

Abstract
The spindle checkpoint ensures accurate chromosome segregation by delaying anaphase in response to misaligned sister chromatids during mitosis. Upon checkpoint activation, Mad2 binds directly to Cdc20 and inhibits the anaphase-promoting complex or cyclosome (APC/C). Cdc20 binding triggers a dramatic conformational change of Mad2. Consistent with an earlier report, we show herein that depletion of p31comet (formerly known as Cmt2) by RNA interference in HeLa cells causes a delay in mitotic exit following the removal of nocodazole. Purified recombinant p31comet protein antagonizes the ability of Mad2 to inhibit APC/CCdc20 in vitro and in Xenopus egg extracts. Interestingly, p31comet binds selectively to the Cdc20-bound conformation of Mad2. Binding of p31comet to Mad2 does not prevent the interaction between Mad2 and Cdc20 in vitro. During checkpoint inactivation in HeLa cells, p31comet forms a transient complex with APC/CCdc20-bound Mad2. Purified p31comet enhances the activity of APC/C isolated from nocodazole-arrested HeLa cells without disrupting the Mad2–Cdc20 interaction. Therefore, our results suggest that p31comet counteracts the function of Mad2 and is required for the silencing of the spindle checkpoint.
Keywords: anaphase-promoting complex, Mad2, mitosis, p31comet , spindle checkpoint
Introduction
During the cell division cycle, cells duplicate their chromosomes once and only once and segregate the sister chromatids evenly in mitosis, thus faithfully transmitting their genetic information to the daughter cells. The replicated chromosomes are held together by the cohesin protein complex (Nasmyth, 2002). At the metaphase–anaphase transition, a large ubiquitin protein ligase called the anaphase-promoting complex or cyclosome (APC/C) mediates the degradation of securin, an inhibitor of separase (Peters, 2002). The active separase then cleaves a subunit of the cohesin complex, Scc1, leading to the dissolution of chromosome cohesion and the onset of sister-chromatid separation (Nasmyth, 2002). To maintain genetic stability, the spindle checkpoint delays the onset of chromosome segregation in response to sister chromatids that have not attached to microtubules emanating from the two opposite poles of the mitotic spindle (biorientation) (Rudner and Murray, 1996; Gorbsky, 2001; Wassmann and Benezra, 2001; Millband et al, 2002; Musacchio and Hardwick, 2002; Yu, 2002; Bharadwaj and Yu, 2004). A single kinetochore that is not captured by microtubules and not under tension activates this checkpoint (Li and Nicklas, 1995; Rieder et al, 1995; Nicklas, 1997). APC/C is a critical target of the spindle checkpoint (Peters, 2002; Yu, 2002). Inhibition of APC/C by the checkpoint causes the stabilization of securin, inhibition of separase, and a delay in sister-chromatid separation (Peters, 2002).
Progress has been made toward understanding the mechanism by which the spindle checkpoint inhibits APC/C in response to spindle defects (Millband et al, 2002; Yu, 2002). In vertebrates, Mad2 and BubR1 act synergistically to inhibit APC/C in vitro through their direct binding to Cdc20 (Li et al, 1997; Fang et al, 1998a; Hwang et al, 1998; Kim et al, 1998; Wassmann and Benezra, 1998; Sudakin et al, 2001; Tang et al, 2001; Fang, 2002). Furthermore, BubR1 and Mad2 can assemble into a single mitotic checkpoint complex (MCC) containing BubR1, Bub3, Mad2, and Cdc20 in vivo (Hardwick et al, 2000; Fraschini et al, 2001; Sudakin et al, 2001; Millband and Hardwick, 2002). It is unclear whether Mad2 and BubR1 act exclusively in the context of MCC or they also form independent BubR1–Bub3–Cdc20 and Mad2–Cdc20 complexes to inhibit APC/C in living cells. Nevertheless, it has been firmly established that the Mad2–Cdc20 interaction is absolutely required for the proper execution of the spindle checkpoint (Fang et al, 1998a; Hwang et al, 1998; Kim et al, 1998; Dobles et al, 2000; Michel et al, 2001).
Recent structural data have provided insights into the Mad2–Cdc20 interaction (Luo et al, 2000, 2002; Sironi et al, 2002). Mad2 recognizes a short conserved peptide motif within Cdc20 (Luo et al, 2000, 2002). Binding of the Cdc20 peptide to Mad2 triggers a dramatic structural rearrangement of Mad2, which involves the shuffling of several β strands in the main β sheet of Mad2 (Luo et al, 2000, 2002). Interestingly, Mad1 contains a small Mad2-binding motif similar to that of Cdc20, and Mad1 binding triggers the same large conformational change of Mad2 as does Cdc20 (Luo et al, 2000, 2002; Sironi et al, 2002). Mad1 is also required for the kinetochore localization of Mad2 (Chen et al, 1998; Chung and Chen, 2002; Luo et al, 2002). Very recently, we have shown that Mad2 possesses two natively folded conformations at equilibrium, referred to as N1 and N2 (Luo et al, 2004). N2-Mad2 resembles the structure of the Mad1- or Cdc20-bound Mad2 and is more active in APC/C inhibition. The Mad2-binding domain of Mad1 facilitates the N1–N2 conformational switch of Mad2 in vitro. Taken together, these results suggest that, upon checkpoint activation, Mad1 recruits Mad2 to unattached kinetochores and facilitates the formation of N2-Mad2 and thus the establishment of the Mad2–Cdc20 interaction (Chen et al, 1998; Chung and Chen, 2002; Luo et al, 2002). The structural studies on the Mad1–Mad2 and Mad2–Cdc20 complexes have revealed another intriguing feature of these interactions. The dissociation of Mad1–Mad2 and Mad2–Cdc20 complexes requires the partial unfolding of the C-terminal region of Mad2, which imposes a significant energetic barrier on these processes (Luo et al, 2000, 2002; Sironi et al, 2002). This suggests the existence of other factors that may facilitate the disassembly of the Mad2–Cdc20-containing checkpoint complexes when the checkpoint is satisfied.
A novel human Mad2-binding protein called Cmt2 has recently been identified in a yeast two-hybrid screen (Habu et al, 2002). As Cmt2 has been in use for the Charcot–Marie–Tooth disease type 2 (CMT2) gene, we propose to rename this Mad2-binding protein, p31comet, for its comet tail-like cellular localization pattern in mitosis (unpublished results, TH and TM). In HeLa cells, the association of the endogenous p31comet protein with Mad2 coincides with the dissociation of the Mad2–Cdc20 interaction, suggesting that p31comet might play a role in silencing the spindle checkpoint (Habu et al, 2002). Consistent with this notion, we report herein that HeLa cells depleted of p31comet by RNA interference (RNAi) escape from the checkpoint-mediated mitotic arrest with significantly slower kinetics. P31comet blocks the biochemical function of Mad2 in vitro and in Xenopus egg extracts. Strikingly, p31comet only interacts with the Cdc20-bound conformation of Mad2. P31comet binding and Cdc20 binding to Mad2 are not mutually exclusive, indicating that p31comet and Cdc20 bind to different sites on Mad2. Moreover, p31comet, Mad2, and Cdc20 form a ternary complex in vitro and in vivo, and this complex is transiently enriched as cells exit from mitosis. Purified recombinant p31comet protein moderately activates APC/C isolated from nocodazole-arrested HeLa cells without disrupting the Mad2–Cdc20 interaction. Our results indicate that p31comet counteracts the function of Mad2 and is required for spindle checkpoint silencing.
Results
P31comet is required for the inactivation of the spindle checkpoint
To test the hypothesis that p31comet is required for checkpoint inactivation, we examined the effect of RNAi-mediated depletion of p31comet on the mitotic arrest of HeLa cells caused by spindle-damaging agents. Transfection of p31comet-specific siRNA into HeLa cells reduced the protein level of p31comet to 15% of that of the cells that received a control siRNA (Figure 1A). Cells depleted of p31comet did not exhibit any discernable cell cycle phenotype, as judged by FACS (Figure 1C) and cell morphology (data not shown). This indicates that p31comet at 15% of its endogenous concentration is sufficient for cell cycle progression under normal conditions. If p31comet is required for the inactivation of the spindle checkpoint, downregulation of p31comet is expected to cause a delay in the activation of APC/CCdc20 and consequently a delay in mitotic exit after the removal of spindle-damaging agents. HeLa cells transfected with control or p31comet siRNA were arrested in mitosis with nocodazole. After 18 h of nocodazole treatment, cells were then grown in fresh medium to allow them to exit from mitosis. As shown in Figure 1B, two key APC/C substrates, securin and cyclin B1, were degraded in control cells at around 2 h after the removal of nocodazole. In contrast, securin and cyclin B1 were inefficiently degraded in p31comet RNAi cells, resulting in higher levels of both proteins after the removal of nocodazole as compared to control cells. Consistent with the biochemical readout, FACS analysis revealed that p31comet RNAi cells exited from mitosis with a much slower kinetics as compared to the control cells (Figure 1C). For example, at 1.5 h after the release from nocodazole-mediated mitotic arrest, 62% of the control cells had returned to the G1 phase with a 2N DNA content, while only 35% of the p31comet RNAi cells exited from mitosis. These data suggest that p31comet is required for the efficient inactivation of the spindle checkpoint established by extensive spindle damage. The phenotypes of p31comet RNAi cells are consistent with those described for cells transfected with antisense oligonucleotides against p31comet (Habu et al, 2002), with one minor exception. In the latter case, cells also experience a delay in mitotic exit following the release from nocodazole-mediated mitotic arrest. However, a significant portion of cells treated with antisense p31comet underwent cell death. We do not know the exact reason for the lower incidence of cell death in p31comet RNAi cells following the release from nocodazole-mediated mitotic arrest. It is possible that RNAi and antisense technologies knock down the protein level of p31comet with different kinetics and to different extents, thus resulting in slightly different phenotypes.
Figure 1.
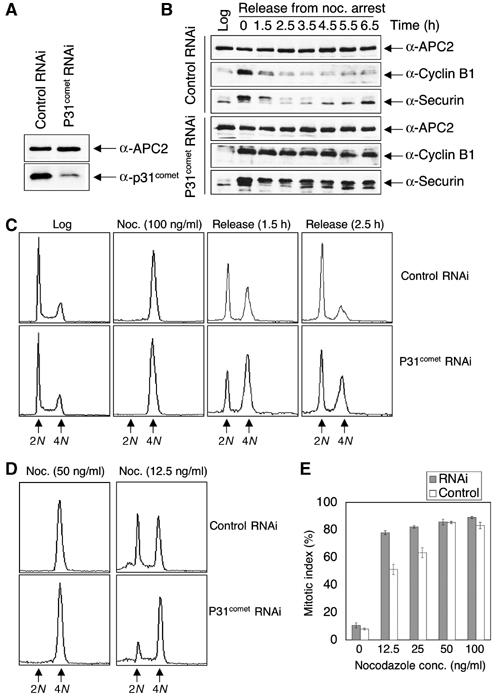
P31comet is required for the inactivation of the spindle checkpoint. (A) HeLa cells transfected with the control or p31comet siRNA duplexes were dissolved in SDS sample buffer, separated on SDS–PAGE, and blotted with anti-APC2 and anti-p31comet antibodies. (B) HeLa cells transfected with control or p31comet siRNA were treated with 100 ng/ml nocodazole for 18 h and released into fresh medium. The total cell lysates of log-phase cells and cell samples taken at the indicated time points were separated on SDS–PAGE and blotted with the indicated antibodies. (C) FACS analysis of some of the cell samples described in (B). The peaks corresponding to 2N and 4N DNA contents are labeled. (D) FACS analysis of HeLa cells that were transfected with control or p31comet siRNA and treated with the indicated concentrations of nocodazole. The peaks corresponding to 2N and 4N DNA contents are labeled. (E) HeLa Tet-on cells transfected with the control or p31comet siRNA were treated with varying concentrations of nocodazole for 16 h and stained with Hoechst 33342. The mitotic indices were determined by directly observing the cells with an inverted fluorescence microscope. The mitotic cells were round and contained condensed DNA, while the interphase cells were flat with decondensed DNA. This experiment was repeated three times and standard deviations are included as error bars.
We next checked the behavior of p31comet RNAi cells in the presence of lower concentrations of nocodazole. As shown in Figure 1D and E, p31comet RNAi cells underwent mitotic arrest more efficiently at lower concentrations of nocodazole (e.g. 12.5 ng/ml). This again is consistent with the notion that p31comet counteracts the spindle checkpoint function of Mad2. Higher levels of p31comet inactivate Mad2 and the spindle checkpoint in living cells (Habu et al, 2002), whereas lower levels of p31comet lead to checkpoint activation at a lower threshold of spindle damage.
P31comet blocks the ability of Mad2 to inhibit APC/CCdc20 in vitro and in Xenopus extracts
Binding of Mad2 to Cdc20 inhibits the ubiquitin ligase activity of APC/C in vitro (Fang et al, 1998a). We next checked whether p31comet blocked the ability of Mad2 to inhibit APC/CCdc20 in a ubiquitination assay reconstituted with purified components (Figure 2A). As shown previously, addition of purified Cdc20 protein to interphase Xenopus APC/C activated its ligase activity using a fragment of human cyclin B1 (residues 1–102) as the substrate (Figure 2B, compare lanes 1 and 2) (Fang et al, 1998b; Kramer et al, 1998). Preincubation of Cdc20 with the dimeric wild-type Mad2 greatly reduced the activity of APC/CCdc20 (Figure 2B, lanes 2 and 3). Addition of increasing amounts of p31comet together with Mad2 partially restored the activity of APC/CCdc20 (Figure 2B, lanes 4–8). As a control, addition of increasing amounts of recombinant human Mob1, a protein that does not interact with Mad2, did not affect the inhibition of APC/CCdc20 by Mad2 (Figure 2B, lanes 9–13). We also performed the APC/C assay in the presence of ΔC-Mad2 and p31comet (Figure 2C). As the C-terminal region of Mad2 is required for the Mad2–Cdc20 interaction, a Mad2 mutant with its C-terminal 10 residues deleted (ΔC-Mad2) was inactive in APC/C inhibition (Figure 2C). Addition of p31comet or Mob1 along with ΔC-Mad2 did not affect the activity of APC/C (Figure 2C). This indicates that p31comet does not have a general APC/C-stimulatory effect. Our data clearly demonstrate that p31comet partially reverses the APC/C-inhibitory activity of Mad2 in vitro.
Figure 2.
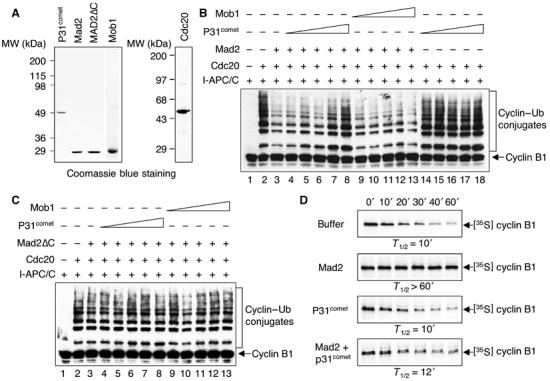
P31comet antagonizes the ability of Mad2 to inhibit the activity of APC/CCdc20. (A) The purified His6-tagged p31comet, Mad2, ΔC-Mad2, Mob1, and Cdc20 proteins were separated on SDS–PAGE and stained with Coomassie blue. (B) The ubiquitination activity of APC/C was assayed with a Myc-tagged N-terminal fragment of human cyclin B1. The reaction mixtures were separated on SDS–PAGE and blotted with the anti-Myc antibody. The positions of the cyclin B1 substrate and the cyclin B1–ubiquitin conjugates are labeled. Incubation of APC/C isolated from interphase Xenopus egg extracts (I-APC/C) with human Cdc20 greatly stimulated the activity of APC/C (compare lanes 1 and 2). The purified Mad2 protein (4 μM) inhibited the activity of APC/CCdc20 using cyclin B1 as a substrate (compare lanes 2 and 3). Addition of increasing amounts of p31comet (250 nM–4 μM) together with Mad2 restored the activity of APC/CCdc20 (compare lane 3 with lanes 4–8). Addition of a control protein Mob1 (250 nM–4 μM) did not have any effect on Mad2-mediated inhibition of APC/CCdc20 (lanes 9–13). Addition of p31comet (4 μM) alone in the absence of Mad2 did not stimulate the activity of APC/CCdc20 (lanes 14–18). (C) Same as in panel B (lanes 1–13) except that the ΔC-Mad2 protein (4 μM) was used in the assays. (D) P31comet reverses Mad2-mediated inhibition of APC/CCdc20 in Xenopus egg extracts. In vitro-translated 35S-labeled full-length human cyclin B1, a known APC/C substrate, was added to mitotic Xenopus egg extracts in the presence of XB buffer, recombinant purified wild-type Mad2, p31comet, or both Mad2 and p31comet. The final concentrations of Mad2 and p31comet were 20 μM. Samples were taken at the indicated time points and analyzed by SDS–PAGE followed by autoradiography. The half-life of cyclin B in each experiment is indicated.
We next tested the effect of p31comet on Mad2-mediated APC/C inhibition in Xenopus egg extracts. Consistent with the earlier reports (Fang et al, 1998a), cyclin B1 was rapidly degraded in mitotic Xenopus egg extracts that contained active APC/CCdc20 (Figure 2D). Addition of dimeric wild-type Mad2 inhibited APC/CCdc20 in these extracts and stabilized cyclin B1 (Figure 2D) (Fang et al, 1998a). Addition of p31comet together with Mad2 to these extracts again partially restored degradation of cyclin B1 (Figure 2D). As a control, p31comet alone did not perturb cyclin B1 degradation in these extracts (Figure 2D). Therefore, p31comet also blocked the APC/C-inhibitory activity of Mad2 in Xenopus egg extracts.
P31comet selectively binds to the Cdc20-bound conformation of Mad2
It has been previously shown that bacterially expressed wild-type Mad2 protein exists as two species: a monomer and a dimer (Fang et al, 1998a). Interestingly, only the dimeric form of Mad2 is active in inhibiting APC/C in Xenopus egg extracts (Fang et al, 1998a). A point mutant of Mad2, Mad2R133A, exists exclusively as a monomer (Sironi et al, 2001). Surprisingly, we have recently shown that Mad2R133A also exists as two distinct monomeric conformers, referred to as N1 and N2, which can be separated by anion exchange chromatography (Luo et al, 2004). N2-Mad2R133A is much more potent in APC/C inhibition in Xenopus extracts (Luo et al, 2004). The structures of both conformers have been determined by NMR spectroscopy (Figure 3A). The structure of N2-Mad2 resembles that of the Cdc20-bound form of Mad2, thus explaining why N2-Mad2 is more active in APC/C inhibition. Our NMR studies also indicate that the wild-type Mad2 monomer exhibits the N1 conformation, whereas the Mad2 dimer is in the N2 conformation. More interestingly, the Mad2-binding domain of Mad1 facilitates the structural conversion from N1-Mad2 to N2-Mad2 in vitro. These data suggest that the N1–N2 conformational change of Mad2 is required for its APC/C-inhibitory activity, and this structural transition might be regulated by other spindle checkpoint proteins, such as Mad1.
Figure 3.
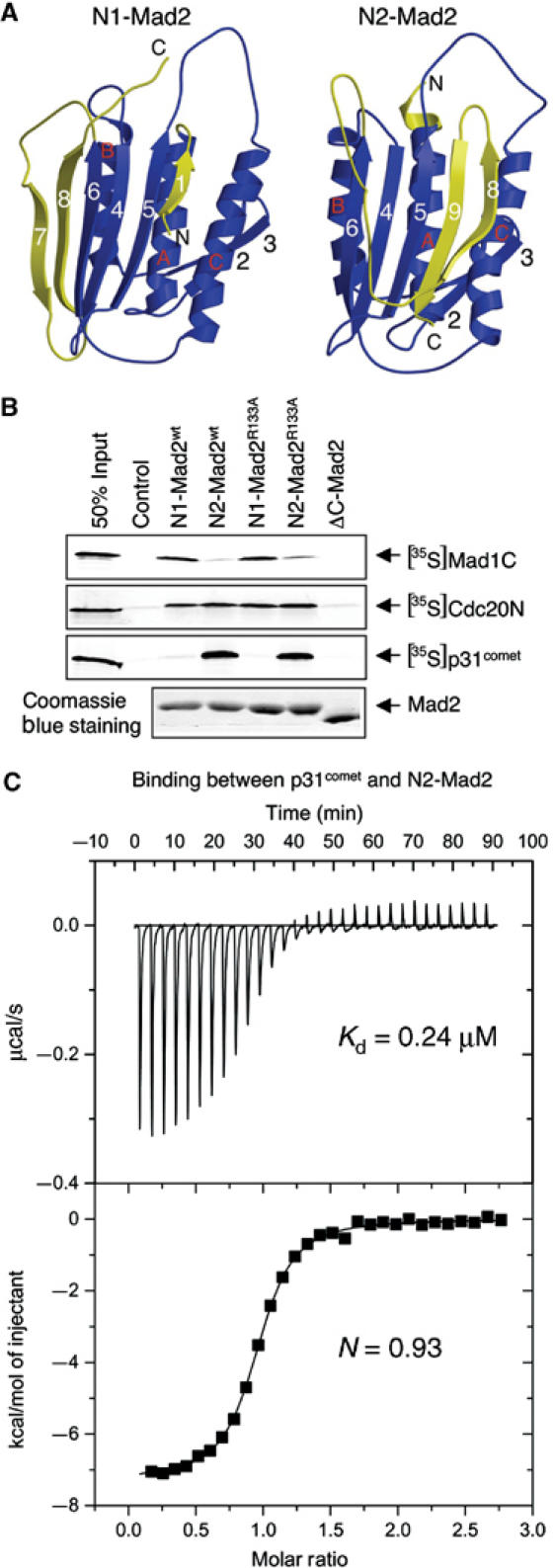
P31comet selectively binds to the N2 conformation of Mad2. (A) Ribbon drawing of the structures of the N1 and N2 forms of Mad2. The structural elements of Mad2 that undergo major changes between the N1 and N2 conformers are colored yellow, while the rest of the structural elements are in blue. The strands are numbered 1–8, while the helices are labeled A–C in the N1-Mad2 structure. The secondary structure elements of the N2-Mad2 are labeled in a similar manner with the exception of β9, which is unstructured in N1-Mad2. The structures were generated with Molscript and Raster3D. (B) Various forms of His6-Mad2 were bound to Ni2+-NTA beads and incubated with 35S-labeled Mad1C, Cdc20N, and p31comet proteins. Empty beads were used as a control, and 50% of the input protein used in each binding reaction was loaded for comparison. After washing, the proteins retained on beads were analyzed by SDS–PAGE followed by autoradiography. The Mad2 proteins bound to the beads were analyzed by SDS–PAGE followed by Coomassie blue staining. (C) ITC analysis of the binding between p31comet and N2-Mad2R133A. The dissociation constant (Kd) and the stoichiometry (N) of binding are indicated.
As the interaction between Mad2 and p31comet is regulated during the cell cycle (Habu et al, 2002), we tested whether the conformational change of Mad2 regulates the Mad2–p31comet interaction. Various purified His6-tagged Mad2 proteins were immobilized on Ni2+-NTA beads and incubated with 35S-labeled p31comet, Cdc20, or Mad1 proteins translated in reticulocyte lysate. After washing, the proteins bound to beads were analyzed by SDS–PAGE followed by autoradiography. As shown in Figure 3B, p31comet was selectively retained on beads containing N2-Mad2wt dimer or N2-Mad2R133A monomer. There was no detectable binding between p31comet and N1-Mad2 (Figure 3B). In contrast, N1-Mad2wt and N1-Mad2R133A bound more tightly to the C-terminal domain of Mad1 (Mad1C) as compared to N2-Mad2, while both N1 and N2 forms of Mad2 bound equally well to the N-terminal fragment (residues 1–174) of Cdc20 (Figure 3B). Therefore, these Mad2-binding proteins exhibit different binding preferences toward the N1 and N2 forms of Mad2. P31comet preferably binds to N2-Mad2. We do not yet fully understand why Mad1 prefers N1-Mad2 because the binding of N1-Mad2 to Mad1 is a complicated process (Luo et al, 2004). However, this is very likely due to a difference in binding kinetics between Mad1 and the two forms of Mad2.
To confirm the findings of these qualitative binding assays, we used isothermal calorimetry titration (ITC) to measure the binding affinity between p31comet and Mad2 quantitatively (Figure 3C). The dissociation constant (Kd) of the p31comet–N2-Mad2R133A complex was determined to be 0.24 μM (Figure 3C), while there was no detectable binding between p31comet and N1-Mad2 (data not shown). Thus, the ITC experiment demonstrates that p31comet only binds to the N2 form of Mad2. Furthermore, the stoichiometry of binding between p31comet and Mad2 was very close to 1:1. The native molecular weight of the Mad2wt–p31comet or the Mad2R133A–p31comet complexes was estimated to be around 60 kDa by gel filtration chromatography (data not shown). These data indicate that p31comet selectively forms a simple 1:1 heterodimer with N2-Mad2 with high affinity. This finding was further confirmed by NMR studies (Figure 4). Addition of p31comet caused dramatic line broadening and disappearance of most HSQC signals of N2-Mad2R133A due to the formation of the 60 kDa p31comet–Mad2 complex (Figure 4A). In contrast, at a final concentration of 80 μM, addition of p31comet did not alter the pattern of the 1H–15N HSQC spectrum of N1-Mad2R133A, demonstrating that there is no binding even at high concentrations (Figure 4B). Thus, the Kd between p31comet and N1-Mad2 is greater than 80 μM. This in turn indicates that p31comet binds to N2-Mad2 preferably with a selectivity greater than 330-fold.
Figure 4.
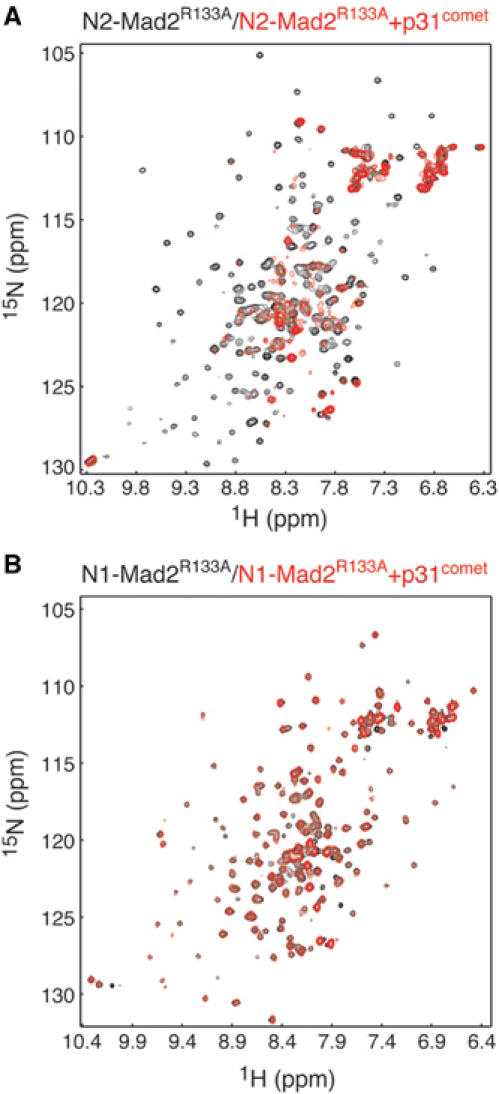
P31comet selectively binds to N2-Mad2 but not to N1-Mad2. (A) Overlay of the 1H–15N HSQC spectra of the free 15N-labeled N2-Mad2R133A (in black) and the mixture of 60 μM 15N-labeled N2-Mad2R133A and 80 μM unlabeled p31comet (in red). (B) Overlay of the 1H–15N HSQC spectra of the free 15N-labeled N1-Mad2R133A (in black) and the mixture of 60 μM 15N-labeled N1-Mad2R133A and 80 μM unlabeled p31comet (in red).
We next tested whether p31comet only interacts with N2-Mad2 in living cells. As the C-terminal region of Mad2 is also required for the N1–N2 structural transition, ΔC-Mad2 exclusively adopts the N1 conformation and cannot undergo the N1–N2 conversion (data not shown). We cotransfected plasmids encoding Myc-tagged Mad2 or ΔC-Mad2 and HA-tagged p31comet into HeLa cells and performed co-immunoprecipitation experiments. As shown in Figure 5A, a significant portion of HA-p31comet was precipitated with anti-Myc beads when the Myc-tagged Mad2 was coexpressed. The Myc-tagged Mad2 protein was also detected in the anti-HA immunoprecipitates when coexpressed with HA-p31comet (Figure 5A). As expected, p31comet did not interact with ΔC-Mad2 in this assay (Figure 5A). The expression levels of Mad2, ΔC-Mad2, and p31comet in the lysates were similar in these samples (Figure 5B). Therefore, p31comet does not interact with ΔC-Mad2 in living cells, presumably due to the inability of ΔC-Mad2 to adopt the N2 conformation.
Figure 5.
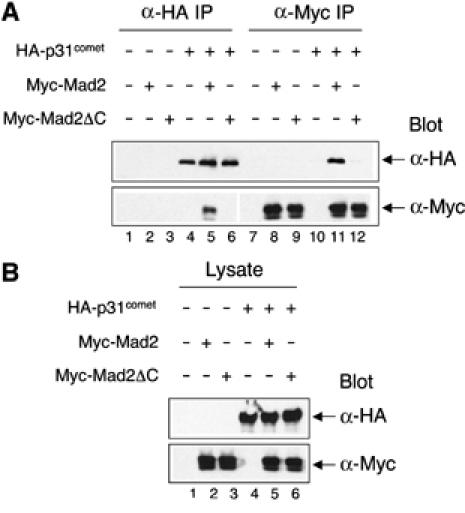
P31comet does not bind to ΔC-Mad2 in living cells. (A) HeLa Tet-on cells were transfected with the indicated plasmids and lysed. The resulting lysates were immunoprecipicated with anti-Myc or anti-HA beads and the immunoprecipitates were then blotted with anti-Myc or anti-HA. (B) The cell lysates in (A) were blotted with anti-Myc or anti-HA.
Cdc20 and p31comet bind to distinct sites on Mad2
As described above, p31comet blocks the ability of Mad2 to inhibit APC/CCdc20 in vitro and in Xenopus egg extracts. P31comet also selectively binds to the N2 conformation of Mad2 with high affinity. The most straightforward model to explain these findings is that p31comet competes with Cdc20 for binding to the N2 conformer of Mad2. We tested this possibility directly. As shown in Figure 6A, the full-length Cdc20 was selectively retained on glutathione–agarose beads bound to GST-Mad2. Surprisingly, addition of His6-tagged p31comet did not affect the binding of Cdc20 (Figure 6A). This suggested that Mad2, Cdc20, and p31comet might transiently form a ternary complex. We also performed the binding experiment in the reverse order. The preformed Mad2–Cdc20 complex also associated with 35S-labeled p31comet (Figure 6B). We next measured the binding affinities among Mad2, p31comet, and Cdc20 using ITC (Figure 6). A Cdc20 peptide (Cdc20P1) containing the Mad2-binding motif of Cdc20 bound to N2-Mad2R133A with a dissociation constant (Kd) of 0.65 μM (Figure 6C and D). Consistent with the qualitative beads pull-down experiments, Cdc20P1 bound to the Mad2–p31comet complex with a similar affinity (Kd=0.69 μM) (Figure 6D). Thus, these data indicate that binding of p31comet to Mad2 does not prevent Mad2 from binding to Cdc20. Mad2, Cdc20, and p31comet can indeed form a ternary complex. This also indicates that p31comet and Cdc20 use distinct binding mode to interact with Mad2, and they bind to different sites on Mad2. It should be mentioned that the Mad2–Cdc20P1 affinity measured in this study was somewhat lower than, but similar to, the affinity between Mad2R133A and a longer Cdc20 peptide (Kd=0.1 μM) reported by Sironi et al (2002). This difference is very likely due to the use of a longer Cdc20 peptide in their experiment (Sironi et al, 2002).
Figure 6.
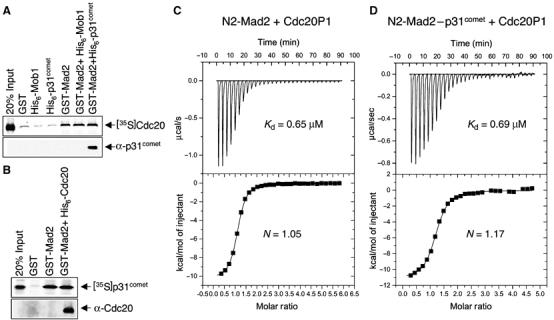
Mad2, Cdc20, and p31comet form a ternary complex in vitro. (A) GST-Mad2 was bound to glutathione–agarose beads and incubated with 35S-labeled full-length human Cdc20 in the presence of 20 μg of His6-tagged Mob1 or p31comet proteins. GST, His6-Mob1, or His6-p31comet alone was used as negative controls and 20% of the input 35S-labeled Cdc20 protein used in the binding reaction was loaded for comparison. After washing, the proteins retained on beads were analyzed by SDS–PAGE followed by autoradiography. The samples were also blotted with α-p31comet. (B) GST-Mad2 or the GST-Mad2–Cdc20 complex was bound to glutathione–agarose beads and incubated with 35S-labeled p31comet. GST was used as a negative control and 20% of the input 35S-labeled p31comet protein used in the binding reaction was loaded for comparison. After washing, the proteins retained on beads were analyzed by SDS–PAGE followed by autoradiography. The samples were also blotted with α-Cdc20. (C) ITC analysis of the binding between N2-Mad2R133A and Cdc20P1. The dissociation constant (Kd) and the stoichiometry (N) of binding are indicated. (D) ITC analysis of the binding between the N2-Mad2R133A–p31comet complex and Cdc20P1. The dissociation constant (Kd) and the stoichiometry (N) of binding are indicated.
P31comet transiently interacts with Mad2–Cdc20-containing complexes during checkpoint inactivation
To determine whether and when Mad2, Cdc20, and p31comet form a ternary complex in vivo, we performed co-immunoprecipitation experiments on endogenous proteins in lysates of HeLa cells undergoing spindle checkpoint inactivation. Treatment of HeLa cells with nocodazole activated the spindle checkpoint and arrested cells in mitosis, as evidenced by the accumulation of cyclin B1 and securin proteins (Figure 7A). The protein levels of cyclin B1 and securin dropped sharply at 2 h following the removal of nocodazole, indicating that these cells recovered from nocodazole-mediated checkpoint arrest and exited from mitosis at this juncture (Figure 7A). Thus, inactivation of the spindle checkpoint occurred between 1 and 2 h after the removal of nocodazole. As p31comet does not bind to Cdc20 directly (data not shown), an interaction between p31comet and Cdc20 in the lysate would indicate the formation of Mad2–Cdc20–p31comet-containing protein complexes. We therefore performed immunoprecipitation with an anti-Cdc20 antibody and blotted the immunoprecipitates with antibodies against p31comet, Mad2, and APC3 (Cdc27) (Figure 7A). Cdc20 formed protein complexes with APC/C and Mad2 in nocodazole-arrested cells (Figure 7C). At 1 h after release from nocodazole arrest, binding between Mad2 and Cdc20 became weaker, whereas the interaction between APC/C and Cdc20 remained constant (Figure 7C, compare lanes 3 and 4). Consistent with our in vitro binding data, p31comet was present in the anti-Cdc20 immunoprecipitates from nocodazole-arrested cells (Figure 7C), indicating that p31comet, Mad2, and Cdc20 can indeed form ternary complexes in vivo. Compared with nocodazole-arrested cells, more p31comet was found in the Cdc20 immunoprecipitates from cells at 1 h after release from nocodazole arrest (Figure 7C, compare lanes 3 and 4). This increased association between Cdc20 and p31comet preceded the activation of APC/C (Figure 7B). This suggests that more p31comet became associated with Cdc20, presumably bridged through Mad2, in cells undergoing checkpoint inactivation. Consistent with earlier findings (Habu et al, 2002), the timing of the indirect binding between p31comet and Cdc20 correlated well with the dissociation of the Mad2–Cdc20-containing protein complexes. These data suggest that p31comet transiently interacts with checkpoint protein complexes containing Mad2, Cdc20, and APC/C during the inactivation of the spindle checkpoint. It is worth mentioning that p31comet also forms a ternary complex with Mad1 and Mad2 (data not shown). However, the Mad1–Mad2–p31comet interaction does not appear to be regulated during the cell cycle (data not shown).
Figure 7.
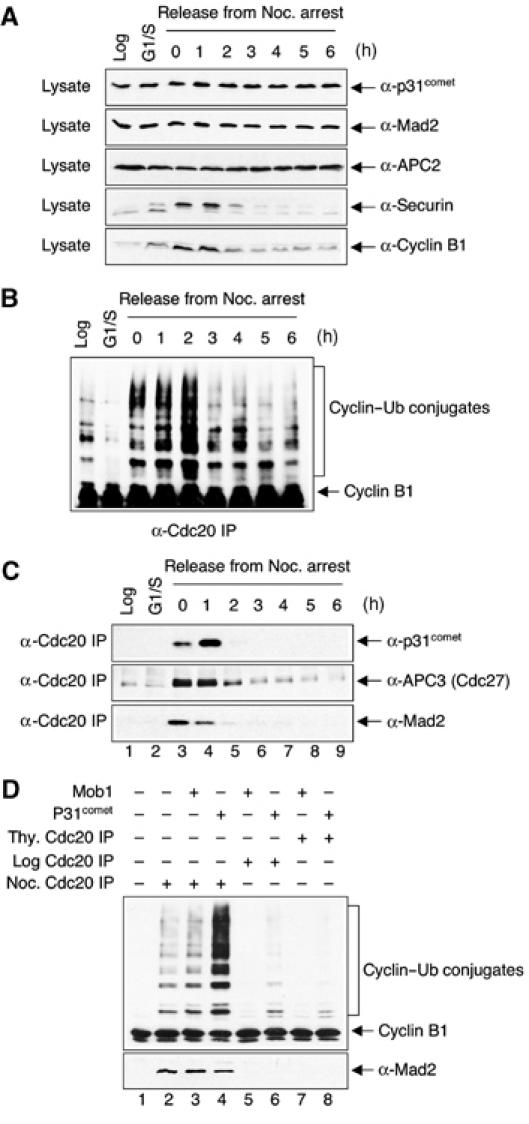
P31comet transiently associates with the APC/C–Cdc20–Mad2 complex during checkpoint inactivation in HeLa cells. (A) HeLa cells were treated with 100 ng/ml nocodazole for 18 h and released into fresh medium. The total cell lysates of log-phase cells (Log), cells arrested at the G1/S boundary with thymidine (G1/S), and cell samples taken at the indicated time points after release from the nocodazole-mediated mitotic arrest were separated on SDS–PAGE and blotted with the indicated antibodies. (B) HeLa cells were treated with 100 ng/ml nocodazole for 18 h and released into fresh medium. Samples were taken at the indicated time points after release from the nocodazole-mediated mitotic arrest. APC/CCdc20 was immunoprecipitated with α-Cdc20 beads from lysates of log-phase cells (Log), cells arrested at the G1/S boundary with thymidine (G1/S), and cell samples after release from the nocodazole-mediated mitotic arrest and assayed for cyclin B1 ubiquitination activity. (C) The same cell samples as in (A) were lysed with the NP-40 lysis buffer and immunoprecipitated with anti-Cdc20 antibody. The immunoprecipitates were separated by SDS–PAGE and blotted with indicated antibodies. (D) Cdc20 was immunoprecipitated from log-phase (lanes 5 and 6), thymidine-treated (lanes 7 and 8), or nocodazole-treated (lanes 2–4) HeLa cell lysates. The immunoprecipitates were incubated with purified recombinant p31comet (4 μM) or Mob1 (4 μM) proteins. After washing, the beads were assayed for cyclin B1 ubiquitination activity (top panel) and blotted with anti-Mad2 (bottom panel). Anti-Mob1 immunoprecipitates from the nocodazole-treated cell lysate were included as the negative control (lane 1).
P31comet enhances the activity of Mad2-inhibited APC/C from HeLa cells without disrupting the Mad2–Cdc20 interaction
On the one hand, we have clearly demonstrated that p31comet binding to Mad2 does not interfere with the binding between Mad2 and Cdc20. Instead, Mad2, Cdc20, and p31comet can form a ternary complex in vitro and in vivo. On the other hand, it is also clear that addition of recombinant purified p31comet protein to the in vitro APC/C ubiquitination assay partially reverses the inhibition of APC/CCdc20 by Mad2. This suggests that p31comet forms a transient complex with APC/C–Cdc20–Mad2 to restore the activity of APC/CCdc20. To test this hypothesis more directly, we immunoprecipitated Cdc20 from log-phase, thymidine-treated, or nocodazole-treated HeLa cells. As shown in Figure 7C, the Cdc20 immunoprecipitates from log-phase or thymidine-treated cells did not contain detectable amounts of APC/C, whereas the Cdc20 immunoprecipitates from nocodazole-arrested cells contained APC/C, Mad2, and a small amount of p31comet. Consistent with earlier reports (Fang et al, 1998a), APC/C isolated from nocodazole-arrested HeLa cells was largely inactive (Figure 7D, lane 2). Addition of purified recombinant p31comet protein greatly enhanced the ubiquitin ligase activity of APC/C (Figure 7D, lanes 2 and 4), while addition of the irrelevant Mob1 protein did not significantly affect the APC/C activity (Figure 7D, lanes 2 and 3). Interestingly, after incubation with p31comet, the amount of Mad2 bound to Cdc20 was only slightly reduced (Figure 7D, bottom panel, lane 4). Therefore, p31comet alone is not sufficient to break up the Mad2–Cdc20 interaction. It forms a transient complex with APC/C, Cdc20, and Mad2 to stimulate the activity of APC/C.
Discussion
Biochemical and genetic data have established a critical role of Mad2 in the spindle checkpoint. For example, inactivation of Mad2 in mammalian cells through gene deletion, antibody injection, or RNAi abolishes the checkpoint, causing chromosome mis-segregation, premature onset of anaphase, and the inability of these cells to undergo mitotic arrest in the presence of nocodazole (Gorbsky et al, 1998; Dobles et al, 2000; Michel et al, 2001; Luo et al, 2004). Furthermore, yeast mutant cells harboring mutations in the Mad2-binding motif of Cdc20 have a defective spindle checkpoint and fail to arrest in the presence of spindle-damaging agents (Hwang et al, 1998; Kim et al, 1998). Overexpression of a Cdc20 mutant with its Mad2-binding motif mutated in HeLa cells promotes mitotic exit in the presence of nocodazole more efficiently than does the ectopic expression of the wild-type Cdc20 (data not shown). These results indicate that the Mad2–Cdc20 interaction is essential for the proper execution of the spindle checkpoint. Consistent with this notion, the Mad2–Cdc20 interaction is enhanced in checkpoint-active cells (Fang et al, 1998a; Zhang and Lees, 2001). It is not exactly clear how the spindle checkpoint promotes the formation of Mad2–Cdc20 checkpoint complexes. According to one model, upon checkpoint activation, Mad2 is recruited to the unattached kinetochores via the Mad1–Mad2 interaction (Yu, 2002). As Cdc20 is also localized at the kinetochores, the close proximity of Mad2 and Cdc20 might promote the formation of the Mad2–Cdc20 checkpoint complexes (Yu, 2002). Mad1 might also convert Mad2 to a conformation more amenable for Cdc20 binding, thus facilitating the interaction between Mad2 and Cdc20 (Yu, 2002). Consistent with this notion, we have recently shown that Mad2 exists in two conformations, one of which (N2-Mad2) is more active in inhibiting APC/CCdc20 (Luo et al, 2004). Mad1 facilitates the formation of the N2 conformer of Mad2 in vitro. Binding of Mad2 to Cdc20, either as a binary complex or in the context of a larger MCC, leads to the inhibition of APC/C (Fang et al, 1998a; Sudakin et al, 2001; Tang et al, 2001; Fang, 2002; Yu, 2002).
Little is known about the mechanism of checkpoint inactivation and the disassembly of the Mad2–Cdc20-containing checkpoint complexes (Yu, 2002). Mad2 only localizes to unattached kinetochores (Howell et al, 2000; Hoffman et al, 2001). It is thus possible that, upon microtubule attachment and the establishment of kinetochore tension, the absence of Mad2 at the kinetochores decreases the efficiency of the formation of the Mad2–Cdc20 checkpoint complexes. The dissociation of these complexes may then lead to the activation of APC/CCdc20. It is unclear whether the breakup of these checkpoint complexes is a spontaneous process or is assisted by an active mechanism. However, the existence of an active mechanism is suggested by recent structural studies. The Mad2-binding region of Cdc20 inserts as a central strand in a large β sheet (Luo et al, 2000, 2002; Sironi et al, 2002). The subsequent structural rearrangement of the C-terminal region of Mad2 then locks in the Mad2-binding region of Cdc20 (Luo et al, 2000, 2002; Sironi et al, 2002). The dissociation of the Mad2–Cdc20 complex requires the partial unfolding of Mad2, that is, the unfolding of strands β8 and β9 (Luo et al, 2002; Sironi et al, 2002). This allows the C-terminal region of Mad2 to flip open, thus releasing Cdc20. This mode of Mad2–ligand binding has been referred to as the ‘safety belt' mechanism (Sironi et al, 2002). As the partial unfolding of Mad2 imposes a kinetic barrier on the breakup of the Mad2–Cdc20 complex, this mechanism may prolong the lifetime of the Mad2-containing checkpoint complexes. On the other hand, the unassisted breakup of the Mad2–Cdc20 complexes might be too slow for the inactivation of the spindle checkpoint and the onset of anaphase during the normal cell cycle. This suggests that the disassembly of the inhibitory checkpoint complexes might be assisted by active mechanisms.
P31comet might be a part of one such active mechanism for checkpoint inactivation for the following observations presented in this study (Figure 8). First, RNAi-mediated depletion of p31comet in HeLa cells slowed down the kinetics of mitotic exit following the removal of spindle-damaging agents. This indicates that p31comet is required for checkpoint inactivation. Second, p31comet negatively regulates the function of Mad2 in the spindle checkpoint in vitro and in vivo, consistent with Mad2 being the critical target of p31comet during checkpoint inactivation. Third, p31comet selectively recognizes and binds to the Cdc20-bound conformation of Mad2. This striking behavior of p31comet lends strong support to the notion that p31comet is responsible for the inactivation of Mad2 after termination of the checkpoint signal. Third, p31comet transiently interacts with Mad2–Cdc20–APC/C-containing protein complexes in checkpoint-active cells, and this interaction is enriched during mitotic exit. Finally, purified p31comet partially activates APC/CCdc20 isolated from nocodazole-arrested HeLa cells. These results support a role of p31comet in the activation of Mad2-inhibited APC/CCdc20 during spindle checkpoint silencing (Figure 8).
Figure 8.
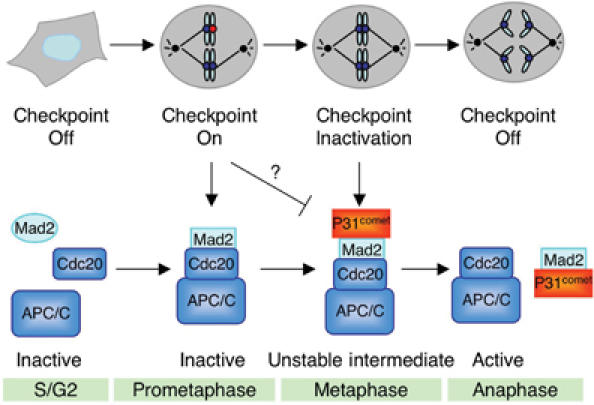
Different forms of APC/C during checkpoint activation and inactivation and the proposed role of p31comet (see Discussion for details).
P31comet is not sufficient to break up the Mad2–Cdc20 interaction. It is possible that p31comet is not directly involved in the disassembly of the Mad2–Cdc20-containing spindle checkpoint complexes. It may simply offset the APC/C-inhibitory effect of Mad2 through the formation of APC/C–Cdc20–Mad2–p31comet complex. The degradation of securin and cyclin B1 then initiates a cascade of events that ultimately lead to the disassembly of the checkpoint complexes. However, it is equally possible that p31comet does play a rather direct role in the breakup of the Mad2–Cdc20 complexes. Although not sufficient, it might be required for this process. Conceivably, p31comet might collaborate with other cofactors, such as chaperones, to disassemble Mad2–Cdc20 complexes. In the future, it will be interesting to identify additional p31comet-interacting proteins. Given its remarkable binding selectivity toward N2-Mad2, it will also be interesting to use p31comet as a ‘sensor' to monitor the formation of the Mad2-containing checkpoint complexes in living cells.
Materials and methods
Tissue culture, RNAi, flow cytometry, and microscopy
HeLa Tet-on cells (Clontech) were grown in Dulbecco's modified Eagle's medium (DMEM; Invitrogen) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and 100 μg/ml penicillin and streptomycin. At 40–50% confluency, the cells were transfected with the appropriate siRNA oligonucleotides. The p31comet-siRNA oligonucleotides were chemically synthesized and contained sequences corresponding to nucleotides 108–130 of the p31comet coding region. The siRNA duplex corresponding to nucleotides 648–670 of human Mst1 gene was used as a control. The Mst1 siRNA did not result in any reduction of the protein levels of either Mst1 or p31comet. The annealing of the siRNAs and subsequent transfection of the RNA duplexes into HeLa cells were performed exactly as described (Elbashir et al, 2001). After 24 h of transfection, cells were treated with 100 ng/ml nocodazole for 16 h and released into fresh medium. Samples were taken at indicated time points, fixed with 70% ethanol, stained with propidium iodide, and analyzed by flow cytometry (FACS). A portion of the cells were dissolved directly in SDS sample buffer, sonicated, boiled, and separated by SDS–PAGE followed by immunoblotting with antibodies against APC2, cyclin B1 (Santa Cruz), and securin. To determine the mitotic index, the transfected cells treated with various concentrations of nocodazole were stained with Hoechst 33342, and directly observed with a Zeiss Axiovert 200M inverted fluorescence microscope using a × 40 objective. Round cells with condensed DNA were counted as mitotic cells.
Immunoprecipitation and immunoblotting
HeLa Tet-on cells were transfected with pCS2-HA-p31comet, pCS2-Myc-Mad2, or pCS2-Myc-ΔC-Mad2 using the Effectene reagent (Qiagen) according to the manufacturer's protocols. Cells were lysed with the NP-40 lysis buffer (50 mM Tris–HCl, pH 7.7, 150 mM NaCl, 0.5% NP-40, 1 mM DTT, 10% glycerol, 0.5 μM okadaic acid, and 10 μg/ml each of leupeptin, pepstatin, and chymostatin). The lysates were cleared by centrifuging for 30 min at 4°C at top speed in a microcentrifuge to make the high-speed supernatants. Anti-HA and anti-Myc monoclonal antibodies (Boehringer Mannheim) were covalently coupled to Affi-Prep Protein A beads (Bio-Rad) and incubated with the HeLa cell supernatants for 2 h at 4°C. The beads were then washed five times with the NP-40 lysis buffer. The proteins bound to the beads were dissolved in SDS sample buffer, separated by SDS–PAGE, and blotted with anti-HA or anti-Myc antibodies at a concentration of 1 μg/ml. For immunoprecipitation of the endogenous proteins, the supernatants of HeLa Tet-on cells at various cell cycle stages were obtained as described above. Affinity-purified anti-Cdc20 polyclonal antibodies were covalently coupled to Affi-Prep Protein A beads (Bio-Rad) and incubated with the HeLa cell supernatants for 2 h at 4°C. The beads were then washed five times with the NP-40 lysis buffer. The proteins bound to the beads were dissolved in SDS sample buffer, separated by SDS–PAGE, and blotted with antibodies against APC3 (Cdc27), p31comet, or Mad2.
Expression and purification of recombinant p31comet, Mad2, Mob1, and Cdc20 proteins
The coding region of p31comet was cloned into pQE30 (Qiagen) and the resulting plasmid was transformed into the Escherichia coli strain M15[pREP4] for protein expression. The coding region of human Mob1 was cloned into pET28 (Novagen) and the resulting plasmid was transformed into BL21(DE3)pLysS for protein expression. The N-terminally His6-tagged p31comet and Mob1 proteins were isolated on Ni2+-NTA beads (Qiagen) and further purified on a Superdex 75 gel filtration column (Amersham). The recombinant wild-type Mad2, Mad2R133A, and ΔC-Mad2 proteins were expressed and purified as described (Luo et al, 2004). In addition, the coding region of Mad2 was also subcloned into pGEX-4T1 and the resulting plasmid was transformed into BL21. The resulting GST-Mad2 protein was purified using glutathione–agarose beads (Amersham). For the production of human Cdc20 protein, a recombinant baculovirus encoding Cdc20 fused at its N-terminus with a His6 tag was constructed using the Bac-to-Bac system (Invitrogen). Expression and purification of Cdc20 in Sf9 cells were performed as per the manufacturer's protocols.
In vitro protein binding, ubiquitination, and degradation assays
To assay the binding between human Mad2, p31comet, Mad1, and Cdc20 proteins, the C-terminal fragment of Mad1 (residues 481–718; Mad1C), the N-terminal fragment of Cdc20 (residues 1–174; Cdc20N), and p31comet were translated in reticulocyte lysate in the presence of [35S]methionine. Purified His6-tagged Mad2 proteins in either N1 or N2 form were bound to Ni2+-NTA beads (Qiagen), incubated with 35S-labeled Mad1C, Cdc20N, or p31comet proteins, and washed three times with TBS containing 0.05% Tween. The proteins retained on the beads were analyzed by SDS–PAGE followed by autoradiography. To assay the binding among Mad2, p31comet, and Cdc20, GST-Mad2 fusion protein was bound to glutathione–agarose beads, incubated with 35S-labeled full-length Cdc20 protein in the presence of His6-tagged p31comet or Mob1 proteins, and washed three times with TBS containing 0.05% Tween. Glutathione–agarose beads bound with GST were used as controls. The proteins retained on the beads were analyzed by SDS–PAGE followed by autoradiography. Alternatively, GST-Mad2 fusion protein was bound to glutathione–agarose beads and incubated with His6-Cdc20 protein expressed and purified from Sf9 cells. After washing, the beads were incubated with 35S-labeled p31comet and washed three times with TBS containing 0.05% Tween. The proteins retained on the beads were analyzed by SDS–PAGE followed by autoradiography. Cyclin ubiquitination and degradation assays were performed as described (Tang et al, 2001).
Isothermal titration calorimetry (ITC) and NMR spectroscopy
ITC was performed with a VP-ITC titration calorimeter (MicroCal Inc.) at 20°C. Calorimetric measurements were carried out with protein samples (p31comet and the N2 form of Mad2R133A) purified as described above and a synthetic peptide corresponding to residues 124–136 of human Cdc20. For each titration experiment, 2 ml of 10–20 μM protein solution (Mad2R133A, p31comet, or the p31comet–Mad2R133A complex) in a buffer containing 50 mM NaH2PO4, pH 6.8, 300 mM KCl, and 0.5 mM TCEP was added to the calorimeter cell. The Cdc20 peptide (0.4–0.6 mM) or the Mad2R133A protein (0.2 mM) in the exact same buffer was injected with 30 portions of 8 μl with an injection syringe. Binding parameters were evaluated using the Origin software package provided with the instrument. NMR experiments were carried out at 30°C on a Varian Inova 600 MHz spectrometer equipped with four channels and pulsed field gradients. Samples contained 80 μM of unlabeled p31comet and 60 μM of 15N-labeled N1-Mad2R133A or N2-Mad2R133A in the NMR buffer (50 mM phosphate, pH 6.8, 300 mM KCl, and 1 mM DTT).
Acknowledgments
We thank Hui Zou for anti-securin antibody and Minghua Wen for assistance with protein expression. HY is the Michael L Rosenberg Scholar in Biomedical Research. This work was supported by the National Institutes of Health (GM61542 to HY and a KO1 award to XL), the Robert A Welch Foundation (to HY and JR), the Ministry of Education, Culture, Sports, Science and Technology of Japan (COE Research) (to TM), the Packard Foundation (to HY), and the WM Keck Foundation (to HY).
References
- Bharadwaj R, Yu H (2004) The spindle checkpoint, aneuploidy, and cancer. Oncogene 23: 2016–2027 [DOI] [PubMed] [Google Scholar]
- Chen RH, Shevchenko A, Mann M, Murray AW (1998) Spindle checkpoint protein Xmad1 recruits Xmad2 to unattached kinetochores. J Cell Biol 143: 283–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung E, Chen RH (2002) Spindle checkpoint requires Mad1-bound and Mad1-free Mad2. Mol Cell Biol 13: 1501–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobles M, Liberal V, Scott ML, Benezra R, Sorger PK (2000) Chromosome missegregation and apoptosis in mice lacking the mitotic checkpoint protein Mad2. Cell 101: 635–645 [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411: 494–498 [DOI] [PubMed] [Google Scholar]
- Fang G (2002) Checkpoint protein BubR1 acts synergistically with Mad2 to inhibit anaphase-promoting complex. Mol Cell Biol 13: 755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G, Yu H, Kirschner MW (1998a) The checkpoint protein MAD2 and the mitotic regulator CDC20 form a ternary complex with the anaphase-promoting complex to control anaphase initiation. Genes Dev 12: 1871–1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G, Yu H, Kirschner MW (1998b) Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol Cell 2: 163–171 [DOI] [PubMed] [Google Scholar]
- Fraschini R, Beretta A, Sironi L, Musacchio A, Lucchini G, Piatti S (2001) Bub3 interaction with Mad2, Mad3 and Cdc20 is mediated by WD40 repeats and does not require intact kinetochores. EMBO J 20: 6648–6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbsky GJ (2001) The mitotic spindle checkpoint. Curr Biol 11: R1001–R1004 [DOI] [PubMed] [Google Scholar]
- Gorbsky GJ, Chen RH, Murray AW (1998) Microinjection of antibody to Mad2 protein into mammalian cells in mitosis induces premature anaphase. J Cell Biol 141: 1193–1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habu T, Kim SH, Weinstein J, Matsumoto T (2002) Identification of a Mad2-binding protein, CMT2, and its role in mitosis. EMBO J 21: 6419–6428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardwick KG, Johnston RC, Smith DL, Murray AW (2000) MAD3 encodes a novel component of the spindle checkpoint which interacts with Bub3p, Cdc20p, and Mad2p. J Cell Biol 148: 871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DB, Pearson CG, Yen TJ, Howell BJ, Salmon ED (2001) Microtubule-dependent changes in assembly of microtubule motor proteins and mitotic spindle checkpoint proteins at PtK1 kinetochores. Mol Cell Biol 12: 1995–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BJ, Hoffman DB, Fang G, Murray AW, Salmon ED (2000) Visualization of Mad2 dynamics at kinetochores, along spindle fibers, and at spindle poles in living cells. J Cell Biol 150: 1233–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang LH, Lau LF, Smith DL, Mistrot CA, Hardwick KG, Hwang ES, Amon A, Murray AW (1998) Budding yeast Cdc20: a target of the spindle checkpoint. Science 279: 1041–1044 [DOI] [PubMed] [Google Scholar]
- Kim SH, Lin DP, Matsumoto S, Kitazono A, Matsumoto T (1998) Fission yeast Slp1: an effector of the Mad2-dependent spindle checkpoint. Science 279: 1045–1047 [DOI] [PubMed] [Google Scholar]
- Kramer ER, Gieffers C, Holzl G, Hengstschlager M, Peters JM (1998) Activation of the human anaphase-promoting complex by proteins of the CDC20/Fizzy family. Curr Biol 8: 1207–1210 [DOI] [PubMed] [Google Scholar]
- Li X, Nicklas RB (1995) Mitotic forces control a cell-cycle checkpoint. Nature 373: 630–632 [DOI] [PubMed] [Google Scholar]
- Li Y, Gorbea C, Mahaffey D, Rechsteiner M, Benezra R (1997) MAD2 associates with the cyclosome/anaphase-promoting complex and inhibits its activity. Proc Natl Acad Sci USA 94: 12431–12436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Fang G, Coldiron M, Lin Y, Yu H, Kirschner MW, Wagner G (2000) Structure of the Mad2 spindle assembly checkpoint protein and its interaction with Cdc20. Nat Struct Biol 7: 224–229 [DOI] [PubMed] [Google Scholar]
- Luo X, Tang Z, Rizo J, Yu H (2002) The Mad2 spindle checkpoint protein undergoes similar major conformational changes upon binding to either Mad1 or Cdc20. Mol Cell 9: 59–71 [DOI] [PubMed] [Google Scholar]
- Luo X, Tang Z, Xia G, Wassmann K, Matsumoto T, Rizo J, Yu H (2004) The Mad2 spindle checkpoint protein has two distinct natively folded states. Nat Struct Mol Biol 11: 338–345 [DOI] [PubMed] [Google Scholar]
- Michel LS, Liberal V, Chatterjee A, Kirchwegger R, Pasche B, Gerald W, Dobles M, Sorger PK, Murty VV, Benezra R (2001) MAD2 haplo-insufficiency causes premature anaphase and chromosome instability in mammalian cells. Nature 409: 355–359 [DOI] [PubMed] [Google Scholar]
- Millband DN, Campbell L, Hardwick KG (2002) The awesome power of multiple model systems: interpreting the complex nature of spindle checkpoint signaling. Trends Cell Biol 12: 205–209 [DOI] [PubMed] [Google Scholar]
- Millband DN, Hardwick KG (2002) Fission yeast Mad3p is required for Mad2p to inhibit the anaphase-promoting complex and localizes to kinetochores in a Bub1p-, Bub3p-, and Mph1p-dependent manner. Mol Cell Biol 22: 2728–2742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musacchio A, Hardwick KG (2002) The spindle checkpoint: structural insights into dynamic signalling. Nat Rev Mol Cell Biol 3: 731–741 [DOI] [PubMed] [Google Scholar]
- Nasmyth K (2002) Segregating sister genomes: the molecular biology of chromosome separation. Science 297: 559–565 [DOI] [PubMed] [Google Scholar]
- Nicklas RB (1997) How cells get the right chromosomes. Science 275: 632–637 [DOI] [PubMed] [Google Scholar]
- Peters JM (2002) The anaphase-promoting complex. Proteolysis in mitosis and beyond. Mol Cell 9: 931–943 [DOI] [PubMed] [Google Scholar]
- Rieder CL, Cole RW, Khodjakov A, Sluder G (1995) The checkpoint delaying anaphase in response to chromosome monoorientation is mediated by an inhibitory signal produced by unattached kinetochores. J Cell Biol 130: 941–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudner AD, Murray AW (1996) The spindle assembly checkpoint. Curr Opin Cell Biol 8: 773–780 [DOI] [PubMed] [Google Scholar]
- Sironi L, Mapelli M, Knapp S, Antoni AD, Jeang KT, Musacchio A (2002) Crystal structure of the tetrameric Mad1-Mad2 core complex: implications of a ‘safety belt' binding mechanism for the spindle checkpoint. EMBO J 21: 2496–2506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sironi L, Melixetian M, Faretta M, Prosperini E, Helin K, Musacchio A (2001) Mad2 binding to Mad1 and Cdc20, rather than oligomerization, is required for the spindle checkpoint. EMBO J 20: 6371–6382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudakin V, Chan GK, Yen TJ (2001) Checkpoint inhibition of the APC/C in HeLa cells is mediated by a complex of BUBR1, BUB3, CDC20, and MAD2. J Cell Biol 154: 925–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Bharadwaj R, Li B, Yu H (2001) Mad2-independent inhibition of APCCdc20 by the mitotic checkpoint protein BubR1. Dev Cell 1: 227–237 [DOI] [PubMed] [Google Scholar]
- Wassmann K, Benezra R (1998) Mad2 transiently associates with an APC/p55Cdc complex during mitosis. Proc Natl Acad Sci USA 95: 11193–11198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassmann K, Benezra R (2001) Mitotic checkpoints: from yeast to cancer. Curr Opin Genet Dev 11: 83–90 [DOI] [PubMed] [Google Scholar]
- Yu H (2002) Regulation of APC-Cdc20 by the spindle checkpoint. Curr Opin Cell Biol 14: 706–714 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lees E (2001) Identification of an overlapping binding domain on Cdc20 for Mad2 and anaphase-promoting complex: model for spindle checkpoint regulation. Mol Cell Biol 21: 5190–5199 [DOI] [PMC free article] [PubMed] [Google Scholar]