

Abstract
Myc synergizes with Ras and PI3-kinase in cell transformation, yet the molecular basis for this behavior is poorly understood. We now show that Myc recruits TFIIH, P-TEFb and Mediator to the cyclin D2 and other target promoters, while the PI3-kinase pathway controls formation of the preinitiation complex and loading of RNA polymerase II. The PI3-kinase pathway involves Akt-mediated phosphorylation of FoxO transcription factors. In a nonphosphorylated state, FoxO factors inhibit induction of multiple Myc target genes, Myc-induced cell proliferation and transformation by Myc and Ras. Abrogation of FoxO function enables Myc to activate target genes in the absence of PI3-kinase activity and to induce foci formation in primary cells in the absence of oncogenic Ras. We suggest that the cooperativity between Myc and Ras is at least in part due to the fact that Myc and FoxO proteins control distinct steps in the activation of an overlapping set of critical target genes.
Keywords: cyclin D2, FoxO, Myc, Ras, PI3-kinase
Introduction
The c-myc proto-oncogene encodes an evolutionary conserved transcription factor, Myc, which can both activate and repress transcription (Levens, 2003). Recent genome-wide analyses show that Myc binds to up to 11% of all genes directly and controls transcription of a broad variety of genes involved in cellular growth control, cell cycle progression, DNA repair and metabolism (Fernandez et al, 2003; Mao et al, 2003; Orian et al, 2003). Surprisingly, on most target genes, Myc is a weak transcriptional activator. Also, while Myc-induced changes in chromatin organization can be detected at most binding sites of Myc, only a minority of neighboring genes shows changes in mRNA levels (Frank et al, 2001). One potential explanation is that Myc may confer a permissive state on chromatin, allowing activation by other cis-acting factors.
Myc cooperates with an activated allele of Ras in cellular transformation and in tumorigenesis in transgenic animals (Land et al, 1983; Orsulic et al, 2002). Ras encodes a small GTPase that is mutated in many human tumors. In its activated state, Ras controls distinct signaling pathways, including the Raf/MEK-kinase pathway, the RalGDS pathway and the PI3-kinase pathway. While multiple studies have identified effector pathways that mediate transformation by Ras in different biological systems (Rodriguez-Viciana et al, 1997; Hamad et al, 2002), it is much less clear as to which effector pathway(s) of Ras are critical for cooperativity with Myc.
The cooperation between Myc and Ras extends to less complex biological systems, such as the ability to drive proliferation in the absence of external growth factors or in an anchorage-independent manner (Land et al, 1983; Leone et al, 1997). A number of recent observations show that Myc specifically cooperates with active PI3-kinase in deregulating cell proliferation. For example, Myc and PI3-kinase drive proliferation of human mammary epithelial cells and fibroblasts that have been immortalized by expression of hTERT and SV40largeT, both in low serum and in soft agar (Zhao et al, 2003). Importantly, cooperation under these circumstances is not due to abrogation of Myc-induced apoptosis by active PI3-kinase, since the presence of SV40largeT inactivates p53 and abrogates Myc-induced apoptosis in these cells (Wei et al, 2003). Similarly, Myc and PI3-kinase promote progression of NIH3T3 cells through the G1 phase in response to PDGF, arguing that related mechanisms operate both in rodent and in human cells (Jones and Kazlauskas, 2001).
PI3-kinase is a lipid kinase that is activated in response to stimulation of receptor tyrosine kinases and G-protein-coupled receptors (Vanhaesebroeck et al, 2001). PI3-kinase generates several second messenger molecules, most notably phosphatidylinositol-3,4,5-triphosphate (PIP3). PIP3 activates a number of downstream effector molecules, including the protein kinase Akt. Akt in turn phosphorylates a number of target molecules, including Bad, a pro-apoptotic protein localized at the mitochondrial membrane (Datta et al, 1997), the YAP transcriptional coactivator (Basu et al, 2003) and the FoxO family of forkhead proteins (Brunet et al, 1999).
Activated PI3-kinase and Akt stimulate cell growth and proliferation, similar to effects observed in response to enhanced expression of Myc (Vanhaesebroeck et al, 2001). This suggests that both PI3-kinase and Myc may regulate a common set of cellular processes. One potential explanation is the suggestion that PI3-kinase might act upstream of Myc to control some aspects of Myc activity. For example, PI3-kinase has been suggested to control Myc stability through phosphorylation of threonine 58 (Sears et al, 2000). However, Myc and PI3-kinase also cooperate under conditions where no PI3-kinase-dependent alterations in Myc levels occur (see below); therefore, control of Myc stability cannot be the sole reason for cooperativity between both signaling pathways.
We have previously identified cyclin D2 as a target gene of Myc (Bouchard et al, 1999; 2001). cyclin D2 expression is also downstream of PI3-kinase (Banerji et al, 2001). We have now used a panel of antibodies directed against general transcription factors to analyze the assembly of an active transcriptional complex on the cyclin D2 promoter. We show that Myc and PI3-kinase strongly synergize in transcriptional activation of cyclin D2, since PI3-kinase, through FoxO forkhead proteins, controls formation of the preinitiation complex. In contrast, Myc recruits TFIIH, P-TEFb and Mediator to promote clearance of RNA polymerase II, both on cyclin D2 and on all other target genes tested. Constitutively active mutants of FoxO3a that cannot be phosphorylated and inactivated by Akt repress multiple target genes of Myc that are involved in cell proliferation, and block Myc-dependent proliferation and transformation. Conversely, dominant-negative alleles of FoxO proteins allow activation of Myc target genes in the absence of PI3-kinase activity and foci formation of primary cells in the absence of Ras. Our findings provide a model to explain how Myc and PI3-kinase cooperate in cell transformation.
Results
Expression of cyclin D2 depends on external growth factors in multiple cells (e.g., Matsushime et al, 1991; Sicinski et al, 1996); growth factor-dependent activation of cyclin D2 expression requires PI3-kinase activity (Banerji et al, 2001). We have previously shown that cyclin D2 is a target gene of Myc (Bouchard et al, 1999). Therefore, we tested whether the requirement for PI3-kinase activity could be explained by the need to stimulate expression of c-myc. To do so, we stimulated serum-starved RAT1 cells by addition of 10% fetal calf serum (FCS) either in the presence of a solvent control (DMSO) or in the presence of LY294002, an inhibitor of PI3-kinase (Figure 1A). As expected, addition of LY294002 abolished the induction of cyclin D2 expression; in contrast, PI3-kinase activity was not required for stimulation of c-myc expression by FCS.
Figure 1.
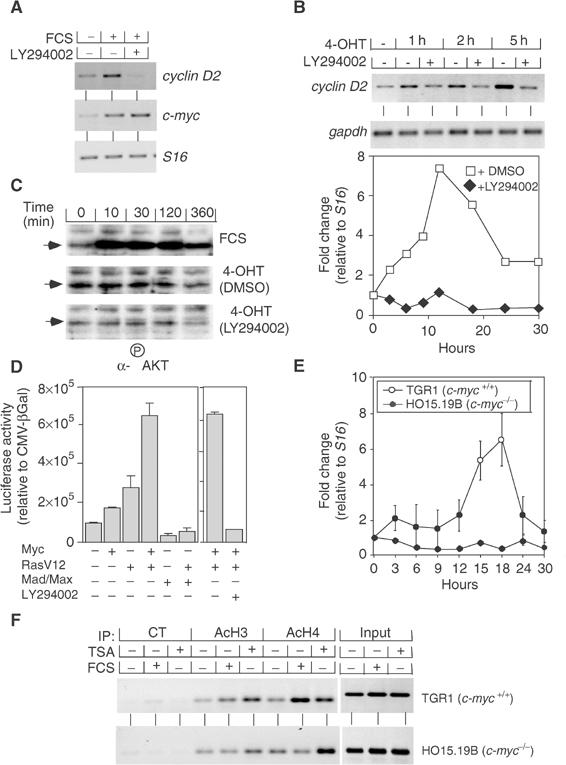
Myc and Ras cooperate in activation of the cyclin D2 gene. (A) Effect of PI3-kinase inhibition on cyclin D2 and c-myc expression in RAT1 cells. Cells were starved in 0.1% FCS and subsequently either left untreated or stimulated by addition of 10% FCS (3 and 18 h for c-myc and cyclin D2 expression, respectively). DMSO or LY294002 was added 1 h prior to treatment with FCS. Shown are the results of RT–PCR assays using primers specific for cyclin D2, c-myc and S16. (B) Inhibition of PI3-kinase abolishes Myc-dependent induction of the cyclin D2 gene. Mouse fibroblasts expressing a MycER chimera were starved, pretreated with LY294002 as described above, and stimulated by addition of 200 nM 4-OHT for the indicated times. Shown are a representative gel of RT–PCR assays using primers specific for cyclin D2 and gapdh (upper panel), and a quantitation of the results from an independent experiment (lower panel). Data were plotted as fold induction of cyclin D2 expression relative to a S16 control. (C) Residual PI3-kinase activity is present in serum-starved MycER fibroblasts. The panels show immunoblots using an antibody directed against phosphorylated Akt of cells stimulated by addition of FCS, 4-OHT in the presence of DMSO or 4-OHT in the presence of LY294002 for the indicated times. (D) Synergistic activation of the cyclin D2 promoter by Myc and Ras. The panels show the results of transient transfection assays in NIH3T3 cells using a 2.3 kb fragment of the murine cyclin D2 promoter. CMV expression plasmids encoding human Myc, RasV12 or Mad-1 and Max as well as LY294002 were used as indicated. (E) Myc is required for growth factor-induced expression of cyclin D2. RAT1c-myc+/+ (TGR1) and RAT1c-myc−/− (HO15.19B) cells were serum-starved and re-stimulated by addition of 10% FCS for the indicated times. The panel shows a quantitation of three independent RT–PCR assays; data are plotted as fold induction of cyclin D2 expression relative to a S16 control. (F) Myc is required for growth factor-induced acetylation at the cyclin D2 locus. The panels show ChIP assays using the indicated antibodies from either RAT1c-myc+/+ (TGR1) or RAT1c-myc−/− (HO15.19B) cells either left untreated or treated with FCS or TSA (both for 6 h). ‘CT' designates a control antibody. Precipitated DNA samples were amplified with primers specific for the E-box domains of rat cyclin D2 promoter (see Supplementary Figure 1 for primer localization).
To test whether constitutive expression of Myc replaces the requirement for PI3-kinase in cyclin D2 induction, we repeated the experiment in fibroblasts that express a MycER chimeric protein (Bouchard et al, 1999). Activation of Myc by addition of 4-hydroxy-tamoxifen (4-OHT) rapidly induced expression of cyclin D2 in the presence of DMSO (Figure 1B). Addition of LY294002 completely abolished induction of cyclin D2 by Myc, demonstrating that PI3-kinase activity is required to render cells permissive for Myc-induced expression of cyclin D2.
In these experiments, we noted that addition of LY294002 suppressed induction of cyclin D2 in response to activation of Myc; therefore, either activation of Myc stimulates PI3-kinase activity or, alternatively, serum-starved cells contain residual PI3-kinase activity that is suppressed by addition of LY294002. To distinguish between both possibilities, we performed immunoblots with antibodies directed against phosphorylated Akt, which is phosphorylated in response to PI3-kinase activation (Figure 1C) (Alessi et al, 1996). The results showed that addition of FCS, but not activation of Myc, strongly stimulated PI3-kinase activity and revealed the presence of residual phosphorylation on Akt in serum-starved cells, which was suppressed by addition of LY294002.
Consistent with the requirement for both Myc and active PI3-kinase in cyclin D2 activation, Myc alone failed to activate significantly a fragment of the cyclin D2 promoter that contains the Myc-binding site in transient transfection assays (Bouchard et al, 1999) (Figure 1D). However, we observed a strong synergy between oncogenic Ras (RasV12) and Myc in promoter activation. The small activation observed upon expression by RasV12 alone could be suppressed by coexpression of Mad proteins, demonstrating that it depends on endogenous Myc activity. Inhibition of PI3-kinase completely abolished the synergy between Myc and Ras in promoter activation, consistent with the observations for the endogenous gene.
We also considered the possibility that activation of PI3-kinase might bypass the requirement for Myc in the regulation of the cyclin D2 expression. To test this, we measured the induction of cyclin D2 expression upon addition of growth factors in a clone of RAT1 cells (TGR1) and in a derivative cell, in which the c-myc gene has been deleted (HO15.19B) (Mateyak et al, 1997) (Figure 1E). In RAT1c-myc+/+ cells, addition of FCS induced expression of cyclin D2; in contrast, cyclin D2 expression was blocked in RAT1c-myc−/− cells, consistent with an essential role of Myc in cyclin D2 expression (Knoepfler et al, 2002). Myc activates cyclin D2 expression at least in part through the recruitment of a histone acetylase complex (Bouchard et al, 2001). Consistent with this view, growth factor-induced histone acetylation occurred in RAT1c-myc+/+, but not in RAT1c-myc−/− cells; in contrast, addition of trichostatin A (TSA), an inhibitor of histone deacetylases, induced histone acetylation in both cell lines to a similar extent (Figure 1F). Taken together, our data show that both Myc and active PI3-kinase are required for activation of cyclin D2 expression.
PI3-kinase has been implicated in the control of Myc protein stability (Sears et al, 2000); therefore, our observations might reflect degradation of Myc upon addition of LY294002. However, immunoblotting revealed no change in the amount of MycER protein in the presence of 4-OHT, LY294002 or PD98059, an inhibitor of MAP-kinases; endogenous Myc was undetectable in these serum-starved cells under any of these conditions (Figure 2A and data not shown). Further, chromatin immunoprecipitations (ChIPs) revealed that, upon activation, MycER bound to the cyclin D2 promoter regardless of whether LY294002 was present or not and that activation of MycER triggered histone H4 acetylation both in the presence and absence of LY294002 (Figure 2B). Finally, both microarray experiments and RT–PCR analyses of individual genes showed that inhibition of PI3-kinase activity does not generally inhibit transactivation by Myc; for example, Myc-dependent induction of nucleolin expression was unimpaired by addition of LY294002 (data not shown). We concluded that inhibition of PI3-kinase does not inhibit cyclin D2 expression through controlling Myc stability or function, but is required independently of Myc.
Figure 2.
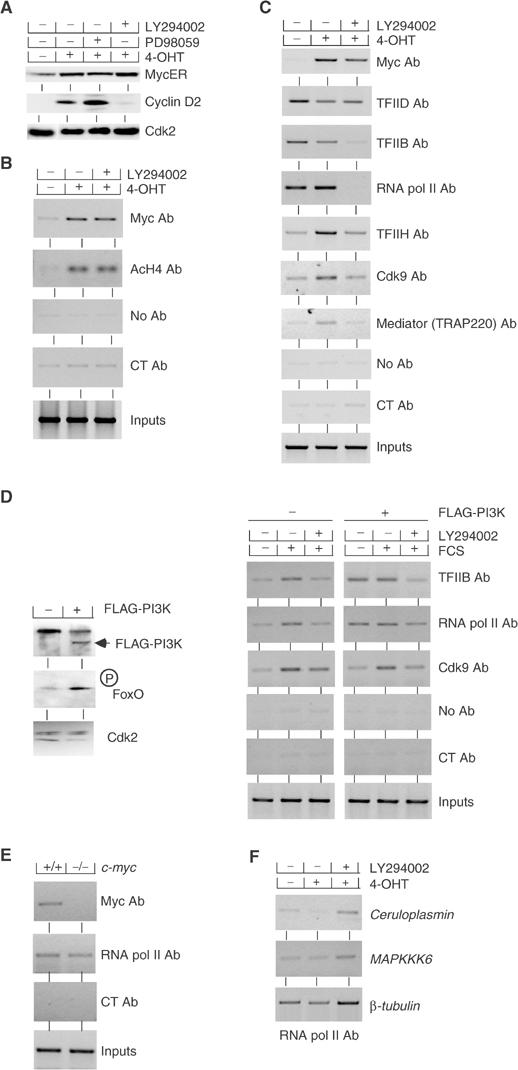
Myc and PI3-kinase are independently required for activation of cyclin D2 transcription. (A) Effect of PI3-kinase inhibition on cyclin D2 and MycER expression in mouse MycER fibroblasts. Cells were starved in 0.1% FCS and subsequently either left untreated or stimulated by addition of 200 nM 4-OHT for 6 h. DMSO or the indicated inhibitors was added 1 h prior to treatment with 4-OHT. Shown are immunoblots documenting the expression levels of MycER, cyclin D2 and Cdk2 as a loading control. (B) Inhibition of PI3-kinase does not affect Myc binding and Myc-mediated histone acetylation at the cyclin D2 promoter. Shown are ChIP assays using the indicated antibodies and primers recognizing the E-boxes of mouse cyclin D2 with chromatin isolated from mouse MycER fibroblasts grown under the same conditions as described above. ‘CT' designates control antibodies. (C) Myc and PI3-kinase control distinct steps in the activation of the cyclin D2 promoter. The panels document ChIP assays performed with chromatin isolated from MycER cells using the indicated antibodies and primers for E-boxes (for Myc and Cdk9 IPs) or the transcription start site of mouse cyclin D2 locus (for the other IPs). The experiment was carried out as in panels A and B. The position of the primers used is indicated in Supplementary Figure 1. (D) Active PI3-kinase promotes loading of RNA polymerase II and TFIIB, but not Cdk9, to the cyclin D2 promoter. The left panels show immunoblots of either control cell pools or pools infected with a retrovirus expressing activated, FLAG-tagged Myr-PI3-Kinase; the antibodies used are indicated. Cdk2 was used as a control for equal loading. The right panels document ChIP assays using the indicated antibodies and primers specific for the transcription start site of mouse cyclin D2 locus from either control cells or cells expressing PI3-kinase. Cells were serum-starved and subsequently re-stimulated by addition of 10% FCS either in the presence of DMSO or LY294002 as indicated. (E) The loading of RNA polymerase II on cyclin D2 promoter is independent of Myc. ChIP assays were performed with exponentially growing RAT1c-myc+/+ (TGR1) and RAT1c-myc−/− (HO15.19B) cells using the indicated antibodies and primers for E-boxes (for Myc IP) or the transcription start site of mouse cyclin D2 locus (for RNA polymerase II IP). (F) LY294002 does not generally inhibit loading of RNA polymerase. The panels document ChIP assays using antibodies directed against RNA polymerase II using primers specific for the transcription start site of the indicated genes. The same chromatin as shown in panel C was used for the experiment. Controls established that the signal shown is specific; there was no amplification in control reactions using either no antibody or a control antibody (not shown).
In order to understand how Myc and PI3-kinase cooperate in cyclin D2 activation, we used a panel of different antibodies to distinguish different steps of promoter activation. We used antibodies against TFIID, TFIIB and RNA polymerase II to measure assembly of the preinitiation complex. In addition, we used antibodies against TFIIH, the common TRAP220 subunit of the TRAP, CRSP, DRIP and ARC Mediator complexes and the Cdk9 subunit of P-TEFb to analyze subsequent steps in cyclin D2 promoter activation. The localization of the primers used in these experiments is shown in Supplementary Figure 1. Activation of Myc had no effect on the loading of RNA polymerase, TFIID or TFIIB to the promoter (Figure 2C), demonstrating that assembly of the preinitiation complex occurs independently of Myc. Loading of RNA polymerase and of TFIIB, but not TFIID, was highly sensitive to inhibition of PI3-kinase, demonstrating that PI3-kinase activity is required for assembly of the preinitiation complex. Activation of Myc recruited TFIIH, Cdk9 and TRAP220 to the cyclin D2 promoter. Inhibition of PI3-kinase activity abolished Myc-induced loading of TFIIH, TRAP220 and Cdk9, most likely because these complexes only bind stably to promoters in the presence of RNA polymerase. We note that under these conditions, Mediator and RNA polymerase behave as distinct entities, not as a holoenzyme, similar to observations made upon activation of the heat shock factor in Drosophila (Park et al, 2001). Several attempts to use antibodies against different phosphorylated residues of RNA polymerase to extend these results yielded inconsistent results (data not shown).
To exclude that these results reflect a specific property of the MycER system, we expressed a constitutively active form of PI3-kinase (Myr-FLAG-p110α; Zhao et al, 2003) in murine fibroblasts (Figure 2D). As a control for PI3-kinase activity, we measured the phosphorylation of the forkhead transcription factor FoxO3a, using antibodies that specifically recognize phosphorylated FoxO3a. We then analyzed binding of RNA polymerase, TFIIB and Cdk9 to the cyclin D2 promoter in control cells and in cell expressing constitutively active PI3-kinase upon addition of growth factors, either in the presence of DMSO or LY294002 (Figure 2D). Addition of growth factors stimulated binding of RNA polymerase, TFIIB and Cdk9 to the cyclin D2 promoter in control cells, suggesting that both formation of the preinitiation complex and clearance of the polymerase on the cyclin D2 promoter are stimulated by growth factors. Activated PI3-kinase stimulated binding of TFIIB and RNA polymerase, but not of Cdk9, to the cyclin D2 promoter in serum-starved cells; these effects are complementary to those observed upon activation of Myc. Inhibition of PI3-kinase activity abrogated the serum-induced increase in binding of RNA polymerase, TFIIB and Cdk9 to the cyclin D2 promoter, analogous to the observations in cells expressing MycER.
To exclude formally a role of Myc proteins in binding of RNA polymerase to the cyclin D2 promoter, we compared binding of RNA polymerase to the cyclin D2 promoter in exponentially growing RAT1-c-myc+/+ and RAT1-c-myc−/− cells (Figure 2E). In both cell types, RNA polymerase was bound to the cyclin D2 promoter. We concluded that endogenous c-myc does not regulate binding of RNA polymerase to the cyclin D2 promoter. As an additional control to show that formation of the preinitiation complex on the cyclin D2 gene is specifically regulated through the PI3-kinase pathway, we measured the effect of LY294002 on the binding of RNA polymerase II to the core promoter of several genes that are either induced by inhibition of PI3-kinase (ceruloplasmin, mapkkk6, cbl-b and foxb30; Ramaswamy et al, 2002) or unaffected by PI3-kinase (β-tubulin) (Figure 2F and data not shown). As expected, addition of LY294002 did not inhibit loading of RNA polymerase to the promoter of these genes. We concluded that LY294002 does not cause a general inhibition of RNA polymerase loading. Rather, our data indicate that there is a specific requirement for PI3-kinase activity for the formation of the preinitiation complex on the cyclin D2 promoter, which may apply to a larger subset of Myc target genes (see below).
These results might reflect a particular mode of Myc action at the cyclin D2 promoter or might reflect a general property of Myc. We therefore repeated these experiments analyzing six different target genes of Myc in a similar manner; the choice of these genes is arbitrary but includes some of the most intensely studied target genes. On all genes, RNA polymerase was present at the promoter independently of Myc, and activation of Myc did not enhance binding of RNA polymerase (data not shown and Figure 3A). With few exceptions, Myc stimulated loading of TFIIH, Cdk9 and TRAP220 to its target genes. The data strongly suggest that Myc acts in the same manner on many, if not all, of its target genes.
Figure 3.
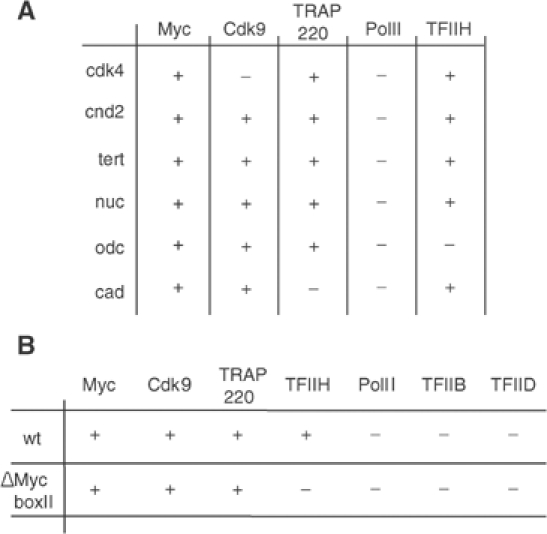
Myc-dependent loading of general transcription factors. (A) Myc recruits TFIIH, TRAP220 and Cdk9 to multiple target genes. The panel shows a summary of ChIP assays performed with the indicated antibodies. MycER fibroblasts were cultured as described in Figure 2A–C. Primers spanning the E-boxes and the transcription start sites of the indicated genes were used in the analysis. The +/− symbols designate whether a change in promoter occupancy of the indicated factor was observed upon activation of Myc; the threshold was arbitrarily set to 1.5. (B) Recruitment of TFIIH, but not of TRAP220 and Cdk9, by Myc depends on the integrity of MycBoxII (MBII). Shown is the summary of parallel ChIP assays from mouse fibroblasts expressing either a WTMycER chimera or a ΔMBIIMycER chimeric protein. Symbols are used as in panel A.
Activation of cyclin D2 requires MycBoxII, a domain that recruits a histone acetylase complex containing TRRAP and the catalytic subunits Gcn5 (McMahon et al, 2000) or Tip60 (Frank et al, 2003) to the cyclin D2 promoter (Bouchard et al, 2001). However, TRRAP-independent pathways of gene activation have been described, raising the possibility that MycBoxII may only be required on specific promoters (Nikiforov et al, 2002). We wondered therefore whether recruitment of TFIIH, Cdk9 and TRAP220 to the cyclin D2 promoter requires MycBoxII (Figure 3B). Recruitment of TFIIH depended on an intact MycBoxII, suggesting that TFIIH is recruited in response to Myc-induced histone acetylation. In contrast, both Cdk9 and TRAP220 were also recruited by a ΔMycBoxIIMycER protein, potentially reflecting direct interactions of Myc with the P-TEFb and Mediator complexes (Eberhardy and Farnham, 2002). Identical results were obtained for the other target genes of Myc analyzed in Figure 3A (data not shown).
We sought to identify the downstream transcription factors that mediate the effect of PI3-kinase on cyclin D2 expression. Transcription factors that are regulated by PI3-kinase include NF-κB and the FoxO transcription factors; both have been implicated in the control of cyclin D transcription (Huang et al, 2001; Ramaswamy et al, 2002; Schmidt et al, 2002). Whereas NF-κB activity is stimulated by PI3-kinase, FoxO factors are excluded from the nucleus upon phosphorylation by PI3-kinase. To address the role of NF-κB in cyclin D2 expression, we expressed a constitutively active form of IκBα, in which two serine residues that are critical for phosphorylation and degradation have been mutated to alanine residues (IκBαA2) in murine MycER fibroblasts (Brockman et al, 1995). To interfere with Akt-mediated inactivation of FoxO factors, we expressed either wild-type FoxO3a or a mutated allele of FoxO3a, in which three PI3-kinase-dependent phosphorylation sites have been mutated to alanine (FoxO3a(A3)) (Brunet et al, 1999). Neither expression of IκBαA2 nor of a wild-type allele of FoxO3a inhibited expression of cyclin D2; in contrast, expression of FoxO3a(A3) abolished basal and Myc-induced transcription of the cyclin D2 gene (Figure 4A). RT–PCR analysis and immunoblotting confirmed that the failure of IκBαA2 and FoxO3a to inhibit cyclin D2 expression was not due to failure to be expressed in these cells (see Supplementary Figure 2 and data not shown). In addition, expression of IκBαA2, FoxO3a or FoxO3a(A3) sensitized cells to apoptosis, demonstrating that all proteins were biologically active (data not shown).
Figure 4.
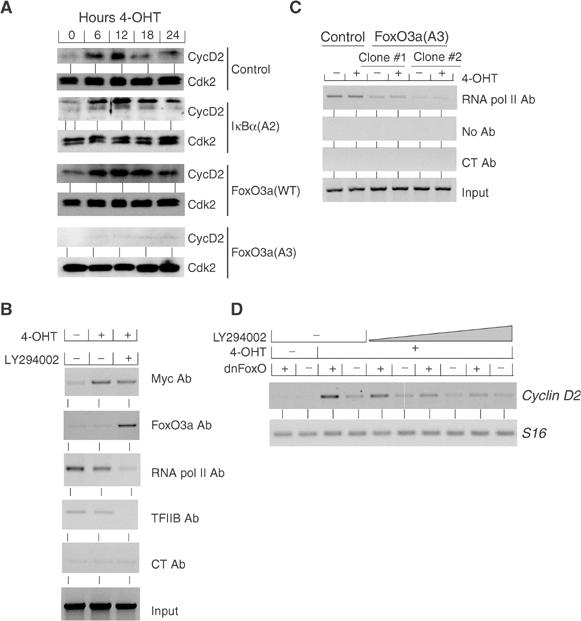
FoxO3 proteins mediate regulation of cyclin D2 expression by PI3-kinase. (A) Shown are immunoblots documenting the expression of cyclin D2 and Cdk2 (as a loading control) in representative clones of IκBα(A2)-, FoxO3a(WT)- and FoxO3a(A3)-expressing mouse MycER fibroblasts. Serum-starved cells were treated with 4-OHT for the indicated times. (B) Endogenous FoxO3a binds to the cyclin D2 promoter upon inhibition of PI3-kinase. Mouse MycER fibroblasts grown under the conditions as described in Figure 2A–C were analyzed by ChIP assays using the indicated antibodies. (C) Nonphosphorylatable FoxO3a (FoxO3a(A3)) blocks the loading of RNA polymerase II on the mouse cyclin D2 promoter. ChIP assays were performed with quiescent and 4-OHT-stimulated control- or FoxO3a(A3)-infected clones of mouse MycER cells using the indicated antibodies and primers for the transcription start site of mouse cyclin D2. (D) Dominant-negative FoxO enhances activation of cyclin D2 expression by Myc. Expressions of cyclin D2 and S16 as a control were analyzed by RT–PCR in pools of mouse MycER fibroblasts infected either with control virus or a retrovirus expressing a dominant-negative allele of FoxO (dnFoxO). Serum-starved cells were treated with 4-OHT for 6 h in the presence of increasing concentrations of LY294002 (5–50 μM).
RT–PCR analysis showed that two FoxO proteins, FoxO1 and FoxO3a, were expressed in murine MycER fibroblasts, with FoxO3a being the predominant gene (data not shown). We therefore used antibodies directed against FoxO3a to test whether endogenous FoxO proteins are bound at the cyclin D2 promoter (Figure 4B). ChIPs revealed that endogenous FoxO3a bound to the cyclin D2 promoter in vivo when PI3-kinase activity was inhibited. Binding of TFIIB and RNA polymerase II to the cyclin D2 promoter inversely correlated to that of FoxO proteins, consistent with the notion that FoxO proteins mediate inhibition of cyclin D2 expression by LY294002 (see below).
In clones expressing FoxO3a(A3), binding of RNA polymerase to the cyclin D2 promoter was reduced relative to control cells even in the absence of LY294002 (Figure 4C). Importantly, activation of Myc in clones expressing FoxO3a(A3) did not restore loading of RNA polymerase to the level observed in control cells, further illustrating the inability of Myc to affect preinitiation complex formation.
To test whether endogenous FoxO proteins are required for inhibition of cyclin D2 under conditions of limiting PI3-kinase activity, we expressed a dominant-negative allele of FoxO (Medema et al, 2000). In pools of cells expressing dnFoxO, expression of cyclin D2 was enhanced upon activation of Myc relative to control cells, both in the absence and, importantly, in the presence of increasing concentrations of LY294002 (Figure 4D). Taken together, the results show that nonphosphorylated FoxO proteins are necessary and sufficient to mediate inhibition of cyclin D2 transcription upon inhibition of PI3-kinase.
We wondered whether cyclin D2 is the only target gene of Myc that is repressed by FoxO proteins; therefore, we compared the recently published comprehensive list of FoxO target genes (Ramaswamy et al, 2002) to the set of Myc target genes (http://www.myccancergene.org/). There was a significant overlap between both sets of genes. Indeed, induction of multiple target genes of Myc, including cyclin D2, cdk4, odc, cyclin E2 (B Amati, personal communication), jvt-1 and cad, was strongly inhibited by FoxO3a(A3) (Figure 5A). FoxO3a did not inhibit induction of nucleolin, demonstrating that FoxO3a does not affect Myc function in general.
Figure 5.
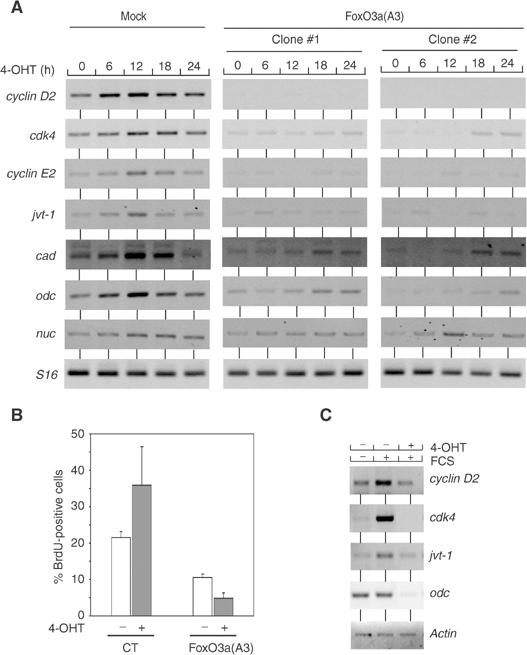
Nonphosphorylatable FoxO3a blocks Myc function in proliferation. (A) Nonphosphorylatable FoxO3a blocks induction of multiple target genes of Myc in MycER fibroblasts. The panels show RT–PCR assays performed with clones of control- and FoxO3a(A3)-infected mouse MycER cells. Quiescent cells were stimulated with 4-OHT for the indicated times. Primers used were specific for the indicated genes. (B) Nonphosphorylatable FoxO3a blocks Myc-induced cell cycle entry. RAT1-MycER cells were either infected with control virus or virus expressing FoxO3a(A3). Shown is the percentage of BrdU incorporating RAT1-MycER cells before and after stimulation with 4-OHT. (C) Nonphosphorylatable FoxO3a (FoxO3a(A3)) blocks growth factor-dependent induction of Myc target genes. Serum-starved RAT1 FoxO3a(A3)-ER cells were either left untreated or pretreated with 4-OHT (1 h) in order to activate FoxO3a(A3)-ER and stimulated with FCS (for 18 h). Shown are RT–PCR assays using primers specific for the indicated Myc target genes and actin (as a control).
Consistent with an inhibitory effect of FoxO proteins on multiple target genes of Myc, retroviral expression of FoxO3a(A3) blocked cell cycle entry by Myc in the mouse fibroblasts used here (data not shown) and in RAT1-MycER cells (Figure 5B), showing that FoxO3a(A3) dominantly blocks Myc function in cell proliferation.
To test whether FoxO proteins specifically inhibit Myc-induced expression of these genes or whether they have a more general role in growth factor-mediated induction of expression, we stimulated RAT1 cells that express a hormone(ER)-inducible allele of FoxO3a(A3) by addition of FCS either in the absence or presence of 4-OHT (Tran et al, 2002) (Figure 5C). Activation of FoxO inhibited growth factor-induced expression of odc, cdk4 and, to a lesser degree, jvt-1. Taken together, the data show that Myc and the FoxO pathway together mediate the induction of a set of G1 regulatory genes in response to serum growth factors.
Finally, we wondered whether the requirement for phosphorylation of FoxO proteins in Myc-dependent gene expression can account for the requirement for oncogenic Ras or PI3-kinase in the transformation of primary cells by Myc. To test this, we infected primary mouse embryo fibroblasts (MEFs) with retroviruses encoding Myc, RasV12 or Myc and RasV12, either alone or together with different alleles of FoxO proteins. Expression of FoxO3a had no effect on the formation of foci by the cooperative action of Myc and RasV12 in these assays (Supplementary Figure 3). In contrast, expression of FoxO3a(A3) abrogated the formation of foci by both oncogenes, demonstrating that phosphorylation of FoxO proteins is required for the cooperative transformation of primary cells by Myc and RasV12 (Figure 6A). Primary MEFs were also transformed by Myc in cooperation with activated Akt, arguing that active Akt can replace oncogenic Ras in these assays; as observed for transformation by Myc and Ras, expression of FoxO3a(A3) abrogated transformation of primary MEFs by Myc and activated Akt (Supplementary Figure 3).
Figure 6.
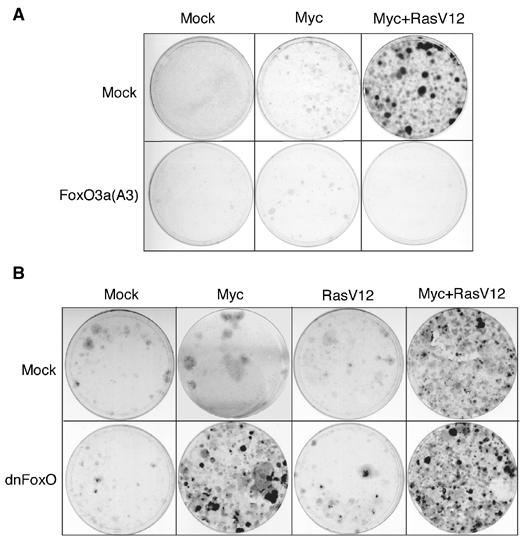
Role of FoxO proteins in primary cell transformation. (A) Nonphosphorylatable FoxO3a blocks Myc/RasV12 collaboration in transformation of primary MEFs. Primary cultures of wild-type MEFs were infected sequentially with retroviruses encoding Myc, or Myc and RasV12, and either pBabePuro-FoxO3a(A3) or empty pBabePuro. At 2 days after the last infection, MEFs were selected with puromycin and cells were allowed to grow for 2 weeks prior to crystal violet staining. (B) Focus formation assay in primary MEFs demonstrating that dominant-negative FoxO (dnFoxO) and Myc cooperate to form foci in the absence of oncogenic Ras. The experiment was carried out as in panel A with retroviruses encoding a dominant-negative allele of FoxO (pBabePuro-dnFoxO).
Expression of dominant-negative FoxO did not significantly affect focus formation by Myc and RasV12 (data not shown). Importantly, expression of dominant-negative FoxO enhanced the formation of foci by Myc in primary cells to a similar extent as oncogenic Ras (Figure 6B; a quantitation of multiple experiments is shown in Supplementary Figure 4). In contrast, dominant-negative FoxO did not cooperate with RasV12 in focus formation of primary MEFs. Also, dominant-negative FoxO did not further enhance colony formation in the presence of both Myc and RasV12. We concluded that the requirement for Akt-mediated phosphorylation of FoxO proteins accounts at least in part for the requirement for oncogenic Ras in the transformation of primary cells by Myc and Ras.
Discussion
In this report, we show that Myc and PI3-kinase cooperate in activation of the cyclin D2 promoter and provide a mechanistic explanation for this behavior. Our findings provide a potential model for understanding the cooperation of Myc and oncogenic Ras during cell transformation.
Our model is based on four findings: First, Myc controls specific steps in the activation of its target genes in a stereotypical manner. Specifically, we show that Myc has no effect on formation of the preinitiation complex and loading of RNA polymerase, but recruits TFIIH, P-TEFb and Mediator to the promoter on all target genes that we tested. We also show that both MycBoxII-dependent and -independent pathways cooperate in the induction of the cyclin D2 gene: recruitment of TFIIH depends on MycBoxII, and may therefore be an indirect consequence of TRAPP-mediated histone acetylation. Recruitment of P-TEFb and Mediator does not depend on MycBoxII. The findings are consistent with a recent report showing that both TRAPP-dependent and -independent mechanisms contribute to gene activation by Myc and suggest that recruitment of P-TEFb and Mediator defines such TRAPP-independent mechanisms (Nikiforov et al, 2002). Our data do not address the issue whether P-TEFb and Mediator are recruited through direct binding to Myc. However, a number of recent reports have demonstrated that both cyclin T1, a component of P-TEFb, and Cdk8, a subunit of Mediator, bind to the Myc transactivation domain in vitro (Eberhardy and Farnham, 2002; Kanazawa et al, 2003). Together, the data are consistent with a view that these interactions contribute to gene activation in vivo. Importantly, our findings explain why Myc often acts as a weak transcriptional activator both in reporter assays and on its target genes, and why binding of Myc in vivo is often not sufficient for gene activation (Frank et al, 2001; Fernandez et al, 2003; Orian et al, 2003). We propose that the formation of the preinitiation complex limits transcriptional activation on genes or under conditions in which Myc acts as a poor activator.
Second, formation of the preinitiation complex and loading of RNA polymerase to the cyclin D2 promoter are controlled by PI3-kinase. This requirement is specific, since inhibition of PI3-kinase does not inhibit loading of RNA polymerase on several control genes. The requirement for PI3-kinase activity is due to the fact that nonphosphorylated FoxO proteins repress formation of the preinitiation complex; phosphorylation by PI3-kinase removes FoxO proteins from the cyclin D2 promoter. PI3-kinase is not generally required for Myc-dependent gene activation, making it unlikely that Myc function and/or stability per se are affected by lack of PI3-kinase.
Third, FoxO proteins and Myc coordinately regulate a set of genes that includes several key regulators of cell proliferation and that is critical for Myc-induced proliferation. From these observations, we propose that the cooperation between Myc and PI3-kinase in controlling cell proliferation results from the fact that FoxO proteins and Myc control sequential steps in the assembly of active promoter complexes on a group of target genes that is critical for proliferation.
FoxO proteins activate transcription through direct binding to an insulin-response element, IRS. They also repress transcription of a distinct set of target genes; we show here that there is a significant overlap between the group of genes repressed by FoxO proteins and Myc-induced genes. Data presented in this paper (Figure 4C) and in a previous report (Ramaswamy et al, 2002) show that FoxO proteins are also bound in vivo to repressed target genes. However, FoxO proteins do not repress genes through binding to an IRS and may be not through direct binding to DNA, since a point mutant in the DNA-binding domain, which disrupts binding to IRS elements and transcriptional activation by FoxO proteins, does not disrupt transcriptional repression (Ramaswamy et al, 2002). Potentially, therefore, FoxO proteins are recruited to targets of repression at least in part through interaction with other DNA-binding factors; a similar model has recently been proposed for the regulation of the p21Cip1 promoter by FoxO proteins, where binding is mediated partly through complex formation with Smad proteins (Seoane et al, 2004). A full understanding of the regulation of Myc target genes by FoxO factors will therefore require the identification of the mechanism of recruitment of FoxO proteins to these genes.
Fourth, a dominant-negative allele of FoxO can replace oncogenic Ras in focus formation assays in primary MEFs, demonstrating that the PI3-kinase-dependent phosphorylation of FoxO proteins is sufficient to account for this activity of oncogenic Ras during primary cell transformation.
Our data are similar to a recent report demonstrating that Akt strongly accelerates B-lymphomagenesis by Myc (Wendel et al, 2004). In these assays, Akt acts through mTOR and eIF4e, and enhanced expression of eIF4e substitutes for Akt. Since eIF4e is a downstream target gene of Myc, it is possible that enhanced eIF4e activity is responsible for the enhanced focus formation that we observe in the presence of a dominant-negative allele of FoxO. We are currently testing this hypothesis.
However, several observations demonstrate that phosphorylation of FoxO proteins is not the only function of oncogenic Ras in cooperation with Myc: For example, human mammary epithelial cells expressing TERT and Myc require PI3-kinase for anchorage-independent growth and Akt cannot substitute for PI3-kinase in this assay (Wei et al, 2003; Zhao et al, 2003). Similarly, human fibroblasts expressing SV40largeT, TERT and Myc require oncogenic Ras for tumor formation in vivo, and smallt, which enhances PI3-kinase activity, cannot replace Ras in these assays (Wei et al, 2003). While these differences might potentially be due to the different cell types used, it seems more likely that they reflect additional functions of oncogenic Ras during cooperative tumor formation with Myc. It is possible that Myc cooperates with other Ras-dependent pathways in a manner similar to that described here in controlling other aspects of cellular transformation, such as anchorage-independent growth and tumor formation in nude mice. We suggest, therefore, that a comprehensive analysis of the sets of genes regulated by Myc in cooperation with specific Ras-dependent signals will give further mechanistic insights into the process of cooperation between both oncogenes.
Materials and methods
Cell culture and transient transfections
Rat and mouse cell lines expressing MycER™ ΔMycBoxIIMycER™ and FoxO3a(A3)ER™ proteins have been described previously (Bouchard et al, 1999; 2001; Tran et al, 2002). Rat TGR1 (c-myc+/+) and HO15.19B (c-myc−/−) cells were a kind gift from John Sedivy and were cultured as described (Mateyak et al, 1997). Cells that were 70% confluent were serum-starved in 0.1% fetal calf serum (Gibco) for 96 h, and then either left untreated or incubated with 200 nM 4-OHT (Sigma), 100 ng/ml trichostatin A (Calbiochem) or 10% FCS for the indicated times. LY294002 (50 μM; Calbiochem) or a solvent control (DMSO; Merck) was added to the cells 1 h prior to further treatment. Primary MEFs were a kind gift from Daniel Peeper and maintained as described previously (Peeper et al, 2001).
Transient transfections into NIH3T3 cells were performed using a standard CaPO4 protocol. A 2.3 kb fragment of the mouse cyclin D2 as a reporter plasmid as well as the CMV expression vectors encoding Myc, Max and Mad have been described previously (Bouchard et al, 1999). LY294002 (at 50 μM) was added to the transfected cells 4 h before harvesting.
Retroviral infection
Retroviral pBabeBleo vectors expressing human FoxO proteins (FoxO3a(WT), FoxO3a(A3) and dominant-negative FoxO4) and constitutively active human IκBα (IκBαA2) were constructed by subcloning from pECE-FoxO3a, pECE-FoxO3a(A3) and Rc/CMV-IκBαA2, respectively (Brockman et al, 1995; Schmidt et al, 2002). Retroviral supernatants were generated by transient transfections of Phoenix cells and used to infect MycER™ cells. Infected cells were selected with 40 μg/ml zeocin (Invitrogen). Resistant clones were picked and expanded for further analysis. pBabePuro-FoxO3a and pBabePuro-FoxO3a(A3) have been described previously (Schmidt et al, 2002).
Chromatin immunoprecipitation
ChIP assays were performed as described previously (Bouchard et al, 2001) using the following antibodies: anti-acetylated histones H3 (#06-599) and H4 (#06-866) (Upstate), anti-Myc (N-262; sc-764), anti-TFIID (SI-1, sc-273), anti-TFIIB (C-18; sc-225), anti-TFIIH p89 (S-19; sc-293), anti-cdk9 (H-169; sc-8338), anti-TRAP220 (M-255; sc-8998), anti-FoxO3a (H-144; sc-11351) and anti-cyclin A (C-19; sc-596) as control antibodies (all Santa Cruz); anti-RNA polymerase II (8WG16; gift from Dirk Eick). Immunoprecipitated DNA samples were amplified by PCR (MWG Biotech) using RedTaq polymerase (Gibco) and primers specific for the E-boxes and for the transcription start site of the indicated promoters. All ChIPs were performed 6 h after stimulation with the indicated agents. For each promoter, PCR reactions were performed with different numbers of cycles or with dilution series of input DNA to determine the linear range of the amplification; all results shown fall within this range. The PCR signals were quantified using NIH Image software. The results shown in Figure 3 represent the average of two independent experiments. Primer sequences specific for the mouse and rat loci are available on request.
RT–PCR
A 3 μg portion of total RNA isolated with the RNeasy Mini Kit (Qiagen) was reverse-transcribed with Superscript II RT (Gibco) according to the manufacturer's instruction. PCR experiments were performed with RedTaq polymerase; primer sequences are available on request. Since the time course of induction varied between Myc stimulation and stimulation by FCS, RNA was harvested 6 h after activation of Myc or 18 h after addition of FCS.
Immunoblotting
The following antibodies were used: anti-cyclin D2 (MS-221-P1; Neomarkers), anti-cdk2 (M2; sc-163); anti-ERα (M-20; sc-542) (all from Santa Cruz Biotechnology), anti-FoxO3a (#06-951; Upstate) and anti-phosphoT32FoxO3a (#9464; Cell Signalling).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Acknowledgments
We acknowledge support from the Association for International Cancer Research (AICR, to CB and ME), the European Commission (ME), the Deutsche Forschungsgemeinschaft (ME), the FCT (PRAXIS XXI/BPD/22127/99 to AB) and the Dutch Scientific Organization (NOW 901-28-092 to RM). We thank Bianca Jebavy and Rob Klompmaker for expert technical assistance, Julian Downward for the CMV-RasV12 vector, William Hahn for the retrovirus expressing Myr-FLAG-p110α, Dirk Eick for the kind gift of antibody 8WG16, Eric Lam for the wild-type FoxO3a retrovirus, Anne Brunet for the Rat-1 FoxO3a(A3)-ER cell line, and Ulf Rapp, Rolf Müller and members of the laboratory for critical comments on the manuscript. We are grateful to Daniel Peeper for kindly providing primary MEFs, pLZRS-HA-Myc-IRES-GFP and pBabeHygro-RasV12 constructs.
References
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 6541–6551 [PMC free article] [PubMed] [Google Scholar]
- Banerji L, Glassford J, Lea NC, Thomas NS, Klaus GG, Lam EW (2001) BCR signals target p27(Kip1) and cyclin D2 via the PI3-K signalling pathway to mediate cell cycle arrest and apoptosis of WEHI 231 B cells. Oncogene 20: 7352–7367 [DOI] [PubMed] [Google Scholar]
- Basu S, Totty NF, Irwin MS, Sudol M, Downward J (2003) Akt phosphorylates the Yes-associated protein, YAP, to induce interaction with 14-3-3 and attenuation of p73-mediated apoptosis. Mol Cell 11: 11–23 [DOI] [PubMed] [Google Scholar]
- Bouchard C, Dittrich O, Kiermaier A, Dohmann K, Menkel A, Eilers M, Luscher B (2001) Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev 15: 2042–2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Thieke K, Maier A, Saffrich R, Hanley-Hyde J, Ansorge W, Reed S, Sicinski P, Bartek J, Eilers M (1999) Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J 18: 5321–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman JA, Scherer DC, McKinsey TA, Hall SM, Qi X, Lee WY, Ballard DW (1995) Coupling of a signal response domain in I kappa B alpha to multiple pathways for NF-kappa B activation. Mol Cell Biol 15: 2809–2818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96: 857–868 [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231–241 [DOI] [PubMed] [Google Scholar]
- Eberhardy SR, Farnham PJ (2002) Myc recruits P-TEFb to mediate the final step in the transcriptional activation of the cad promoter. J Biol Chem 277: 40156–40162 [DOI] [PubMed] [Google Scholar]
- Fernandez PC, Frank SR, Wang L, Schroeder M, Liu S, Greene J, Cocito A, Amati B (2003) Genomic targets of the human c-Myc protein. Genes Dev 17: 1115–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SR, Parisi T, Taubert S, Fernandez P, Fuchs M, Chan HM, Livingston DM, Amati B (2003) MYC recruits the TIP60 histone acetyltransferase complex to chromatin. EMBO Rep 4: 575–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank SR, Schroeder M, Fernandez P, Taubert S, Amati B (2001) Binding of c-Myc to chromatin mediates mitogen-induced acetylation of histone H4 and gene activation. Genes Dev 15: 2069–2082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamad NM, Elconin JH, Karnoub AE, Bai W, Rich JN, Abraham RT, Der CJ, Counter CM (2002) Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev 16: 2045–2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Ohtani K, Iwanaga R, Matsumura Y, Nakamura M (2001) Direct trans-activation of the human cyclin D2 gene by the oncogene product Tax of human T-cell leukemia virus type I. Oncogene 20: 1094–1102 [DOI] [PubMed] [Google Scholar]
- Jones SM, Kazlauskas A (2001) Growth-factor-dependent mitogenesis requires two distinct phases of signalling. Nat Cell Biol 3: 165–172 [DOI] [PubMed] [Google Scholar]
- Kanazawa S, Soucek L, Evan G, Okamoto T, Peterlin BM (2003) c-Myc recruits P-TEFb for transcription, cellular proliferation and apoptosis. Oncogene 22: 5707–5711 [DOI] [PubMed] [Google Scholar]
- Knoepfler PS, Cheng PF, Eisenman RN (2002) N-myc is essential during neurogenesis for the rapid expansion of progenitor cell populations and the inhibition of neuronal differentiation. Genes Dev 16: 2699–2712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land H, Parada LF, Weinberg RA (1983) Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304: 596–602 [DOI] [PubMed] [Google Scholar]
- Leone G, DeGregori J, Sears R, Jakoi L, Nevins JR (1997) Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature 387: 422–426 [DOI] [PubMed] [Google Scholar]
- Levens DL (2003) Reconstructing MYC. Genes Dev 17: 1071–1077 [DOI] [PubMed] [Google Scholar]
- Mao DY, Watson JD, Yan PS, Barsyte-Lovejoy D, Khosravi F, Wong WW, Farnham PJ, Huang TH, Penn LZ (2003) Analysis of Myc bound loci identified by CpG island arrays shows that Max is essential for Myc-dependent repression. Curr Biol 13: 882–886 [DOI] [PubMed] [Google Scholar]
- Mateyak MK, Obaya AJ, Adachi S, Sedivy JM (1997) Phenotypes of c-Myc-deficient rat fibroblasts isolated by targeted homologous recombination. Cell Growth Differ 8: 1039–1048 [PubMed] [Google Scholar]
- Matsushime H, Roussel MF, Ashmun RA, Sherr CJ (1991) Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell 65: 701–713 [DOI] [PubMed] [Google Scholar]
- McMahon SB, Wood MA, Cole MD (2000) The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-Myc. Mol Cell Biol 20: 556–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM (2000) AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 404: 782–787 [DOI] [PubMed] [Google Scholar]
- Nikiforov MA, Chandriani S, Park J, Kotenko I, Matheos D, Johnsson A, McMahon SB, Cole MD (2002) TRRAP-dependent and TRRAP-independent transcriptional activation by Myc family oncoproteins. Mol Cell Biol 22: 5054–5063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orian A, Van Steensel B, Delrow J, Bussemaker HJ, Li L, Sawado T, Williams E, Loo LW, Cowley SM, Yost C, Pierce S, Edgar BA, Parkhurst SM, Eisenman RN (2003) Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev 17: 1101–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE (2002) Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell 1: 53–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JM, Werner J, Kim JM, Lis JT, Kim YJ (2001) Mediator, not holoenzyme, is directly recruited to the heat shock promoter by HSF upon heat shock. Mol Cell 8: 9–19 [DOI] [PubMed] [Google Scholar]
- Peeper DS, Dannenberg JH, Douma S, te Riele H, Bernards R (2001) Escape from premature senescence is not sufficient for oncogenic transformation by Ras. Nat Cell Biol 3: 198–203 [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Nakamura N, Sansal I, Bergeron L, Sellers WR (2002) A novel mechanism of gene regulation and tumor suppression by the transcription factor FKHR. Cancer Cell 2: 81–91 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Khwaja A, Marte BM, Pappin D, Das P, Waterfield MD, Ridley A, Downward J (1997) Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell 89: 457–467 [DOI] [PubMed] [Google Scholar]
- Schmidt M, de Mattos SF, van der Horst A, Klompmaker R, Kops GJ, Lam EW, Burgering BM, Medema RH (2002) Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol Cell Biol 22: 7842–7852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR (2000) Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 14: 2501–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane J, Le HV, Shen L, Anderson SA, Massague J (2004) Integration of smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell 117: 211–223 [DOI] [PubMed] [Google Scholar]
- Sicinski P, Donaher JL, Geng Y, Parker SB, Gardner H, Park MY, Robker RL, Richards JS, McGinnis LK, Biggers JD, Eppig JJ, Bronson RT, Elledge SJ, Weinberg RA (1996) Cyclin D2 is an FSH-responsive gene involved in gonadal cell proliferation and oncogenesis. Nature 384: 470–474 [DOI] [PubMed] [Google Scholar]
- Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ Jr, DiStefano PS, Chiang LW, Greenberg ME (2002) DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science 296: 530–534 [DOI] [PubMed] [Google Scholar]
- Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, Woscholski R, Parker PJ, Waterfield MD (2001) Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem 70: 535–602 [DOI] [PubMed] [Google Scholar]
- Wei W, Jobling WA, Chen W, Hahn WC, Sedivy JM (2003) Abolition of cyclin-dependent kinase inhibitor p16Ink4a and p21Cip1/Waf1 functions permits Ras-induced anchorage-independent growth in telomerase-immortalized human fibroblasts. Mol Cell Biol 23: 2859–2870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J, Lowe SW (2004) Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature 428: 332–337 [DOI] [PubMed] [Google Scholar]
- Zhao JJ, Gjoerup OV, Subramanian RR, Cheng Y, Chen W, Roberts TM, Hahn WC (2003) Human mammary epithelial cell transformation through the activation of phosphatidylinositol 3-kinase. Cancer Cell 3: 483–495 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4