

Abstract
The amyloid-β (Aβ) peptide of Alzheimer’s disease (AD) forms polymorphic fibrils on the micrometer scale and molecular scale. Various fibril growth conditions have been identified to cause polymorphism, but the intrinsic amino acid sequence basis for this polymorphism has been unclear. Several single-site mutations in the center of the Aβ sequence cause different disease phenotypes and fibrillization properties. The E22G (Arctic) mutant is found in familial AD and forms protofibrils more rapidly than wild-type Aβ. Here, we use solid-state NMR spectroscopy to investigate the structure, dynamics, hydration and morphology of Arctic E22G Aβ40 fibrils. 13C, 15N-labeled synthetic E22G Aβ40 peptides are studied and compared with wild-type and Osaka E22Δ Aβ40 fibrils. Under the same fibrillization conditions, Arctic Aβ40 exhibits a high degree of polymorphism, showing at least four sets of NMR chemical shifts for various residues, while the Osaka and wild-type Aβ40 fibrils show a single or a predominant set of chemical shifts. Thus, structural polymorphism is intrinsic to the Arctic E22G Aβ40 sequence. Chemical shifts and inter-residue contacts obtained from 2D correlation spectra indicate that one of the major Arctic conformers has surprisingly high structural similarity with wild-type Aβ42. 13C-1H dipolar order parameters, 1H rotating-frame spin-lattice relaxation times and water-to-protein spin diffusion experiments reveal substantial differences in the dynamics and hydration of Arctic, Osaka and wild-type Aβ40 fibrils. Together, these results strongly suggest that electrostatic interactions in the center of the Aβ peptide sequence play a crucial role in the three-dimensional fold of the fibrils, and by inference, fibril-induced neuronal toxicity and AD pathogenesis.
Graphical Abstract

Introduction
Insoluble amyloid fibrils formed by a variety of proteins underlie many neurodegenerative diseases, principal among which are Alzheimer’s disease (AD), Parkinson’s disease, Huntington’s disease, and Creutzfeldt-Jakob disease 1,2. Elucidating the molecular structures and oligomeric assemblies of amyloid proteins 3–10 is the first step in designing therapeutic compounds against these diseases. For AD, two central events in its pathogenesis are the aggregation of two proteins in the brain: the small amyloid-β (Aβ) peptide of 37–43 residues that forms extracellular plaques and the hyperphosphorylated tau protein that forms intracellular neurofibrillary tangles. Electron microscopy (EM) and solid-state NMR (SSNMR) spectroscopy have been used extensively to characterize Aβ fibrils 11–18, and show that Aβ structures are highly polymorphic on the micron scale and the molecular scale 19,20. On the micron scale, Aβ aggregates can appear as striated ribbons, twisted fibrils, or spherical aggregates, and have variable periodicity of twists and variable widths. On the nanometer scale, SSNMR structures of wild-type (WT) Aβ40 fibrils show a common strand-loop-strand motif that forms hydrogen bond in a parallel in-register fashion 21 along the fibril axis to form a cross-β structure. However, each repeat motif of the cross-β structure can contain two or three molecules with two- or three-fold rotational symmetries along the fibril axis, whose intermolecular interfaces differ significantly 12,13. Local torsion angles show moderate variations among different structures, and the registry between the two β-strands of each peptide can shift 22. Further, a brain-derived and seeded Aβ40 sample exhibits a three-fold symmetric structure that differs from the in vitro three-fold structure in the β-strand length and subunit intermolecular packing 16. These structural variations have so far been largely attributed to fibril growth conditions such as shear forces, pH, temperature and peptide concentration 23–25.
While fibril growth conditions can affect Aβ structure, recent experimental data indicate that the amino acid sequence also plays a significant role in Aβ polymorphism, with single- or double-residue changes able to dramatically alter the fibril structure. An atomic structural model of Aβ42, which is less abundant than Aβ40 but has higher toxicity and morphological heterogeneity 26,27 has recently been reported 15. With only two extra residues, Ile41 and Ala42, the protein adopts a completely different, S-shaped triple parallel β-sheet structure 15,28, stabilized by an intramolecular K28–A42 salt bridge. The chemical shifts of recombinant Aβ42 support the distinct β-strand distributions in the Aβ42 and Aβ40 sequences 29. Another example of the sequence dependence of Aβ fibril structures comes from a high-resolution structure of the E22 deletion (E22Δ) mutant, Osaka, of Aβ40 14. A large number of distance and chemical shift constraints showed that this mutant adopts an intricate two-fold symmetric “cinnamon roll”-like structure that is stabilized by an intermolecular E3–K28 salt bridge 14,30. Finally, a D23N mutant of Aβ40 was found to adopt both parallel and antiparallel packing, in contrast to the parallel-only structures of wild-type Aβ40 fibrils 31,32.
The Aβ structural diversity opens the important questions of whether this polymorphism correlates with and even causes differences in neuronal toxicities, binding stoichiometries of imaging agents, and AD symptoms 16,25,33, and what the intrinsic molecular reasons are beyond fibril growth conditions for this polymorphism. Insights into these questions may be gained by comparisons of mutant Aβ fibrils with each other and with the wild-type fibrils. Many single-site mutations exist within the 43 residues of the Aβ sequence that are related to familial and early-onset AD 34. Interestingly, most of these mutations are clustered in the center of the Aβ sequence, away from the secretase cleavage sites, in a short segment of A21, E22 and D23. These are the Flemish A21G 35, Osaka E22Δ 36, Italian E22K 37, Arctic E22G 38, Dutch E22Q 39, and Iowa D23N 40. Among these mutants, the Arctic E22G Aβ shows fast protofibril formation in vitro and in vivo, and causes fibril morphologies distinct from those of wild-type Aβ40 18,38,41–44. Given the highly charged nature of this segment and the occurrence of multiple charge-altering mutations, we wonder whether electrostatic interactions in this non-β region play an important role in shaping the three-dimensional fold of Aβ.
In this study, we combine magic-angle-spinning (MAS) SSNMR with EM, fluorescence spectroscopy, and structural stability measurements to characterize the structure, dynamics, and fibrillization properties of Arctic Aβ40 fibrils. Synthetic site-specifically labeled E22G Aβ40 is examined using 2D correlation SSNMR techniques. To determine whether the observed conformations are intrinsic to the amino acid sequence or dominated by fibrillization conditions, we also measured the spectra of wild-type and Osaka E22Δ Aβ40 fibrils. Under the same fibrillization conditions, we found that Arctic Aβ40 exhibits at least four conformations while Osaka and wild-type Aβ40 adopt a single or a predominant conformation. Moreover, the chemical shifts of a major Arctic conformer bear close resemblance to reported Aβ42 chemical shifts 15,28,29, suggesting similarities in the β-strand arrangements of these two fibrils. 13C-1H dipolar order parameters, 1H relaxation times, and water-to-protein 1H spin diffusion experiments indicate differences in the mobility and hydration of the three fibrils. These results provide insights into how electrostatic interaction in the center of the Aβ peptide dictates the three-dimensional fold 45 and consequently the fibrillization properties of this peptide.
Materials and Methods
Synthesis and purification of Aβ(1–40) peptides
The peptides were synthesized using a Biotage Initiator+ Alstra automated microwave peptide synthesizer on preloaded HMPB-Chemmatrix resin (0.1 mmol scale). Fmoc deprotection was performed at 70ºC using 0.1 M HOBt in 4-methyl piperidine-DMF (1:4) for 5 min. The resin was then washed with DMF (3×). Coupling reactions were performed using Nα-Fmoc amino acids (5 equiv, 0.5 M), HCTU (4.95 equiv, 0.5 M) and DIEA (10 equiv, 0.5 M) in DMF. All couplings were doubly performed for 5 min at 75ºC except for His (25 min double coupling at RT) and Arg residues (5 min triple coupling at 75ºC). After completion of synthesis, the resin was successively washed with DMF (3 ×) and DCM (3 ×) and dried. For isotope labeled residues, the first coupling was performed using the isotopically labeled Nα-Fmoc amino acids (1.5 equiv, 0.5 M), HCTU (1.45 equiv, 0.5 M) and DIEA (3 equiv, 0.5 M) in DMF, followed by the second coupling using unlabeled Nα-Fmoc amino acids (3 equiv, 0.5 M), HCTU (3 equiv, 0.5 M) and DIEA (6 equiv, 0.5 M) in DMF. Peptide cleavage was performed using TFA-DTT-H2O (95:2.5:2.5) for 3 h at RT. The volatiles were removed by a stream of nitrogen gas and the crude peptide was obtained by precipitation with cold diethyl ether. The crude peptide was dissolved in HFIP and purified by RP-HPLC (Fig. S1). A portion of the crude solution was injected into the heated (60ºC) Vydac 300 Å C4 column (214TP1022, 10 μm, 22 × 250 mm) running a flow of 10 ml/min with a linear gradient from 15% to 40% of buffer B over 25min (buffer A: 0.1% TFA in H2O; buffer B: 0.1% TFA in 99% CH3CN in H2O). Peptide mass was analyzed using MALDI (Shimadzu Axima) and electrospray ionization mass spectrometry (Qtrap 3200, ABSCI EX).
Fibrillization
Lyophilized wild-type, E22G, E22Δ Aβ40 peptides were dissolved at 5 mg/ml in HFIP and evaporated into a peptide film. The peptide films were first suspended at 10 mg/ml in DMSO, briefly vortexed and sonicated for 5 minutes, then diluted to 0.2 mg/ml in 10 mM sodium phosphate buffer, pH 7.2. The solution was shaken at 900 rpm at 37ºC for at least 72 h to form fibrils. This fibrillization method is consistent with our in vivo biological experiments on Arctic Aβ 46,47. To investigate the effect of the solution condition on fibril structure, and to enable comparison with literature results, we also prepared the Osaka and Arctic fibrils using a second method, in which the peptide films were first suspended to 2 mg/ml in 10 mM NaOH and then quickly diluted to 0.2 mg/ml in the dilution buffer, whose final composition is 10 mM H3PO4-NaOH, pH 7.4, and 100 mM NaCl 14,48. We denote the two Osaka samples prepared in this way as Osaka DMSO and Osaka NaOH. All SSNMR spectra shown here for Arctic and wild-type Aβ40 are from the DMSO preparation, but similar data for the NaOH preparation were also acquired.
Electron microscopy
Wild-type, Arctic, and Osaka Aβ40 fibrils (0.2 mg/ml) were adsorbed onto freshly glow-discharged, 200-mesh formvar/carbon-coated copper grids (Ted Pella) and negative-stained with a 2% (w/v) uranyl acetate solution. Images were taken on a Philips/FEI Tecnai F20 electron microscope at 80 kV.
Thioflavin-T fluorescence monitoring of fibril formation kinetics
Thioflavin-T (10 μM) was added to a freshly prepared solution of wild-type, Arctic, and Osaka Aβ40 (0.2 mg/ml) in 10 mM sodium phosphate buffer, pH 7.2. Fluorescence measurements at an excitation wavelength of 485 nm and an emission wavelength of 538 nm were taken every 5 min in a SpectraMax M5 (Molecular Devices) plate reader set to 37ºC with agitation (automix linear shaking) between readings.
Guanidine hydrochloride denaturation
One volume of Aβ fibril sample (0.2 mg/ml) was added to three volumes of concentrated guanidine hydrochloride (GdnHCl) to obtain the desired final concentration (1–6 M GdnHCl) and incubated for 2 h at 22ºC. The fibrils were then centrifuged at 100,000 × g for 1 h, rinsed with PBS, and ultracentrifuged again. The pellet was resuspended in 4 × LDS sample buffer, diluted, and then resolved by SDS-PAGE and stained with Coomassie dyes.
Solid-state NMR spectroscopy
All fibrils were slowly dried in a desiccator to 60–75% water by mass. About 40 mg of these hydrated fibrils were spun into 3.2 mm MAS rotors. Solid-state NMR experiments were conducted on a Bruker Avance II 800 MHz (18.8 Tesla) spectrometer and an Avance 900 MHz (21.1 Tesla) spectrometer using triple-resonance HCN probes. Typical radiofrequency (rf) field strengths were 63–83 kHz for 1H, 50–63 kHz for 13C, and 36–45 kHz for 15N. 13C chemical shifts were externally referenced to the methylene signal of adamantane at 40.48 ppm on the DSS-scale. 15N chemical shifts were referenced to the methionine amide resonance (127.88 ppm) of the model peptide N-formyl-Met-Leu-Phe-OH (formyl-MLF) 49,50 on the liquid ammonia scale.
One-dimensional (1D) 13C and 15N cross-polarization (CP) spectra were measured on 800 and 900 MHz NMR spectrometers under 10.5, 12, and 16 kHz MAS. 1H-13C and 1H-15N contact times were 1 and 2 ms, respectively. The spectra were measured at 273 K and 298 K. Two-dimensional (2D) 13C-13C correlation spectra were measured with 50 ms dipolar-assisted rotational resonance (DARR) mixing 51 for resonance assignment and 1.5 s 1H-driven spin diffusion (PDSD) for detecting long-range correlations. 2D 15N-13C heteronuclear correlation (HETCOR) spectra were measured in two ways: a rotational echo double resonance (REDOR) transfer with a mixing time of 0.8 – 1.0 ms allowed the observation of one-bond 15N-13C cross peaks 49,52, while 2D N(CA)CX 53 spectra allowed the detection of 15N to sidechain 13C cross peaks. The N(CA)CX experiment used a 2 ms 1H-15N CP contact time, a 7 ms 15N-13C CP contact time, and a 30–50 ms 13C DARR mixing period. The 15N-13C CP used 15N and 13C spin-lock fields of 27.8 kHz and 38.3 kHz, respectively, and the 13C carrier frequency was set at 55 ppm. 1H decoupling field strengths were 93 kHz during 15N-13C CP and 83 kHz during acquisition.
To measure Aβ40 hydration, we conducted a water-to-protein 1H spin diffusion experiment 54–58. Following 1H excitation, a 1H T2 filter of 0.6–1.2 ms was applied to suppress the 1H magnetization of the rigid protein while retaining 80–90% of the 1H magnetization of mobile water. A 1H mixing time was then applied to transfer the remaining water 1H polarization to the protein and then to protein 13C via CP for detection. The spin diffusion mixing times were varied from 0 to 196 ms to obtain a magnetization transfer buildup curve. The experiment was conducted at 277 K. The 1D experiment was also extended to 2D by adding a 13C evolution period and a 50 ms DARR mixing period to resolve the peptide 13C signals 59. For the water-edited 2D experiment, the 1H mixing time was fixed to 4 ms to detect only well hydrated protein residues.
To investigate peptide dynamics, we measured 13C-1H dipolar couplings using the dipolar-chemical shift correlation (DIPSHIFT) experiment 60. The samples were spun at 7 kHz at 298 K, and a frequency-switched Lee-Goldburg (FSLG) sequence 61 was applied with a transverse field strength of 80 kHz to remove the 1H-1H dipolar coupling during the evolution period. The FSLG scaling factor was measured on formyl-MLF to be 0.577 for the Osaka and wild-type Aβ40 samples and 0.590 for the Arctic Aβ40 sample. Apparent 13C-1H dipolar couplings were obtained by fitting the t1-dependent intensities, then divided by the scaling factor to obtain the true couplings. Order parameters were calculated by dividing the true couplings with the rigid-limit value of 22.7 kHz. DIPSHIFT time curves were simulated using a FORTRAN program 62,63, available online at meihonglab.mit.edu/software. An empirical 1H T2 relaxation factor was applied to the best-fit curves to reproduce the observed asymmetry in the dipolar oscillation.
1H rotating-frame spin-lattice (T1ρ) relaxation times, which are indicative of microsecond molecular motions 64, were measured with 13C detection at 298 K and 260 K under 10.5 kHz MAS using a Lee-Goldburg (LG) spin-lock pulse sequence 65. The spin lock time varied from 0 to 50 ms, followed by a 0.3 ms Hartman-Hahn CP period. The tilted effective 1H LG spin-lock field was 62.5 kHz, thus the characteristic timescale probed by this experiment is 16 μs.
To compare the chemical shifts of the three Aβ40 fibrils, we calculated the chemical shift differences for each residue using the following equation:
| (1) |
where ξ and κ denote different fibrils, n is the number of 13C chemical shifts available for each residue, and the factor of 2.5 scales the 15N chemical shift relative to the 13C chemical shifts due to their relative gyromagnetic ratios. All 13C chemical shifts are reported on the DSS scale (Table S1, S2). Literature values for chemical shifts referenced to TMS were converted to DSS-referenced values by adding 2.0 ppm.
To evaluate the pairwise similarities for the eleven Aβ fibrils, we conducted a correlation analysis by calculating the Pearson product-moment correlation coefficient. The chemical shifts of four atoms, the backbone amide nitrogen, C′, Cα, Cβ, are included. For each strain, the chemical shifts of identical atoms were combined, to form a dataset. The correlation coefficients between equivalent datasets were then averaged for each pair of strains.
Results
Comparison of Aβ40 fibril formation kinetics, fibril stability and morphology
WT and mutant Aβ40 peptides were assembled into fibrils by incubating at 37°C with constant agitation. Shaking both accelerates fibril formation by fragmenting the fibrils and exposing new ends for the addition of monomers and promotes the formation of equilibrium structures 32. Fibrillization kinetics were monitored using the amyloid-binding dye Thioflavin T (ThT) (Fig. 1a). The three peptides show distinct fibril formation rates. The E22G and E22Δ Aβ40 prepared in DMSO formed at much faster rates than the wild-type peptide. In particular, the Osaka peptide fibrillized on the second to minute timescale and was complete by the first time point of the measurement, which was conducted within 2–3 minutes of sample preparation. This is manifested by the high initial ThT fluorescence value and the rapid disappearance of the peptide signals in the 1H solution NMR spectra (Fig. S2). In comparison, wild-type Aβ40 showed a pronounced lag phase and fibrillized in ~20 hours with greater kinetic stochasticity. For each sample the final increase in fluorescence intensity was significantly different, indicating different extents of binding and/or the environment of the bound ThT dye molecules. The Arctic E22G mutant fibrillized more slowly than the Osaka peptide, but still more rapidly than WT. These observations are in good agreement with literature reports of the kinetics of Arctic Aβ fibrils 38,66–68
Figure 1.

Fibrillization of wild-type, Arctic and Osaka Aβ40 peptides. (A) Osaka and Arctic Aβ40 peptides form fibrils much more rapidly than the wild-type (WT) peptide. Kinetics of fibril formation (0.2 mg/ml in 10 mM sodium phosphate buffer, pH 7.2) was measured by the amyloid-binding dye ThT (10 μM). n=5, mean±SEM. (B) Negative stain TEM images of WT and mutant Aβ40 fibrils. Arctic Aβ40 fibrils display significant twists with variable periodicities and more pronounced entanglement than wild-type and Osaka fibrils. Scale bar = 100 nm. (C) Mutant fibrils have altered stability to denaturation by GdnHCl. Fibrils were incubated with increasing concentrations (1 – 6 M) of GdnHCl and then ultracentrifuged at 100,000 × g to pellet any fibrils that resisted denaturation, which were resolved by SDS-PAGE and Coomassie stain.
We usually first dissolved the peptide in DMSO to prepare fibrils because this method produced fibrils with biological activity in mouse inoculation 69. However we also wanted to prepare the Osaka peptide by dissolving in NaOH to match the published SSNMR structure 14. The use of NaOH for initial peptide solubilization moderately slowed down fibril formation, but the rates are still much faster than wild-type Aβ40. There was also a large change in the final intensity between the DMSO and NaOH treated samples; because the peptide sequence is identical in both experiments, we conclude that the two methods give rise to distinct fibril conformations and/or morphologies. Negatively stained TEM images (Fig. 1b) show that the wild-type and Osaka fibrils are relatively straight, while the Arctic fibrils display significant twists with a distribution of periodicities and are more entangled than the wild-type and Osaka fibrils.
The mutant Aβ40 fibrils not only exhibit different fibrillization kinetics and morphologies from the wild-type fibrils, but also have different thermodynamic stabilities. To investigate the fibril stabilities, we incubated the fibrils with increasing concentrations of guanidine hydrochloride and quantified the amount of insoluble fibrils remaining after a two-hour equilibration time (Fig. 1c). We take the midpoint of the solubilization curve as a proxy for relative thermodynamic stability. Arctic E22G fibrils with varying concentrations of GdnHCl showed fibril loss at lower concentrations of GdnHCl than wild-type fibrils. In contrast, the Osaka E22Δ fibrils are more stable against GdnHCl solubilization than the wild-type peptide. The increase in stability also depended on the method of solubilization.
Together, these experiments indicate that the Arctic E22G mutant forms fibrils with a lower activation energy than the WT peptide and adopt a final solid-state structure with lower stability than WT. In comparison, under the same DMSO-solubilized initial conditions, the Osaka mutant forms fibrils with a lower activation energy than WT, but achieves a final conformation that has greater stability than WT. Moreover, both the kinetics and the thermodynamics of the fibril formation process depend on the initial state of the peptide since NaOH-solubilized Osaka peptide formed fibrils more slowly than DMSO-solubilized peptide, and it achieved a final state of greater thermodynamic stability. Finally, the differences in fluorescence intensity of the final states suggest that each sample achieved a different final state. To determine the structural bases for these differences we next turned to SSNMR.
13C and 15N chemical shifts of E22G, E22Δ and wild-type Aβ40 fibrils
1D 13C and 15N CP-MAS spectra (Fig. 2) give qualitative information about the structural homogeneity and similarity of the three Aβ40 fibrils. All samples show sharp 13C and 15N signals, with the Osaka DMSO fibrils having the most resolved spectra. The Arctic spectra have similar intrinsic linewidths as the wild-type and Osaka spectra, as seen by resolved sidechain signals, but contain more peaks, thus giving more overlapped Cα and CO regions. The 15N spectrum of Arctic Aβ40 shows multiple glycine resonances while the Osaka and wild-type Aβ40 exhibit only one G38 signal. The spectral overlap is the smallest for Osaka Aβ40 and the highest for Arctic Aβ40. Spectra measured from 255 K to 298 K showed little differences in intensities and linewidths, indicating that all fibrils are mostly immobilized at room temperature (data not shown).
Figure 2.

(a) Amino acid sequences of the three Aβ40 peptides. Isotopically labeled residues are shown in red. (b–c) 1D 13C (b) and 15N (c) CP-MAS spectra of Arctic, Osaka, and wild-type Aβ40 fibrils. Spectra were measured on 800 and 900 MHz spectrometers at 273 K under 16 kHz MAS. Assignments obtained from 2D spectra are indicated. All three fibrils show narrow intrinsic linewidths, but the number of peaks and the chemical shifts differ significantly.
2D 13C-13C DARR and 15N-13C correlation spectra fully resolved and permitted the assignment of the chemical shifts of the labeled peptides, and revealed extensive polymorphism of the E22G Aβ40 fibrils: multiple resonances for each labeled residue are seen in both the DARR and N(CA)CX spectra (Fig. 3). F19 exhibits two sets of chemical shifts, A21 and V36 exhibit three, while I32, L34 and G38 have four sets of chemical shifts (Table S1). The 13C chemical shifts vary by up to 3 ppm for each site while 15N chemical shifts vary by as little as 1 ppm for F19 and as large as 20 ppm for L34. The chemical shift differences are much larger than the 13C linewidths of 0.6–0.9 ppm and the 15N linewidths of less than 2 ppm. The concentration ratio of the four conformers is about 3 : 3 : 2 : 2 and is similar for all residues, as estimated from the integrated peak intensities in the 50 ms DARR spectrum, thus even the two minor conformers are not negligible in concentration. We denote the two sets of chemical shifts with the highest intensities as Arctic I and II. No inter-residue cross peaks were obtained to relate the multiple conformations of one residue to another, and we assume that the conformer I chemical shifts in each residue belong to the same molecule, and all conformer II chemical shifts belong together to another molecule.
Figure 3.

2D correlation spectra of Arctic Aβ40 (E22G) fibrils. (a) 2D 13C-13C DARR spectrum with 50 ms mixing. For simplicity, assignments are given with lower-case letters for various carbons, and F19 aromatic carbons are designated by “r”. ssb denotes spinning sidebands. (b) 2D N(CA)CX 15N-13C correlation spectrum. Two to four sets of chemical shifts (denoted by subscripts I – IV) are identified for all labeled residues, indicating extensive structural polymorphism. Ellipses guide the eye for chemical shift multiplicity. The DARR spectrum was processed using a QSINE window function with SSB = 2.5, and were plotted with Topspin parameters lev0 = 4, toplev = 90 and nlev = 16. 2D N(CA)CX spectrum was processed using a Gaussian window function with LB = −10 Hz and GB = 0.05, and was plotted using lev0 = 5, toplev = 80 and nlev =16.
To determine whether the E22G Aβ40 polymorphism is intrinsic to this amino acid sequence or due to suboptimal fibrillization conditions, we measured 2D spectra of wild-type and E22Δ Aβ40 fibrils prepared using the same protocol (Fig. 4, Fig. S3). In dramatic contrast to the E22G mutant, the E22Δ spectra show a single set of cross peaks, while the wild-type spectra show predominantly one species, with one or two additional conformers observed for residues I32, L34 and V36. The major conformer of wild-type Aβ40 represents 50–70% of the whole population. The Osaka and wild-type spectra are therefore much better resolved than the E22G Aβ40 spectra. Arctic and wild-type Aβ40 prepared in DMSO and NaOH solutions showed no spectral differences (Fig. S4), indicating that their conformations are independent of the solvent. Moreover, 13C and 15N spectra (Fig. S4a, b) of independently prepared batches of Arctic Aβ40 fibrils are indistinguishable, demonstrating that the structural polymorphism is reproducible. For Osaka Aβ40, initial peptide solubilization in NaOH before fibrilization in buffer resulted in observable chemical shift differences from the DMSO solubilized sample and a modest degree of chemical shift heterogeneity (Fig. S3d, Fig. S5). Taken together, these results indicate that the pronounced structural polymorphism of Arctic E22G Aβ40 is an intrinsic property of this amino acid sequence rather than an artifact of fibril growth conditions.
Figure 4.

Comparison of the chemical shifts of the three Aβ40 fibrils. (a–c) 2D 13C-13C DARR spectra with 50 ms mixing for (a) Arctic Aβ40 (E22G), (b) wild-type Aβ40, and (c) Osaka Aβ40 (E22Δ). (d–f) 2D 15N-13C correlation spectra for (d) Arctic Aβ40 (E22G), (e) wild-type Aβ40, and (f) Osaka Aβ40 (E22Δ). A single set of chemical shifts is observed for Osaka Aβ40, the wild-type fibril shows modest polymorphism from I32 to V36, while the Arctic Aβ40 fibril has the largest number of polymorphs. All DARR spectra were processed using QSINE window functions with SSB = 2.5 and plotted using lev0 = 4, toplev = 90 and nlev = 16. 2D 15N-13C correlation spectra were processed using LB = −10 Hz and GB = 0.05, and were plotted using lev0 = 5, toplev = 90 and nlev = 12.
The 13C and 15N chemical shifts vary substantially among the three fibrils studied here and also differ to varying degrees from literature chemical shifts of other Aβ fibrils. Our Osaka DMSO chemical shifts are in excellent agreement with those reported by Schütz and coworkers 14, while our wild-type Aβ40 chemical shifts have moderately good agreement with the two-fold and three-fold in vitro wild-type fibrils 19. In Arctic Aβ40, I32 and V36 have characteristic β-strand chemical shifts, A21 and L34 show typical random coil chemical shifts, while F19 and G38 exhibit mixed strand and coil chemical shifts. Interestingly, the chemical shifts of the two major Arctic conformers show unexpected agreement with Aβ42 chemical shifts. For example, the A21 Cα and 15N chemical shift of Arctic I and II are within 1–2 ppm of the chemical shifts of Aβ42 fibrils 15,29 (Table S2). The Arctic conformer I L34 Cδ2 chemical shift of 20.9 ppm matches the reported Aβ42 values but is more than 2 ppm upfield of the Osaka and wild-type Aβ40 chemical shifts 29. The Arctic conformer I L34 15N chemical shift (111.5 ppm) matches that of Aβ42 but is 12 ppm upfield of the 15N chemical shifts of Aβ40 fibrils 15. Arctic G38 chemical shifts show the largest dispersion, 3–5 ppm, among all labeled residues. When we compared the G38 15N and 13CO chemical shifts, which are sensitive to both local conformation and electrostatic effects, to literature values, we find that Arctic conformer II resembles the brain-derived Aβ40 16, while no other conformers agree well with any Aβ peptides.
To obtain quantitative measures of fibril structural similarities, we calculated per-residue average chemical shift differences between the Arctic I conformer and various published Aβ structures, and between our wild-type Aβ40 and reported Aβ structures (Fig. 5a, b). The comparison quantifies the similarity between Arctic Aβ40 and wild-type Aβ42, with differences of less than 0.6–1.0 ppm at F19, A21, I32, and L34 29. Near the C terminus, at V36 and G38, the chemical shift differences increase, as expected since the additional two residues in Aβ42 form part of an extra β-strand that cannot exist in Aβ40 fibrils 15. The chemical shifts of our wild-type Aβ40 fibrils are similar to those of both two-fold and three-fold symmetric in vitro WT fibrils, with slightly better agreement with the three-fold fibrils, principally at I32 12,13. Our Osaka DMSO and Osaka NaOH fibrils exhibit non-negligible chemical shift differences, but the Osaka DMSO chemical shifts are identical within experimental uncertainty to the chemical shifts of recombinant Osaka fibrils 14. To compare all the Aβ fibrils reported in the literature and the samples studied here, we computed the Pearson product-moment correlation coefficient (Fig. 5e, Fig. S6). A total of eleven Aβ fibril samples were analyzed, and four chemical shifts, 15N, C′, Cα, and Cβ, were included in the correlation coefficient calculation. The 2D heat maps confirm that Arctic I has the highest correlation with WT Aβ42, and our Osaka DMSO data has excellent correlation with the literature Osaka structure. The 2D correlation map also revealed that the WT Aβ40 sample in this study has significant similarity to the Osaka NaOH sample.
Figure 5.

Per-residue average chemical shift differences between different Aβ fibrils, calculated using equation 1. 13C chemical shifts were referenced to DSS for the calculation. (a) Chemical shift differences of Arctic conformer I from published wild-type Aβ40 and Aβ42 data. (b) Chemical shift differences of wild-type Aβ40 studied here from published wild-type Aβ40 and Aβ42 data. (c) Chemical shift differences between Osaka DMSO and Osaka NaOH studied here. (d) Chemical shift differences of Osaka DMSO from recombinant Osaka Aβ40. The very small differences indicate that the synthetic Osaka fibrils have the same structure as fibrils obtained from recombinant peptide 14. (e) 2D heat map representing the average pairwise Pearson product-moment correlation coefficients between the chemical shifts of Aβ fibrils. Eleven different Aβ fibrils, including five studied here and six literature cases, are compared. The white cross in each row indicates the strain with the highest correlation coefficient. Arctic conformer I has the highest correlation with WT-42 2MXU. PDB codes are given where available to simplify notation. WT-42 corresponds to the study of Colvin et al 29, Osaka 2MVX corresponds to the study of Schutz et al 70, WT 2M4J is the study of Lu et al 16, WT 2LMP/Q is that of Paravastu et al 12, and WT 2LMN/O is that of Petkova et al 13.
Mobility of Aβ40 fibrils
To obtain information about the amplitude and rates of motion of the Aβ40 fibrils, we measured 13C-1H dipolar order parameters (SCH) and NMR relaxation times. Representative 13C-1H dipolar dephasing data from the 2D DIPSHIFT spectra are shown in Fig. 6a. The backbone Cα-Hα dipolar order parameters are at least 0.90, indicating that fast motions are largely absent, while sidechain order parameters range from 0.24 to 0.50 (Fig. S7). Interestingly, Osaka DMSO and wild-type Aβ40 exhibit similar order parameters that are smaller than the Arctic Aβ40 SCH values for both the backbone and sidechains. However, the Arctic sample exhibits larger intensity asymmetry in the dipolar oscillation, indicating more pronounced microsecond timescale motion 71,72.
Figure 6.

Mobilities of Aβ40 fibrils. (a) Representative 13C-1H dipolar dephasing curves for Arctic, wild-type and Osaka DMSO Aβ40 fibrils. Best-fit couplings and the resulting order parameters (SCH) are given in each panel. All fibrils show mostly immobilized backbones but significant sidechain motions. The data were obtained at 298 K under 7 kHz MAS. (b) Representative 13C-detected 1H T1ρ relaxation data of the three Aβ40 fibrils. Top row: Arctic E22G Aβ40 shows longer T1ρ than wild-type and Osaka Aβ40 at 298 K. Second to fourth rows: temperature dependence of the 1H T1ρ. Arctic Aβ40 shows shorter T1ρ at lower temperature, while wild-type and Osaka Aβ40 exhibit longer T1ρ’s at lower temperature. These indicate that the Arctic Aβ40 sidechains have faster microsecond motions than the other two fibrils.
13C-resolved 1H T1ρ relaxation times reveal further differences in the microsecond motions of the three fibrils. At 298 K, Arctic E22G Aβ40 shows the longest 1H T1ρ’s while wild-type Aβ40 has the shortest relaxation times (Fig. 6b, Table S3). To determine whether these relaxation rates reflect motions faster or slower than the ~16 μs characteristic timescale of our T1ρ experiment, we measured the temperature dependence of T1ρ. Interestingly, decreasing the temperature to 260 K shortened the relaxation times of Arctic Aβ40 residues while lengthening those of wild-type Aβ40 (Fig. S8). Thus, Arctic Aβ40 motion occurs on timescales faster than ~16 μs at ambient temperature while wild-type Aβ40 motion occurs in the slow limit relative to the characteristic time. Osaka Aβ40 T1ρ displays a mixed temperature dependence: the F19 sidechain exhibits slow motional behavior while I32 and L34 sidechains show fast-limit motional characteristics.
Together, these order parameters and relaxation times indicate that, while all three Aβ samples are relatively rigid, there are non-negligible differences in the peptide motion. Wild-type Aβ40 has larger motional amplitudes than the other two peptides but much slower motions, while Arctic E22G Aβ40 has the smallest amplitudes for fast motions but has the fastest motion on the microsecond time scale.
Hydration and inter-residue contacts of Aβ40 fibrils
Water contact and long-range inter-residue correlations provide useful constraints for the three-dimensional structures of Aβ fibrils. Representative 1D and 2D 13C-detected water-to-protein 1H spin diffusion spectra are shown in Fig. 7 and Fig. S9. Compared with the equilibrium spectra measured with 100 ms 1H mixing, the Osaka fibrils have the highest initial intensities at 4 ms among the three samples, indicating that the Osaka fibrils are the most hydrated. Both the wild-type and Osaka fibrils exhibit similar spectral envelopes at 4 ms as the 100 ms spectra, indicating relatively uniform hydration for these fibrils, but the Arctic sample displays a significantly different intensity pattern at 4 ms compared to 100 ms, with the A21 signals dominating the spectrum, indicating that A21 is preferentially hydrated over other residues. Consistently, when the water-to-protein 1H spin diffusion is measured as a function of the mixing time, the buildup curves (Fig. 7d–f, Fig. S5c) show that water polarization transfer to Arctic and wild-type peptides reached equilibrium at ~100 ms, while the Osaka Aβ40 fibrils intensities have equilibrated at ~50 ms, indicating that the Osaka fibrils have a larger water-exposed surface area. Moreover, wild-type and Osaka fibrils have uniform buildup rates across the different labeled residues, with a narrow intensity distribution at 4 ms, while Arctic Aβ40 fibrils exhibit a broader intensity distribution of 10–25% at 4 ms, indicating heterogeneous hydration, consistent with the conformational polymorphism of the Arctic Aβ fibrils.
Figure 7.

Different hydration behaviors of the three Aβ40 fibrils. (a–c) 13C-detected water-to-protein 1H spin diffusion spectra at 100 ms and 4 ms for (a) Arctic (E22G) Aβ40, (b) wild-type Aβ40, and (c) Osaka (E22Δ) Aβ40. The Osaka fibrils show the highest initial intensity. The Arctic initial intensity distribution deviates from the equilibrium intensity more than the wild-type peptide fibrils. (d–f) Water-to-protein 1H spin diffusion buildup curves for (d) Arctic Aβ40, (e) wild-type Aβ40, and (f) Osaka Aβ40. Vertical dashed line and shaded horizontal bars guide the eye for the magnetization equilibration times and the initial buildup rates. Osaka Aβ40 has the shortest equilibration time, indicating the highest hydration, while Arctic Aβ40 has the largest distribution of buildup rates.
Using a 1.5 s mixing 13C-13C PDSD experiment, we looked for long-range correlations among the labeled residues. In the Arctic spectrum, the F19 sidechain shows several cross peaks to the I32 sidechain of conformer I but no cross peaks to any other residues, while the Osaka spectrum shows F19 cross peaks with L34 and V36 sidechains (Fig. 8). No F19 cross peaks were observed for the wild-type sample. Fig. 9 compares four Aβ fibril structures solved using solid-state NMR, with special emphasis on the hydrophobic contacts between F19 and the last ten residues of the peptide. The two-fold and three-fold wild-type Aβ40 structures exhibit intermolecular F19 – L34 contacts: the F19 ring of one peptide packs against the L34 sidechain of a different peptide with a short distance of ~4 Å 12,13. A D23–K28 salt bridge is present in the two-fold structure and the three-fold brain-derived structure, but absent in the three-fold in vitro structure 13,16,25. A subtly different two-fold structural model 22 has also been reported using fibrils formed by Aβ(M1–40), which contains an exogenous N-terminal methionine. This model (not shown here) differs in the intramolecular sidechain packing and intermolecular contacts between the two cross-β subunits. In the published structure of Osaka Aβ40 fibrils derived from recombinant protein, F19 and L34 form intramolecular contacts of ~ 4 Å, which lock the turn from F19 to L34 14,73. Compared to these Aβ40 structures, Aβ42 has a distinct pattern of long F19–L34 distances and short F19–I32 distances of ~4 Å 15. Our observed F19 sidechain - L34 Cδ2 cross peaks in the Osaka PDSD spectrum are fully consistent with the reported Osaka Aβ40 structure 70, while the absence of F19–L34 cross peaks in conjunction with the observed F19–I32 cross peaks in our Arctic PDSD spectrum rules out the wild-type Aβ40 and Osaka Aβ40 structures as possible folds for any of the Arctic conformations. However, the observed F19–I32 cross peaks of Arctic conformer I is consistent with the Aβ42 fold. Fig. 9 also shows that the A21 methyl group faces inward toward the other strand in the two wild-type Aβ40 structures, but points outward to water in the Osaka Aβ40 and Aβ42 structures. The preferential hydration of A21 observed in our Arctic sample is thus also consistent with the Aβ42 fold.
Figure 8.
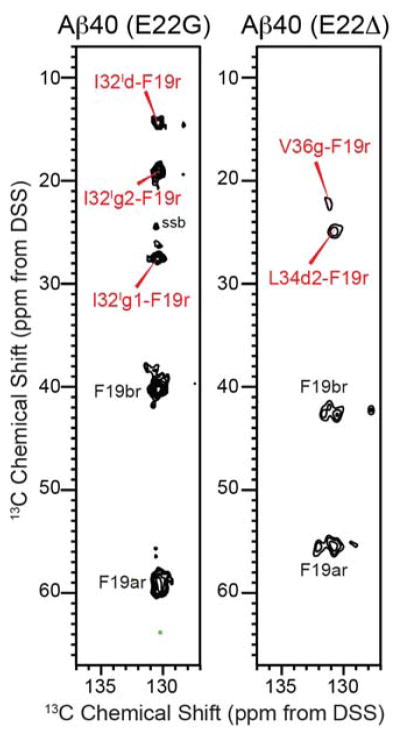
Inter-residue contacts of the Ab40 fibrils from 1.5 s mixing 2D 13C-13C PDSD spectra. In the aromatic region, Arctic Ab40 (E22G) shows several F19–I32 cross peaks for conformer I while Osaka Ab40 (E22D) shows F19–L34 and F19–V36 cross peaks. The unresolved aromatic carbons of F19 are denoted as F19r.
Figure 9.

Comparison of four published structures of wild-type and mutant Aβ fibrils. The sidechains of residues labeled in this study are shown as red and green sticks. Key charged residues that are known to be important for the three-dimensional fold of the peptides are shown in blue. (a) Two-fold in vitro structure of wild-type Aβ40 (PDB: 2LMN) 13. (b) Three-fold in vitro structure of wild-type Aβ40 (PDB: 2LMQ) 12. (c) Osaka Aβ40 (E22Δ) structure 14. (d) Wild-type Aβ42 structure (PDB: 2MXU) 15. The shortest F19–I32 and F19–L34 distances are indicated in the structures. Note that the distances in (a) and (b) are intermolecular, while distances in (c) and (d) are intramolecular.
Discussion
These chemical shift data indicate that the E22G mutant of Aβ40 is highly polymorphic, and this polymorphism is intrinsic to this particular amino acid sequence. At least four distinct sets of chemical shifts are resolved, with population ratios of about 3 : 3 : 2 : 2, thus the minor forms are not negligible. This molecular structural polymorphism is reproducible from one sample preparation to another, is unaffected by the initial solvent used in the fibrilization (Fig. S4), and occurs despite shaking during fibril growth, which tends to promote self-seeding and conformational homogeneity 23,32, thus the Arctic molecular structure polymorphism is robust. Under the same fibrilization condition, the E22Δ Aβ40 fibrils, with initial solubilization in DMSO, exhibited a single set of chemical shifts that are identical within experimental uncertainty to the published Osaka chemical shifts 14, while wild-type Aβ40 is dominated by one major conformer. Therefore, the Arctic structural polymorphism is not an artifact of sample preparation but is due to the mutation of the anionic Glu to Gly at residue 22. The chemical shift heterogeneity of the Arctic fibrils is qualitatively consistent with the presence of both straight and twisted fibrils with variable twist periodicities in the TEM images (Fig. 1), which in turn agrees well with a detailed TEM and AFM study by Antzutkin and coworkers, who resolved and quantitatively analyzed 4–5 types of Arctic fibril morphologies with different extents of twists and heights. They further measured the mass-per-length of four of the polymorphs and found them to correspond to 2, 4 and 6 peptide molecules per 0.48 nm repeat of the fibrils 66, suggesting that the molecular-level polymorphism of Arctic Aβ may manifest as different numbers of cross-β subunits.
The Arctic Aβ40 polymorphism is discrete, as shown by the much narrower linewidths of each conformer compared to the chemical shift differences between conformers. This property differs from that of membrane peptides and proteins, which usually exhibit a single broad peak per site, indicating continuous Gaussian distributions of conformations 59,74, The discreteness of the Arctic structural polymorphism can be attributed to the quasi-crystalline nature of the fibrils, which preserve and amplify the structure homogeneity within each fibril but leave different fibrils able to adopt different structures that started from a specific monomer or nucleus structure. The narrow linewidths of both the “monomorphic” Osaka Aβ40 and each conformer of the polymorphic Arctic Aβ40 also indicate that Aβ fibrils obtained from synthetic peptides without seeding can have inherently homogeneous structures that are comparable to the homogeneity of fibrils obtained from recombinant proteins 14,17,29.
Since our main motivation in this study is to understand the origins of Aβ polymorphism, we did not try to reduce or eliminate the Arctic structural polymorphism by seeding, and it remains to be seen whether repeated rounds of seeding can render the Arctic fibril structures more homogeneous as it has for other Aβ peptides 32,75. Even without a unique structure, chemical shifts and other lines of evidence constrain the structures of the major Arctic conformer I and rule out unlikely structural models. Since different chemical shifts cannot correspond to the same structures, our measured chemical shifts (Fig. 5, Table S2) indicate that Arctic conformer I differs from all published Aβ40 structures so far, but bears significant resemblance to wild-type Aβ42. This chemical shift similarity is supported by the observed F19–I32 contact, which is absent in all Aβ40 structures but present in the Aβ42 structure, and the preferential hydration of A21, which is inward-facing in wild-type Aβ40 structures but outward-facing in Osaka Aβ40 and wild-type Aβ42 structures (Fig. 9). Other biophysical and biological data have previously alluded to similar physical properties of Arctic Aβ40 and Aβ42 fibrils. Both Arctic Aβ40 and wild-type Aβ42 fibrils have faster and more heterogeneous growth than wild-type Aβ40 66, and both Arctic Aβ40 and wild-type Aβ42 fibrils appear in TEM and AFM images as more twisted and entangled than wild-type Aβ40 fibrils 18,66.
The hydration data provide further insights into the overall fold of Arctic Aβ40. The water-protein spin diffusion rates reflect the water-exposed surface area of the fibrils. Ignoring the unstructured N-terminal loop, the external surface area of the fibril core differs for the four Aβ structures (Fig. 9): the approximate cross-sectional areas are 5 × 3 nm for the two-fold wild-type Aβ40, 7 × 7 × 7 nm for the three-fold wild-type Aβ40, 6 × 3 nm for Osaka Aβ40, and a compact 4.5 × 3.5 nm for Aβ42. The smaller dimension of the Osaka structure is consistent with the observed fast water 1H spin diffusion to the Osaka Aβ40, while the large dimension of the three-fold Aβ40 structure is consistent with the slow water magnetization transfer to the wild-type Aβ40. Therefore, the slow water-protein spin diffusion of the Arctic sample suggests a large fibril cross section. If the Arctic conformer I has a similar subunit structure to Aβ42, then our data suggest the existence of multiple cross-β subunits in the Arctic I fibril. For the 4.5 × 3.5 nm S-shape triple-β structure of Aβ42 reported by Ishii and coworkers, corresponding TEM images showed fibril diameters ranging from 4.5–6.0 nm for thin fibrils to 6–13 nm for thicker ones. Thus, while a single subunit was proposed for the structure, a dimer model cannot be excluded 15. Earlier mass-per-length (MPL) data of Aβ42 fibrils showed a broad distribution of 13–50 kD/nm with a maximum at 19 kD/nm 76, also suggesting that Aβ42 fibrils have multiple cross-β subunits, with the dimer being dominant. Interestingly, this Aβ42 MPL distribution is much broader than reported wild-type Aβ40 fibrils 12,25, indicating more pronounced polymorphism, which is also seen for the chemical-shift-similar Arctic Aβ40 fibrils. Indeed, during the publication of this study, an atomic-resolution SSNMR structure of Aβ42 was reported that revealed a dimer as the repeat motif of the cross-β structure 77.
In addition to water on the external fibril surface, X-ray fiber diffraction data and MD simulations suggest the presence of water inside the Aβ fibril core. Comparison of the calculated diffraction patterns for solid versus hollow cylinders with the measured diffraction pattern suggests that the three-fold in-vitro structure of wild-type Aβ40 should contain a hollow water-filled core 78. MD simulations suggest that this central cavity may have dimensions ranging from 1.6 to 2.3 nm 79, and water molecules may diffuse from the fibril ends into the core and may localize in the turn region to solvate the D23–K28 salt bridge 80,81. The current water-to-protein 1H spin diffusion experiment does not explicitly distinguish external versus internal water; but under the condition of our experiments, the external water may be preferentially detected because of its larger quantity. Interior water cavities have also been reported in other amyloid fibrils, including residues 105–115 of transthyretin 6, the SH3 domain of PI3 kinase 82, and a poly-L-glutamine peptide of huntingtin 83, thus they may be a general phenomenon of interest. Additional more specialized experiments will be necessary to differentiate external and internal water molecules.
The polymorphism of E22G Aβ40 is also consistent with the enhanced microsecond motions of the peptide compared to the wild-type counterpart and the Osaka mutant. Our data indicate that the motional amplitudes in the Arctic fibrils are small for very fast motions but larger for microsecond motions, as seen by the DIPSHIFT intensity asymmetry. Perhaps more importantly, the motional rates are faster when residue 22 is occupied by Gly than if it is occupied by the negatively charged Glu, or if Ala21 is followed directly by Asp23. The replacement of a charged Glu by Gly at residue 22 likely has the simultaneous effect of perturbing a stabilizing electrostatic interaction and conferring greater conformational flexibility to the peptide backbone. Our data here do not indicate the relative importance of these two factors for polymorphism, but comparison with literature suggests that both factors play a role. An intermolecular K16–E22 salt bridge has been observed in WT fibrils prepared quiescently 25 while absent in antiparallel fibrils of the D23N mutant 31. However, a charged E22 may impact the fibril structure at the nucleation stage, by restricting the initial conformational ensemble, without manifesting a permanent salt bridge in the mature fibril. The fact that the charge-removing D23N mutation at the next residue caused the coexistence of parallel and antiparallel fibrils 31,32, which is a dramatic departure from the parallel-only fibrils of the WT peptide, also suggests that removing an electrostatic charge in this region increases the conformational heterogeneity. For the D23N mutant, the parallel fibrils resemble the WT parallel fibril structure, which differ from the E22G mutant, where none of the four polymorphs resembles any existing Aβ40 structures based on the chemical shifts. Thus E22 appears to exert a more significant effect than D23 on the final three-dimensional fold of the fibril. This is generally consistent with the fact that E22 harbors the largest number of single-site mutations in Aβ peptides 34.
Conclusions
These solid-state NMR data show that mutation of Glu at residue 22 to Gly causes pronounced structural polymorphism and larger-amplitude microsecond motions in Aβ40 fibrils. 13C and 15N chemical shifts indicate that at least four distinct molecular conformations exist in this Arctic mutant, none of which resembles the wild-type and other mutant Aβ40 structures determined so far. However, chemical shifts and inter-residue contacts of one of the Arctic conformers have significant resemblances to those of Aβ42 fibrils, suggesting similar structures. Comparison with the wild-type peptide and with a E22 deletion mutant indicate that the amino acid sequence with the lowest charge density in the region of residues 21–23, which is the Arctic Ala-Gly-Asp, has the highest degree of structural polymorphism and the fastest microsecond motions, while the amino acid sequence with the highest charge density in this region, which is the wild-type Ala-Glu-Asp, displays low polymorphism and the slowest microsecond motion. The intermediate sequence, adopted by the Osaka mutant, Ala-Asp, has a single structure and intermediate motional properties between the wild-type and Arctic Aβ40. These results suggest that electrostatic interactions in this non-β region of the Aβ peptide, together with conformational flexibility, are crucial for determining the number of possible β-strand arrangements and the exact three-dimensional folds of this family of amyloid fibrils 73. Interactions of these structurally distinct fibrils with other cellular macromolecules can conceivably lead to different AD phenotypes.
Supplementary Material
Acknowledgments
The SSNMR part of the work at MIT is supported by National Institutes of Health grants GM088204 and P01 AG002132. 900 MHz NMR spectra were measured at the MIT/Harvard Center for Magnetic Resonance, which is supported by NIH grant EB002026. W.F.D., M.N., S.B.P and J.S are supported by grants from the National Institutes of Health (AG02132 and AG10770) and T32 training grant HL07731 (M.N.). J.S. was supported by a fellowship from the Glenn Foundation and the New Investigator Research Grant (NIRG) of the Alzheimer’s Association. We thank Dr. Y. Wu for measuring the 1H solution NMR spectra.
Footnotes
Supporting Information Available:
Additional NMR spectra, tables, and sample characterization data include:
Peptide purification and early fibrilization data (Figures S1 and S2);
Arctic Aβ chemical shifts and their comparison with other Aβ fibrils (Tables S1 and S2);
Comparative 1D and 2D spectra of Aβ fibrils (Figures S3, S4, S5);
Residue-specific heat maps of chemical shift differences (Figure S6);
Additional order parameter and relaxation data (Figures S7, S8, Table S3);
2D water-edited spectra (Figure S9);
This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Tanzi RE, Bertram L. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J, Selkoe DJ. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 3.Wasmer C, Lange A, Van Melckebeke H, Siemer AB, Riek R, Meier BH. Science. 2008;319:1523–1526. doi: 10.1126/science.1151839. [DOI] [PubMed] [Google Scholar]
- 4.Meier BH, Bockmann A. Curr Opin Struct Biol. 2015;30:43–49. doi: 10.1016/j.sbi.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 5.Helmus JJ, Surewicz K, Surewicz WK, Jaroniec CP. J Am Chem Soc. 2010;132:2393–2403. doi: 10.1021/ja909827v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fitzpatrick AW, Debelouchina GT, Bayro MJ, Clare DK, Caporini MA, Bajaj VS, Jaroniec CP, Wang L, Ladizhansky V, Muller SA, MacPhee CE, Waudby CA, Mott HR, De Simone A, Knowles TP, Saibil HR, Vendruscolo M, Orlova EV, Griffin RG, Dobson CM. Proc Natl Acad Sci USA. 2013;110:5468–5473. doi: 10.1073/pnas.1219476110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Debelouchina GT, Bayro MJ, Fitzpatrick AW, Ladizhansky V, Colvin MT, Caporini MA, Jaroniec CP, Bajaj VS, Rosay M, MacPhee CE, Vendruscolo M, Maas WE, Dobson CM, Griffin RG. J Am Chem Soc. 2013;135:19237–19247. doi: 10.1021/ja409050a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su Y, Sarell CJ, Eddy MT, Debelouchina GT, Andreas LB, Pashley CL, Radford SE, Griffin RG. J Am Chem Soc. 2014;136:6313–6325. doi: 10.1021/ja4126092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VM, George JM, Rienstra CM. Nat Struct Mol Biol. 2016;23:409–415. doi: 10.1038/nsmb.3194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heise H, Hoyer W, Becker S, Andronesi OC, Riedel D, Baldus M. Proc Natl Acad Sci USA. 2005;102:15871–15876. doi: 10.1073/pnas.0506109102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tycko R. Annu Rev Phys Chem. 2011;62:279–299. doi: 10.1146/annurev-physchem-032210-103539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paravastu AK, Leapman RD, Yau WM, Tycko R. Proc Natl Acad Sci USA. 2008;105:18349–18354. doi: 10.1073/pnas.0806270105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Petkova AT, Yau WM, Tycko R. Biochemistry. 2006;45:498–512. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schutz AK, Vagt T, Huber M, Ovchinnikova OY, Cadalbert R, Wall J, Guntert P, Bockmann A, Glockshuber R, Meier BH. Angew Chem Int Ed Engl. 2015;54:331–335. doi: 10.1002/anie.201408598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao YL, Ma BY, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, Ishii Y. Nat Struct Mol Biol. 2015;22:499–U497. doi: 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lopez del Amo JM, Schmidt M, Fink U, Dasari M, Fandrich M, Reif B. Angew Chem Int Edit. 2012;51:6136–6139. doi: 10.1002/anie.201200965. [DOI] [PubMed] [Google Scholar]
- 18.Antzutkin ON. Magn Reson Chem. 2004;42:231–246. doi: 10.1002/mrc.1341. [DOI] [PubMed] [Google Scholar]
- 19.Tycko R. Prot Sci. 2014;23:1528–1539. doi: 10.1002/pro.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Meinhardt J, Sachse C, Hortschansky P, Grigorieff N, Fandrich M. J Mol Biol. 2009;386:869–877. doi: 10.1016/j.jmb.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Antzutkin ON, Balbach JJ, Leapman RD, Rizzo NW, Reed J, Tycko R. Proc Natl Acad Sci USA. 2000;97:13045–13050. doi: 10.1073/pnas.230315097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bertini I, Gonnelli L, Luchinat C, Mao JF, Nesi A. J Am Chem Soc. 2011;133:16013–16022. doi: 10.1021/ja2035859. [DOI] [PubMed] [Google Scholar]
- 23.Qiang W, Kelley K, Tycko R. J Am Chem Soc. 2013;135:6860–6871. doi: 10.1021/ja311963f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petkova AT, Buntkowsky G, Dyda F, Leapman RD, Yau WM, Tycko R. J Mol Biol. 2004;335:247–260. doi: 10.1016/j.jmb.2003.10.044. [DOI] [PubMed] [Google Scholar]
- 25.Petkova AT, Leapman RD, Guo Z, Yau WM, Mattson MP, Tycko R. Science. 2005;307:262–265. doi: 10.1126/science.1105850. [DOI] [PubMed] [Google Scholar]
- 26.Masuda Y, Uemura S, Ohashi R, Nakanishi A, Takegoshi K, Shimizu T, Shirasawa T, Irie K. Chem Bio Chem. 2009;10:287–295. doi: 10.1002/cbic.200800411. [DOI] [PubMed] [Google Scholar]
- 27.Jarrett JT, Berger EP, Lansbury PT. Biochemistry. 1993;32:4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 28.Parthasarathy S, Inoue M, Xiao YL, Matsumura Y, Nabeshima Y, Hoshi M, Ishii Y. J Am Chem Soc. 2015;137:6480–6483. doi: 10.1021/jacs.5b03373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Colvin MT, Silvers R, Frohm B, Su Y, Linse S, Griffin RG. J Am Chem Soc. 2015;137:7509–7518. doi: 10.1021/jacs.5b03997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huber M, Ovchinnikova OY, Schutz AK, Glockshuber R, Meier BH, Bockmann A. Biomol NMR Assign. 2015;9:7–14. doi: 10.1007/s12104-013-9535-x. [DOI] [PubMed] [Google Scholar]
- 31.Qiang W, Yau WM, Luo Y, Mattson MP, Tycko R. Proc Natl Acad Sci USA. 2012;109:4443–4448. doi: 10.1073/pnas.1111305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qiang W, Yau WM, Tycko R. J Am Chem Soc. 2011;133:4018–4029. doi: 10.1021/ja109679q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mathis CA, Wang YM, Holt DP, Huang GF, Debnath ML, Klunk WE. J Med Chem. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- 34.Benilova I, Karran E, De Strooper B. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 35.Hendriks L, Vanduijn CM, Cras P, Cruts M, Vanhul W, Vanharskamp F, Warren A, Mcinnis MG, Antonarakis SE, Martin JJ, Hofman A, Van Broeckhoven C. Nat Genet. 1992;1:218–221. doi: 10.1038/ng0692-218. [DOI] [PubMed] [Google Scholar]
- 36.Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, Wada Y, Yoshioka E, Nishizaki T, Watanabe Y, Mori H. Ann Neurol. 2008;63:377–387. doi: 10.1002/ana.21321. [DOI] [PubMed] [Google Scholar]
- 37.Bugiani O, Giaccone G, Rossi G, Mangieri M, Capobianco R, Morbin M, Mazzoleni G, Cupidi C, Marcon G, Giovagnoli A, Bizzi A, Di Fede G, Puoti G, Carella F, Salmaggi A, Romorini A, Patruno GM, Magoni M, Padovani A, Tagliavini F. Arch Neurol. 2010;67:987–995. doi: 10.1001/archneurol.2010.178. [DOI] [PubMed] [Google Scholar]
- 38.Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, Naslund J, Lannfelt L. Nat Neurosci. 2001;4:887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 39.Van Broeckhoven C, Haan J, Bakker E, Hardy JA, Van Hul W, Wehnert A, Vegter-Van der Vlis M, Roos RA. Science. 1990;248:1120–1122. doi: 10.1126/science.1971458. [DOI] [PubMed] [Google Scholar]
- 40.Grabowski TJ, Cho HS, Vonsattel JPG, Rebeck GW, Greenberg SM. Ann Neurol. 2001;49:697–705. doi: 10.1002/ana.1009. [DOI] [PubMed] [Google Scholar]
- 41.Englund H, Sehlin D, Johansson AS, Nilsson LN, Gellerfors P, Paulie S, Lannfelt L, Pettersson FE. J Neurochem. 2007;103:334–345. doi: 10.1111/j.1471-4159.2007.04759.x. [DOI] [PubMed] [Google Scholar]
- 42.Johansson AS, Berglind-Dehlin F, Karlsson G, Edwards K, Gellerfors P, Lannfelt L. FEBS J. 2006;273:2618–2630. doi: 10.1111/j.1742-4658.2006.05263.x. [DOI] [PubMed] [Google Scholar]
- 43.Cheng IH, Palop JJ, Esposito LA, Bien-Ly N, Yan FG, Mucke L. Nat Med. 2004;10:1190–1192. doi: 10.1038/nm1123. [DOI] [PubMed] [Google Scholar]
- 44.Lord A, Kalimo H, Eckman C, Zhang XQ, Lannfelt L, Nilsson LN. Neurobiol Aging. 2006;27:67–77. doi: 10.1016/j.neurobiolaging.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 45.Lam AR, Teplow DB, Stanley HE, Urbanc B. J Am Chem Soc. 2008;130:17413–17422. doi: 10.1021/ja804984h. [DOI] [PubMed] [Google Scholar]
- 46.Stohr J, Watts JC, Mensinger ZL, Oehler A, Grillo SK, DeArmond SJ, Prusiner SB, Giles K. Proc Natl Acad Sci USA. 2012;109:11025–11030. doi: 10.1073/pnas.1206555109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watts JC, Condello C, Stohr J, Oehler A, Lee J, DeArmond SJ, Lannfelt L, Ingelsson M, Giles K, Prusiner SB. Proc Natl Acad Sci USA. 2014;111:10323–10328. doi: 10.1073/pnas.1408900111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finder VH, Vodopivec I, Nitsch RM, Glockshuber R. J Mol Biol. 2010;396:9–18. doi: 10.1016/j.jmb.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 49.Hong M, Griffin RG. J Am Chem Soc. 1998;120:7113–7114. [Google Scholar]
- 50.Rienstra CM, Tucker-Kellogg L, Jaroniec CP, Hohwy M, Reif B, McMahon MT, Tidor B, Lozano-Perez T, Griffin RG. Proc Natl Acad Sci USA. 2002;99:10260–10265. doi: 10.1073/pnas.152346599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Takegoshi K, Nakamura S, Terao T. Chem Phys Lett. 2001;344:631–637. [Google Scholar]
- 52.Hong M. J Biomol NMR. 1999;15:1–14. doi: 10.1023/a:1008334204412. [DOI] [PubMed] [Google Scholar]
- 53.Baldus MP, AT, Herzfeld J, Griffin RG. Mol Phys. 1998;95:1197–1207. [Google Scholar]
- 54.Kumashiro KK, Schmidt-Rohr K, Murphy OJI, Ouellette KL, Cramer WA, Thompson LK. J Am Chem Soc. 1998;120:5043–5051. [Google Scholar]
- 55.Luo W, Hong M. J Am Chem Soc. 2006;128:7242–7251. doi: 10.1021/ja0603406. [DOI] [PubMed] [Google Scholar]
- 56.Ader C, Schneider R, Seidel K, Etzkorn M, Becker S, Baldus M. J Am Chem Soc. 2009;131:170–176. doi: 10.1021/ja806306e. [DOI] [PubMed] [Google Scholar]
- 57.White PB, Wang T, Park YB, Cosgrove DJ, Hong M. J Am Chem Soc. 2014;136:10399–10409. doi: 10.1021/ja504108h. [DOI] [PubMed] [Google Scholar]
- 58.Williams JK, Hong M. J Magn Reson. 2014;247:118–127. doi: 10.1016/j.jmr.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liao SY, Fritzsching KJ, Hong M. Protein Sci. 2013;22:1623–1638. doi: 10.1002/pro.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Munowitz MG, Griffin RG, Bodenhausen G, Huang TH. J Am Chem Soc. 1981;103:2529–2533. [Google Scholar]
- 61.Bielecki A, Kolbert AC, Levitt MH. Chem Phys Lett. 1989;155:341–346. [Google Scholar]
- 62.Hong M, Gross JD, Rienstra CM, Griffin RG, Kumashiro KK, Schmidt-Rohr K. J Magn Reson. 1997;129:85–92. doi: 10.1006/jmre.1997.1242. [DOI] [PubMed] [Google Scholar]
- 63.Hong M. J Magn Reson. 1999;139:389–401. doi: 10.1006/jmre.1999.1805. [DOI] [PubMed] [Google Scholar]
- 64.Rothwell WP, Waugh JS. J Chem Phys. 1981;74:2721–2732. [Google Scholar]
- 65.Cady SD, Hong M. J Biomol NMR. 2009;45:185–196. doi: 10.1007/s10858-009-9352-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Norlin N, Hellberg M, Filippov A, Sousa AA, Grobner G, Leapman RD, Almqvist N, Antzutkin ON. J Struct Biol. 2012;180:174–189. doi: 10.1016/j.jsb.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Betts V, Leissring MA, Dolios G, Wang R, Selkoe DJ, Walsh DM. Neurobiol Dis. 2008;31:442–450. doi: 10.1016/j.nbd.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cloe AL, Orgel JP, Sachleben JR, Tycko R, Meredith SC. Biochemistry. 2011;50:2026–2039. doi: 10.1021/bi1016217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stohr J, Condello C, Watts JC, Bloch L, Oehler A, Nick M, DeArmond SJ, Giles K, DeGrado WF, Prusiner SB. Proc Natl Acad Sci USA. 2014;111:10329–10334. doi: 10.1073/pnas.1408968111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schutz AK, Habenstein B, Luckgei N, Bousset L, Sourigues Y, Nielsen AB, Melki R, Bockmann A, Meier BH. Biomol NMR Assign. 2014;8:349–356. doi: 10.1007/s12104-013-9515-1. [DOI] [PubMed] [Google Scholar]
- 71.Cobo MF, Achilles A, Reichert D, Deazevedo ER, Saalwächter K. J Magn Reson. 2012;221:85–96. doi: 10.1016/j.jmr.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 72.deAzevedo ER, Saalwachter K, Pascui O, de Souza AA, Bonagamba TJ, Reichert D. J Chem Phys. 2008;128:104505. doi: 10.1063/1.2831798. [DOI] [PubMed] [Google Scholar]
- 73.Schledorn M, Meier BH, Bockmann A. Front Mol Biosci. 2015;2:14. doi: 10.3389/fmolb.2015.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Su Y, Hong M. J Phys Chem B. 2011;115:10758–10767. doi: 10.1021/jp205002n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ravotti F, Walti MA, Guntert P, Riek R, Bockmann A, Meier BH. Biomol NMR Assign. 2016 doi: 10.1007/s12104-016-9682-y. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 76.Antzutkin ON, Leapman RD, Balbach JJ, Tycko R. Biochemistry. 2002;41:15436–15450. doi: 10.1021/bi0204185. [DOI] [PubMed] [Google Scholar]
- 77.Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev IV, Rosay M, Donovan KJ, Michael B, Wall JS, Linse S, Griffin RG. J Am Chem Soc. 2016 doi: 10.1021/jacs.6b05129. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McDonald M, Box H, Bian W, Kendall A, Tycko R, Stubbs G. J Mol Biol. 2012;423:454–461. doi: 10.1016/j.jmb.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miller Y, Ma B, Nussinov R. J Am Chem Soc. 2011;133:2742–2748. doi: 10.1021/ja1100273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Buchete NV, Tycko R, Hummer G. J Mol Biol. 2005;353:804–821. doi: 10.1016/j.jmb.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 81.Prade E, Bittner HJ, Sarkar R, Lopez Del Amo JM, Althoff-Ospelt G, Multhaup G, Hildebrand PW, Reif B. J Biol Chem. 2015;290:28737–28745. doi: 10.1074/jbc.M115.675215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bayro MJ, Maly T, Birkett NR, MacPhee CE, Dobson CM, Griffin RG. Biochemistry. 2010;49:7474–7484. doi: 10.1021/bi100864t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Perutz MF, Finch JT, Berriman J, Lesk A. Proc Natl Acad Sci U S A. 2002;99:5591–5595. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.