

Abstract
The actin filament-severing functionality of gelsolin resides in its N-terminal three domains (G1–G3). We have determined the structure of this fragment in complex with an actin monomer. The structure reveals the dramatic domain rearrangements that activate G1–G3, which include the replacement of interdomain interactions observed in the inactive, calcium-free protein by new contacts to actin, and by a novel G2–G3 interface. Together, these conformational changes are critical for actin filament severing, and we suggest that their absence leads to the disease Finnish-type familial amyloidosis. Furthermore, we propose that association with actin drives the calcium-independent activation of isolated G1–G3 during apoptosis, and that a similar mechanism operates to activate native gelsolin at micromolar levels of calcium. This is the first structure of a filament-binding protein bound to actin and it sets stringent, high-resolution limitations on the arrangement of actin protomers within the filament.
Keywords: actin, apoptosis, familial amyloidosis, gelsolin, X-ray crystallographic structure
Introduction
Cell locomotion requires the spatial and temporal control of actin filament assembly and disassembly at the leading edge (Pollard et al, 2000). Gelsolin is an actin filament capping and severing protein that enhances the rate of cell migration (Cunningham et al, 1991). The control of gelsolin activity during cell movement has a number of elements: gelsolin is sequestered by phosphatidylinositol 4,5-bisphosphate (PIP2) at the cell membrane and held in an inactive state; hydrolysis of PIP2 releases gelsolin into the cytoplasm; calcium activates free gelsolin to allow it to cap and sever actin filaments; and, selected filaments are uncapped by a PIP2-rich membrane to allow actin polymerization to proceed and to render gelsolin inactive (Janmey and Stossel, 1987; Allen, 2003). Branching of newly uncapped filaments through a mechanism that involves arp2/3 amplifies propulsive actin polymerization (Falet et al, 2002), while gelsolin-capped filaments are marked for rapid depolymerization by cofilin (Ressad et al, 1998).
The relative importance of these gelsolin activities appears to vary among cell types. Platelet activation initially releases gelsolin via a transient increase in hydrolysis of PIP2, accompanied by an elevation in calcium ion concentration, which leads to filament severing (Hartwig, 1992). Subsequent accumulation of PIP2 in membranes causes filament uncapping and elongation (Hartwig et al, 1995). More typically, EGF-induced motility proceeds through enhanced hydrolysis of PIP2 by phospholipase C, which releases membrane-bound gelsolin and enhances actin filament severing at the leading edge (Chou et al, 2002). In accord with this observation, cells with increased PIP2 have a phenotype similar to the gelsolin-null phenotype (Yamamoto et al, 2001).
In a calcium-free environment, gelsolin exists as a compact, inert arrangement of six related domains, G1–G6 (Burtnick et al, 1997). On binding calcium, metamorphosis of gelsolin to an activated structure entails the opening at least of three identifiable latches (tail latch, G1–G3 latch and G4–G6 latch) to expose actin-binding surfaces on G2, G1 and G4, respectively (Robinson et al, 1999; Choe et al, 2002). Tryptophan fluorescence experiments show that gelsolin undergoes two or, possibly, three conformational changes characterized by calcium binding with Kd values of 0.1, 7 and, controversially, 0.3 μM (Kinosian et al, 1998; Lin et al, 2000). These sites have been assigned responsibility for opening the tail and the G4–G6 latches, although there is disagreement as to which calcium level is required for which latch (Pope et al, 1995; Kinosian et al, 1998; Lin et al, 2000). Equilibrium dialysis experiments also have identified two or three high-affinity sites (Pope et al, 1995; Lin et al, 2000). New evidence from synchrotron footprinting experiments confirms the tail latch to be released at micromolar calcium concentrations, and identifies 3–6 further calcium ions that bind at around 100 μM and are thought necessary to open the G1–G3 and G4–G6 latches (Kiselar et al, 2003). In vitro, gelsolin capping and severing activities occur at a low level under conditions similar to those in a resting cell, where the cytoplasmic Ca2+ concentration is approximately 0.2 μM (Kinosian et al, 1998). The rate of gelsolin severing accelerates beyond that at resting calcium concentrations in response to calcium transients.
Protein crystallography has identified a conserved Ca2+-binding site in each gelsolin domain studied to date (McLaughlin et al, 1993; Burtnick et al, 1997; Choe et al, 2002; Kazmirski et al, 2002). These six variable affinity sites are termed type-2 Ca2+-binding sites and are responsible for the structural changes that activate gelsolin. Each type-2 calcium ion acts both to disrupt the structure of calcium-free gelsolin and to stabilize the activated form. Of particular note are the sites within G2 and G6, which are well situated to cooperate in opening the tail latch. Two other Ca2+-binding sites, termed type-1 sites, are sandwiched at the actin:G1 and the actin:G4 interfaces, respectively, and likely moderate the affinity of activated gelsolin for actin (Choe et al, 2002). In these sites, coordination of the Ca2+ ion involves residue Glu167 from two different actin protomers along with analogous contacts from G1 and G4.
Secreted gelsolin prevents elevation of the viscosity of extracellular fluids by severing actin filaments that are released as a result of cell death or injury (Haddad et al, 1990). A hereditary mutation in gelsolin of Asp187 to Asn or Tyr in sufferers of Finnish-type familial amyloidosis (FAF) exposes the Arg172 to Ala173 peptide bond to proteolysis by furin during transport through the trans-Golgi network (Chen et al, 2001). A subsequent extracellular proteolytic event generates a gelsolin fragment, residues 173–243, which self-assembles into amyloid fibrils. Amyloid accumulation effects a range of neuropathies, ophthalmic disorders and dermatological abnormalities.
Appreciation of the mechanism of disintegration of FAF gelsolin has evolved in parallel with knowledge of the conformation and function of normal gelsolin. The structure of calcium-free gelsolin revealed that both the mutation site and primary cleavage site reside in G2, and demonstrated that Asp187 contributes to the stability of G2 through a series of electrostatic interactions (Burtnick et al, 1997). Later, it was realized that proteolysis requires an activated form of FAF gelsolin; the binding of Ca2+ to Asp187 protects normal gelsolin from cleavage (Zapun et al, 2000; Chen et al, 2001; Robinson et al, 2001; Kazmirski et al, 2002). Intriguingly, however, overexpression of furin can lead to detectable attack even on normal gelsolin (Huff et al, 2003).
G1–G3 as an independent fragment of gelsolin is biologically relevant, being generated from the intact protein during apoptosis via the actions of caspase-3, -7 and -9 (Kothakota et al, 1997; Azuma et al, 2000). Free of the three C-terminal domains, G1–G3 is relieved of regulation by Ca2+ and proceeds to dismantle the actin cytoskeleton unchecked. This outcome is countered by full-length gelsolin and its C-terminal half, G4–G6, which display antiapoptotic activity by blocking voltage-dependant anion channels, inhibiting mitochondrial membrane potential loss and cytochrome c release (Koya et al, 2000; Kusano et al, 2000). In addition, complexes that involve caspases, gelsolin and PIP2 are able to block cleavage of gelsolin and inhibit apoptosis (Azuma et al, 2000). Gelsolin-deficient cells display a retarded onset of apoptosis, while transient overexpression of G1–G3 induces apoptosis (Kothakota et al, 1997). Clearly, gelsolin plays a multifaceted role in the control and execution of apoptosis, many details of which remain to be discovered.
Atomic coordinates currently are available for intact gelsolin in the calcium-free state (Burtnick et al, 1997) and for the actin complexes with gelsolin fragments G1 (McLaughlin et al, 1993; Irobi et al, 2003) and G4–G6 (Choe et al, 2002). Absent from this set is the structure of the activated F-actin recognition unit, G2–G3. The present work amends this deficiency by detailing the architecture of the apoptotic fragment of gelsolin, G1–G3, bound to an actin monomer. A novel compact arrangement of G2–G3 is revealed. Calcium ions occupy type-2 sites in G1 and G3, but the corresponding site in G2 is vacant. A complete set of conformations for the resting and activated domains of gelsolin, together with published calcium-binding data, allows proposal of schemes for the overall activation of gelsolin and for its actin-modulating activities.
Results
G1–G3:actin structure
X-ray diffraction data from crystals grown from a solution of equine plasma gelsolin in complex with rabbit skeletal actin in which proteolysis of the gelsolin had occurred led to determination of the structure of G1–G3:actin at a resolution of 3.0 Å (Table I). In this structure, G1 is observed binding to its established site on actin (McLaughlin et al, 1993) between subdomains 1 and 3 (Figure 1A). The polypeptide chain, in line with its sequence similarity to the WH2 domain family of proteins (Irobi et al, 2003), then extends up, and is tightly associated with, the face of actin to allow G2 to contact actin subdomain 2. Finally, the G2–G3 linker positions G3 to bind back to actin subdomain 1. G2 and G3, respectively, mask 290 and 309 Å2 of the surface of actin. In turn, the G2–G3 contact area comprises 815 Å2 on each domain. The arrangement of G1–G3 differs strikingly from that observed for G4–G6 in its complex with actin (Figure 1B; Robinson et al, 1999; Choe et al, 2002). The disulfide bridge between Cys188 and Cys201, found in calcium-free plasma gelsolin (Burtnick et al, 1997), is absent under the reducing conditions employed for crystallization. The loss of this disulphide bond does not significantly alter the structure of G2, and the distance between the Cα positions of residues Cys188 and Cys201 is 3.8 and 4.1 Å in the oxidized and reduced forms, respectively. Four calcium ions are associated with G1–G3:actin. A type-1 Ca2+ is sandwiched at the G1:actin interface and a second Ca2+ is associated with the actin-bound ATP. Type-2 calcium ions, which instigate the requisite conformational changes to activate gelsolin (Robinson et al, 1999; Choe et al, 2002), are found at their expected positions in each of G1 and G3, but not in G2.
Table 1.
Data collection and refinement statistics for G1–G3:actin
| Wavelength (Å) | 1.0052 |
| Space group | P3121 |
| Unit cell | a=b=145.25, c=129.95 Å, α=β=90°, γ=120° |
| Resolution range (Å) | 20.0–3.0 (3.11–3.00) |
| Unique reflections | 31 101 (3093) |
| Redundancy | 2.9 (3.9) |
| Completeness (%) | 97.5 (98.5) |
| Average I/σ | 21.3 (3.2) |
| Rmerge (%)a | 5.9 (38.3) |
| Rfactor (%)b | 22.5 (31.0) |
| Rfree (%)c | 25.9 (43.0) |
| Non-hydrogen atoms (G1–G3, actin) (ATP, calcium ions, waters) | 5480 (2740, 2740) (31, 4, 24) |
| G1–G3, modeled as residues | 26–371 |
| Actin, modeled as residues | 5–39, 50–200, 204–365, 375 |
| Mean derived B-factor (residual B-factors) (Å2), G1–G3, actin, ATP | 85.9 (32.9), 70.0 (31.5), 58.3 (32.8) |
| R.m.s. deviation bonds (Å) | 0.014 |
| R.m.s. deviation angles (deg) |
1.40 |
| a Rmerge (∣I−〈I〉∣/〈I〉). | |
| b Rfactor (∥Fo∣−∣Fc∥/∣Fo∣). | |
| c Based on 5% of the data. | |
Figure 1.
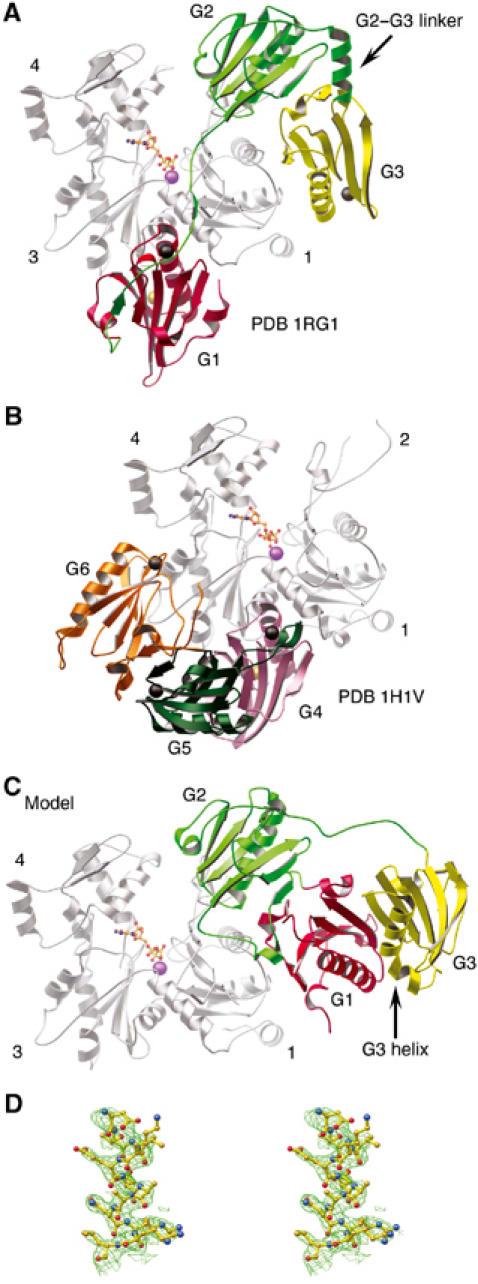
The structure of the G1–G3:actin complex. (A) A schematic representation of the G1–G3:actin complex. The gelsolin domains are colored: G1, red; G2, green; G3, yellow. This scheme is preserved in subsequent figures unless explicitly stated. Actin, with subdomains 1, 3 and 4 indicated, is colored gray. ATP is shown as a ball-and-stick representation with its associated Ca2+ in purple. Type-1 and type-2 Ca2+ ions are depicted as gold and black spheres, respectively. (B) The structure of the G4–G6:actin complex (PDB 1H1V; Choe et al, 2002) for comparison. Gelsolin domains are colored: G4, pink; G5, dark green; G6, orange. (C) Model of Ca2+-free G1–G3 interacting with actin: obtained by taking the structure of G1–G3 excised from Ca2+-free, inactive gelsolin (PDB 1D0N; Burtnick et al, 1997) and positioned on actin, in accord with the overlaying of G2 onto the structure presented in (A). (D) Stereo view of a representative portion of the 2Fo–Fc electron density map contoured at 1σ.
Comparison with Ca2+-free G1–G3
The two halves of the β-sheet that runs continuously from G1 to G3 to seal the G1–G3 latch in calcium-free gelsolin (Figure 1C) are separated during transformation to the active form (Figure 1A). This mirrors the opening of the G4–G6 latch during the activation of the C-terminal half of gelsolin (Robinson et al, 1999). These latches open to reveal actin-binding surfaces on G1 and G4, respectively. The rupture of the G1–G3 sheet appears to be triggered by calcium binding to G3, straightening its kinked long helix and releasing its interactions with G1. G1 then translates away from G2, made possible by displacement of strand A′ (residues 137–141) from the edge of the core β-sheet of G2, to extend the reach of the G1–G2 linker to approximately 30 Å. In contrast, the G2–G3 linker shortens through the adoption of a helical conformation. A portion of the final electron density map around this region is shown in Figure 1D. The edge of the β-sheet exposed in G2 by extraction of the A′ strand forms the center of a new interface with G3.
Discussion
Calcium activation
The observation that two calcium ions bind exclusively within activated G1–G3 agrees with the biochemical data. Equilibrium dialysis experiments identified a high-affinity calcium site within isolated G1–G3 and a second, low-affinity site in G1, which becomes high affinity on binding actin (Weeds et al, 1995; Pope et al, 1997).
Conspicuously absent is a calcium ion in the type-2 site of G2. All of the requisite ligating residues are present (Choe et al, 2002), and Ca2+ is able to bind to this isolated domain in solution (Huff et al, 2003). We conclude that calcium binding by G2 during normal activation is a transient event, important in undermining the calcium-free structure, but that the ion is released by the final activated structure. The cadmium-bound structure of isolated G2 shows that Asp259, from the G2–G3 linker, moves to participate in the coordination sphere (Kazmirski et al, 2002). We propose that this represents the midpoint in the activation process. This conformation is then sufficiently close to the activated one to allow G3 to dock with G2 and, simultaneously, release the calcium ion.
Two high-affinity Ca2+-binding sites have been identified in G4–G6 (Pope et al, 1995). Based on structural considerations, we previously assigned these as type-2 binding sites in G4 and G6, and also located a third Ca2+ bound to G5 (Choe et al, 2002). Now, with the present structure in hand, we propose a multilevel scheme for activation of gelsolin through occupation of all six type-2 Ca2+-binding sites (Figure 2A). Level A, approximately 0.2 μM Ca2+ (Pope et al, 1995): Ca2+ occupies the G6 site, weakening the tail latch. This calcium ion straightens the G6 helix and destabilizes the G4–G6 latch. At this level, we suggest that gelsolin retains its global structural integrity, but that the tail helix dynamically moves between attached and detached states. Level B, approximately 2 μM Ca2+ (Pope et al, 1995): Ca2+ binds to G4, producing a small shift in the relative positions of G4 and G5. This movement further weakens the G4–G6 latch, dynamically allowing it to open and close, while fully releasing the tail latch. Level C, approximately 100 μM Ca2+ (Kiselar et al, 2003): Ca2+ binding to G5 stabilizes the G4–G5 interface and locks G4–G6 into an open state. Simultaneously, Ca2+ binding to G3 straightens its long helix and opens the G1–G3 latch, revealing the cryptic Ca2+-binding site on G2. The latter is a high-affinity site (Chen et al, 2001) (Kd=0.7 μM), which cooperatively binds Ca2+ and drives G2–G3 toward an active conformation. Achievement of the activated state coincides with the release of the G2-bound calcium ion. Level D, in excess of 0.6 mM Ca2+ (Zapun et al, 2000): Finally, the G1 site is occupied, cocking G1 in a position, through the G1–G2 linker, to strike and sever F-actin.
Figure 2.
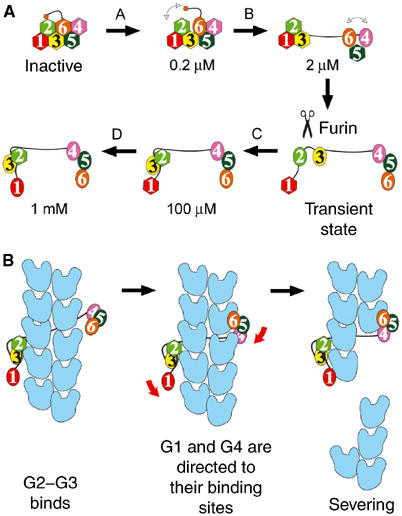
Calcium-induced activation of gelsolin and the severing of F-actin. (A) Levels of calcium activation. Ca2+-free gelsolin domains are shown as hexagons and calcium-bound domains are depicted as ovals. Calcium ion concentrations are indicated for each step. The scissors represent the stage at which FAF gelsolin is cleaved. (B) The sequence of events during severing of actin by fully activated gelsolin. Actin protomers are shown in blue.
The foregoing scheme is based on claims of two high-affinity Ca2+-binding sites in gelsolin (Pope et al, 1995; Kinosian et al, 1998). Different conditions of pH and temperature led to three high-affinity sites being observed (Lin et al, 2000). Under such conditions, we suggest that Ca2+ binding to G2 and G6 occurs cooperatively at Level A. However, the major conformational changes in G1–G3 are still to be expected to occur at Level C, when Ca2+ binding to G3 releases the G1–G3 latch (Figure 2A).
PIP2 inactivation
PIP2 is a general second messenger in the regulation of actin-binding proteins. Its presence favors actin polymerization, and often instigates large conformational changes within actin modulators, such as in the cases of WASp, ezrin and vinculin (Sechi and Wehland, 2000). PIP2 is able to release gelsolin both from actin monomers and from capped filaments (Janmey and Stossel, 1987). The PIP2-binding sites on gelsolin are located within residues 135–142, 160–172 and 621–634 (Janmey et al, 1992; Yu et al, 1993; Cunningham et al, 2001; Feng et al, 2001). Residues 132–140 and 161–172 form integral parts of domains G1 and G2, respectively, in both the active (Figure 3A) and inactive (Figure 3B) forms of the protein. As such, G1 and G2 are likely to undergo PIP2-induced inter- rather than intradomain conformational changes. Gelsolin residues 168–170 also contain an actin-binding determinant (Puius et al, 2000); it may be that, at least in part, PIP2 releases gelsolin from an actin filament by direct competition for the actin-binding residues (Burtnick et al, 1997). Support for this idea is found in ADF/cofilin family proteins, which also bind to PIP2 with a site that overlaps with their actin-binding site (Moriyama et al, 1992).
Figure 3.
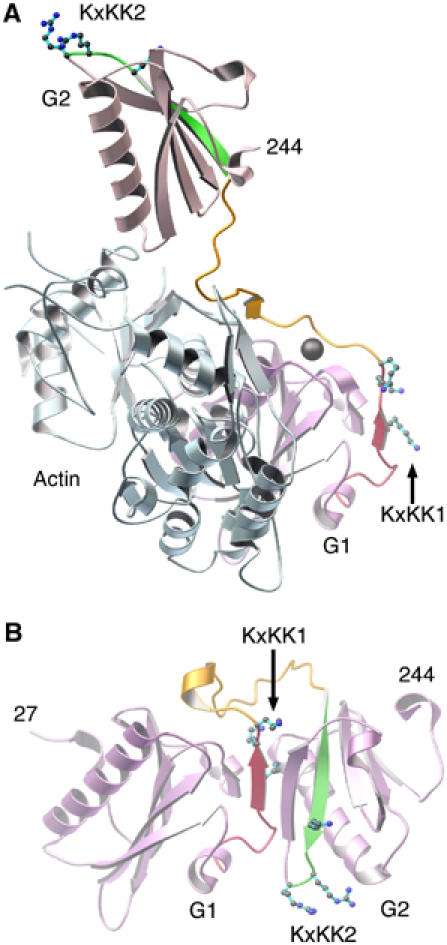
Regions of gelsolin involved in binding PIP2. (A) Activated G1 and G2 (pink) bound to actin (pale blue) as observed in the G1–G3:actin structure. PIP2-binding regions 132–140 and 161–172 are highlighted in red and green, respectively. Lysine residues from the KxKK motifs, within these regions, are drawn and labeled KxKK1 and KxKK2, respectively. The hydrophobic-rich G1–G2 linker, residues 141–160, is colored orange. The G1 type-2 calcium ion is drawn as a gray sphere. (B) G1–G2 excised from the Ca2+-free inactive form of gelsolin (PDB 1D0N; Burtnick et al, 1997). Colors are as in (A).
Phospholipase Cβ2 presents a KxxxKxKK motif that directs the enzyme to PIP2-rich surfaces (Simoes et al, 1995). Similarly, each PIP2-binding region in gelsolin contains a KxKK sequence (Figure 3). These lysine residues are exposed in the activated, actin-bound structures of G1–G3 and G4–G6 and may govern initial contacts with PIP2. In contrast, the majority of these lysine residues lie buried in the structure of calcium-free whole gelsolin. Therefore, the determinants of the initial interactions between gelsolin and PIP2 are expected to be different in the presence and absence of Ca2+ (Lin et al, 1997).
The G1–G2 linker (residues 143–154) is largely hydrophobic (eight hydrophobic residues, two basic residues and a serine). It separates the two PIP2-binding regions discussed above, following distinctly different paths prior to and after calcium activation (Figure 3), and thus is a strong candidate to be involved in PIP2-induced interdomain conformational change. The linker makes intimate contact with actin in the G1–G3:actin complex, creating a 410 Å2 interaction interface. Through competition for this surface, possibly by internalizing the hydrophobic residues from the G1–G2 linker and positioning the basic residues (KxKK) appropriately at its surface, PIP2 may dislodge the type-2 calcium ion from the G1:actin interface and release gelsolin from actin (Figure 3; Weeds et al, 1995; Choe et al, 2002).
Implications for the structure of the actin filament
An important application of the G1–G3:actin structure is that it provides a stringent test for actin filament models. The structure dictates that actin subdomains 1 and 2 should be on the outside of the filament, ready for G2–G3 to initiate simultaneous contact with two longitudinally adjacent actin protomers. We have constructed a model of an ADP-actin filament (ADP model) by overlaying the structure of rhodamine-modified ADP-actin (Otterbein et al, 2001) onto each actin protomer in the Holmes model for F-actin (Holmes et al, 1990). Overlaying of actin subdomains 1 and 2 of the G1–G3:actin structure onto the homologous subdomains of a barbed-end actin protomer in the ADP model results in G2 lying in close proximity to a second actin (Figure 4A).
Figure 4.

Model of a gelsolin-capped filament. (A) The ADP model of the actin filament (four protomers are drawn in blue and gray), with G1–G3:actin (1RGI) and G4–G6:actin (G4, pink; G5, dark green; G6, orange; PDB 1H1V; Choe et al, 2002) overlaid onto the barbed end. Purple spheres mark Asp371 of G3 and Met412 of G4, a gap of 63.1 Å to be bridged by the G3–G4 linker, which is modeled in purple. (B) A second view of the gelsolin-capped model filament, looking directly at the capped, barbed end. Three actin protomers are depicted. (C) The G1:actin structure (red:blue) with subdomain 2 of a second actin protomer from the ADP model drawn in black. The arrow indicates a steric clash between the long helix of G1 and the ADP helix of actin subdomain 2. (D) The tail latch. G2 (green) and the C-terminal tail (residues 736–755; orange) from inactive gelsolin positioned on the ADP model by overlaying G2 in (A). Only one actin from the filament is shown (gray). In this position, the C-terminal tail obscures the actin-binding site on G2.
A more detailed inspection of the model reveals that the long helix of G2 lies close to the binding site on actin occupied by the analogous helix of G1, but on a different actin protomer (Figure 5A and B). A peptide consisting of amino-acid residues in the G2 helix binds to actin in a manner similar to the helix from G1 (Van Troys et al, 1996). We have suggested from sequence alignments (Choe et al, 2002) that Arg221 in G2 is the functional equivalent of Asp109 bound to a calcium ion in G1, and is able to form a salt bridge to actin residue Glu167. These residues are close and pointing at each other in the model (Figure 5B). Furthermore, the G1–actin interface is centered on Ile103, which binds to the hydrophobic patch between actin subdomains 1 and 3 (Figure 5A). A similarly located residue, Leu211, is available on G2 (Figure 5B), and resides within a triplet of residues (210–212) that has been shown by mutational analysis to mediate binding to F-actin (Puius et al, 2000). In this model, Arg221 and Leu211 lie 6.5 and 4.8 Å, respectively, from actin residues Glu167 and Leu349. A second actin-binding determinant (Puius et al, 2000) is contained within gelsolin residues 168–170. Arg168 is within 9.1 Å of actin Asp25 and 7.2 Å from actin residue Ser348, while Arg169 lies a distance of 8.5 Å from actin residue Ser350. Taken together, the ADP model and the G1–G3:actin structure provide details of the interactions of G2 with actin that concur strongly with predictions from earlier data.
Figure 5.
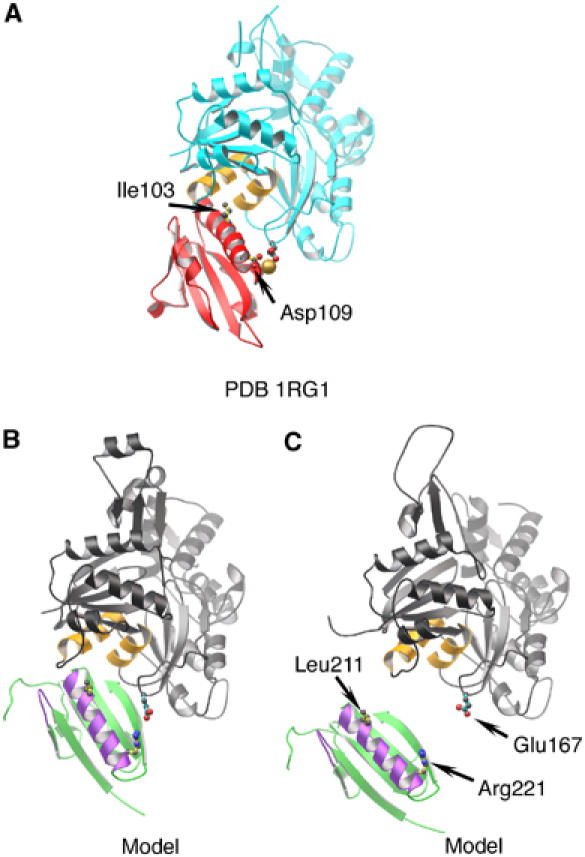
Comparison of the G1 (structure) and G2 (model) interfaces with actin. (A) G1:actin structure (from 1RGI). G1 is red, with Ile103 and Asp109 drawn. The type-1 calcium ion is shown as a gold sphere. Actin is depicted cyan, with two relevant hydrophobic helices painted orange and residue Glu167 indicated. (B) The G2:actin interaction within the model presented in Figure 4A. G2 is shown in green, with F-actin contact regions in purple (Van Troys et al, 1996; Puius et al, 2000) and residues Leu211 and Arg221 indicated. An actin protomer is depicted in gray, with the two hydrophobic helices in orange and residue Glu167 drawn in. (C) G2 docked onto the EM model for the open conformation of an actin filament (Belmont et al, 1999).
Docking of G1–G3:actin onto the electron microscope-based (EM) reconstruction of an actin filament (Belmont et al, 1999) by overlaying all four subdomains of actin provides essentially identical results to those described for the ADP model. Hence, both the Holmes model and EM reconstruction appear to provide legitimate approximations to the orientations of actin protomers within F-actin. Despite the global correctness of the discussed models, neither model adequately explains a number of observations, for example, inhibition of actin polymerization by rhodamine modification (Otterbein et al, 2001) or filament severing by trisoxazole macrolide toxins (Klenchin et al, 2003). Both these agents occupy the same region on actin. It is apparent that changes must occur within the actin protomer upon polymerization, which include the DNase I-binding loop approaching the rhodamine-binding pocket.
Controversy exists as to the state of the nucleotide-binding cleft in ADP-actin filaments. EM reconstructions have indicated the cleft to be relatively open in ADP protomers within F-actin (Sablin et al, 2002). However, crystallographic studies have demonstrated a closed state for ADP-G-actin (Otterbein et al, 2001). Docking of G1–G3:actin onto an EM reconstruction of the actin filament (Belmont et al, 1999) by overlaying subdomains 1 and 2 of actin reveals a dislocation of G2 from the surface of the filament (Figure 5C). Therefore, the present structure supports the closed state for the actin protomer bound to G1 and, by analogy, G4, at the end of a gelsolin-severed and capped filament. The insertion of the long helices from G1 and G4 between respective actin subdomains 1 and 3 confers the closed conformation on these two actins. Interestingly, the angle of the G2 long helix in our model is more parallel to the axis of the filament than observed for G1 and G4 (Figure 4A). This glancing angle may allow the G2-bound actin to be somewhat more flexible and probably establishes an important difference between side binding (G2) and severing (G1, G4) domains of gelsolin. Gelsolin is known to induce a conformational change in actin protomers that is propagated along the filament (Orlova et al, 1995). This rearrangement potentially could be initiated by G2 binding to the filament. Therefore, it is possible that gelsolin-capped filaments may contain actin protomers of more than one conformation.
Actin severing by gelsolin
With confidence established in models of the actin filament, predictions can be made about the mechanism for severing. Superimposition of the G1–G3:actin and G4–G6:actin (Choe et al, 2002) structures onto two actins in the ADP model offers a representation of a gelsolin-capped actin filament (Figure 4A and B). This model places G2–G3 at the junction between two longitudinally neighboring actin protomers, in confirmation of the original prediction of Pope et al (1991) and discounts more conservative suggestions for rearrangement of the gelsolin domains on activation (McLaughlin et al, 1993; Burtnick et al, 1997; Renoult et al, 2001). Both G1 and G4 provide steric hindrance to actin monomers joining the capped barbed end (Figure 4C). The model substantiates the tail latch hypothesis (Burtnick et al, 1997). When G2 from the present structure and from calcium-free gelsolin are superimposed, the C-terminal helix of calcium-free gelsolin occupies the space that would be filled by actin in the gelsolin-capped filament model (Figure 4D). In addition, G4 and G6, from calcium-free gelsolin, both encounter minor clashes with the filament (not shown). Therefore, the C-terminal tail must be removed from its blocking position on the surface of G2 and the two halves of gelsolin dissociated from each other before G2 is able to initiate contact with the side of an actin filament.
Electron micrographs of G2–G6 bound to F-actin point to the G3–G4 linker following a path from the actin to which the long helix of G2 docks around the filament to the next actin protomer in the short-pitch left-handed helical representation of F-actin (McGough et al, 1998). X-ray structures allow this orientation, in which the linker can be modeled to follow the short way around the filament as depicted in Figure 4A, but also are consistent with the long route around the filament to place G4 on an actin protomer that would lie one position below that indicated in Figure 4A (not shown). A construct consisting of G2–G3 and 80% of the G3–G4 linker (residues 150–406) is efficiently able to cap actin filaments (Sun et al, 1994). Hence, the G3–G4 linker takes an active part in the capping function of gelsolin by assisting to obscure the surface of the barbed end of a capped filament (Figure 4B).
The severing activity of gelsolin generally is thought to be vested in G1–G3, but G2–G6 also exhibits a low level of activity, with respective efficiencies of 87 and 17% compared to full-length gelsolin (Way et al, 1989). Cooperative binding of a pair of G1–G3 molecules at positions across the filament axis from each other explains the high degree of retention of severing activity by this isolated half of gelsolin (Selden et al, 1998). Geometric constraints preclude two G2–G6 molecules from acting together in such a manner (Figure 2B). As such, severing data obtained from gelsolin truncates are misleading, and both halves of gelsolin, within whole gelsolin, should be viewed to contain potent actin-severing domains.
The G1–G3:actin structure supports many features of the severing model proposed by Pope et al (1991) in the absence of structural data. Severing of actin by fully activated gelsolin can be summarized in three steps (Figure 2B). First, the G2–G3 structural unit binds to the side of a filament. Next, the G1–G2 linker and, in an independent direction, a combination of the 40-residue G3–G4 linker and G6 wrap themselves over the surface of the filament to direct G1 and G4, respectively, toward their binding sites. Finally, a coordinated pincer movement of G1 and G4 causes catastrophic steric strain in the two actins below those to which G1 and G4 are bound to sever the filament.
Severing by G1–G3 during apoptosis
During apoptosis, G1–G3 is produced through cleavage of cytoplasmic gelsolin by several caspases (Kothakota et al, 1997; Azuma et al, 2000). This fragment then participates in preparing the cell for death by dismantling its actin-based architecture in a manner unregulated by calcium. G1–G3 has been studied extensively in vitro, where it is able to sever actin filaments in the absence of calcium ions (Selden et al, 1998). Despite the lack of absolute control by Ca2+ of severing, calcium ions do regulate the structure and activity of G1–G3. The hydrodynamic volume of G1–G3 decreases, protection against proteolysis increases, and tryptophan fluorescence increases (Pope et al, 1997; Selden et al, 1998) on addition of Ca2+. The structure of isolated G1–G3 in the absence of calcium ions is unknown; however, the high levels of Ca2+ required to open the G1–G3 latch (Kiselar et al, 2003) led us to propose that isolated calcium-free G1–G3 adopts a structure similar to that observed for G1–G3 in calcium-free whole gelsolin (Burtnick et al, 1997).
Superimposing G2 in G1–G3 from calcium-free whole onto G2 in G1–G3:actin produces no steric clashes with actin (Figure 1C). This calcium-free conformation of G1–G3 may be expected to bind to the filament with a similar affinity to G2–G3, which exhibits Kd values for binding to F-actin of 2.0 and 3.6 μM, respectively (Way et al, 1992), in the presence and absence of 0.1 mM CaCl2. Once G2 is bound to the side of a filament, the G1–G2 linker will exchange its gelsolin contacts for actin interactions, zipping down the face of actin in the direction of the barbed end of the filament, driving G1 into its binding site to dislodge the actin below. Simultaneously with the activation of the G1–G2 linker, the actin-bound G2 will acquire the G2–G3 interface observed in G1–G3:actin, abrogating the need for the transiently bound calcium ion proposed earlier to act during activation of intact isolated gelsolin. Hence, we propose that actin association provides the impetus to activate G1–G3 in the absence of calcium, and that this is the mechanism that is responsible for dismantling of the actin cytoskeleton during apoptosis.
A construct of gelsolin that lacks the C-terminal 23 amino-acid residues also overcomes its absolute control by calcium ions and severs actin filaments in their absence (Lin et al, 2000). Furthermore, the increased affinity of the G1 type-2 calcium-binding site in the presence of actin (Weeds et al, 1995) indicates that actin binding induces conformational changes in gelsolin. Therefore, we suggest that at cytoplasmic Ca2+ concentrations beyond those needed to open the tail latch (0.2–2 μM), but below the levels extant in blood plasma (2 mM), actin-induced gelsolin conformational changes are able to replace rearrangements induced by occupancy of low-affinity Ca2+-binding sites, enabling cytoplasmic gelsolin to sever filaments.
Implications for FAF
In calcium-free gelsolin, the cryptic furin cleavage site (Chen et al, 2001) between Arg172 and Ala173 lies protected in the B strand of the core β-sheet of G2, flanked by the A and A′ strands (Figure 6B, purple). A salt bridge between Asp187 and Lys166 helps stabilize the β-sheet (Burtnick et al, 1997). On activation of gelsolin, this connection is undone to allow Lys166 to interact with Glu263 from the G2–G3 linker, and the G2–G3 linker itself forms several new contacts with the A–B loop (Figure 6A). The net result is placement of the furin-sensitive site at the center of the G2–G3 interface, shielding it from proteolytic attack.
Figure 6.
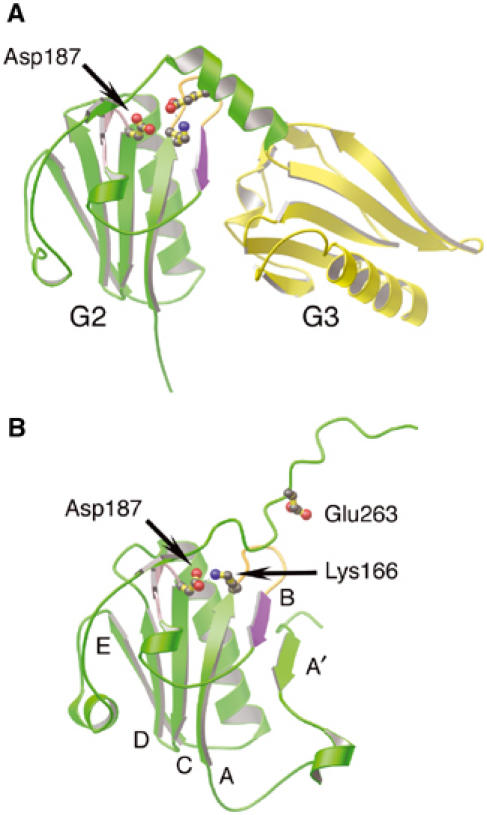
Regions of gelsolin implicated in FAF. (A) Ribbon representation of G2 (green) and G3 (yellow) excised from the G1–G3:actin complex structure (1RGI). The loop preceding the C strand (pink), the A–B loop (orange) and the B strand (purple), containing the protease-sensitive site of FAF gelsolin (Arg172–Ala173), are highlighted. Residue Lys166 is depicted forming a salt bridge to Glu263, and the locus of FAF mutation, residue Asp187, is drawn. (B) Ribbon representation of G2 (green) from inactive gelsolin (PDB 1D0N; Burtnick et al, 1997). The FAF mutation residue, Asp187, is depicted forming an ion pair with Lys166. Residue Glu263 is also drawn. The β-strands are identified by letter. Mutation of Asp187, we propose, results in trapping G2–G3 between active (A) and inactive (B) states, increasing accessibility of the critical bond in the B strand (purple).
We advocate that transient binding of the type-2 calcium ion to G2 mediates conversion between the inactive and activated states. The FAF mutation site, Asp187, has been shown to ligate this ion and stabilize G2 in the wild-type protein (Kazmirski et al, 2002; Huff et al, 2003). Mutation of Asp187 to Asn or Tyr prevents G2 from binding the transient calcium ion and, therefore, would alter the kinetics of interchange between the two protective states. We propose that the molecule will spend more time, possibly becoming stranded, between conformations, rendering the cleavage site available for proteolysis (Figure 2A).
Materials and methods
Purification, crystallization and data collection
Horse plasma gelsolin was purified as previously (Burtnick et al, 1997) and actin was prepared from rabbit skeletal muscle acetone powder (Spudich and Watt, 1971). Gelsolin, in 25 mM Tris–HCl, 1 mM EDTA, pH 8.0, was incubated for 5 min in the presence of 2 mM CaCl2 at room temperature. Then actin, in 2 mM Tris–HCl, 0.2 mM ATP, 0.2 mM CaCl2, 1 mM dithiothreitol, pH 7.6, was added to the activated gelsolin to a molar excess of 2:1. The resulting solution was incubated overnight at 4°C, followed by gel filtration chromatography using Sephacryl S300 (Amersham Biosciences). The complex was concentrated to 10 mg/ml, estimated spectrophotometrically with an absorption coefficient at 280 nm of 1.25 (ml/mg)/cm.
Crystals were grown at 4°C using the hanging drop vapor diffusion method. The protein solution was combined in a 1:1 ratio (v/v) with a well solution consisting of 2% PEG 8000, 100 mM sodium acetate and 2 mM CaCl2, pH 4.7. Prior to X-ray diffraction analysis, crystals were transferred to a cryoprotectant solution (25% glycerol, 10% PEG 8000, 0.4 M NaCl, 100 mM sodium acetate, 5 mM CaCl2, pH 4.7) and flash-frozen in liquid nitrogen. SDS–PAGE analysis of washed crystals demonstrated that proteolysis of the original gelsolin:actin complex had occurred, and that the crystals were of a half-gelsolin:actin complex. Diffraction data from a frozen crystal (100 K) were collected at beamline ID-29, ESRF, Grenoble. These data were indexed, integrated and scaled using DENZO and SCALEPACK (Otwinowski and Minor, 1997).
Structure determination and refinement
Structural analysis was initiated by molecular replacement using G1:actin (McLaughlin et al, 1993) as the search model in the program AMORE (CCP4, 1994). The solution was unambiguous, and the resulting phase information allowed positioning of G2 and G3 in electron density maps, confirming the crystals to be of a G1–G3:actin complex. The model was refined and a bulk solvent correction applied in CNS (Brünger et al, 1998) with extra attention given to the free R-factor to monitor the correct progress of refinement. The last two cycles of model refinement utilized TLS refinement in the program REFMAC5 prior to isotropic B-factor refinement, after setting all B-factors to 40 Å2 (CCP4, 1994). The coordinates were refined as nine TLS groups: one each for the four subdomains of actin, one each for the three domains of G1–G3, and one each for the G1–G2 and G2–G3 linker regions. The implementation of this treatment immediately lowered the Rfree value by 0.6 and improved the electron density maps. Derived B-factors were calculated in the program TLSANL (CCP4, 1994). Omit maps were referred to at each cycle of model building (Brünger et al, 1998). The unoccupied calcium site in G2 was carefully inspected. No density was present at this site in the 3σ contoured Fo–Fc electron density difference map. Attempts to place either a calcium or sodium ion at this site resulted in the ion being moved away from the site during refinement. Finally, water molecules were added at positions that fulfilled the following three conditions: a peak in the Fo–Fc electron density difference map contoured at 4σ; a peak in the omit map contoured at 1σ; a water at the corresponding position in other gelsolin or actin high-resolution structures.
The quality of the final model was assessed in PROCHECK (CCP4, 1994). Two residues from gelsolin (Ala256 and Asp259) lie in the disallowed region of a Ramachandran plot. Figures were prepared using the program MOLSCRIPT (Kraulis, 1991). The model of the ADP-actin filament (ADP model) was produced by superimposing the crystal structure of ADP-actin (Otterbein et al, 2001) onto the actin filament model obtained from fiber diffraction data (Holmes et al, 1990). The model of the gelsolin-capped filament was generated by superimposing the actins from G1–G3:actin and G4–G6:actin (Choe et al, 2002) onto subdomains 1 and 2 of the two barbed end actins in the ADP model or of the EM reconstruction model (Belmont et al, 1999).
Coordinates
The coordinates and merged structure factors of the G1–G3:actin complex have been deposited in the Protein Data Bank under accession code 1RGI.
Acknowledgments
We acknowledge the ESRF for provision of synchrotron radiation. We thank the Heart and Stroke Foundation of BC and Yukon (LDB) and the Swedish Medical Science Research Council (RCR) for financial support. KN thanks the Swedish Foundation for International Cooperation in Research and Higher Education (STINT) for support through a grant to Dr U Lindberg.
Competing interests statement The authors declare that they have no competing financial interests.
References
- Allen PG (2003) Actin filament uncapping localizes to ruffling lamellae and rocketing vesicles. Nat Cell Biol 5: 972–979 [DOI] [PubMed] [Google Scholar]
- Azuma T, Koths K, Flanagan L, Kwiatkowski D (2000) Gelsolin in complex with phosphatidylinositol 4,5-bisphosphate inhibits caspase-3 and -9 to retard apoptotic progression. J Biol Chem 275: 3761–3766 [DOI] [PubMed] [Google Scholar]
- Belmont LD, Orlova A, Drubin DG, Egelman EH (1999) A change in actin conformation associated with filament instability after Pi release. Proc Natl Acad Sci USA 96: 29–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Crosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Burtnick LD, Koepf EK, Grimes J, Jones EY, Stuart DI, McLaughlin PJ, Robinson RC (1997) The crystal structure of plasma gelsolin: implications for actin severing, capping, and nucleation. Cell 90: 661–670 [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) Collaborative Computing Project 4, The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Chen CD, Huff ME, Matteson J, Page L, Phillips R, Kelly JW, Balch WE (2001) Furin initiates gelsolin familial amyloidosis in the Golgi through a defect in Ca(2+) stabilization. EMBO J 20: 6277–6287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe H, Burtnick LD, Mejillano M, Yin HL, Robinson RC, Choe S (2002) The calcium activation of gelsolin: insights from the 3 Å structure of the G4–G6/actin complex. J Mol Biol 324: 691–702 [DOI] [PubMed] [Google Scholar]
- Chou J, Stolz DB, Burke NA, Watkins SC, Wells A (2002) Distribution of gelsolin and phosphoinositol 4,5-bisphosphate in lamellipodia during EGF-induced motility. Int J Biochem Cell Biol 34: 776–790 [DOI] [PubMed] [Google Scholar]
- Cunningham CC, Stossel TP, Kwiatkowski DJ (1991) Enhanced motility in NIH 3T3 fibroblasts that overexpress gelsolin. Science 251: 1233–1236 [DOI] [PubMed] [Google Scholar]
- Cunningham CC, Vegners R, Bucki R, Funaki M, Korde N, Hartwig JH, Stossel TP, Janmey PA (2001) Cell permeant polyphosphoinositide-binding peptides that block cell motility and actin assembly. J Biol Chem 276: 43390–43399 [DOI] [PubMed] [Google Scholar]
- Falet H, Hoffmeister KM, Neujahr R, Italiano JE Jr, Stossel TP, Southwick FS, Hartwig JH (2002) Importance of free actin filament barbed ends for Arp2/3 complex function in platelets and fibroblasts. Proc Natl Acad Sci USA 99: 16782–16787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Mejillano M, Yin HL, Chen J, Prestwich GD (2001) Full-contact domain labeling: identification of a novel phosphoinositide binding site on gelsolin that requires the complete protein. Biochemistry 40: 904–913 [DOI] [PubMed] [Google Scholar]
- Haddad JG, Harper KD, Guoth M, Pietra GG, Sanger JW (1990) Angiopathic consequences of saturating the plasma scavenger system for actin. Proc Natl Acad Sci USA 87: 1381–1385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig JH (1992) Mechanisms of actin rearrangements mediating platelet activation. J Cell Biol 118: 1421–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig JH, Bokoch GM, Carpenter CL, Janmey PA, Taylor LA, Toker A, Stossel TP (1995) Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell 82: 643–653 [DOI] [PubMed] [Google Scholar]
- Holmes KC, Popp D, Gebhard W, Kabsch W (1990) Atomic model of the actin filament. Nature 347: 44–49 [DOI] [PubMed] [Google Scholar]
- Huff ME, Page LJ, Balch WE, Kelly JW (2003) Gelsolin domain 2 Ca2+ affinity determines susceptibility to furin proteolysis and familial amyloidosis of Finnish type. J Mol Biol 334: 119–127 [DOI] [PubMed] [Google Scholar]
- Irobi E, Burtnick LD, Urosev D, Narayan K, Robinson RC (2003) From the first to the second domain of gelsolin: a common path on the surface of actin? FEBS Lett 552: 86–90 [DOI] [PubMed] [Google Scholar]
- Janmey PA, Lamb J, Allen PG, Matsudaira PT (1992) Phosphoinositide-binding peptides derived from the sequences of gelsolin and villin. J Biol Chem 267: 11818–11823 [PubMed] [Google Scholar]
- Janmey PA, Stossel TP (1987) Modulation of gelsolin function by phosphatidylinositol 4,5-bisphosphate. Nature 325: 362–364 [DOI] [PubMed] [Google Scholar]
- Kazmirski SL, Isaacson RL, An C, Buckle A, Johnson CM, Daggett V, Fersht AR (2002) Loss of a metal-binding site in gelsolin leads to familial amyloidosis-Finnish type. Nat Struct Biol 9: 112–116 [DOI] [PubMed] [Google Scholar]
- Kinosian HJ, Newman J, Lincoln B, Selden LA, Gershman LC, Estes JE (1998) Ca2+ regulation of gelsolin activity: binding and severing of F-actin. Biophys J 75: 3101–3109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselar JG, Janmey PA, Almo SC, Chance MR (2003) Visualizing the Ca2+-dependent activation of gelsolin by using synchrotron footprinting. Proc Natl Acad Sci USA 100: 3942–3947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenchin VA, Allingham JS, King R, Tanaka J, Marriott G, Rayment I (2003) Trisoxazole macrolide toxins mimic the binding of actin-capping proteins to actin. Nat Struct Biol 10: 1058–1063 [DOI] [PubMed] [Google Scholar]
- Kothakota S, Azuma T, Reinhard C, Klippel A, Tang J, Chu K, McGarry TJ, Kirschner MW, Koths K, Kwiatkowski DJ, Williams LT (1997) Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science 278: 294–298 [DOI] [PubMed] [Google Scholar]
- Koya RC, Fujita H, Shimizu S, Ohtsu M, Takimoto M, Tsujimoto Y, Kuzumaki N (2000) Gelsolin inhibits apoptosis by blocking mitochondrial membrane potential loss and cytochrome c release. J Biol Chem 275: 15343–15349 [DOI] [PubMed] [Google Scholar]
- Kraulis PJ (1991) A program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24: 946–950 [Google Scholar]
- Kusano H, Shimizu S, Koya RC, Fujita H, Kamada S, Kuzumaki N, Tsujimoto Y (2000) Human gelsolin prevents apoptosis by inhibiting apoptotic mitochondrial changes via closing VDAC. Oncogene 19: 4807–4814 [DOI] [PubMed] [Google Scholar]
- Lin KM, Mejillano M, Yin HL (2000) Ca2+ regulation of gelsolin by its C-terminal tail. J Biol Chem 275: 27746–27752 [DOI] [PubMed] [Google Scholar]
- Lin KM, Wenegieme E, Lu PJ, Chen CS, Yin HL (1997) Gelsolin binding to phosphatidylinositol 4,5-bisphosphate is modulated by calcium and pH. J Biol Chem 272: 20443–20450 [DOI] [PubMed] [Google Scholar]
- McGough A, Chiu W, Way M (1998) Determination of the gelsolin binding site on F-actin: implications for severing and capping. Biophys J 74: 764–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin PJ, Gooch JT, Mannherz HG, Weeds AG (1993) Structure of gelsolin segment 1–actin complex and the mechanism of filament severing. Nature 364: 685–692 [DOI] [PubMed] [Google Scholar]
- Moriyama K, Yonezawa N, Sakai H, Yahara I, Nishida E (1992) Mutational analysis of an actin-binding site of cofilin and characterization of chimeric proteins between cofilin and destrin. J Biol Chem 267: 7240–7244 [PubMed] [Google Scholar]
- Orlova A, Prochniewicz E, Egelman EH (1995) Structural dynamics of F-actin: II. Cooperativity in structural transitions. J Mol Biol 245: 598–607 [DOI] [PubMed] [Google Scholar]
- Otterbein LR, Graceffa P, Dominguez R (2001) The crystal structure of uncomplexed actin in the ADP state. Science 293: 708–711 [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 24: 307–326 [DOI] [PubMed] [Google Scholar]
- Pollard TD, Blanchoin L, Mullins RD (2000) Molecular mechanisms controlling actin filament dynamics in nonmuscle cells. Annu Rev Biophys Biomol Struct 29: 545–576 [DOI] [PubMed] [Google Scholar]
- Pope B, Maciver S, Weeds A (1995) Localization of the calcium-sensitive actin monomer binding site in gelsolin to segment 4 and identification of calcium binding sites. Biochemistry 34: 1583–1588 [DOI] [PubMed] [Google Scholar]
- Pope B, Way M, Weeds AG (1991) Two of the three actin-binding domains of gelsolin bind to the same subdomain of actin. Implications of capping and severing mechanisms. FEBS Lett 280: 70–74 [DOI] [PubMed] [Google Scholar]
- Pope BJ, Gooch JT, Weeds AG (1997) Probing the effects of calcium on gelsolin. Biochemistry 36: 15848–15855 [DOI] [PubMed] [Google Scholar]
- Puius YA, Fedorov EV, Eichinger L, Schleicher M, Almo SC (2000) Mapping the functional surface of domain 2 in the gelsolin superfamily. Biochemistry 39: 5322–5331 [DOI] [PubMed] [Google Scholar]
- Renoult C, Blondin L, Fattoum A, Ternent D, Maciver SK, Raynaud F, Benyamin Y, Roustan C (2001) Binding of gelsolin domain 2 to actin. An actin interface distinct from that of gelsolin domain 1 and from ADF/cofilin. Eur J Biochem 268: 6165–6175 [DOI] [PubMed] [Google Scholar]
- Ressad F, Didry D, Xia GX, Hong Y, Chua NH, Pantaloni D, Carlier MF (1998) Kinetic analysis of the interaction of actin-depolymerizing factor (ADF)/cofilin with G- and F-actins. Comparison of plant and human ADFs and effect of phosphorylation. J Biol Chem 273: 20894–20902 [DOI] [PubMed] [Google Scholar]
- Robinson RC, Choe S, Burtnick LD (2001) The disintegration of a molecule: the role of gelsolin in FAF, familial amyloidosis (Finnish type). Proc Natl Acad Sci USA 98: 2117–2118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson RC, Mejillano M, Le VP, Burtnick LD, Yin HL, Choe S (1999) Domain movement in gelsolin: a calcium-activated switch. Science 286: 1939–1942 [DOI] [PubMed] [Google Scholar]
- Sablin EP, Dawson JF, VanLoock MS, Spudich JA, Egelman EH, Fletterick RJ (2002) How does ATP hydrolysis control actin's associations? Proc Natl Acad Sci USA 99: 10945–10947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechi AS, Wehland J (2000) The actin cytoskeleton and plasma membrane connection: PtdIns(4,5)P(2) influences cytoskeletal protein activity at the plasma membrane. J Cell Sci 113 (Part 21): 3685–3695 [DOI] [PubMed] [Google Scholar]
- Selden LA, Kinosian HJ, Newman J, Lincoln B, Hurwitz C, Gershman LC, Estes JE (1998) Severing of F-actin by the amino-terminal half of gelsolin suggests internal cooperativity in gelsolin. Biophys J 75: 3092–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simoes AP, Reed J, Schnabel P, Camps M, Gierschik P (1995) Characterization of putative polyphosphoinositide binding motifs from phospholipase C beta 2. Biochemistry 34: 5113–5119 [DOI] [PubMed] [Google Scholar]
- Spudich JA, Watt S (1971) The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin–troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem 246: 4866–4871 [PubMed] [Google Scholar]
- Sun HQ, Wooten DC, Janmey PA, Yin HL (1994) The actin side-binding domain of gelsolin also caps actin filaments. Implications for actin filament severing. J Biol Chem 269: 9473–9479 [PubMed] [Google Scholar]
- Van Troys M, Dewitte D, Goethals M, Vandekerckhove J, Ampe C (1996) Evidence for an actin binding helix in gelsolin segment 2; have homologous sequences in segments 1 and 2 of gelsolin evolved to divergent actin binding functions? FEBS Lett 397: 191–196 [DOI] [PubMed] [Google Scholar]
- Way M, Gooch J, Pope B, Weeds AG (1989) Expression of human plasma gelsolin in Escherichia coli and dissection of actin binding sites by segmental deletion mutagenesis. J Cell Biol 109: 593–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Way M, Pope B, Weeds AG (1992) Evidence for functional homology in the F-actin binding domains of gelsolin and alpha-actin: implications for the requirements of severing and capping. J Cell Biol 119: 835–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeds AG, Gooch J, McLaughlin P, Pope B, Bengtsdotter M, Karlsson R (1995) Identification of the trapped calcium in the gelsolin segment 1-actin complex: implications for the role of calcium in the control of gelsolin activity. FEBS Lett 360: 227–230 [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Hilgemann DH, Feng S, Bito H, Ishihara H, Shibasaki Y, Yin HL (2001) Phosphatidylinositol 4,5-bisphosphate induces actin stress-fiber formation and inhibits membrane ruffling in CV1 cells. J Cell Biol 152: 867–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Lin SC, Morrison-Bogorad M, Atkinson MA, Yin HL (1993) Thymosin beta 10 and thymosin beta 4 are both actin monomer sequestering proteins. J Biol Chem 268: 502–509 [PubMed] [Google Scholar]
- Zapun A, Grammatyka S, Deral G, Vernet T (2000) Calcium-dependent conformational stability of modules 1 and 2 of human gelsolin. Biochem J 350 (Part 3): 873–881 [PMC free article] [PubMed] [Google Scholar]