

Abstract
Neocortical projection neurons, which segregate into six cortical layers according to their birthdate, have diverse morphologies, axonal projections and molecular profiles, yet they share a common cortical regional identity and glutamatergic neurotransmission phenotype. Here we demonstrate that distinct genetic programs operate at different stages of corticogenesis to specify the properties shared by all neocortical neurons. Ngn1 and Ngn2 are required to specify the cortical (regional), glutamatergic (neurotransmitter) and laminar (temporal) characters of early-born (lower-layer) neurons, while simultaneously repressing an alternative subcortical, GABAergic neuronal phenotype. Subsequently, later-born (upper-layer) cortical neurons are specified in an Ngn-independent manner, requiring instead the synergistic activities of Pax6 and Tlx, which also control a binary choice between cortical/glutamatergic and subcortical/GABAergic fates. Our study thus reveals an unanticipated heterogeneity in the genetic mechanisms specifying the identity of neocortical projection neurons.
Keywords: laminar specification, neocortex, Neurogenins, neurotransmitter phenotype
Introduction
Neuronal diversity in the neocortex is striking, with the human neocortex subdivided into more than 40 tangential areas and six radial layers, each characterized by unique neuronal morphologies, cytoarchitectures, axonal projections and molecular identities (Job and Tan, 2003). During development, neocortical neurons are generated sequentially, with multipotent progenitors in the dorsal telencephalon initially giving rise to neurons in the cortical preplate, followed by lower-layer (layers V/VI) and finally upper-layer (layers II–IV) neurons of the cortical plate (Caviness, 1982). In addition to their unique properties, cortical projection neurons share several essential properties, including their regional identity, as defined by common gene-expression profiles in progenitors and neurons, and use of glutamate as an excitatory neurotransmitter. They can be distinguished from cortical interneurons, which are born and differentiate in the ventral telencephalon, reach the neocortex by tangential migration, express distinct ventral-specific regional markers and use GABA as a neurotransmitter (Parnavelas et al, 2000). A question that remains unanswered is whether a single genetic pathway specifies all common features of neocortical neurons, or whether distinct genetic programs are sequentially activated to specify the features that are both common and unique to projection neuron populations in each cortical layer.
The known molecular determinants of cortical identity include three homeodomain (HD) transcription factors, Lhx2, Emx2 and Pax6, that act either alone or in combination to pattern the telencephalon and establish a cortical territory (Bulchand et al, 2001; Monuki et al, 2001; Muzio et al, 2002). Pax6 and Emx2 are also required to establish regional identities along the tangential axis of the neocortex, setting up territories that are thought to prefigure the formation of cortical areas (Bishop et al, 2000; Mallamaci et al, 2000). In contrast, the molecules involved in specifying laminar fates and a glutamatergic neurotransmitter phenotype remain virtually unexplored. Given the central role that Pax6 plays in cortical development, it is of particular interest that Pax6 directly activates Ngn1 and Ngn2, two highly related basic–helix–loop–helix (bHLH) transcription factors (Scardigli et al, 2003). Ngns have been implicated in multiple cell fate choices in the nervous system, including the selection of neural progenitors, specification of neuronal phenotype at the expense of glial cell fates, and choice of neuronal differentiation programs (Bertrand et al, 2002). In the telencephalon, Ngns are specifically expressed in cortical and not subcortical progenitors, where they specify the regional identity of the earliest-born preplate neurons in the neocortex (Fode et al, 2000). Here we examine the function of Ngn1, Ngn2, Pax6 and Tlx in specifying the cortical regional identity, glutamatergic neurotransmitter phenotype and laminar-specific properties of neurons in the cortical plate.
Results
Ngn1 and Ngn2 specify a glutamatergic, cortical phenotype and repress GABAergic, subcortical genes
We analyzed the role of Ngn1 and Ngn2 in specifying the identity of neocortical neurons. To evaluate specification defects resulting from Ngn mutations at the early stage of corticogenesis, we profiled and compared gene expression in wild-type versus Ngn1 and Ngn2 single-, and Ngn1;Ngn2 double-mutant cortices at embryonic day (E) 13.5. Hybridization of total cortical cDNA to Affymetrix microarrays revealed a significant number of up- and downregulated genes in all genotypes, except in Ngn1 mutants, which were not investigated further (Figure 1A).
Figure 1.
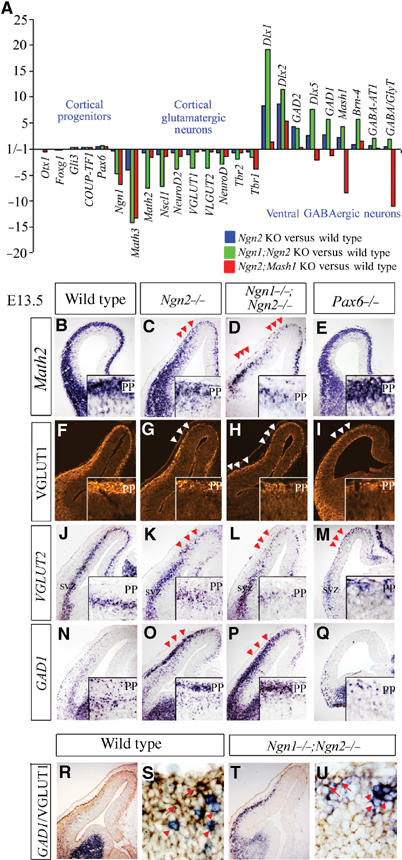
Gene profiling reveals a global shift of neuronal phenotype from cortical, glutamatergic to subcortical, GABAergic in E13.5 Ngn mutants. (A) Expression profiling in E13.5 cortices using Affymetrix microarrays, showing fold differences in gene expression, comparing Ngn2 (blue bar), Ngn1;Ngn2 (green bar) and Ngn2;Mash1 (red bar) mutants to wild type. (B–E) Expression of Math2 in PP and early-born CP neurons was reduced in Ngn2 and Ngn1;Ngn2 mutants (arrowheads, C, D). Expression of VGLUT1 protein in the PP (F–I) and VGLUT2 transcripts (J–M) in the SVZ was reduced in Ngn2, Ngn1;Ngn2 and Pax6 mutants (arrowheads, G-I, K–M). (N–Q) GAD1 was ectopically expressed in the PP/SVZ of Ngn2 and Ngn1;Ngn2 mutants (arrowheads, O, P). (R–T) Double staining of GAD1 RNA (blue) and VGLUT1 protein (brown) in wild-type (R, S) and Ngn1;Ngn2 (T, U) mutants showing that most cells express either the glutamatergic (arrows) or the GABAergic (arrowheads) marker. PP, preplate; SVZ, subventricular zone.
Affymetrix data and RNA in situ hybridization for nonrepresented genes revealed no deregulation of genes normally expressed in dorsal telencephalic progenitors (Gli3, Pax6, Emx2, Lhx2, Foxg1, Otx1, Tlx, COUP-TFI) in Ngn mutants (Figure 1A, Supplementary Figure S1). In contrast, several transcription factors specifically expressed by cortical neurons (Math2, Nscl1, NeuroD, NeuroD2, Tbr1, Tbr2) were reduced in Ngn2 mutant cortices, and more severely downregulated in Ngn1;Ngn2 mutants (Figure 1A–D). Vesicular glutamate transporter1 (VGLUT1) and VGLUT2, which load glutamate into synaptic vesicles (Fremeau et al, 2001), were also downregulated in Ngn mutants (Figure 1A). In E13.5 telencephalic sections, VGLUT genes were specifically expressed in dorsal neurons that use glutamate as a neurotransmitter, with VGLUT1 protein predominant in preplate (PP) and cortical plate (CP) neurons (Figure 1F), whereas VGLUT2 was transiently expressed in differentiating cortical neurons in the subventricular zone (SVZ; Figure 1J). In Ngn2 mutants, VGLUT1 protein and VGLUT2 transcript levels were reduced in dorsomedial and not lateral cortical neurons, likely due to compensation by Ngn1, which persists in lateral domains (Fode et al, 2000), whereas defects extended throughout the cortex of Ngn1;Ngn2 double mutants (Figure 1F–H and J–L). Ngns are thus required to activate cortical- and glutamatergic-specific differentiation programs in early-born CP neurons, likely acting downstream of cortical patterning genes, which are normally expressed in Ngn mutants, and cannot compensate for the loss of Ngn activity.
Mash1 is upregulated in Ngn mutant cortical progenitors, and was previously linked to the ectopic expression of subcortical genes Dlx1 and GAD1 in PP neurons (Fode et al, 2000). Gene profiling at E13.5 revealed a more extensive upregulation of subcortical genes in Ngn2 and Ngn1;Ngn2 mutants that included ventral telencephalic transcription factors (Mash1, Dlx1, Dlx2, Dlx5, Brn4), biosynthetic enzymes for GABA (glutamic acid decarboxylase 1 (GAD1), GAD2) and GABA transporters (GABA transporter 1 (GABA-T1), GABA and glycine transporter (GABA/glyT)), suggesting that many, and possibly all, components of a subcortical, GABAergic differentiation program were ectopically activated in Ngn mutant cortical neurons (Figure 1A). Consistent with a switch of neurotransmitter phenotype, GAD1 was ectopically expressed in the PP/CP and SVZ of Ngn2 and Ngn1;Ngn2 mutants (Figure 1N–P). The ectopic ventral-like neurons were misspecified neurons of cortical origin, and not subcortical neurons that had inappropriately migrated into the cortex, based on previous explant and migration studies (Fode et al, 2000; Chapouton et al, 2001). Moreover, GAD1 transcripts and VGLUT1 protein were for the most part detected in complementary sets of cortical neurons in both wild-type and Ngn1;Ngn2 mutant cortices (Figure 1R–U), suggesting that cortical neurons choose between a glutamatergic and GABAergic phenotype.
To determine the extent to which ectopic Mash1 expression was responsible for specification defects in Ngn mutant cortices, we profiled gene expression in E13.5 Ngn2;Mash1 double mutants. A similar reduction in transcription of cortical-specific neuronal markers was observed in Ngn2 and Ngn2;Mash1 mutants, with the exception of Math3 (Figure 1A; compare Ngn2 versus WT (blue bars) against Ngn2;Mash1 versus WT (red bars)). In Ngn2;Mash1 mutants, the loss of Math2 transcripts was restricted to rostromedial domains (Figure 2L), where Ngn1 was no longer expressed. Thus, the loss of neurons with a cortical character in Ngn mutants occurs independently of the upregulation of Mash1, suggesting that the Ngns directly activate a cortical, glutamatergic differentiation pathway.
Figure 2.
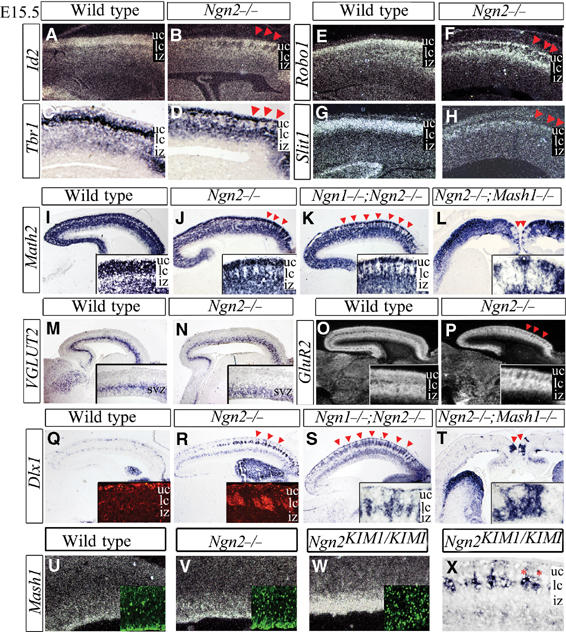
Change in regional identity and neurotransmitter phenotype of early-born CP neurons in E15.5 Ngn mutants. E15.5 expression of cortical neuronal markers Id2 (A, B), Tbr1 (C, D), Robo1 (E, F), Slit1 (G, H) and Math2 (I–L), showing a correctly specified upper layer in Ngn2 and Ngn1;Ngn2 mutants above distinct gaps in lower CP expression (arrowheads, B, D, F, H, J, K) and in the medial cortex of Ngn2;Mash1 mutants (arrowheads, L). (M–P) The glutamatergic marker VGLUT2 was appropriately expressed in newly born neurons in the SVZ of wild-type (M) and Ngn2 mutants (N), whereas GluR2 was expressed in the IZ and CP of wild-type cortices (O), but displayed distinct gaps in the lower CP of Ngn2 mutants (arrowheads, P). (Q–T) Dlx1 transcripts and protein (insets) were detected in marginal zone and SVZ interneurons, and were ectopic in lower CP clusters in Ngn2 and Ngn1;Ngn2 mutants (arrowheads, R, S), and in medial clusters in Ngn2;Mash1 mutants (arrowheads, T). (U–W) Mash1 transcript and protein (insets) levels were moderately and strongly upregulated in Ngn2 (V) and Ngn2KIM1 mutants (W), respectively. (X) Ectopic Dlx1+ clusters were in the deep CP in Ngn2KIM1 mutants (asterisks). uc, upper CP; lc, lower CP; iz, intermediate zone; svz, subventricular zone.
Dlx1, Dlx2 and Brn4 remained elevated to varying extents in Ngn2;Mash1 cortices as compared to wild type (Figure 1A), with the ectopic expression of ventral markers restricted to rostromedial domains (Figure 2T). Ngns therefore repress ventral-specific genes in at least a partially Mash1-independent manner. In contrast, ectopic expression of Dlx5, GAD1, GAD2, GABA-T1 and GABA/glyT in Ngn2 mutant cortices was mostly or strictly dependent on the presence of Mash1 (Figure 1A), suggesting that ectopic activation of these genes was largely due to derepression of Mash1. Ngn1/2 thus specify regional and neurotransmitter phenotypes via multiple mechanisms, including activation of dorsal, cortical-specific genetic pathway(s), and repression of ventral telencephalic programs that are both Mash1 dependent and independent (Figure 8).
Figure 8.
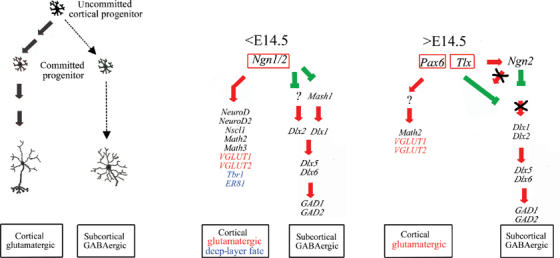
Two-phase model of neuronal fate specification in the neocortex. A schematic model of cellular and genetic cascades underlying early and late stages of neuronal phenotype specification in the neocortex. The early phase (<E14.5) is dependent on Ngn1/2, which induces expression of cortical, glutamatergic and lower-layer specification programs, while simultaneously repressing the differentiation of GABAergic neurons. The late phase of neuronal fate specification (>E14.5) occurs in the absence of Ngn1/2 activity, and is dependent on synergistic interactions between Tlx and Pax6, which specify a cortical and glutamatergic identity while repressing a GABAergic phenotype.
Ngns are required to specify early- and not later-born CP neurons
We extended our analysis of Ngn mutants to mid-corticogenesis (E15.5), when lower-layer neurons have differentiated and are migrating to the CP. As at earlier stages, telencephalic patterning genes (Emx2, Lhx2, Tlx, Pax6) were normally expressed in Ngn2 and Ngn1;Ngn2 mutants, suggesting a correct regional identity of progenitors (data not shown). To assess neuronal identities, we analyzed the expression of CP-specific markers Id2, Robo1, Slit1, Math2 and Tbr1 (Figure 2A–K). In the rostral cortex of Ngn2 mutants, where Ngn1 expression was selectively lost, and throughout the cortex of Ngn1;Ngn2 mutants, gaps in expression of all cortical markers were observed in the deep CP (Figure 2B, D, F, H, J and K). The loss of cortical-specific gene expression was not due to a loss of neurons, as pan-neuronal markers such as SCG10 were unperturbed (data not shown), and cell death, as assessed by TUNEL, was not elevated in E15.5 Ngn mutants (Supplementary Figure S2).
Strikingly, gaps in cortical-specific gene expression were not observed in the superficial CP or intermediate zone (IZ) of Ngn mutants, zones that contain more recently generated CP neurons, suggesting that only early-born CP neurons are misspecified. Consistent with this, CP neurons differentiating at E15.5 in Ngn2 and Ngn1;Ngn2 mutants acquired their correct glutamatergic phenotype, as assessed by normal levels of VGLUT2, which transiently labels glutamatergic neurons migrating through the SVZ (Figure 2M–N). Transcripts for the glutamate receptor GluR2 were also maintained in the IZ and upper CP of E15.5 Ngn2 mutants, and were only lost in clusters of lower-layer CP neurons (Figure 2O and P). In a complementary manner, ectopic expression of Dlx1 (Figure 2Q–S) and GAD1 (data not shown) was confined to large aggregates in the lower CP of Ngn mutants (Figure 2A–K), and was excluded from the more recently generated, superficial layer of the CP. Thus, in contrast with early-born CP neurons, neurons differentiating during mid-corticogenesis acquire a normal identity in the absence of Ngn function.
Changing Mash1 responsiveness of cortical progenitors
The misspecification of early- and not later-born CP neurons in Ngn mutants was surprising, given that Ngn expression persists throughout corticogenesis (data not shown). Since derepression of Mash1 contributes to cortical misspecification in Ngn mutants at E13.5 (Fode et al, 2000), we speculated that Mash1 might not be sufficiently upregulated at later stages to alter the differentiation of cortical progenitors. We tested this using Ngn2KIM1 homozygous mutant embryos in which Ngn2 coding sequences were replaced with Mash1 (Fode et al, 2000; Parras et al, 2002). At E15.5, Mash1 transcript and protein levels were low in wild-type cortical progenitors, slightly elevated in Ngn2 mutants and very significantly elevated in Ngn2KIM1 mutants (Figure 2U–W). However, Dlx1 was ectopically expressed only in small clusters deep within the rostral CP of Ngn2KIM1 mutants (Figure 2X). Thus, increasing Mash1 expression levels was not sufficient to respecify cortical neurons born after E14.5, suggesting that both the dependency of cortical neurons on Ngn1/2 and the responsiveness of cortical progenitors to ectopic Mash1 change over time.
A subset of Ngn mutant early-born neurons contribute to deep cortical layers while others segregate out of the CP
To further examine the fate of early-born, misspecified neurons, we analyzed Ngn mutant cortices at E18.5, a stage when neurogenesis is complete, although CP neurons are still migrating to their final destination. Strikingly, the cortical-specific marker Math2 was for the most part uniformly expressed throughout the CP in E18.5 Ngn2 and Ngn1;Ngn2 mutant cortices (Figure 3A–D), in contrast to defects at E15.5 (Figure 2I–K). Moreover, in E18.5 Ngn mutants, misspecified Dlx1+ (Figure 3E–J) and GAD1+ (data not shown) neurons segregated out of the CP, aggregating instead in small clusters in the germinal zone (GZ), rostral marginal zone (MZ) and flanking the IZ/lower CP border. Thus, by E18.5, the Ngn mutant CP was comprised almost exclusively of neurons with their correct regional identities.
Figure 3.
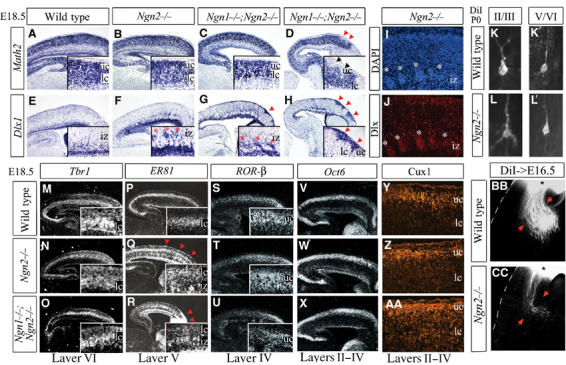
Misspecification of lower-layer CP neurons in E18.5 Ngn mutants. (A–D) Math2 was uniformly expressed in the CP and IZ of wild-type (A), Ngn2 (B) and Ngn1;Ngn2 (C) mutants, with the exception of small superficial gaps in Ngn1;Ngn2 mutants (uc, arrowheads, D). (E–H) Dlx1 was expressed in cortical interneurons in the GZ, MZ and CP (E), and in ectopic clusters in the IZ (asterisks) and superficial CP (arrowheads) of Ngn2 (F) and Ngn1;Ngn2 (G, H) mutants. (I) DAPI and (J) Dlx immunostaining showed that abnormal cellular aggregates (asterisks) beneath the CP were Dlx+. (K–L) Retrograde labeling of P0 cortical neurons in layers II/III (K, L) and V/VI (K′, L′) revealed similar immature neuronal morphologies in wild-type (K, K′) and Ngn2 mutant (L, L′) cortices, with sparsely branched apical and basal dendritic processes (also see Supplementary Figure S4). (M–O) Layer VI expression of Tbr1 was reduced in the rostral CP in Ngn2 mutants (N), and throughout the Ngn1;Ngn2 mutant cortex (O). (P–R) ER81 expression was reduced and disorganized (arrowheads) in rostral layer V in Ngn2 mutants (Q), and throughout the Ngn1;Ngn2 mutant layer V (R). (S–U) Layer IV expression of RORβ was normal in Ngn2 (T) and Ngn1;Ngn2 (U) mutants. Ectopic RORβ (U) and ER81 (Q, R) were observed in clusters in the IZ and MZ of Ngn2 and Ngn1;Ngn2 mutants, correlating with migration defects of a subset of early-born neurons. Oct6 transcripts (V–X) and Cux1 protein (Y, Z, AA) were expressed in layers II–IV in all genotypes. (BB, CC) Anterograde tracing revealed fewer corticofugal projections in E16.5 Ngn2 mutants (arrows, CC). uc, upper CP; lc, lower CP; iz, intermediate zone.
To determine whether any early-born cortical neurons remained in the CP of Ngn mutants, neuronal lamination was assessed with histological stains at P0, revealing that lower layers were indeed present in Ngn2 and Ngn1;Ngn2 mutants, but were significantly thinner in double mutants (Figure 4A–F). To determine whether histologically identifiable lower layers were composed of neurons born at the correct time, we used birthdating (Caviness, 1982; Caviness et al, 1995). BrdU was administered at different embryonic stages (Supplementary Figure S3), followed by an assessment at P0 of the laminar position of darkly labeled nuclei, marking neurons born at the time of BrdU injection (black bars, Figure 4G), and lightly labeled nuclei, representing neurons derived from progenitors that had undergone additional cell divisions (gray bars, Figure 4G). Labeling at E12 revealed a peak accumulation of strong BrdU+ nuclei in layer VI of wild-type cortices (top row, Figure 4G). In Ngn mutants, there was an increased number of heavily labeled neurons blocked in the GZ/IZ, and in the MZ of Ngn1;Ngn2 mutants (top row, Figure 4G), corresponding to sites of aggregation of misspecified Dlx1+/GAD1+ neurons (Figure 3F–J), and confirming that many early-born, misspecified neurons were not integrated in the CP. However, not all Ngn mutant neurons born at E12 migrated aberrantly, as a significant number of BrdU+ neurons were correctly positioned in layer VI, likely corresponding to the subset of lower-layer CP neurons with a correct regional identity and neurotransmitter phenotype at E15.5 (Figure 2A–P).
Figure 4.
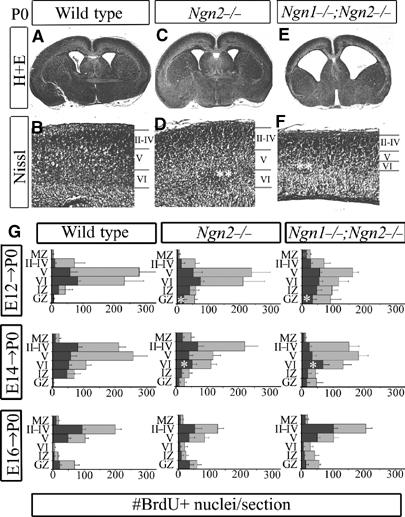
Aberrant location of lower-layer CP neurons in Ngn mutants. (A–F) Histological analyses at P0 revealed neurogenesis defects primarily in lower layers of Ngn1;Ngn2 mutants (asterisks, F), a disorganization of layer VI in Ngn2 mutants (asterisks, D), but a correctly laminated CP in both genotypes. (G) Graphical representations of BrdU birthdating studies showing the distribution of P0 cortical neurons labeled with BrdU at different times (E12, E14, E16). Cortices were divided into six bins corresponding to MZ, layers II–IV, layer V, layer VI, IZ and GZ. Darkly labeled nuclei (black bar) and lightly labeled nuclei (gray bars) in each bin were counted. Asterisks label the increased number of cells generated at E12 in the Ngn2 and Ngn1;Ngn2 mutant GZ, and the skewed distribution of neurons labeled at E14 in Ngn2 and Ngn1;Ngn2 mutants. MZ, marginal zone; GZ, germinal zone.
A BrdU pulse at E14 primarily marked the genesis of layer IV and V neurons, but in Ngn2 and Ngn1;Ngn2 mutants there was a clear decline in the number of darkly stained nuclei in the lower CP, and instead an accumulation of cells in the GZ (middle row, Figure 4G). This suggested that a subset of CP neurons destined for layers IV–V aggregated in abnormally deep positions in Ngn2 and Ngn1;Ngn2 mutant CPs. In contrast, the laminar position of upper-layer neurons was largely unperturbed in Ngn mutants, as assessed by birthdating at E16, which in mutant as well as control brains resulted in a clear bias towards darkly stained nuclei localizing to upper layers II–IV (bottom row, Figure 4G). Specification defects resulting in abnormal laminar localization in Ngn2 and Ngn1;Ngn2 mutants were thus largely restricted to cortical neurons born between E12 and E14, but a subset of early-born neurons acquired a correct regional identity and populated deep cortical layers.
Disruptions in laminar specification restricted to lower layers in Ngn mutants
The correct specification and layering of a subset of lower-layer neurons in the Ngn mutant CP allowed us to examine whether these neurons had their correct laminar phenotypes based on molecular markers. At E18.5, Tbr1 was expressed at high levels in layer VI in wild-type cortices, but at diminished levels in the rostral cortex of Ngn2 mutants and throughout the cortex of Ngn1;Ngn2 double mutants (Figure 3M–O). ER81, which is expressed in layer V, was lost in small rostral gaps in layer V of Ngn2 mutants, and was more globally disturbed throughout layer V in Ngn1;Ngn2 mutants (Figure 3P–R). In contrast, layer II–IV markers RORβ (Figure 3S–U), Oct6 (Figure 3V–X) and Cux1 (Figure 3Y, Z and AA) labeled correctly positioned neurons in all genotypes, indicating that upper-layer neurons were correctly specified. Anterograde tracing of descending corticofugal projections of layer V/VI (Koester and O'Leary, 1993) revealed strongly reduced axonal numbers in Ngn2 mutant versus wild-type cortices at E16.5, which were consistent with selective specification defects of lower-layer neurons (Figure 3BB and CC). Lower-layer neurons in the Ngn mutant CP thus present both molecular and axonal projection defects.
Neuronal migration in the cortex is not complete until postnatal day (P) 7, such that unequivocal conclusions about laminar phenotypes in Ngn mutants could not be made from embryonic analyses. We thus examined laminar specification in the rare Ngn2 single-mutant pups that survived the first postnatal week, but could not analyze laminar identities in the complete absence of Ngn activity because all Ngn1;Ngn2 mutants died at birth. At P5, the overall size of the Ngn2 mutant neocortex was smaller, but all the six cortical layers were clearly identified by cell morphology (Figure 5O–P). Analysis of molecular markers revealed striking defects in layer VI, with expression of Slit1 (Figure 5A and B), Tbr1 (Figure 5C and D) and Id2 (Figure 5M and N) strongly reduced in the rostromedial CP (where Ngn1 is lost) of P5–P7 Ngn2 mutants. Layer V defects were more modest, likely due to persistence of Ngn1, although the distribution of both Robo1 (Figure 5E and F) and ER-81 (Figure 5G and H) transcripts was clearly disorganized in the rostral cortex of P7 Ngn2 mutants. In contrast, layer IV expression of ROR-β (Figure 5I and J) and layer II/III expression of Oct6 (Figure 5K and L) and Id2 (Figure 5M and N) appeared normal in postnatal Ngn2 mutants. We performed several additional tests to confirm that, in Ngn2 mutants, lower-layer neurons did not lose gene expression due to apoptosis, and that upper-layer neurons had their correct molecular and cellular properties (i.e. barrel field formation in layer IV, synaptic zinc in layer II/III axon terminals, upper-layer contribution to callosal projections; Figure 5Q and R; Supplementary Figure S5).
Figure 5.
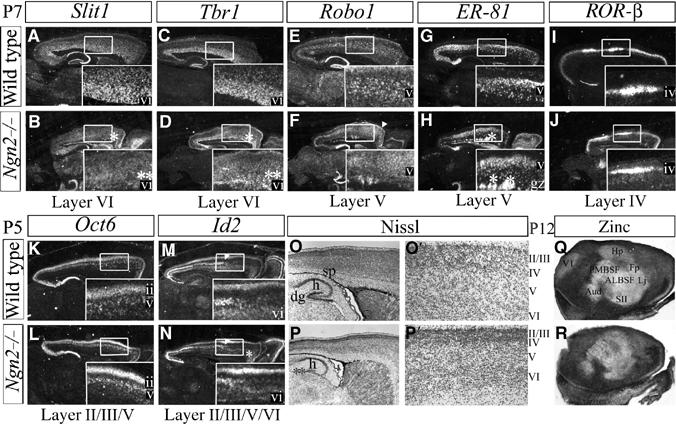
Postnatal laminar defects restricted to deep CP layers in Ngn2 mutants. At P7, layer VI expression of Slit1 (A, B) and Tbr1 (C, D) was severely reduced in the rostral cortex of Ngn2 mutants (asterisks, B, D). Layer V expression of Robo1 (E, F) and ER81 (G, H) demarcated a thinner, disorganized layer, and ectopic expression in the IZ of Ngn2 mutants (asterisks, H). Layer IV expression of RORβ at P7 (I, J), layer II/III/V expression of Oct6 at P5 (K, L) and layer II/III expression of Id2 (rostral limit marked by arrowhead) at P5 (M, N) were unaffected in Ngn2 mutants (J, L, N). Id2 expression was lost in Ngn2 mutant layer VI (asterisks, N). (O, P) Nissl staining of P5 wild-type (O, O′) and Ngn2 mutant (P, P′) cortices revealed no defects in cortical layering, but neurogenesis defects were evident in the SP (*) and dentate gyrus (**; P) of Ngn2 mutants. (Q, R) P12 cortices stained for synaptic zinc, a marker of axon terminals of glutamatergic projection neurons in layers II/III and VI, were normally excluded by layer IV in P12 Ngn2 mutants (R). gz, germinal zone; h, hippocampus; dg, dentate gyrus; sp, subplate; ALBSF, antero-lateral barrel subfield; PMBSF, posterio-medial barrel subfield; Hp, hindpaw; Fp, forepaw; Lj, lower jaw; SII, secondary somatosensory cortex; Aud, primary auditory cortex; V1, primary visual cortex.
Pax6 specifies a cortical, glutamatergic phenotype and represses subcortical phenotypes at mid-corticogenesis
Given that Ngn1/2 were not required to specify neuronal phenotypes in mid–late corticogenesis, we hypothesized that an Ngn-independent genetic program operated during a second phase of neuronal fate specification. Previous studies had suggested that mutations in Pax6 and the orphan nuclear receptor Tlx specifically affected development of upper cortical layers, but molecular analyses were limited (Tarabykin et al, 2001; Land and Monaghan, 2003). These two factors also cooperate genetically to establish the cortical–subcortical boundary (Stenman et al, 2003), prompting us to investigate whether genetic interactions between Pax6 and Tlx were required to specify regional, neurotransmitter and/or laminar phenotypes of upper-layer cortical neurons.
During early corticogenesis (E12.5–E13.5), Ngn2 was expressed in Pax6 mutant cortical progenitors, except in a small lateral territory (data not shown; Toresson et al, 2000; Yun et al, 2001), allowing us to assess how Pax6 functions independently of the Ngns. The regional identity of Pax6 mutant cortical neurons was correctly specified at E13.5, as assessed by Math2 expression (Figure 1E) and Tbr1 (Stoykova et al, 2000). Despite this, VGLUT1 and VGLUT2 expression was strongly reduced in Pax6 mutants (Figure 1I and M), indicating that these neurons were not glutamatergic. Pax6 mutant cortical neurons were also not GABAergic, as GAD1 was not ectopically expressed (Figure 1Q), likely due to Ngn-mediated repression of GABAergic pathways at this stage (Figure 1A). Thus, Pax6 is not required to specify a cortical (regional) identity or suppress GABAergic differentiation pathways in early-born neurons, but does participate in the specification of a glutamatergic phenotype.
We next assessed cortical and glutamatergic identities in Pax6 and Tlx mutants at E15.5. No specification defects were observed in cortical progenitors or neurons in Tlx single mutants at this stage (data not shown). There was a clear decrease in Ngn2 transcript levels in the rostral GZ of Pax6 and Pax6;Tlx mutants, but other cortical-specific progenitor markers were normally expressed (Figure 6B and C; Stoykova et al, 2000; Toresson et al, 2000; Muzio et al, 2002). Expression of cortical neuronal markers Math2 and Tbr1 was also clearly reduced in newly generated neurons migrating through the IZ of Pax6 and Pax6;Tlx mutants, and VGLUT2 expression was reduced in the SVZ of Pax6 and Pax6;Tlx mutants (Figure 6D–L). Thus, specification of both a cortical regional identity and glutamatergic neurotransmission phenotype was perturbed in neurons generated during mid-corticogenesis in Pax6 and Pax6;Tlx mutants, in contrast to Ngn mutants where neuronal specification defects were rescued by E15.5.
Figure 6.

Defects in neuronal specification at mid-corticogenesis (E15.5) in Pax6;Tlx mutants. The rostral CP of Pax6 and Pax6;Tlx mutants was visibly thinner, and there was an overall reduction in the size of the Pax6;Tlx cortex. (A–C) GZ expression of Ngn2 was reduced in the rostral GZ of Pax6 (arrowheads, B) and Pax6;Tlx mutants (arrowheads, C). (D–I) Math2 (D–F) and Tbr1 (G–I) were expressed in the IZ and CP, and were reduced throughout the IZ and rostral CP of Pax6 (arrowheads, E, H) and Pax6;Tlx mutants (arrowheads, F, I). (J–L) VGLUT2 expression in the SVZ was reduced in Pax6 (arrowheads, K) and Pax6;Tlx (arrowheads, L) mutants. (M–O) Dlx1 was expressed in migrating interneurons in the MZ and SVZ (M), and a massive, ectopic upregulation of Dlx1 was detected in the SVZ/IZ of Pax6 (asterisks, N) and Pax6;Tlx mutants (asterisks, O). gz, germinal zone; uc, upper CP; lc, lower CP; iz, intermediate zone; svz, subventricular zone.
Many Pax6 mutant CP neurons born from E14 onwards do not migrate appropriately, becoming trapped in an expanded SVZ (Caric et al, 1997). Neurons trapped in the SVZ of E15.5 Pax6 and Pax6;Tlx mutants were misspecified and ectopically expressed Dlx1 (Figure 6N–O) and GAD1 (data not shown), suggesting that neurons born during mid-corticogenesis acquired a subcortical phenotype, in striking contrast to the normal regional identity of neurons generated at E13.5. Thus, Pax6 is required to specify a cortical identity and glutamatergic neurotransmitter phenotype in CP neurons generated in mid-corticogenesis, while simultaneously repressing a ventral, GABAergic fate. Although Pax6 regulates Ngn expression at E15.5, the requirement for Pax6 in specifying cortical identities must be independent of Ngn regulation, since CP neurons born during mid-corticogenesis are correctly specified in Ngn mutants.
Pax6 and Tlx cooperate in the specification of late-born cortical neurons
To assess the specification of late-born cortical neurons, we examined E18.5 Pax6 and Tlx single- and double-mutant cortices. In Pax6 and Pax6;Tlx mutants, Math2 labeled a thinner CP, and the IZ was almost completely devoid of Math2 transcripts, suggesting that many CP neurons generated during mid–late corticogenesis did not acquire their appropriate cortical identity or migrate into the CP (Figure 7A–D). In Tlx mutants, small gaps in Math2 expression were observed in the GZ, suggesting that a small subset of late-born CP neurons were not correctly specified (Figure 7B). The misspecified neurons instead acquired a subcortical identity as Dlx1 (Figure 7E–H) and GAD1 (data not shown) were expressed in a complementary manner, in small ectopic patches in the Tlx mutant GZ and large ectopic bands spanning the GZ/IZ in Pax6 and Pax6;Tlx mutants. Thus, Tlx and Pax6 both act at late stages of corticogenesis to promote a cortical identity and suppress a subcortical GABAergic phenotype.
Figure 7.
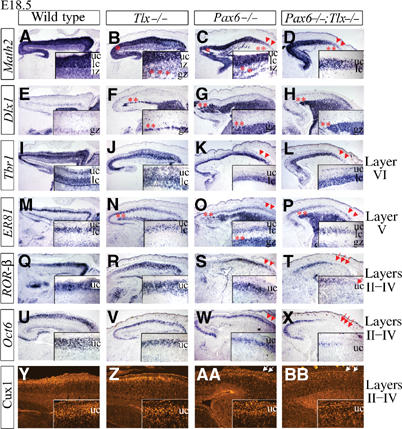
Pax6 and Tlx cooperate during late corticogenesis (E18.5) to specify neuronal phenotypes. (A–D) Math2 expression in the CP and IZ was lost in small clusters in Tlx mutant IZ, and in large regions of the IZ in Pax6 and Pax6;Tlx mutants (asterisks, insets, B–D), but was maintained in the upper CP (arrowheads, C, D). (E–H) Dlx1 was expressed in migrating cortical interneurons in the GZ, MZ and CP (E), in small ectopic clusters in the GZ of Tlx mutants, and in a grossly expanded GZ in Pax6 and Pax6;Tlx mutants (asterisks, F–H). (I–L) Layer VI expression of Tbr1 was maintained in all genotypes (arrowheads, K, L). (M–P) ER81 was expressed in layer V in all genotypes (arrowheads, O, P) and ectopically expressed in the IZ of Tlx mutants, and SVZ/IZ of Pax6 and Pax6;Tlx mutants (asterisks, N–P). (Q–T) Layer IV expression of ROR-β was lost in rostral CP of Pax6;Tlx mutants (arrows, T), but maintained in Pax6 mutants (arrowheads, S). Layers II–IV expression of Oct6 (U–X) and Cux1 (Y-BB) was detected, albeit at decreased levels, in Pax6 mutants (arrowheads, W; arrows, AA), but was not expressed in rostral CP of Pax6;Tlx double mutants (arrows, X, BB). gz, germinal zone; uc, upper CP; lc, lower CP; iz, intermediate zone.
We next examined laminar identities of CP neurons in Pax6 and Tlx mutants. In contrast to Ngn mutants, no defects were detected in lower layers V and VI, with Tbr1 expressed in a distinct layer VI (Figure 7I–L) and ER81 in layer V (Figure 7M–P) in all genotypes. In contrast, the layer IV marker RORβ was not detectable in the rostral cortex of Pax6;Tlx mutants, while it was expressed in Pax6 and Tlx single mutants (Figure 7Q–T). Similarly, two markers of layers II–IV, Oct6 (Figure 7U–X) and Cux1 (Figure 7Y–BB), were completely absent in the upper layers of the rostral cortex of Pax6;Tlx double mutants, but were detectable, although at reduced levels, in Tlx and Pax6 single mutants. Thus, Pax6 and Tlx must both be mutated to lose upper-layer marker expression, suggesting that they cooperate genetically to specify the identity of late-born neurons in the cortex.
Discussion
We have identified several genetic determinants that operate in a biphasic manner to specify the regional (cortical), neurotransmission (glutamatergic) and laminar identity of neocortical neurons. Early in cortical development, Ngn1 and Ngn2 function redundantly to specify regional and glutamatergic phenotypes common to all neocortical projection neurons, and participate in specifying the laminar identity of deep-layer neurons (Figure 8). In mid–late corticogenesis, as Ngn function becomes dispensable, Pax6 and Tlx begin to function both independently and synergistically to specify cortical identity and neurotransmitter choice by later-born neurons in upper layers (Figure 8). Interestingly, both the Ngns and Pax6/Tlx are required not only to activate cortical-specific traits but also to repress an alternative subcortical, GABAergic differentiation program. Taken together, these data reveal an unexpected degree of heterogeneity in the genetic mechanisms underlying neuronal specification during corticogenesis, and highlight the highly coordinated manner in which common and diverse aspects of neuronal phenotype are specified.
Distinct genetic programs specify features common to early- and late-born cortical neurons
Despite their inherent diversity, neocortical projection neurons share several essential characteristics, including their regional identity and use of glutamate as a neurotransmitter. A priori, we anticipated that such uniformity would arise through the activation of a common genetic cascade. On the contrary, we found that distinct genetic programs specify shared regional and neurotransmitter properties during early and late stages of corticogenesis. In early stages, Ngn activity is absolutely required to induce a cortical differentiation program in a subset of deep-layer neurons. The ability of the Ngns to function in cortical progenitors to specify the properties of postmitotic neurons is consistent with previous results that suggest that neuronal identities are specified at the progenitor cell stage in the neocortex (Job and Tan, 2003). However, given that some early-born neurons do acquire their correct regional and neurotransmitter identities in Ngn mutants, additional regulatory genes may normally act in parallel with the Ngns to specify lower-layer phenotypes. Furthermore, Ngn2 is not sufficient to specify a dorsal cortical identity in subcortical progenitors, suggesting that Ngns must interact with as yet unidentified cofactors to activate cortical-specific programs of gene transcription (Parras et al, 2002).
In contrast to their limited roles early, Pax6 and Tlx are absolutely required to activate cortical- and glutamatergic-specific differentiation programs in CP neurons during mid–late corticogenesis. We hypothesize that Emx2 also functions during mid–late corticogenesis, primarily in the caudal cortex, where neuronal specification defects are less severe in Pax6 and Pax6;Tlx mutants. Consistent with this, the cortex is converted to striatal-like tissue in the absence of both Pax6 and Emx2 (Muzio et al, 2002). Importantly, although Ngns are downstream of Pax6 and Emx2 in mid–late cortical development, as shown by the loss of Ngn1 and Ngn2 transcripts in the Pax6;Emx2 mutant pallium, and the direct transcriptional activation of Ngn2 by Pax6 in the cortex (Stoykova et al, 2000; Muzio et al, 2002; Scardigli et al, 2003; Stenman et al, 2003), they are not mediating the specification function of these genes at these stages.
A binary switch between cortical/glutamatergic and subcortical/GABAergic phenotypes
We uncovered a binary switch between glutamatergic and GABAergic phenotypes, which is regulated by distinct genetic pathways at different stages of corticogenesis. Interestingly, in the spinal cord, differentiating neurons must also choose between a glutamatergic and GABAergic fate, and the homeodomain transcription factors Tlx1 and Tlx3 promote an excitatory neurotransmission phenotype, while inhibiting an alternative GABAergic fate (Cheng et al, 2004).
Coinciding with the shift from early Ngn-dependent to mid/late Pax6-dependent neuronal fate specification is a change in the competence of progenitor cells to respond to elevated levels of Mash1. That is, whereas early Ngn mutant cortical progenitors activate GABAergic differentiation pathways in response to an upregulation of Mash1, at E15.5, Ngn2 mutant progenitors no longer activate a GABAergic pathway in the same context. Presumably, this is not due to a loss of competence of late cortical progenitors to undergo GABAergic differentiation, since cortical progenitors are respecified in E15.5 Pax6 mutants, but rather due to a shift in the genetic control of this binary decision, from Ngn/Mash1 to Pax6 dependent. Mechanistically, we showed that Ngn1 and Ngn2 inhibit a subcortical identity and GABAergic phenotype through the simultaneous repression of Mash1-dependent and Mash1-independent regulatory pathways (Figure 8). Components of a Mash1-dependent pathway have been previously identified, with Mash1 capable of turning on Dlx1, which in turn induces Dlx5/6 and GAD target genes (Fode et al, 2000; Letinic et al, 2002; Stuhmer et al, 2002), but the existence of a Mash1-independent pathway involved in specifying subcortical identities has only been inferred from the maintenance of a GABAergic phenotype in the ventral telencephalon of Mash1 mutants (Casarosa et al, 1999; Horton et al, 1999).
This study and others have identified several genes, including Gli3, Pax6, Emx2, Tlx and Ngn1/Ngn2, which are all required to repress GABAergic differentiation in cortical progenitors (Theil et al, 1999; Fode et al, 2000; Stoykova et al, 2000; Muzio et al, 2002). Given the multitude and complexity of these genetic mechanisms, it is not surprising that the vast majority of GABAergic cortical interneurons are derived from the ventral subpallium in mice (Anderson et al, 2002). However, we have found that Mash1 is expressed in a significant number of cortical progenitors (C Schuurmans and F Guillemot, unpublished), suggesting that Mash1 may indeed play a role in the rodent cortex, but that this function may be distinct from its capacity to activate a GABAergic differentiation program. Interestingly, in humans, the majority of cortical interneurons appear to be dorsally derived, and cortical progenitors express several genes that are normally at least partially repressed in mouse, including Mash1, suggestive of a role for Mash1 in specifying GABAergic interneurons in the human cortex (Letinic et al, 2002). In future, it will be interesting to investigate whether the production of GABAergic neurons in the human neocortex reflects changes during evolution in the expression or function of Ngns, Pax6 and/or other genes that repress subcortical GABAergic fates in rodents.
Ngns participate in specifying the laminar identity of lower-layer cortical neurons
Here, we showed that Ngn1/2 are essential for the specification of laminar identity in a subset of deep-layer neurons. Similarly, we hypothesize that Pax6 and Tlx act synergistically to specify the laminar identity of upper-layer neurons, but, because there was a complete loss of layer II–IV neurons in the rostral cortex of Pax6;Tlx double mutants, we cannot ascertain whether Pax6 and Tlx are directly required to activate an upper-layer-specific differentiation program, or if the loss of upper-layer markers is secondary to the transformation of these neurons to a ventral-like phenotype. Despite this, from our studies, we come to the unexpected conclusion that early and late phases of neuronal specification in the neocortex are regulated by independent genetic mechanisms, and that the specification of laminar identity, an aspect of neuronal phenotype that changes during cortical development, is directly coupled to the acquisition of common regional and neurotransmission properties, at least in lower-layer neurons. A possible consequence of the existence of independent programs of neuronal specification for different phases of corticogenesis is that evolutionary changes in deep and upper layers could be uncoupled through alterations in the genetic program controlling only one of the two populations. A clear example of such uncoupling is the expansion of supragranular layers II/III in primates (Kornack and Rakic, 1998), possibly resulting from a specific modification of upper-layer genetic programs (Tlx/Pax6) without changing lower-layer programs (Ngn) in the primate cortex, an exciting prediction that deserves further study.
Materials and methods
Maintenance and genotyping of mice
Ngn2lacZ, Ngn1, Mash1, Ngn2KIM1, Pax6 (Sey) and Tlx mutant lines were maintained as single and double heterozygotes, with mutants generated and genotyped as previously described (Ma et al, 1998; Parras et al, 2002; Stenman et al, 2003).
Microarray hybridization
Cortices were dissected from E13.5 wild-type, Ngn2, Ngn1, Ngn2;Mash1 and Ngn1;Ngn2 mutants. RNA was extracted with Trizol reagent, total RNA from three cortices was pooled to average genetic variation and 10 μg RNA was used to generate probes, which were hybridized to U74A and U74B chips following standard Affymetrix guidelines. The experiment was performed twice for single mutants and once for double mutants. Gene accession numbers are listed in Supplementary data. Normalization of data and comparative analysis were performed with GeneChip software.
RNA in situ hybridization
Brains were dissected from staged embryos, fixed overnight in 4% paraformaldehyde (PFA), impregnated in 20% sucrose and embedded in OCT (Tissue-Tek). Sections (10 μm) were cut using a cryostat. Nonradioactive and radioactive RNA in situ hybridization and double in situ/immunohistochemistry were previously described (Gradwohl et al, 1996; Fode et al, 2000). Probe sources are described in Supplementary data.
Immunohistochemistry
Brains were fixed for 2 h in 4% PFA and processed for frozen sectioning as above. Sections were blocked for 1 h in 10% normal goat serum/PBS/0.1% Triton X-100 (PBT), and then incubated overnight at 4°C in primary antibody diluted in blocking solution. Sections were washed with PBT, exposed for 1 h to secondary antibodies and washed again. Primary antibodies included mouse monoclonal anti-Mash1 (1:1), rabbit polyclonal pan-Dll antibody (1/500, Grace Panganiban), rabbit polyclonal VGLUT1/2 (1/500; SySy) and rabbit polyclonal anti-Cux1 (1/250, H Tang and JM Cunningham).
Histology and BrdU birthdating
Brains were fixed in Bouin's fixative, transferred to 70% ethanol and processed for paraffin sectioning as described (Rhinn et al, 1998). For histology, brains were stained with cresyl violet. For birthdating, pregnant females were injected intraperitoneally with 100 μg/g BrdU on E12, E14 and E16, killed at P0, processed for wax sectioning and immunostained with anti-BrdU as described (Gradwohl et al, 1996). Darkly stained BrdU+ nuclei were considered born at the time of injection, and lightly stained nuclei derived from progenitors that had undergone additional cell divisions. For quantification, the cortex was divided into bins corresponding to CP layers I (MZ), II–IV, V, VI, IZ and GZ. Two brains/genotype and six sections/brain were counted and averaged.
Axonal tracing and zinc staining
Dissected brains were fixed overnight in 4% PFA. Corticofugal projections were labeled by placing a DiI crystal in the dorsal cortex as described (Seibt et al, 2003). Brains were incubated in 4% PFA for 6 weeks, embedded in 3% agar, and 100 μm thick vibratome sections were cut and analyzed. To chelate and stain synaptic zinc, mice were administered sodium selenite (5 mg/ml intraperitoneal) 60 min prior to dissection, and cortical hemispheres were sectioned and processed as described (see Supplementary data for details; Danscher, 1982).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Material
Acknowledgments
We thank Vasso Episkopou, Magdalena Götz, Qiufu Ma, Pierre Mattar and Ruth Slack for critical reading, and acknowledge David Anderson, Silvia Arber, Magdalena Götz, Gérard Gradwohl, Peter Gruss, Jane Johnson, Ryoichiro Kageyama, Grace Panganiban, Qiufu Ma, Ronald Evans and John Rubenstein for reagents, Philippe Kastner and the microarray service of the IGBMC for Affymetrix experiments, Thomas Ding and Bruno Weber for histology, and Didier Hentsch and Jean-Luc Vonesh for imaging assistance. CS was supported by fellowships from the HFSP and MRC of Canada and is now a CIHR New Investigator and AHFMR scholar. This work was supported by research grants QLG3-CT-2000-01471 and QLR-2000-0072 from the European Commission, and by l'Association pour la Recherche sur le Cancer, and the French Ministere de l'Enseignement et de la Recherche to FG, by an HFSP grant to FG and KC, by CIHR operating grant MOP-44094 to CS, and by institutional funds from INSERM, CNRS and Hôpital Universitaire de Strasbourg.
References
- Anderson SA, Kaznowski CE, Horn C, Rubenstein JL, McConnell SK (2002) Distinct origins of neocortical projection neurons and interneurons in vivo. Cereb Cortex 12: 702–709 [DOI] [PubMed] [Google Scholar]
- Bertrand N, Castro DS, Guillemot F (2002) Proneural genes and the specification of neural cell types. Nat Rev Neurosci 3: 517–530 [DOI] [PubMed] [Google Scholar]
- Bishop KM, Goudreau G, O'Leary DD (2000) Regulation of area identity in the mammalian neocortex by Emx2 and Pax6. Science 288: 344–349 [DOI] [PubMed] [Google Scholar]
- Bulchand S, Grove EA, Porter FD, Tole S (2001) LIM-homeodomain gene Lhx2 regulates the formation of the cortical hem. Mech Dev 100: 165–175 [DOI] [PubMed] [Google Scholar]
- Caric D, Gooday D, Hill RE, McConnell SK, Price DJ (1997) Determination of the migratory capacity of embryonic cortical cells lacking the transcription factor Pax-6. Development 124: 5087–5096 [DOI] [PubMed] [Google Scholar]
- Casarosa S, Fode C, Guillemot F (1999) Mash1 regulates neurogenesis in the ventral telencephalon. Development 126: 525–534 [DOI] [PubMed] [Google Scholar]
- Caviness VS Jr (1982) Neocortical histogenesis in normal and reeler mice: a developmental study based upon [3H]thymidine autoradiography. Brain Res 256: 293–302 [DOI] [PubMed] [Google Scholar]
- Caviness VS Jr, Takahashi T, Nowakowski RS (1995) Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci 18: 379–383 [DOI] [PubMed] [Google Scholar]
- Chapouton P, Schuurmans C, Guillemot F, Gotz M (2001) The transcription factor neurogenin 2 restricts cell migration from the cortex to the striatum. Development 128: 5149–5159 [DOI] [PubMed] [Google Scholar]
- Cheng L, Arata A, Mizuguchi R, Qian Y, Karunaratne A, Gray PA, Arata S, Shirasawa S, Bouchard M, Luo P, Chen CL, Busslinger M, Goulding M, Onimaru H, Ma Q (2004) Tlx3 and Tlx1 are post-mitotic selector genes determining glutamatergic over GABAergic cell fates. Nat Neurosci 7: 510–517 [DOI] [PubMed] [Google Scholar]
- Danscher G (1982) Exogenous selenium in the brain. A histochemical technique for light and electron microscopical localization of catalytic selenium bonds. Histochemistry 76: 281–293 [DOI] [PubMed] [Google Scholar]
- Fode C, Ma Q, Casarosa S, Ang SL, Anderson DJ, Guillemot F (2000) A role for neural determination genes in specifying the dorsoventral identity of telencephalic neurons. Genes Dev 14: 67–80 [PMC free article] [PubMed] [Google Scholar]
- Fremeau RT Jr, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH (2001) The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron 31: 247–260 [DOI] [PubMed] [Google Scholar]
- Gradwohl G, Fode C, Guillemot F (1996) Restricted expression of a novel murine atonal-related bHLH protein in undifferentiated neural precursors. Dev Biol 180: 227–241 [DOI] [PubMed] [Google Scholar]
- Horton S, Meredith A, Richardson JA, Johnson JE (1999) Correct coordination of neuronal differentiation events in ventral forebrain requires the bHLH factor MASH1. Mol Cell Neurosci 14: 355–369 [DOI] [PubMed] [Google Scholar]
- Job C, Tan SS (2003) Constructing the mammalian neocortex: the role of intrinsic factors. Dev Biol 257: 221–232 [DOI] [PubMed] [Google Scholar]
- Koester SE, O'Leary DD (1993) Connectional distinction between callosal and subcortically projecting cortical neurons is determined prior to axon extension. Dev Biol 160: 1–14 [DOI] [PubMed] [Google Scholar]
- Kornack DR, Rakic P (1998) Changes in cell-cycle kinetics during the development and evolution of primate neocortex. Proc Natl Acad Sci USA 95: 1242–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land PW, Monaghan AP (2003) Expression of the transcription factor, tailless, is required for formation of superficial cortical layers. Cereb Cortex 13: 921–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letinic K, Zoncu R, Rakic P (2002) Origin of GABAergic neurons in the human neocortex. Nature 417: 645–649 [DOI] [PubMed] [Google Scholar]
- Ma Q, Chen Z, del Barco Barrantes I, de la Pompa JL, Anderson DJ (1998) neurogenin1 is essential for the determination of neuronal precursors for proximal cranial sensory ganglia. Neuron 20: 469–482 [DOI] [PubMed] [Google Scholar]
- Mallamaci A, Muzio L, Chan CH, Parnavelas J, Boncinelli E (2000) Area identity shifts in the early cerebral cortex of Emx2−/− mutant mice. Nat Neurosci 3: 679–686 [DOI] [PubMed] [Google Scholar]
- Monuki ES, Porter FD, Walsh CA (2001) Patterning of the dorsal telencephalon and cerebral cortex by a roof plate-Lhx2 pathway. Neuron 32: 591–604 [DOI] [PubMed] [Google Scholar]
- Muzio L, DiBenedetto B, Stoykova A, Boncinelli E, Gruss P, Mallamaci A (2002) Conversion of cerebral cortex into basal ganglia in Emx2(−/−) Pax6(Sey/Sey) double-mutant mice. Nat Neurosci 5: 737–745 [DOI] [PubMed] [Google Scholar]
- Parnavelas JG, Anderson SA, Lavdas AA, Grigoriou M, Pachnis V, Rubenstein JL (2000) The contribution of the ganglionic eminence to the neuronal cell types of the cerebral cortex. Novartis Found Symp 228: 129–139, discussion 139–147 [DOI] [PubMed] [Google Scholar]
- Parras CM, Schuurmans C, Scardigli R, Kim J, Anderson DJ, Guillemot F (2002) Divergent functions of the proneural genes Mash1 and Ngn2 in the specification of neuronal subtype identity. Genes Dev 16: 324–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhinn M, Dierich A, Shawlot W, Behringer RR, Le Meur M, Ang SL (1998) Sequential roles for Otx2 in visceral endoderm and neuroectoderm for forebrain and midbrain induction and specification. Development 125: 845–856 [DOI] [PubMed] [Google Scholar]
- Scardigli R, Baumer N, Gruss P, Guillemot F, Le Roux I (2003) Direct and concentration-dependent regulation of the proneural gene Neurogenin2 by Pax6. Development 130: 3269–3281 [DOI] [PubMed] [Google Scholar]
- Seibt J, Schuurmans C, Gradwhol G, Dehay C, Vanderhaeghen P, Guillemot F, Polleux F (2003) Neurogenin2 specifies the connectivity of thalamic neurons by controlling axon responsiveness to intermediate target cues. Neuron 39: 439–452 [DOI] [PubMed] [Google Scholar]
- Stenman J, Yu RT, Evans RM, Campbell K (2003) Tlx and Pax6 co-operate genetically to establish the pallio–subpallial boundary in the embryonic mouse telencephalon. Development 130: 1113–1122 [DOI] [PubMed] [Google Scholar]
- Stoykova A, Treichel D, Hallonet M, Gruss P (2000) Pax6 modulates the dorsoventral patterning of the mammalian telencephalon. J Neurosci 20: 8042–8050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuhmer T, Anderson SA, Ekker M, Rubenstein JL (2002) Ectopic expression of the Dlx genes induces glutamic acid decarboxylase and Dlx expression. Development 129: 245–252 [DOI] [PubMed] [Google Scholar]
- Tarabykin V, Stoykova A, Usman N, Gruss P (2001) Cortical upper layer neurons derive from the subventricular zone as indicated by Svet1 gene expression. Development 128: 1983–1993 [DOI] [PubMed] [Google Scholar]
- Theil T, Alvarez-Bolado G, Walter A, Ruther U (1999) Gli3 is required for Emx gene expression during dorsal telencephalon development. Development 126: 3561–3571 [DOI] [PubMed] [Google Scholar]
- Toresson H, Potter SS, Campbell K (2000) Genetic control of dorsal–ventral identity in the telencephalon: opposing roles for Pax6 and Gsh2. Development 127: 4361–4371 [DOI] [PubMed] [Google Scholar]
- Yun K, Potter S, Rubenstein JL (2001) Gsh2 and Pax6 play complementary roles in dorsoventral patterning of the mammalian telencephalon. Development 128: 193–205 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Material