

Abstract
Optogenetics provides an array of elements for specific biophysical control, while designer chemogenetic receptors provide a minimally invasive method to control circuits in vivo by peripheral injection. We developed a strategy for selective regulation of activity in specific cells that integrates opto- and chemogenetic approaches, and thus allows manipulation of neuronal activity over a range of spatial and temporal scales in the same experimental animal. Light-sensing molecules (opsins) are activated by biologically produced light through luciferases upon peripheral injection of a small molecule substrate. Such luminescent opsins, luminopsins, allow conventional fiber optic use of optogenetic sensors, while at the same time providing chemogenetic access to the same sensors. We describe applications of this approach in cultured neurons in vitro, in brain slices ex vivo, and in awake and anesthetized animals in vivo.
Keywords: Luminopsin, Luciferase, Bioluminescence, Coelenterazine, Optogenetics, Chemogenetics, Neuron, Electrophysiology, Multielectrode array, Behavior
1 Introduction
Currently, there are two major approaches to control activity of genetically defined neurons in the brain of freely behaving animals: chemogenetic approaches that utilize diffusible small molecules [1] and optogenetic approaches that utilize externally delivered light [2]. The two methods have their own distinct merits; optogenetics offers the advantage of temporal precision while chemogenetic approaches offer scalability and ease of application. Combining these two approaches within single molecules complements each other and allows the use of either mode of interrogation in the same brain circuit. Such molecular actuators would allow acute activation with precise time resolution in defined spaces, as well as chronic and noninvasive control of entire populations throughout the brain through the same molecules.
Luminopsins (luminescent opsins or LMOs) were developed to achieve combined chemo- and optogenetic manipulation [3]. They are fusion proteins of a light-emitting luciferase and a light-sensing opsin (see Fig. 1). Application of the luciferase substrate, coelenterazine (CTZ), leads to emission of photons that is sufficient to activate the coupled opsin. We established proof of concept of this technology by using fusion proteins that directly link Gaussia luciferase (GLuc; [4, 5]) to Chlamydomonas channelrhodopsin 2 (ChR2; [6]) or Volvox channelrhodopsin 1 (VChR1; [7]). As for inhibition, we were able to harness bioluminescence from engineered Renilla luciferase (Nano-lantern; [8]) for activation of Natronomonas halorhodopsin [9] (inhibitory luminopsin or iLMO). When these fusion proteins are expressed in neurons, bright bioluminescence from GLuc and Nano-lantern was able to excite or silence a neuronal population, respectively, in cultured neurons in vitro, in brain slices ex vivo, and in awake and anesthetized animals upon application of CTZ. Moreover LMO and iLMO were able to elicit specific motor behavior in awake animals in vivo [unpublished results; also see Society for Neuroscience Meeting Abstracts 2013 (Tung et al., Berglund et al.) and 2014 (Clissold et al., Higashikubo et al.)].
Fig. 1.
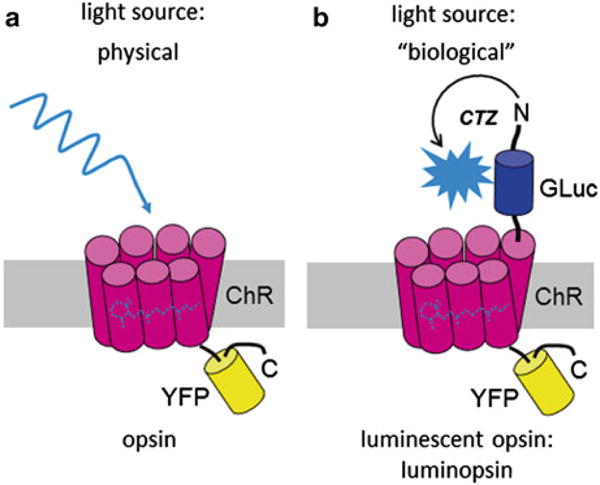
Combined opto- and chemogenetic control with luminopsins. (a) An optogenetic element (here: channelrhodopsin, ChR) can be activated by a physical light source (LED, laser, arc lamp). (b) The same optogenetic element, when fused to a luciferase (here: Gaussia luciferase, Gluc) by a 15-amino acid linker, can also be activated by “biological” light, which is produced when the attached luciferase catalyzes oxidation of the supplied substrate coelenterazine (CTZ). Fused to the C-terminus of the optogenetic element is the fluorescent reporter YFP, allowing identification of cells expressing the element. The optogenetic element can be a channel or a pump, and depending on its biophysical properties can activate (LMO) or inhibit (iLMO) a neuron
LMOs retain the capability for conventional optogenetic control of neuronal activity. Light from a physical source (e.g. arc lamp, laser, LED) through optical fibers can activate the opsin moiety similar to when they are expressed by themselves without luciferase. As chemogenetic probes, however, LMOs have characteristics distinct from conventional chemogenetic probes such as DREADDs [10]. First, chemogenetic access is mediated by opsins instead of G-protein coupled receptors. Thus, there are no requirements for additional signaling pathways for LMOs to function. Second, while there is commonly one designer receptor each for activation and silencing, for example Gq and Gi DREADDs, respectively, LMOs can capitalize on the entire molecular palette of optogenetic actuators, which have a wide range of kinetics and sensitivity, and which can be matched with luciferases with a wide variety of emission spectra. Third, while there is a single designer drug (CNO) for activating both Gq and Gi DREADDs, different luciferases utilize different substrates, making multiplexing feasible. For example, hCTZ, a 2-deoxy derivative of native coelenterazine, which we use as a substrate for our RLuc-based inhibitory LMO (iLMO), cannot be used by Gaussia luciferase, which only emits light with native CTZ. Lastly, the approach is unique in that LMOs integrate opto- and chemogenetic approaches, and thus allow manipulation of neuronal activity over a range of spatial and temporal scales in the same experimental animal.
In this chapter, we will detail critical aspects of working with LMOs for combined opto- and chemogenetic manipulation of neuronal activity. We will address handling and application of the luciferase substrate, simultaneous bioluminescence imaging and electrophysiological and behavioral readouts in vitro and in vivo using LMOs.
2 Materials
Standard equipment and materials used for the application of interest will suffice (tissue culture, single-cell patch clamp recording, multielectrode array recording, brain slice recording, in vivo electrophysiology, behavioral testing). Detailed below are materials we routinely use for tissue culture experiments, and supplies and procedures for preparing the luciferase substrate.
2.1 Cell Culture
Tissue culture plasticware (4-well plates, 24-well plates, 12-well plates).
12 or 18 mm poly-D-lysine-coated glass coverslips.
293T human embryonic kidney fibroblasts.
Rat embryonic cortical tissue for preparing primary neurons.
suppDMEM: Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % fetal bovine serum, 1× Nonessential Amino Acids, 2 mM GlutaMax, 100 U penicillin and 0.1 mg streptomycin per milliliter. Store at 4 °C. Warm to at least room temperature before use.
Serum-NB: Neurobasal Medium (NB) containing 1× B27, 2 mM Glutamax, and 5 % FBS (medium for neuron plating) or 2 % FBS (medium for MEA cultures).
Plain-NB: Neurobasal Medium (NB) containing 1× B27, 2 mM Glutamax without any serum.
All-trans retinal: Prepare, in a dark environment, a 10 mM stock by dissolving 25 mg in 8.8 ml ETOH, aliquot, and store at −80 °C. As needed, prepare a 1:10 dilution in PBS to obtain a working stock of 1 mM, which can be kept at 4 °C and protected from light for a few days. Add to the culture medium directly or after further dilution for a final concentration of 1 μM.
2.2 Coelenterazine
Coelenterazine (CTZ), native as well as analogs (hCTZ, eCTZ, etc.), can be commercially obtained from several sources (Note: they differ in purity).
!CTZ should always be protected from light!
2.3 Solvents for Coelenterazine
Acidified alcohol (ethanol or methanol): 0.06 N HCl in alcohol; add 0.2 ml 3 N HCl to 10 ml alcohol.
Cyclodextrin: CTZ can be solubilized in (2-Hydroxypropyl) β-cyclodextrin, as originally described by Teranishi and Shimomura [11], and used by Naumann et al. in live zebrafish [12]. The amount of cyclodextrin used depends on the amount of CTZ being complexed and can be determined from ref. 11, Fig. 3. After dissolving cyclodextrin in PBS, filter-sterilize and add to CTZ dissolved in a small volume of ethanol. We typically dissolve 250 μg CTZ in 10 μl of ethanol and dilute it with 500 μl of 20 mM β-cyclodextrin to reach a stock concentration of 600 μM CTZ. Stocks are then kept frozen and protected from light until they are ready for use. On the day of the experiment, a stock aliquot is thawed and diluted with aqueous solvent (PBS, saline) to the desired concentration.
NanoFuel Solvent (NanoLight Technology): a proprietary solvent for coelenterazine. Native coelenterazine can be dissolved in a ten times higher concentration compared to 100 % ethanol or methanol. In our experience, NanoFuel-dissolved CTZ emits more bioluminescence than ethanol-dissolved CTZ when applied at the same final concentration. We dissolve CTZ at 50 mM, a concentration higher than the company’s recommendation, to minimize the amount of solvent used and avoid possible unwanted side effects. Native CTZ is dissolved in the solvent (23.6 μl solvent for 500 μg CTZ, resulting in a 50 mM solution) at room temperature. Pipette up and down and vortex to insure complete dissolving. The stock solution is stored in the same tube with a tightly closed lid at −80 °C for continued use up to several months. In the morning of the day of experiments, we make small aliquots of concentrated CTZ (typically 1 μl) and keep them on ice. Just before application, we dilute it with a buffer of choice (HEPES-buffered saline or PBS) to a specified final concentration (typically 1:500 for a 100 μM solution). Unlike the stock solution, CTZ diluted in a buffer can only last several hours at room temperature. Loss of typical yellowish color in a buffer indicates auto-oxidation and loss of activity. The solvent is also compatible with many other coelenterazine analogs that have become available in recent years.
Inject-A-Lume (NanoLight Technology): injectable CTZ specifically designed for in vivo use. Coelenterazine comes in sterile injection vials with a low retention volume and with sterile Fuel-Inject diluent. Fuel-Inject allows one to dissolve CTZ at high concentrations without precipitating CTZ. Inject-a-Lume is available for native CTZ, hCTZ, and eCTZ. Inject-A-Lume is typically stored in the freezer and warmed up prior to use.
Water-soluble coelenterazine (NanoLight Technology): CTZ (native or h) has nontoxic additives for water-solubility and is formulated to be isosmotic and easily secreted by the kidneys. It is safe for repeated intravenous injections and at high concentrations (up to 500 μg/100 μl) achieving very high coelenterazine levels within the body and leading to high bioluminescence signals [13].
Fig. 3.
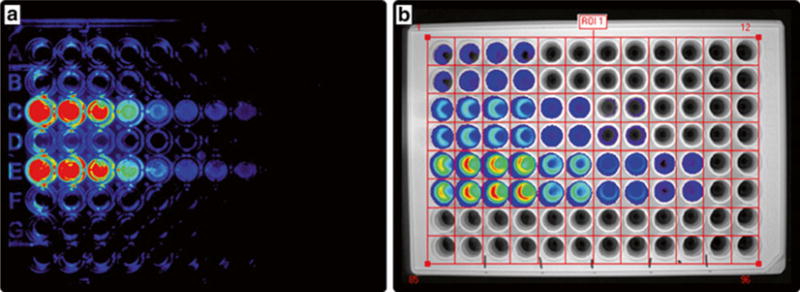
Bioluminescense imaging in 96-well plate format. HEK cells (50,000 per well) transfected with LMOs were plated. Forty-eight hours later bioluminescence intensities were measured after adding different concentrations of CTZ (1–100 μM final concentrations). Images were taken with a LiCor Odyssee Fc using the chemiluminescence channel (a) and with a Caliper IVIS Kinetic system for in vivo imaging (b). Region of Interest (ROI) can be selected and bioluminescence intensity can be quantified
3 Methods
3.1 Cell Culture
While the goal is to use tools for targeted modification of neuronal activity in the intact living brain of a behaving animal, there are several applications in vitro, specifically in cultured cells. These applications range from simply testing constructs or validating viral vector preparations for expression of bioluminescence to studying neuronal networks in long-term cultures to investigating network connections in brain slices. HEK cells provide a convenient heterologous expression system to test and verify new luminopsin constructs, while primary neurons are usually needed to test viral preparations with neuron-specific promoters.
3.1.1 HEK Cells
Grow HEK cells in medium of choice such as suppDMEM at 37 °C and 5 % CO2 in a humidified atmosphere.
Trypsinize cells, count, and adjust to 5 × 104 cells per 0.5 ml culture medium. Using sterile forceps, place uncoated or coated (poly-D-lysine) glass coverslips into as many 24-well size wells as needed. Seed 0.5 ml per well cells onto coverslips. For plate reader assays, seed appropriate number of cells onto a 96-well plate (1–5 × 104 cells per 0.1 ml per well) (also: see Note 1).
The next day, transfect each well with a luminopsin construct (see Notes 2 and 3). It is convenient to set up transfections for 5 wells per construct, and transfect 4 wells per 4-well dish. For best results, change medium 4 h after transfection.
(Optional) One or two days after transfection, the day before electrophysiology experiments, add all-trans retinal to the cultures. Opsins require retinal as a chromophore. This step is not necessary when HEK cells are cultured in serum-supplemented medium, which contains trans-retinal.
At the day of electrophysiology experiments (2–3 days after transfection of DNA) transfer coverslips from tissue culture wells to a recording chamber.
3.1.2 Primary Neurons
Isolate primary cortical or hippocampal neurons (see Note 4) and determine cell number. Adjust cells to 1–2.5 × 105 per 0.5 ml in Serum-NB. Using sterile forceps, place poly-D-lysine-coated glass coverslips (see Note 5) into as many wells as needed (24-well for 12-mm coverslips; 12-well for 18-mm coverslips). Seed 0.5 ml per well cells onto coverslips. Together with the dishes of seeded neurons place a culture tube in the incubator with its lid slightly loosened, containing Plain-NB medium sufficient to add 1 ml per seeded well (pre-equilibrated Plain-NB).
-
Neurons can be transfected with LMOs using one of the following methods:
Lipofection: low efficiency; highest toxicity; ideal when testing constructs in single cells by patch-clamp recording.
Electroporation: moderate efficiency; moderate toxicity; requires special equipment (e.g. Lonza Nucleofector) and a higher number of cells. Typically done before seeding.
Viral transduction: highest efficiency; least toxic; ideal when examining activity of many neurons at a time using a multielectrode array (MEA).
For lipofection, collect 0.5 ml of conditioned culture medium from each well 1 or 2 days after medium change-out in step 3 below (DIV2 or DIV3) and save in a culture tube (we usually collect 2 ml per 4-well dish and save in a separate 15 ml tube for each 4-well dish). Keep tubes in incubator with lid slightly loosened to allow air exchange. Transfect cells in the remaining 0.5 ml medium per well with a luminopsin construct. For transfection of neurons to be used for patch clamping, we use Lipofectamine 2000 according to the manufacturer’s recommendations, except that less of the recommended amount of Lipofectamine and DNA per well is used (per well of a 4-well or 24-well dish: 0.2 μl Lipofectamine, a tenth of the recommended amount; 200 ng plasmid DNA, a quarter of the recommended amount). Again, it is convenient to set up transfections for 5 wells per construct (1 μg DNA/250 μl OptiMEM; 1 μl Lipofectamine/250 μl OptiMEM), and transfect 4 wells per 4-well dish. Change out medium 4 h after transfection with the prewarmed and pre-equilibrated saved medium from above (see Note 6).
For electroporation, we follow the manufacturer’s recommendations with slight modifications. In brief, spin down up to six million cells per reaction in a 1.5 ml tube. Resuspend the cells in 100 μl of the proprietary solution provided or in DPBS with calcium, magnesium, glucose, and pyruvate. Add and mix with 2 μg of LMO DNA. Transfer the cell suspension into a cuvette. After electroporation, using the recommended parameters for primary neurons, add 500 μl prewarmed, CO2-equilibrated low-calcium recovery medium (i.e. RPMI) and transfer the cell suspension into the original tube. Incubate the tube in a CO2 tissue-culture incubator for 5 min before seeding onto coverslips in a well filled with prewarmed, CO2-equilibrated Serum-NB at a density higher than that used for lipofection.
For viral transduction, add high titer virus (>108 infectious units/ml lentivirus, >1011 vg/ml AAV) to the Plain-NB medium used in step 1. Use viral preparations with a multiplicity of infection (MOI) > 10 to ensure near 100 % transduction efficiency (Fig. 2). Depending on the promoter, expression can typically be visualized by fluorescence microscopy 3–4 days after infection with lentivirus, and 7–10 days after infection with AAV.
Regardless of the method of transfection, the next day after plating (day in vitro 1, DIV1), remove Serum-NB medium completely from neurons and replace with 1 ml per well prewarmed and pre-equilibrated Plain-NB medium. Carry out the medium exchange well by well, so as to not leave neurons without medium for more than a few seconds.
Neurons are good for electrophysiological recordings between DIV10 and DIV14. Add ¼ volume of prewarmed and pre-equilibrated Plain-NB every 3–4 days, if needed. Make sure cultures do not evaporate by placing dishes in a humidified incubator or humidity chamber (see Note 7).
(Optional) The day before electrophysiological experiments, add all-trans retinal to the cultures for a final concentration of 1 μM. This step is not necessary if Plain-NB is supplemented with vitamin A-containing B27.
On the day of electrophysiological experiments transfer coverslips from tissue culture wells to the recording chamber. Coverslips should be immediately perfused with recording buffer to minimize amount of time coverslips are exposed to air.
Fig. 2.
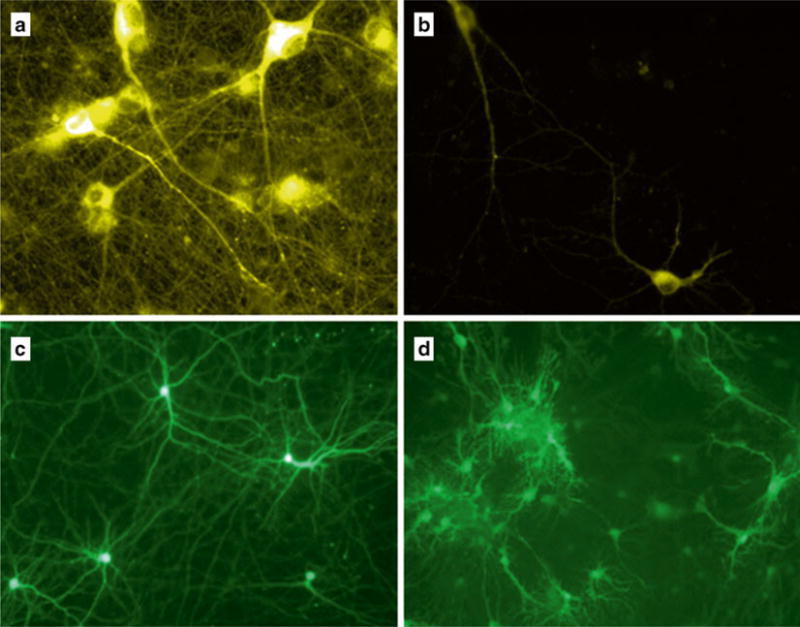
Viral transduction of cultured neurons. Primary rat embryonic day 18 cortical neurons were plated on poly-D-lysine-coated coverslips at equal densities. The next day (DIV1), neurons were transduced with viral vectors. (a, b) The same transgene (CAG-Gluc-VChR1-EYFP) expressed from different vectors (a: AAV, b: lentivirus). (c,d) Lentivirus expressing GLuc-Mac-GFP under control of different promoters (c: human synapsin; d: CAG), with hSyn favoring expression in neurons versus glia
3.2 Bioluminescence Imaging In Vitro
For a simple bioluminescence assessment (i.e. bioluminescence intensity and emission spectrum), luminometers provide the most sensitive and quantitative measurements. They are specifically designed to measure photon emission upon time-controlled substrate injection, usually in a plate format (96-well plate in the basic models, various plate formats in the higher end models). Alternatively, we have utilized bioluminescence imagers designed for in vivo imaging (see below) and obtained satisfactory results.
If none of the above is available, a good option is imaging systems designed for chemiluminescence, such as image documenting systems for Western blots equipped with a cooled CCD camera (e.g. Fuji Film LAS-3000, Li-Cor Odyssey Fc). For high-emission luciferases, even common gel documentation systems can be used (see Fig. 3).
Simultaneous imaging with electrophysiological recordings is addressed in each individual section below.
3.3 Intracellular Recording In Vitro
The effects of LMOs on neuronal firing can be assessed in primary neurons in culture. Concurrent imaging of bioluminescence helps to establish a causal relationship between biological light and activation of LMOs.
Cells are imaged and recorded using an upright or inverted epifluorescence microscope equipped with a 40× 0.8 NA water immersion objective or a 60× 1.35 NA oil immersion objective, respectively, an arc lamp, an electronic shutter, a GFP filter cube, and a cooled CCD camera or sCMOS camera with acquisition software. The recording chamber is constantly superfused with the extracellular solution at ~500 μl/min. Experiments are carried out at room temperature.
Locate an LMO-positive cell by its fluorescent-tag expression.
Obtain whole-cell recording under voltage or current clamp mode.
We routinely measure photocurrent to conventional wide-field photostimulation using the lamp and an appropriate filter cube (e.g. a GFP filter cube for ChR2). The measured photocurrents reflect the total functional surface expression level of LMO of the cell.
CTZ-induced photocurrents can be measured under voltage clamp. Effects of CTZ on spiking can be recorded under current clamp. The effect of inhibitory LMOs should be assessed under constant action potential firing, which can be evoked by a train of brief peri-threshold current injection (note: large current injection can mask subtle effects of CTZ). Immediately before application, reconstitute CTZ in the extracellular solution at 100 μM and bath-apply CTZ solution (~0.5 ml) to a recording chamber (see Note 8 for different options) (Fig. 4). Bioluminescence is imaged concurrently throughout the recording through the camera (see Note 9 for specific settings). To synchronize the two recordings, an exposure signal from the camera can be used to trigger electrophysiological recording. To maximize bioluminescence collection, move the filter turret to an open position before CTZ application. For bioluminescence imaging the microscope needs to be in complete darkness (see Note 10).
Measure photocurrent in response to direct photostimulation by lamp after CTZ application to confirm recording conditions have not changed.
Fig. 4.
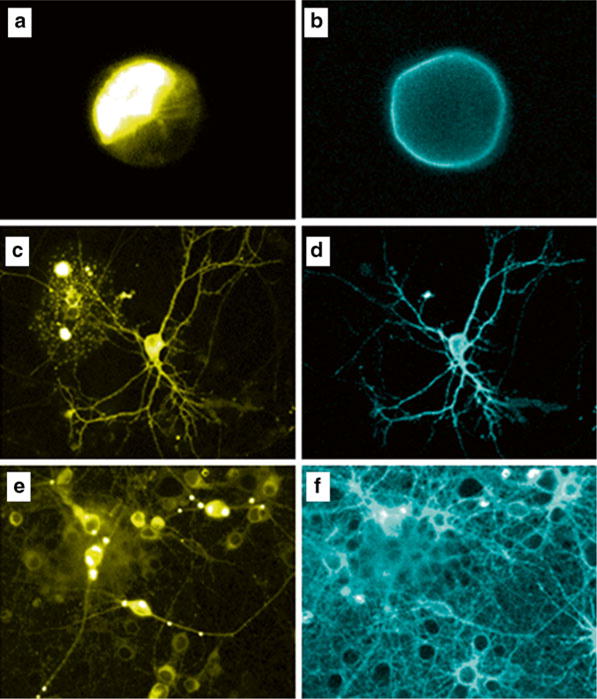
Luminescence in cell culture. HEK cells (a, b), individual neurons (c, d), and neuronal cultures (e, f) transfected (a–d) or transduced (e, f) with LMOs were imaged under fluorescent light (a, c, e) and after adding coelenterazine to the culture (b, d, f)
3.4 Extracellular Recording In Vitro
Recordings from multiple neurons at a time using a multielectrode array (MEA) provides not only a convenient and high-throughput method [14], but also enables analysis on change in network dynamics after activation of LMOs.
Prepare MEAs according to the manufacturer’s recommendations (sterilize, coat) (see Note 11).
Seed MEAs with cortical neurons (for example: 5 × 104/array in 1-well or 1.25 × 104/array in 6-well arrays from MultiChannel Systems).
The next day (DIV1) transduce cultures with AAV or lentivirus with an MOI >10 to achieve close to 100 % transduction (Fig. 5).
Change half-volume of media every 3–4 days.
Spontaneous synchronous bursting activity can be recorded around DIV 14. Insert an MEA into the appropriate headstage and connect to an amplifier. Allow the MEA to equilibrate to achieve a stable baseline of activity.
Optical stimulation can be conducted with an external LED or lamp since the MEAs are optically clear.
To measure effects of CTZ, add CTZ (5 μl per 200 μl culture volume) to reach a final concentration of 10–50 μM. Recording can continue for up to several hours if cultures are kept in a humidified incubator. For simultaneous imaging of bioluminescence, use 1-well MEAs and place the headstage on an inverted microscope stage. Image bioluminescence as described above (Subheading 3.3, step 5).
Fig. 5.
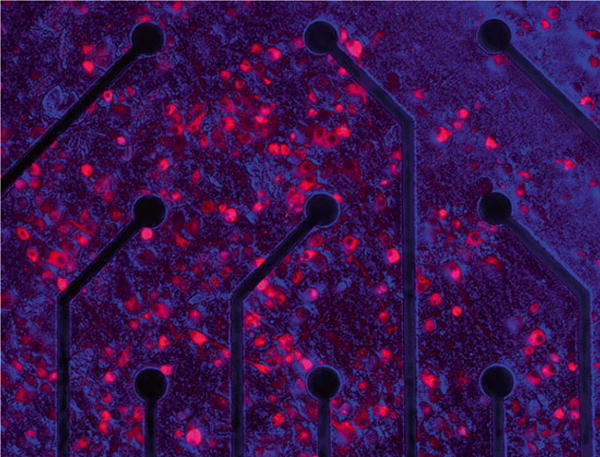
MEA culture expressing iLMO1. Rat embryonic cortical neurons were plated on electrodes in a MEA chamber (Multichannel Systems) and transduced on DIV 2 with lentivirus containing Ubiquitin-NpHR-TagRFP-Rluc (iLMO1). The image was taken on DIV14
3.5 Intracellular Recording Ex Vivo
Brain slice preparations retain innate synaptic connection to some degree. Recordings from LMO-expressing brain slices enable precise control of a given network using conventional optogenetic photostimulation [15] as well as activation of the entire network by the chemical, CTZ, in a more physiologically relevant setting than in cultured cells.
Inject AAV carrying the desired LMO or iLMO gene into the brain region of interest in mice or rats. Wait at least 10 days after injection to allow adequate expression before carrying out experiments.
Prepare acute brain slices (~300 μm thick sections) using a vibratome.
Whole-cell patch clamp recordings are made similar to in vitro recordings using conventional methods. Recordings can be made from cells expressing LMOs or from cells receiving synaptic inputs from LMO-expressing neurons.
LMOs can be activated by photostimulation with an arc lamp, with laser spots for detailed photo-mapping of synaptic inputs, or by adding CTZ to the recording chamber (see above: Subheading 3.3).
For simultaneous imaging of bioluminescence, a camera can be used as described above (Subheading 3.3, step 5). Alternatively, if working with a scanning microscope (i.e. confocal or two-photon microscope), a photodetector (i.e. a photomultiplier tube) can be used to record bioluminescence continuously. Switching to a lower magnification lens will help to collect more photons.
3.6 Animal Studies
For studying neuronal activity and/or behavior in awake animals expressing LMOs, viral vectors (generally AAV) are placed into the brain region of interest by stereotaxic injections as done for other optogenetic and chemogenetic approaches (Fig. 6). The sections below focus on specific usage of LMOs.
Fig. 6.
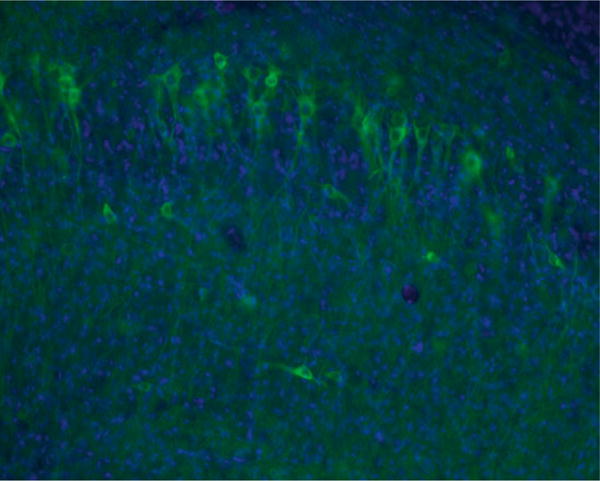
Hippocampal pyramidal cells expressing iLMO2. The dorsal hippocampus in a rat was injected with AAV expressing CamKIIa-NpHR-Nano-Lantern (Venus-Rluc), i.e., iLMO2. The rat was perfused and the brain sectioned 3 weeks after viral injection. Robust expression can be seen in pyramidal cells
3.7 Administration of CTZ In Vivo
For all in vivo experiments, CTZ preparations specifically designed for in vivo applications should be used (i.e. Nanolight Inject-A-Lume or water soluble CTZ). The choice of routes of delivery of CTZ in rodents depends on the goal of the experiment. Concentrations of CTZ should be aimed at final concentrations in the animal of 100–200 μM (see Note 12).
Intracranial injection: Concentrated CTZ in a minute volume can be delivered to a target brain region directly using an injection cannula inserted in the guide cannula (example: 34 ng CTZ in 0.4 μl PBS). Although this causes very rapid action only near the injection site, it may cause irreversible damage to the injection site if injection rate is too fast or large volumes are injected.
-
Intravenous injection: Application of substrate intravenously results in fast onset of effects (rapid rise in spike rate within several seconds of CTZ entering the bloodstream), a peak typically between 20 and 30 s after injection, and a slower decay (several minutes). Volumes for injections should be small (50–75 μl for mice; 200–250 μl for rats).
Retro-orbital injections in mice are an easy and reliable way of intravenous delivery, but usually require brief anesthesia.
Tail vein injections in mice or rats can be done without anesthesia. The animals should be warmed up by a heat lamp to improve circulation. There are single-use catheters available for tail vein injections (see Note 13).
Jugular catheters in mice or rats offer an easy access port for repeated administration of precisely timed injections with defined concentrations (e.g. obtaining a dose-response curve). Note: catheters must be kept patent with bi-weekly flushing of heparin and gentamicin (see Note 14).
Intraperitoneal injection: Application of substrate intraperitoneally results in a slower onset (several minutes) of measurable effects, which subside after a longer period of time (~30 up to 60 min) compared to intravenous injections. This is the route of choice for CTZ administrations repeatedly over long periods of time, for example to test the effects of chronic stimulation or silencing of populations over several months on an animal’s behavior. Example: 200 μg CTZ in 200 μl PBS intraperitoneally into a 25–30 g mouse.
3.8 Bioluminescence Imaging In Vivo
In vivo bioluminescence imaging can be conducted as a means for noninvasive confirmation of LMO expression and delivery of CTZ to a target. Imaging is conducted ideally with a dedicated small animal imager (e.g. IVIS imaging system). Depending on the species and area of expression in the brain, imaging can be conducted through the intact skull (Fig. 7).
Fig. 7.
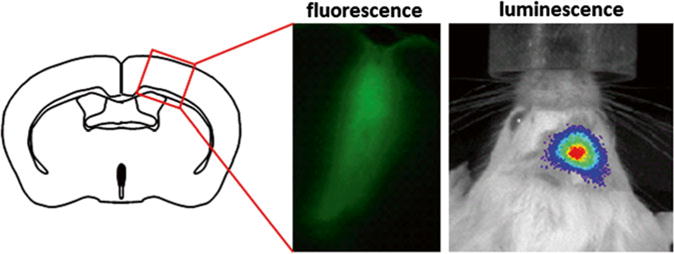
Bioluminescence imaging in vivo. Lentivirus carrying synapsin-LMO1 (Gluc-ChR2-EYFP) was injected into the right premotor cortex of a mouse. A month later mice were imaged in vivo after intravenous injection of CTZ (luminescence), then perfused and brain-sectioned for fluorescence imaging (fluorescence)
If such an imaging system is not available, other systems equipped with a cooled CCD camera or a sCMOS camera can be utilized, although with lower sensitivity. We have had success utilizing a gel image documenting system equipped with a cooled CCD camera (i.e. Fuji Film LAS-3000). In this pre-terminal setting, the skull of the animal was removed for bioluminescence imaging from the rat striatum (see Note 15).
3.9 Extracellular Recording In Vivo
Since luminopsins modulate neuronal activity, their activity can be measured by any conventional means of in vivo extracellular recording in both acute and chronic settings. Implantable electrodes obtained commercially or custom-made electrodes/optrodes (e.g. glass pulled electrodes, cannula-electrodes, or micro-drivable tetrodes such as FlexDrive, http://www.open-ephys.org/flexdrive/; [16]) are especially useful for recording both single-unit and local field potentials of LMO-expressing cells. EEG electrodes can also be utilized depending on the extent of expression, but these approaches would be unable to detect changes at single-unit firing rates.
Luminopsins offer the ability for multiple modes of activation: by direct light stimulation and by chemical substrate. Either mode can be used as a control for the other because the same population of neurons is targeted with the same single molecule. This can be achieved using a cannula-electrode, whereby photostimulation (through a fiber optic inserted through the cannula) and chemical stimulation (through injection of CTZ through the same cannula) can be restricted to only the neurons being recorded from. Cannula-electrodes can be fabricated by attaching guide cannulas to commercially available electrodes using superglue. These can be assembled before the actual implantation surgery to customize distance of the fiber tip/injection cannula from the recording electrodes (Fig. 8).
Fig. 8.

Cannula-electrode. A 16-channel electrode array (left) was superglued to a guide cannula (right). An optical fiber was inserted through the cannula to illuminate cells while recording from them. Subsequent CTZ injections were also given through the same guide cannula
The general procedure for recording responses of LMO-expressing cells to chemical and photostimulation is as follows:
Inject AAV into the rodent brains as described above. The amount of time for adequate expression will be specific to the construct/vector and will need to be empirically determined. We typically wait at least 10 days for expression.
Commercially bought or custom-made electrodes/optrodes should be configured so that the intended target is covered with the appropriate number of recording contacts and is able to be illuminated by a fiber optic (if included).
Stereotaxically implant the electrodes under anesthesia. Correct placement of the electrodes can be determined in real time by optically stimulating while advancing the electrode towards the intended target depth. The cell types being recorded can be determined by analyzing single-unit firing properties. After correct placement of the electrodes, a chronic implant can be created by securing everything to the skull via skull screws and dental acrylic. A chronic implant will allow for multiple injections/stimulations within the same animal: we were able to record up to 3 months after viral injection.
Recordings from implanted electrodes can be carried out in freely moving animals before and after administration of CTZ as described in Subheading 3.7 above. The effect of CTZ can be determined by quantifying changes in the single-unit activity and low frequency power in the local field potential.
Acute recordings can be conducted in head-fixed awake or lightly anesthetized animals. Wait at least 10–15 min after advancing an electrode before collecting data (see Note 16). Then establish a baseline before CTZ injection by recording at least 10–20 min without moving the probe. This is usually sufficient to confirm a stable recording and to capture characteristics of neurons at rest (but: see Note 17). Using an optrode (simultaneous recording and fiber optic light stimulation) allows stimulation before and after CTZ administration: stimulation before is used to confirm electrode location, and after to see if there are any remaining effects on driven activity as well as to confirm that the same units were recorded for the duration of the trial.
3.10 Behavioral Testing
Any behavioral test can be carried out in freely moving animals before and after administration of CTZ, acutely or to assess changes in behavior after chronic stimulation with bioluminescence. Combining behavioral assays with electrophysiological recording and/or bioluminescence imaging in a freely moving animal will offer a very unique application of LMOs.
Acknowledgments
This work was in part supported by grants from NIH (NS079268, R.G.; NS086433, J.T.; MH101525, U.H.), NSF (CBET1464686, U.H.), Duke Institute for Brain Sciences (U.H.), The Michael J. Fox Foundation (C.M.), and The Brain Research Foundation (C.M.).
Footnotes
Rather than transfect cells in 96-well plates, we usually transfect cells in 6-well or 6 cm plates. The next day, transfected cells are trypsinized, and seeded into 96-well plates.
Unlike neurons, the choice of a transfection agent is not so critical for HEK cells. We have used Effectene (Qiagen) or Lipofectamine 2000 (Invitrogen) with equal results.
Quality of DNA is critical for transfection, especially in neurons. Use DNA prepared from endotoxin-free maxi prep kits (e.g. Qiagen EndoFree Plasmid Maxi Kit) instead of mini prep kits.
Primary neurons of high quality can be isolated from freshly harvested embryonic day 18 rat pups, or from tissue pieces shipped the day before by BrainBits, Inc.
Prepare coated coverslips according to standard protocols. Pre-coated coverslips are also commercially available through NeuVitro and BD Biosciences.
For lipofection of neurons replacing the transfection medium with the “preconditioned” medium within 4 h (can be shortened to 2 h) is critical for obtaining healthy neurons for patch clamp recordings.
A humidity chamber can be a plastic Tupperware box or pyrex glass dish containing a glass plate kept just above an inch of water. The glass plate holds the culture dishes. Another glass plate put on top of the container without completely closing it serves as lid.
For bath-application of CTZ, we do not recommend manual pipetting, which will likely disrupt electrophysiological recordings. A better way is to fill a line of Tygon or silicone tubing of small diameter with the CTZ solution, connect a syringe at the other end, and deliver it to the recording chamber through a micro-manifold with flow control (AutoMate Scientific) by manually pushing the plunger of the syringe. We had limited success by localized application with a Picospritzer: it seems CTZ oxidizes inside the glass pipette before the recording starts.
Settings for bioluminescence imaging: (a) cooled CCD camera (for example, CoolSNAP-fx; Photometrics): without any filter cube with 4 by 4 binning. (b) scientific CMOS camera (for example, OptiMOS; QImaging) without binning. For Gaussia luciferase, exposure time is 1–5 s, for Renilla luciferase, 10–20 s.
Bioluminescence imaging should be conducted in complete darkness. This includes taping over all lights from instrument panels, and covering the microscope with light-impenetrable material (plastic, heavy cloth, felt) or keeping the entire room in the dark.
There are three main suppliers of MEAs: MultiChannel Systems (distributed through ALA Scientific), Axion Biosystems, and MED64 (distributed through AutoMate Scientific). We are using the MultiChannel MEAs, specifically the 6-chamber MEA with 9 electrodes per unit, and the 1-chamber MEA with 64 electrodes per unit for concurrent recording and bioluminescence imaging.
Example calculations based on an estimated blood volume for a 30 g mouse of 2.5 ml (molecular weight of native CTZ: 423.46): (1) Injections at 4 mg/kg of CTZ result in 120 μg for a 30 g mouse or a final concentration of ~112 μM CTZ in the bloodstream. (2) Intravenous injection of 50 μl of a 6 mM solution of CTZ results in an estimated final concentration in the bloodstream of ~120 μM. III. Using Nanolight’s instructions for Inject-A-Lume, dissolve one vial CTZ (500 μg) in 150 μl NanoFuel (3.33 mg/ml); injection of 60 μl (200 μg) in a 25–30 g mouse should result in ~200 μM final concentration. Given the small volumes of liquid it is recommended to use a syringe with as little dead space as possible (Insulin syringes in either 0.3 or 0.5 ml sizes).
Instech Laboratories has a selection of catheters, connectors, and vascular access ports for rats and mice (http://www.instechlabs.com/).
We typically push a mixture of 50 μl of heparin (30 U/ml) and 50 μl of Gentamicin (4 mg/ml) in sterile saline twice a week.
Whole animal imaging must be conducted with the animal under anesthesia, which may make it difficult to access peripheral veins for intravenous CTZ delivery due to vasoconstrictive effects of inhaled anesthetics like isoflurane.
The typical amount of wait time prior to starting baseline recording after electrode implantation takes into consideration deformation of tissue and transient depression in response to the mechanical stress. We found it important to be especially aware of this when using an optrode (custom made or commercially obtained). Even small-core fiber optics are large when compared to extracellular electrode tips, including multicontact silicon probes. As a result, the tissue movement and recovery time can be greater. Advance slowly after penetrating the cortical surface, and while looking for responsive cells. After finding light-activated units at the tip of the probe (if using a multielectrode array), wait 5–10 min to see if they are stable, and whether they drift to another contact. Overall, regardless of the recording site, let the electrode sit 20–30 min before starting baseline recordings when using a typical optrode.
Longer times for establishing a baseline may be required when recording in a structure with very high variability or if the intent is to capture the effect of luminopsins on neural or behavioral state transitions, as a certain number of events would be needed.
References
- 1.Sternson SM, Roth BL. Chemogenetic tools to interrogate brain functions. Annu Rev Neurosci. 2014;37:387–407. doi: 10.1146/annurev-neuro-071013-014048. [DOI] [PubMed] [Google Scholar]
- 2.Fenno L, Yizhar O, Deisseroth K. The development and application of optogenetics. Annu Rev Neurosci. 2011;34:389–412. doi: 10.1146/annurev-neuro-061010-113817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berglund K, Birkner E, Augustine GJ, Hochgeschwender U. Light-emitting channelrhodopsins for combined optogenetic and chemical-genetic control of neurons. PLoS One. 2013;8:e59759. doi: 10.1371/journal.pone.0059759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Verhaegen M, Christopoulos TK. Recombinant Gaussia luciferase. Overexpression, purification, and analytical application of a bioluminescent reporter for DNA hybridization. Anal Chem. 2002;74:4378–4385. doi: 10.1021/ac025742k. [DOI] [PubMed] [Google Scholar]
- 5.Tannous BA, Kim D-E, Fernandez JL, et al. Codon-optimized Gaussia luciferase cDNA for mammalian gene expression in culture and in vivo. Mol Ther. 2005;11:435–443. doi: 10.1016/j.ymthe.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 6.Nagel G, Szellas T, Huhn W, et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A. 2003;100:13940–13945. doi: 10.1073/pnas.1936192100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang F, Prigge M, Beyrière F, et al. Red-shifted optogenetic excitation: a tool for fast neural control derived from Volvox carteri. Nat Neurosci. 2008;11:631–633. doi: 10.1038/nn.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saito K, Chang Y-F, Horikawa K, et al. Luminescent proteins for high-speed single-cell and whole-body imaging. Nat Commun. 2012;3:1262. doi: 10.1038/ncomms2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schobert B, Lanyi JK. Halorhodopsin is a light-driven chloride pump. J Biol Chem. 1982;257:10306–10313. [PubMed] [Google Scholar]
- 10.Zhu H, Roth BL. DREADD: a chemo-genetic GPCR signaling platform. Int J Neuropsychopharmacol. 2014;18(1) doi: 10.1093/ijnp/pyu007. pii: pyu007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teranishi K, Shimomura O. Solubilizing coelenterazine in water with hydroxypropyl-BETA-cyclodextrin. Biosci Biotechnol Biochem. 1997;61:1219–1220. [Google Scholar]
- 12.Naumann EA, Kampff AR, Prober DA, et al. Monitoring neural activity with bioluminescence during natural behavior. Nat Neurosci. 2010;13:513–520. doi: 10.1038/nn.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morse D, Tannous BA. A water-soluble coelenterazine for sensitive in vivo imaging of coelenterate luciferases. Mol Ther. 2012;20:692–693. doi: 10.1038/mt.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hales CM, Rolston JD, Potter SM. How to culture, record and stimulate neuronal networks on micro-electrode arrays (MEAs) J Vis Exp. 2010 doi: 10.3791/2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, Peca J, Matsuzaki M, et al. High-speed mapping of synaptic connectivity using photostimulation in Channelrhodopsin-2 transgenic mice. Proc Natl Acad Sci U S A. 2007;104:8143–8148. doi: 10.1073/pnas.0700384104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Voigts J, Siegle JH, Pritchett DL, Moore CI. The flexDrive: an ultra-light implant for optical control and highly parallel chronic recording of neuronal ensembles in freely moving mice. Front Syst Neurosci. 2013;7:8. doi: 10.3389/fnsys.2013.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]