–37 and 48–50 from L1 are omitted for clarity. Side chains are omitted, except for those of prolines 28 and 127 and residues from the aromatic belts. Interchain hydrogen bonds are drawn with dashed lines.
The tilt angles for each strand relative to the barrel axis range from 38 to 42°, as expected for an eight-stranded barrel with a shear number of 10 (Schulz, 2002). However, because of the tilting of the barrel in the bilayer, there are large differences in the exposure of each of the eight strands to the lipidic environment of the membrane. Strands A, B, and H are roughly aligned with the membrane normal (angle ∼20°) and cross the bilayer with 6–8 residues. In contrast, strands D, E, and F are tilted by ∼50° relative to the membrane normal and require roughly 12 residues to span the bilayer (Figure 4).
The protein interior includes a binding pocket for lipid acyl chains
Relative to all previously described β-barrel outer membrane proteins, PagP has an unusual interior: the upper half of the barrel core is distinctly hydrophobic and is devoid of interior waters, while the lower half has a typical hydrophilic interior filled with polar side chains (Figure 3D). These two distinct interiors map to the upper lipid A leaflet and to the lower phospholipid leaflet of the membrane, respectively.
During the course of refinement, difference electron density maps revealed the presence of several LDAO detergent molecules associated with the exterior surface of the barrel, and a single detergent molecule nested within the upper hydrophobic half of the protein core (Figure 3B). The LDAO bound inside the barrel is roughly aligned with the expected membrane normal, and is oriented with its polar head group facing the extracellular region and its acyl tail extending into the protein center. The cavity is lined with hydrophobic residues and extends approximately halfway through the barrel (Figures 3D and 5), corresponding to the region of the protein that is embedded in the outer lipid A leaflet of the membrane. The residues that contact the bound LDAO are highly conserved in the PagP homologues (Figures 5C and 4A). The putative active site residues His33, Asp76, and Ser77 are located on the extracellular surface of the barrel with their side chains oriented towards the central axis of the barrel (Figure 3B), and several solvent molecules are found in a sequestered pocket located near these residues. However, the deeper hydrophobic region of the pocket is completely filled with the detergent acyl chain and does not contain any interior waters.
Figure 5.
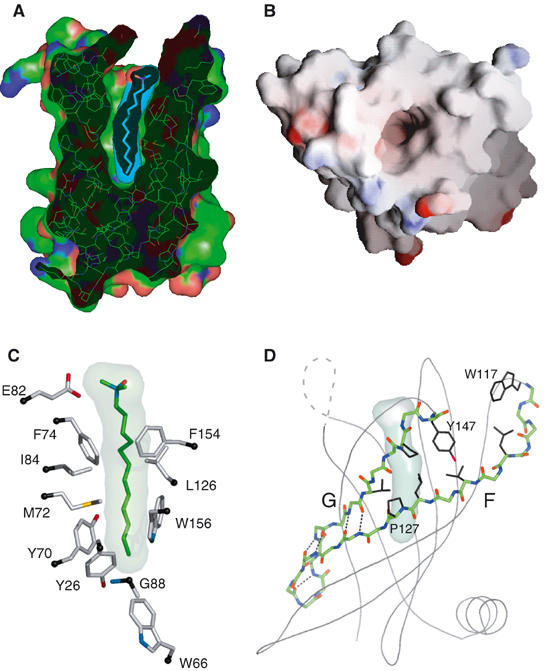
Ligand interactions in PagP. (A) Cut-away view of the protein, showing the inner ligand pocket and the internally bound detergent molecule. (B) Overhead view of the empty barrel in surface representation, colored according to the electrostatic surface potential. (C) Local environment of the internal LDAO detergent. Side-chain Cα carbons are indicated in black. (D) Strands F and G are shown in stick representation, and form main-chain interstrand H bonds only in the bottom part of the barrel. In the upper part of the barrel, the space between the two strands is filled with side chains from apolar residues. Hydrogen bonds are indicated with dashed lines, and the side chains for residues Trp117, Leu122, Val124, Leu126, Pro127 (strand F) and Thr141, Pro144, and Tyr147 (strand G) are shown in black.
Nine water molecules are located in the lower hydrophilic portion of the PagP interior, and this lower part of the protein has features seen in the overall interior of other outer membrane proteins, including an extensive network of hydrogen-bonding and salt-bridge interactions. The interior cores of previously solved structures of outer membrane β-barrel proteins are filled with polar side chains (Schulz, 2002; Wimley, 2002), including eight-stranded members of the family and members with larger barrels, such as the porins. An interesting new example is the NalP autotransporter β-domain, a 12-stranded barrel with a highly charged interior that is filled with an α-helical peptide from the N-terminus of the protein (Oomen et al, 2004).
Some vague similarities to the PagP structure are seen in the outer membrane protein NspA, which has a hydrophilic interior within the transmembrane domain and a bound detergent molecule located at the top of its barrel (Vandeputte-Rutten et al, 2003). However, in this case, the detergent lies across a shallow groove at a position external to the bilayer and remains largely exposed to the solvent. The limited depth of this binding site can be seen in the analysis of the hydrophobicity of inward-facing residues in NspA (Figure 3D).
A hydrocarbon ruler determines palmitate specificity
Based on these structural data, several predictions can be made regarding the mechanism of action of PagP. PagP has a typical polar core only in the lower half of the protein, and a distinctly hydrophobic interior in the region that would be located in the outer lipid A leaflet of the membrane bilayer (Figure 3D). We believe that the LDAO molecule bound in the hydrophobic pocket occupies the palmitate transfer binding site, and hypothesize that internalization of a phospholipid acyl chain occurs during the transfer reaction.
The relevance of the detergent-binding site in PagP is confirmed by data in which LDAO and dodecylphosphocholine (DPC) act as inhibitors of PagP transferase activity, while detergents such as dodecyl-β-D-maltoside (DDM) and CYFOS-7 that cannot fit in the barrel core have no inhibitory effect (Figure 6A–C). The hydrophobic tail of CYFOS-7 terminates with a cyclohexyl group and would not fit into the PagP acyl cavity, while the disaccharide headgroup of DDM would exclude it from the binding site. Molecular modeling suggests that a C16 ligand in place of the shorter LDAO molecule would position the headgroup in proximity to the catalytic Asp76/Ser77 residues.
Figure 6.
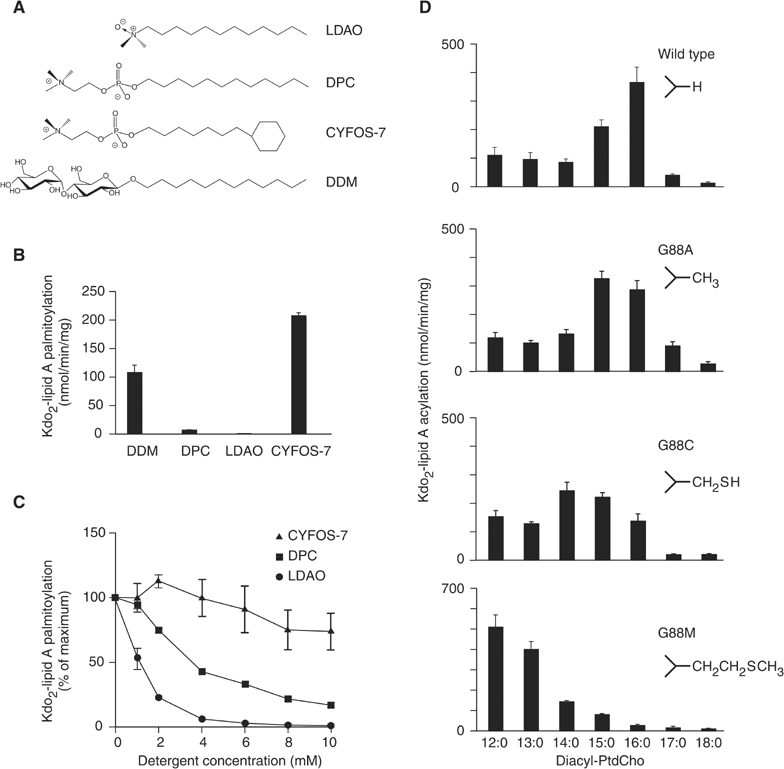
Enzymatic activity of PagP. (A) LDAO and DPC are relatively compact amphiphiles, while CYFOS-7 and DDM have bulky tail and head groups, respectively. (B) PagP is active in CYFOS-7 and DDM, but shows little activity in DPC or LDAO. (C) PagP in 0.25% DDM is inhibited by DPC and LDAO. (D) The PagP donor acyl-chain specificity in phosphatidylcholine (PtdCho) is a function of the amino-acid side chain at position 88. The wild-type protein has Gly88 and preferentially transfers a palmitate group to Kdo2-lipid A. The G88A, G88C, and G88M mutants provide side chains that occupy the acyl-binding pocket of PagP to progressively greater extents, and decrease the specificity of the transferred chain by 1, 2, and 4 methylene equivalents, respectively.
To further assess the biological significance of the hydrophobic pocket, we characterized a series of mutants designed to test the idea that the dimensions of the pocket determine the specificity of the length of the transferred acyl chain. Gly88 is located on strand D near the center of the barrel and forms part of the ‘floor' of the inner cavity. The pro-S hydrogen on the Cα of this residue is oriented directly towards the terminal methyl group of the bound detergent (Figure 5C). A series of mutations, G88A, G88C, and G88M, were tested for substrate specificity (Figure 6D). We observed that PagP mutants with longer amino-acid side chains at position 88 preferentially transferred shorter acyl chains in donor phosphatidylcholines. In effect, a ‘hydrocarbon ruler' allows PagP to discriminate between linear acyl groups with different numbers of carbon atoms.
Determinants of lipid trafficking to the active site
The lipid A acceptor in the PagP transfer reaction is anchored in the outer leaflet of the outer membrane, but the phospholipid donor is normally restricted to the inner leaflet (Kamio and Nikaido, 1976). We presume that translocation of a phospholipid into the outer leaflet is followed by internalization of the sn-1 acyl chain into the PagP interior. The route of entry for a hydrophobic ligand into the PagP interior poses an interesting structural question, since an approach along the long axis of the ligand-binding pocket is not an option here, as it would be with a soluble protein. Instead, the internalization most likely occurs via a lateral route from the membrane phase to the interior of the protein, and the absence of hydrogen bonding between strands A/B and F/G in the upper half of the membrane-embedded region provides two plausible gateways for access of lipid substrates to the β-barrel interior (Figure 4B). The main-chain interactions between strands F and G are disrupted in part by residue Pro127, which is conserved in all known PagP homologues (Figure 4A). In this region, the wall of the barrel is made from nonpolar side-chain–side-chain interactions (Figure 5D), and side-chain reorientations may provide an energetically reasonable pathway for entry and/or exit of ligands to the binding pocket. A second site that may also play a role in providing access to the active site is localized near the β-bulge residue Pro28 on strand A. This residue interrupts the hydrogen bonding with strand B, and causes a change in registration of the H-bonding pattern between strands A and H. This region involves the highly dynamic loop L1 connecting strands A and B (Hwang et al, 2002), and recent NMR studies of PagP dynamics in the noninhibitory detergent CYFOS-7 have revealed a conformational change in the L1 loop and adjacent regions of the β-barrel (Hwang et al, 2004). A multiple sequence alignment of PagP homologues (Figure 4A) shows that the L1 region contains several highly conserved residues and does not contain any insertions or deletions, further supporting an important functional and structural role for this part of the protein.
The requirement of invariant His33 in L1, and Asp76 and Ser77 in L2 for catalysis (Hwang et al, 2002), might suggest that PagP utilizes an acyl-enzyme mechanism characteristic of known serine esterases. However, these putative active site residues are not organized into a catalytic triad in the crystal structure (Figure 3B), nor in the NMR structure in DPC (Figure 3A; Hwang et al, 2002). Given the presence of two possible sites for access of lipid substrates to the β-barrel interior (Figure 4B), the PagP structure is also compatible with a ternary complex mechanism for direct transfer of the palmitoyl group from the donor phospholipid to the endotoxin acceptor. Either mechanism requires a stereospecific interaction with lipopolysaccharide, and PagP contains several highly conserved arginine and lysine residues in the exterior-facing loops L1–L3 (Figure 4A). A structural motif for lipopolysaccharide recognition involving four positively charged amino acids has been identified in lipopolysaccharide-interacting proteins (Ferguson et al, 2000). We expect some variability in the recognition motifs in the PagP family of sequences because there are known variations in both lipopolysaccharide structure and PagP stereoselectivity in different bacterial species. However, the invariant Lys42 in the disordered region of the L1 loop has been mutated to an Ala with the observation that this mutant retains a slow phospholipase activity that is found in the wild-type enzyme, but is defective in its ability to palmitoylate lipid A. This observation is consistent with a role of Lys42 in lipopolysaccharide recognition (EI Lo and RE Bishop, unpublished observations). Clearly, detailed knowledge of the structural transitions that occur in the L1 loop and the surrounding regions (Hwang et al, 2004) will hold the key to elucidating the nature of the interactions of PagP with phospholipids and lipopolysaccharides, and we expect that this insight will ultimately lead to an understanding of the catalytic mechanism.
Structural relationships with the lipocalins
A striking similarity is seen between the structures of PagP and the lipocalins, a widely distributed family of generally soluble lipid-binding proteins, the archetype of which is serum retinol-binding protein (RBP) (Newcomer and Ong, 2000). Both PagP and RBP are eight-stranded antiparallel β-barrels with a deep lipid-binding pocket located inside one end of the barrel and a flanking α-helix at the other end (Figure 7). The structures superpose with the proper alignment of the β-strands (i.e. A–H of PagP superposes with A–H of RBP) with an r.m.s.d. of 2.9 Å over 93 Cα atoms. The structural similarity between OmpA and the lipocalins has been noted (Pautsch and Schulz, 2000), but there are significant differences in the length of the barrels (Figure 2A and B) and in the nature of the protein interiors. The case for homology is strengthened in the case presented here because the PagP and RBP barrels have a higher percentage of alignable positions, and the two proteins have similar inner lipid-binding pockets (Figure 7). Furthermore, in the lipocalins, a long loop spans strands A and B and typically forms a lid that partially closes the ligand-binding site (Flower et al, 2000). It is intriguing that the long L1 loop from PagP is at an equivalent position and is strongly implicated in the function of this enzyme (Hwang et al, 2002, 2004); however, additional structural data will be required to test whether this loop interacts directly with ligand and/or the upper region of the barrel.
Figure 7.
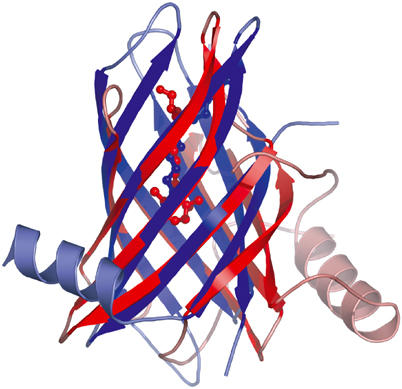
Superposition of the PagP (blue) and retinol-binding protein (red, Protein Data Bank code 1AQB) structures. The LDAO (blue) and retinol (red) ligands are shown in ball-and-stick representation. PagP has an N-terminal α-helix, while most lipocalin structures, including RBP, have a single α-helix at the C-terminus.
We cannot rule out the possibility that this structural similarity represents a case of convergent evolution. However, based on the absence of lipocalins from those bacteria that also lack β-barrel membrane proteins (Bishop, 2000; Ganfornina et al, 2000), which include all non-mycolic acid-producing Gram-positive bacteria and the Archaea (Nikaido, 2003), we cannot currently exclude the hypothesis that PagP and lipocalins share a common ancestor. This hypothesis is supported by the observation that the vast majority of bacterial lipocalins are anchored in the outer membrane as lipoproteins (Bishop et al, 1995; Campanacci et al, 2004), which could represent an intermediate state in the adaptation between membrane-bound and soluble globular domains (Bishop and Weiner, 1996). Outer membrane β-barrel domains should be suitably adapted to occupy both soluble and membrane-bound conformations because both environments are encountered during the outer membrane assembly process (Voulhoux et al, 2003).
Conclusions
PagP is distinguished from other known β-barrel membrane protein structures by the presence of an internal ligand-binding pocket that serves as a hydrocarbon ruler. Other unusual features include a tilting of the barrel axis with respect to the plane of the membrane, and the presence of non-hydrogen-bonded regions between β-strands in the outer leaflet half of the protein that may provide routes for lateral access of lipid substrates to the interior pocket.
Many of these features are also found in the lipocalins, and the lipocalin fold provides a rich scaffold for the design of ligand-binding specificity (Skerra, 2000). As the length of the secondary acyl groups in lipid A analogues is critical for signal transduction through the toll-like receptor 4 pathway (Stover et al, 2004), our finding that the specificity of the PagP hydrocarbon ruler can be altered may provide enzymatic routes for the synthesis of novel endotoxin antagonists. Efforts to develop inhibitors of PagP-catalyzed lipid A palmitoylation may provide novel treatments for bacterial infections.
Materials and methods
Cloning, expression, and purification of PagP
The cloning of the E. coli pagP gene with a deleted leader sequence and a C-terminal His tag was described previously (Hwang et al, 2002). The protein was expressed in E. coli BL21(DE3) cells (Novagen) with isopropyl-1-thio-β-D-galactopyranoside induction (0.2 mM). Cells were harvested, resuspended in 10 mM Tris–HCl (pH 8.0) and ruptured by cavitation in EmulsiFlex-05 (Avestin). Insoluble protein was collected by centrifugation at 26 000 g, and the pellet was resuspended and washed twice with 2% Triton X-100. The protein was solubilized in 6 M GuHCl at a concentration of 5 mg/ml, and rapidly diluted 10-fold into a solution of 0.5% LDAO, and 10 mM Tris–HCl (pH 8.0) at room temperature and stirred for 4 h. The refolded protein was applied to a Poros HQ anion-exchange column in 0.05% LDAO and the bound protein was eluted with a linear gradient of elution buffer (2 M NaCl, 10 mM Tris (pH 8.0)). E. coli B834(DE3) grown in the presence of seleno-L-methionine was used for the expression of SeMet-substituted protein, and protein purification proceeded as with the native protein, except that 1 mM β-mercaptoethanol was included in all buffers.
Crystallization and crystallographic data collection and processing
We were able to produce small crystals of recombinant E. coli PagP that was purified directly from bacterial membranes, as described in Bishop et al (2000). These crystals formed in space group P4122 with unit cell dimensions a=50.46 Å, b=50.46 Å, and c=158.18 Å. However, in this study, we used isomorphous crystals of significantly higher quality that were obtained from protein expressed without the signal peptide and refolded from intracellular inclusion bodies as described above. Crystals were grown in hanging drops by mixing equal volumes of 10 mg/ml protein solution with reservoir solution (30% 2-methyl-2,4-pentanediol, 0.1 M sodium citrate (pH 5.6), 0.2 M ammonium acetate). All diffraction data collections were carried out at 100 K. For the bromine and cesium derivatives, crystals were soaked briefly in mother liquor containing either 1 M NaBr or 1 M CsCl and flash frozen. Diffraction data were collected for the native and derivative crystals at beamline F2 at the Cornell High Energy Synchrotron Source, and processed with MOSFLM and SCALA (CCP4, 1994). Crystals belonged to space group P4122 and contained a single PagP molecule per asymmetric unit. Experimental maps derived from three wavelength bromide MAD data were not interpretable, even when supplemented with the CsCl data. We could not obtain crystals of the pure selenomethionine protein, and attempts at seeding with native protein crystals failed. However, crystals could be obtained by mixing 60% native and 40% selenomethionine protein, and we collected additional diffraction data on these crystals on a rotating anode X-ray source (CuKα). Multiple isomorphous replacement with anomalous scattering (MIRAS) phases were calculated from the NaBr, CsCl, and SeMet derivative data sets with SOLVE/RESOLVE (Terwilliger and Berendzen, 1999), and the resulting electron density maps were interpretable and could be partially traced with the automatic chain-building methods in RESOLVE. Additional model building was performed with O (Jones et al, 1991), and refinement and addition of water molecules were carried out with CNS (Brunger et al, 1998) and REFMAC (Murshudov et al, 1997). Molecular graphics were prepared with PYMOL (DeLano, 2002). The final model consists of residues 7–37 and 48–161 of PagP, with two additional non-native C-terminal residues, Leu162 and Glu163, resulting from the expression plasmid. The following six C-terminal histidines were not observed in the electron density maps.
Activity assays
Diacylphosphatidylcholines (diacyl-PtdChos) were obtained from Avanti Polar Lipids (Alabaster, AL) and detergents were obtained from Anatrace (Maumee, OH). 14C-sodium acetate (57 mCi/mmol) and sn-1,2-di-(16:0-114C)-PtdCho (100 mCi/mmol) were obtained from Perkin-Elmer Life Sciences. Kdo2-lipid A was extracted and purified from the heptose-deficient E. coli strain WBB06 (Brabetz et al, 1997) according to Kanipes et al (2001), and quantified by the purpald assay (Lee and Tsai, 1999). 14C-Kdo2-lipid A was prepared from WBB06 cells grown in 5 ml of medium containing 20 μM (1 μCi/ml) 14C-sodium acetate and grown at 37°C for 5 h until OD600=1.0 before isolation (Kanipes et al, 2001). Assays were performed by thin-layer chromatography as described previously (Bishop et al, 2000), but with 0.25% n-dodecyl-β-D-maltoside in place of Triton X-100 as the supporting detergent, unless indicated otherwise. Mutant proteins were generated with the QuikChange protocol (Stratagene) and purified from membranes (Bishop et al, 2000). Detergent inhibition studies utilized sn-1,2-di-(16:0-114C)-PtdCho at 20 μM (4000 cpm/μl) and 100 μM Kdo2-lipid A. Hydrocarbon ruler experiments utilized 14C-Kdo2-lipid A at 10 μM (∼110 cpm/μl) with sn-1,2-diacyl-PtdChos at 1 mM.
Coordinates
The coordinates of PagP have been deposited in the Protein Data Bank (identification code 1THQ).
Acknowledgments
We thank the members of the Cornell High Energy Synchrotron Source for data collection support and Christian Raetz for helpful advice and discussions. This work was supported by grants from the Canadian Institutes for Health Research to GGP and REB.
References
- Bishop RE (2000) The bacterial lipocalins. Biochim Biophys Acta 1482: 73–83 [DOI] [PubMed] [Google Scholar]
- Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz CR (2000) Transfer of palmitate from phospholipids to lipid A in outer membranes of gram-negative bacteria. EMBO J 19: 5071–5080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop RE, Penfold SS, Frost LS, Holtje JV, Weiner JH (1995) Stationary phase expression of a novel Escherichia coli outer membrane lipoprotein and its relationship with mammalian apolipoprotein D. Implications for the origin of lipocalins. J Biol Chem 270: 23097–23103 [DOI] [PubMed] [Google Scholar]
- Bishop RE, Weiner JH (1996) ‘Outlier' lipocalins more than peripheral. Trends Biochem Sci 21: 127. [PubMed] [Google Scholar]
- Brabetz W, Muller-Loennies S, Holst O, Brade H (1997) Deletion of the heptosyltransferase genes rfaC and rfaF in Escherichia coli K-12 results in an Re-type lipopolysaccharide with a high degree of 2-aminoethanol phosphate substitution. Eur J Biochem 247: 716–724 [DOI] [PubMed] [Google Scholar]
- Brozek KA, Bulawa CE, Raetz CR (1987) Biosynthesis of lipid A precursors in Escherichia coli. A membrane-bound enzyme that transfers a palmitoyl residue from a glycerophospholipid to lipid X. J Biol Chem 262: 5170–5179 [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Cloare GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Campanacci V, Nurizzo D, Spinelli S, Valencia C, Tegoni M, Cambillau C (2004) The crystal structure of the Escherichia coli lipocalin Blc suggests a possible role in phospholipid binding. FEBS Lett 562: 183–188 [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 Suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Dekker N (2000) Outer-membrane phospholipase A: known structure, unknown biological function. Mol Microbiol 35: 711–717 [DOI] [PubMed] [Google Scholar]
- DeLano WL (2002) The PyMOL Molecular Graphics System. San Carlos, CA: DeLano Scientific [Google Scholar]
- Derzelle S, Turlin E, Duchaud E, Pages S, Kunst F, Givaudan A, Danchin A (2004) The PhoP-PhoQ two-component regulatory system of Photorhabdus luminescens is essential for virulence in insects. J Bacteriol 186: 1270–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AD, Welte W, Hofmann E, Lindner B, Holst O, Coulton JW, Diederichs K (2000) A conserved structural motif for lipopolysaccharide recognition by procaryotic and eucaryotic proteins. Struct Fold Des 8: 585–592 [DOI] [PubMed] [Google Scholar]
- Flower DR, North AC, Sansom CE (2000) The lipocalin protein family: structural and sequence overview. Biochim Biophys Acta 1482: 9–24 [DOI] [PubMed] [Google Scholar]
- Fukuoka S, Brandenburg K, Muller M, Lindner B, Koch MH, Seydel U (2001) Physico-chemical analysis of lipid A fractions of lipopolysaccharide from Erwinia carotovora in relation to bioactivity. Biochim Biophys Acta 1510: 185–197 [DOI] [PubMed] [Google Scholar]
- Ganfornina MD, Gutierrez G, Bastiani M, Sanchez D (2000) A phylogenetic analysis of the lipocalin protein family. Mol Biol Evol 17: 114–126 [DOI] [PubMed] [Google Scholar]
- Guo L, Lim KB, Poduje CM, Daniel M, Gunn JS, Hackett M, Miller SI (1998) Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell 95: 189–198 [DOI] [PubMed] [Google Scholar]
- Hwang PM, Bishop RE, Kay LE (2004) The integral membrane enzyme PagP alternates between two dynamically distinct states. Proc Natl Acad Sci USA 101: 9618–9623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang PM, Choy WY, Lo EI, Chen L, Forman-Kay JD, Raetz CR, Privé GG, Bishop RE, Kay LE (2002) Solution structure and dynamics of the outer membrane enzyme PagP by NMR. Proc Natl Acad Sci USA 99: 13560–13565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan S, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kamio Y, Nikaido H (1976) Outer membrane of Salmonella typhimurium: accessibility of phospholipid head groups to phospholipase c and cyanogen bromide activated dextran in the external medium. Biochemistry 15: 2561–2570 [DOI] [PubMed] [Google Scholar]
- Kanipes MI, Lin S, Cotter RJ, Raetz CR (2001) Ca2+-induced phosphoethanolamine transfer to the outer 3-deoxy-D-manno-octulosonic acid moiety of Escherichia coli lipopolysaccharide. A novel membrane enzyme dependent upon phosphatidylethanolamine. J Biol Chem 276: 1156–1163 [DOI] [PubMed] [Google Scholar]
- Kawasaki K, Ernst RK, Miller SI (2004) 3-O-deacylation of lipid A by PagL, a PhoP/PhoQ-regulated deacylase of Salmonella typhimurium, modulates signaling through toll-like receptor 4. J Biol Chem 279: 20044–20048 [DOI] [PubMed] [Google Scholar]
- Koebnik R, Locher KP, Van Gelder P (2000) Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol Microbiol 37: 239–253 [DOI] [PubMed] [Google Scholar]
- Lee CH, Tsai CM (1999) Quantification of bacterial lipopolysaccharides by the purpald assay: measuring formaldehyde generated from 2-keto-3-deoxyoctonate and heptose at the inner core by periodate oxidation. Anal Biochem 267: 161–168 [DOI] [PubMed] [Google Scholar]
- Long SB, Casey PJ, Beese LS (1998) Cocrystal structure of protein farnesyltransferase complexed with a farnesyl diphosphate substrate. Biochemistry 37: 9612–9618 [DOI] [PubMed] [Google Scholar]
- Muroi M, Ohnishi T, Tanamoto K (2002) MD-2, a novel accessory molecule, is involved in species-specific actions of Salmonella lipid A. Infect Immun 70: 3546–3550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Newcomer ME, Ong DE (2000) Plasma retinol binding protein: structure and function of the prototypic lipocalin. Biochim Biophys Acta 1482: 57–64 [DOI] [PubMed] [Google Scholar]
- Nikaido H (2003) Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67: 593–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oomen CJ, Van Ulsen P, Van Gelder P, Feijen M, Tommassen J, Gros P (2004) Structure of the translocator domain of a bacterial autotransporter. EMBO J 23: 1257–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pautsch A, Schulz GE (2000) High-resolution structure of the OmpA membrane domain. J Mol Biol 298: 273–282 [DOI] [PubMed] [Google Scholar]
- Pilione MR, Pishko EJ, Preston A, Maskell DJ, Harvill ET (2004) pagP is required for resistance to antibody-mediated complement lysis during Bordetella bronchiseptica respiratory infection. Infect Immun 72: 2837–2842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston A, Maxim E, Toland E, Pishko EJ, Harvill ET, Caroff M, Maskell DJ (2003) Bordetella bronchiseptica PagP is a Bvg-regulated lipid A palmitoyl transferase that is required for persistent colonization of the mouse respiratory tract. Mol Microbiol 48: 725–736 [DOI] [PubMed] [Google Scholar]
- Raetz CR, Whitfield C (2002) Lipopolysaccharide endotoxins. Annu Rev Biochem 71: 635–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebeil R, Ernst RK, Gowen BB, Miller SI, Hinnebusch BJ (2004) Variation in lipid A structure in the pathogenic yersiniae. Mol Microbiol 52: 1363–1373 [DOI] [PubMed] [Google Scholar]
- Robey M, O'Connell W, Cianciotto NP (2001) Identification of Legionella pneumophila rcp, a pagP-like gene that confers resistance to cationic antimicrobial peptides and promotes intracellular infection. Infect Immun 69: 4276–4286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz GE (2002) The structure of bacterial outer membrane proteins. Biochim Biophys Acta 1565: 308–317 [DOI] [PubMed] [Google Scholar]
- Skerra A (2000) Lipocalins as a scaffold. Biochim Biophys Acta 1482: 337–350 [DOI] [PubMed] [Google Scholar]
- Snijder HJ, Ubarretxena-Belandia I, Blaauw M, Kalk KH, Verheij HM, Egmond MR, Dekker N, Dijkstra BW (1999) Structural evidence for dimerization-regulated activation of an integral membrane phospholipase. Nature 401: 717–721 [DOI] [PubMed] [Google Scholar]
- Stover AG, Da Silva Correia J, Evans JT, Cluff CW, Elliott MW, Jeffery EW, Johnson DA, Lacy MJ, Baldridge JR, Probst P, Ulevitch RJ, Persing DH, Hershberg RM (2004) Structure–activity relationship of synthetic toll-like receptor 4 agonists. J Biol Chem 279: 4440–4449 [DOI] [PubMed] [Google Scholar]
- Tanamoto K, Azumi S (2000) Salmonella-type heptaacylated lipid A is inactive and acts as an antagonist of lipopolysaccharide action on human line cells. J Immunol 164: 3149–3156 [DOI] [PubMed] [Google Scholar]
- Tarshis LC, Proteau PJ, Kellogg BA, Sacchettini JC, Poulter CD (1996) Regulation of product chain length by isoprenyl diphosphate synthases. Proc Natl Acad Sci USA 93: 15018–15023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J (1999) Automated MAD and MIR structure solution. Acta Crystallogr D 55: 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance DE, Vance JE (eds) (2002) Biochemistry of Lipids, Lipoproteins, and Membranes. Amsterdam: Elsevier [Google Scholar]
- Vandeputte-Rutten L, Bos MP, Tommassen J, Gros P (2003) Crystal structure of neisserial surface protein A (NspA), a conserved outer membrane protein with vaccine potential. J Biol Chem 278: 24825–24830 [DOI] [PubMed] [Google Scholar]
- Voulhoux R, Bos MP, Geurtsen J, Mols M, Tommassen J (2003) Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 299: 262–265 [DOI] [PubMed] [Google Scholar]
- Wimley WC (2002) Toward genomic identification of beta-barrel membrane proteins: composition and architecture of known structures. Protein Sci 11: 301–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff TJ, Lin S, Cotter RJ, Dotson GD, Raetz CR (1998) Hydrocarbon rulers in UDP-N-acetylglucosamine acyltransferases. J Biol Chem 273: 32369–32372 [DOI] [PubMed] [Google Scholar]