

Abstract
Most growth factors control cellular functions by activating specific receptor tyrosine kinases (RTKs). While overactivation of RTK signalling pathways is strongly associated with carcinogenesis, it is becoming increasingly clear that impaired deactivation of RTKs may also be a mechanism in cancer. A major deactivation pathway, receptor downregulation, involves ligand-induced endocytosis of the RTK and subsequent degradation in lysosomes. A complex molecular machinery that uses the small protein ubiquitin as a key regulator assures proper endocytosis and degradation of RTKs. Here we discuss evidence that implicates deregulation of this machinery in cancer.
Keywords: Cbl, endocytosis, signal transduction, Tsg101, ubiquitin
Introduction
Growth factors control cell growth, proliferation, differentiation, survival and migration by activating receptors on specific target cells. Most growth factor receptors belong to the receptor tyrosine kinase (RTK) family (Blume-Jensen and Hunter, 2001). Upon ligand binding, cytoplasmic tyrosine residues of the RTK become autophosphorylated and thus provide docking sites for a variety of phosphotyrosine-binding proteins. The specific recruitment of these proteins, which harbour various catalytic and/or scaffolding domains, determines the signalling output. Since deregulation of more than 30 RTKs has been associated with cancer (Blume-Jensen and Hunter, 2001), it is essential to describe how RTKs are activated and deactivated. Increased RTK signalling in cancer is often caused by gene amplifications, increased transcription/translation or mutations that promote ligand-independent autophosphorylation. However, recent data also show that the failure of RTKs to be appropriately deactivated may be a cause of neoplastic growth (Dikic and Giordano, 2003). A major deactivation pathway for RTKs, termed receptor downregulation, involves their ligand-induced internalization by means of endocytosis, followed by degradation in lysosomes (Peschard and Park, 2003). In this mini-review, we will highlight evidence that links defective RTK downregulation to cancer.
Endocytosis of RTKs
Exposure of cells and tissues to a variety of hormones and growth factors results in a reduced number of specific binding sites on the cell surface. This accelerated endocytosis of ligand-bound receptors might be initiated to promote downregulation of activated receptors and the switching off of initiated cellular signalling pathways. The ErbB family of receptor tyrosine kinases, which includes the epidermal growth factor receptor (EGFR) ErbB-1 and the related proteins ErbB-2, ErbB-3 and ErbB-4, is one of the most extensively analysed (Dikic and Giordano, 2003; Peschard and Park, 2003; Shtiegman and Yarden, 2003). Binding of EGF to the EGFR results in dimerization and autophosphorylation of several tyrosine residues in the cytoplasmic tail of the receptor. One of the phosphorylated tyrosines provides a binding site for the SH2 domain of the E3 ubiquitin ligase c-Cbl (Dikic and Giordano, 2003; Peschard and Park, 2003; Shtiegman and Yarden, 2003). In turn, c-Cbl is phosphorylated, and thereby its ubiquitin ligase ability activated. Next to the SH2 domain of c-Cbl is a RING-finger domain, which recruits a ubiquitin-loaded E2 enzyme, and this machinery covalently tags EGFR with ubiquitin (Figure 1). The ability of c-Cbl to interact with more than 40 intracellular proteins also makes this enzyme a complex scaffolding protein. Recently, it has been demonstrated that EGFR and platelet-derived growth factor receptor (PDGFR) are modified by Cbl-mediated mono-ubiquitination at several sites (multi-ubiquitinated), a modification that appears to be necessary and sufficient for endocytosis and degradation of the receptors (Haglund et al, 2003; Mosesson et al, 2003). Interestingly, the RING-finger domain is missing or defective in two oncogenic forms of Cbl, v-Cbl and 70Z-Cbl, and c-Cbl cannot interact with ErbB-3 and -4, two receptors whose ligand-induced downregulation is impaired (Peschard and Park, 2003). In summary, this means that the internalization of RTKs is controlled by two types of reversible modifications, namely phosphorylation and ubiquitination.
Figure 1.
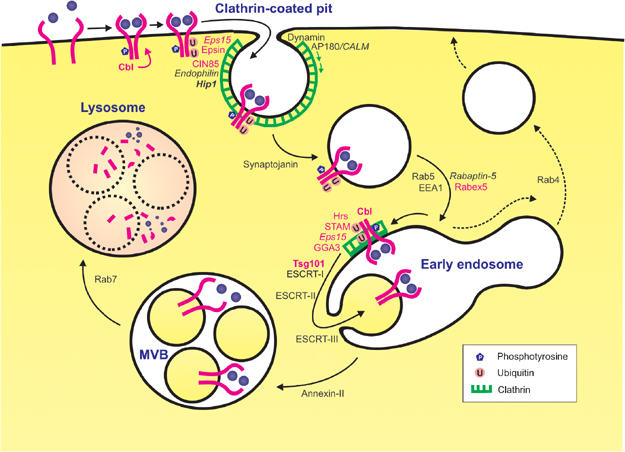
Mechanism of endocytic downregulation of RTKs. The figure illustrates RTK downregulation, with EGFR as an example. Ligand binding triggers receptor autophosphorylation via receptor dimerization. c-Cbl associates with the tyrosine-phosphorylated RTK and mediates its multi-ubiquitination. This facilitates endocytosis of the receptor via clathrin-coated pits. Proteins such as Epsin, Eps15 and Hip1 may be involved in cargo recruitment into clathrin-coated pits. Clathrin polymerization is catalysed by AP-180/CALM, and vesicle formation is facilitated by Dynamin and Endophilin. The endocytic vesicle uncoats (facilitated by Synaptojanin) and fuses with an early endosome, by a mechanism catalysed by the small GTPase Rab5 and its effectors. At the early endosome, the ubiquitinated RTK is sorted into a ‘bilayered' clathrin coat, probably by associating with ubiquitin-binding proteins such as Hrs and STAM. From here, the RTK is sorted into intraluminal vesicles of the endosome, in a process mediated by ESCRT-I (whose subunit Tsg101 binds Hrs as well as ubiquitin), ESCRT-II and ESCRT-III. The RTK is probably dephosphorylated and deubiquitinated during this sorting step. From the early endosome, a multivesicular body (MVB)/endosomal carrier vesicle is formed in an Annexin-II-dependent fashion (Gruenberg and Stenmark, 2004), which ultimately fuses with a late endosome or a lysosome. Intraluminal vesicles with their content are then degraded by lysosomal hydrolases. Ubiquitin-binding/monoubiquitinated proteins are in red, proteins found as oncogenic fusion proteins (see Table I) are in italics, and proteins experimentally implicated in cancer (see Table II) are in bold. A recycling pathway from the early endosome to the plasma membrane, which is followed by non-ubiquitinated membrane proteins, is indicated by dashed arrows. For simplicity, a number of known components of the endocytosis machinery, including specific lipids (Conner and Schmid, 2003), have been omitted from the figure. Note that RTKs can also be endocytosed from other invaginations than clathrin-coated pits, but the significance of this alternative internalization in RTK downregulation is not known (Gonzalez-Gaitan and Stenmark, 2003).
The adaptor protein CIN85 interacts and functions with c-Cbl in EGFR downregulation (Soubeyran et al, 2002). Additionally, CIN85 interacts with a regulatory component of clathrin-coated pits, Endophilin. By analysis of mutants of the various components, both EGFR and the hepatocyte growth factor receptor, Met, have been shown to depend on the assembly of this c-Cbl/CIN85/Endophilin complex at the active receptors in order to accelerate endocytosis and thereby attenuate signalling activity (Petrelli et al, 2002; Soubeyran et al, 2002; Dikic et al, 2003). Clathrin adaptors such as AP-2 and AP-180 mediate clathrin polymerization. AP-2 also functions to incorporate cargo such as transferrin receptors into clathrin-coated pits. However, AP-2 is dispensible for EGFR endocytosis; so alternative clathrin adaptors must recognize this receptor (Nesterov et al, 1999; Motley et al, 2003). The ubiquitin-binding proteins Epsin and Eps15 are possible candidates (Di Fiore et al, 2003). Another candidate is Huntingtin interacting protein 1 (Hip1), which, like AP-180, AP-2 and Epsin, is an endocytic regulator that binds phosphoinositides as well as clathrin (Hyun and Ross, 2004). As Hip1 has been implicated in tumorigenesis (see below), its exact function in endocytosis is under intensive investigation.
Lysosomal sorting of endocytosed RTKs
After internalization, endocytic vesicles uncoat and fuse with early (sorting) endosomes, which results in delivery of the cargo (see Figure 1). Early endosomes can also fuse with other early endosomes, and both hetero- and homotypic endosome fusions (and probably even the biogenesis of endocytic vesicles) are controlled by the small GTPase Rab5 (Zerial and McBride, 2001). In its GTP-bound form, Rab5 recruits a number of effectors to endosome membranes. One of these effectors is Rabaptin-5, which plays an essential role in early endosomal fusion. Rabaptin-5 is complexed to the GDP/GTP exchange factor (GEF) Rabex5, and the Rabaptin-5/Rabex5 complex may function in local activation of Rab5 through a positive-feedback mechanism. Another central Rab5 effector in membrane fusion is the phosphoinositide-binding coiled-coil protein EEA1, which probably tethers endocytic vesicles to early endosomes (Simonsen et al, 1998; Zerial and McBride, 2001).
Upon delivery to the early endosome, the EGFR is sorted to the inner membranes of MVBs (see Figure 1). The molecular mechanism for selection of receptors destined for downregulation seems to be conserved through evolution from yeast to mammals. It involves conjugation of ubiquitin onto the receptor (Dupre et al, 2001; Hicke and Dunn, 2003), and retention of the receptor from the recycling pathway by ubiquitin-binding proteins localized on sorting endosomes, such as the hepatocyte growth factor-regulated tyrosine kinase substrate Hrs (Katzmann et al, 2002; Raiborg et al, 2002; 2003) and the signal-transducing adaptor molecule STAM (Bache et al, 2003b; Kanazawa et al, 2003). Recently, one of the mammalian Golgi-localized, γ-ear-containing, Arf-binding (GGA) proteins, GGA3, has also been implicated in the ubiquitin-sorting machinery at the endosomes (Puertollano and Bonifacino, 2004). Both Hrs and GGA3 bind clathrin, and the former has been found together with STAM, Eps15 and ubiquitinated proteins in a nonbudding ‘bilayered' clathrin coat on endosomes (see Figure 1), which could play a role in RTK sorting (Raiborg et al, 2003). Moreover, Hrs functions as a docking site for the endosomal sorting complex required for transport, ESCRT-I (Bache et al, 2003a; Katzmann et al, 2003), which further ‘escorts' the receptor towards the lysosomes in conjunction with two additional complexes, ESCRT-II and ESCRT-III (Katzmann et al, 2002; Raiborg et al, 2003). The EGFR needs to be ubiquitinated at the endosomal membrane in order to undergo sorting to the lysosomes. Although c-Cbl-dependent ubiquitination is associated with sorting at the plasma membrane, this ubiquitin ligase is also recruited to endosomes where it colocalizes with internalized receptors (Longva et al, 2002; Rowinsky, 2004). The finding that Cbl proteins defective in ubiquitination cause increased recycling of EGFR demonstrates the need of receptor ubiquitination for proper endosomal sorting and degradation (Longva et al, 2002; Rowinsky, 2004).
Endosome to lysosome trafficking regulates signalling output of RTKs
As signalling can continue from the limiting membrane of endosomes after internalization, the need for proper sorting into the intraluminal space of MVBs in order to shut down the signal becomes evident (Dikic and Giordano, 2003). Studies of hrs mutant Drosophila have been especially useful to clarifing the relationship between receptor signalling and endosomal sorting (Lloyd et al, 2002; Jekely and Rorth, 2003). As in Hrs-deficient yeast and mammalian cells, endosomes in hrs mutant Drosophila cells are enlarged and unable to invaginate their limiting membranes, indicating that Hrs regulates the formation of MVBs (Lloyd et al, 2002). hrs mutant Drosophila fails to downregulate the EGFR, PVR and Torso RTKs, which results in enhanced signalling and altered embryonic patterning. In addition, recent work demonstrates that a number of signalling receptors, including RTKs, accumulate in ubiquitin-positive endosomes in hrs mutant Drosophila (Jekely and Rorth, 2003). Together, these reports provide in vivo evidence for the importance of proper sorting of multiple signalling receptors into a functional degradative pathway in order to attenuate signalling. Since enhanced receptor signalling was generally observed in hrs mutant Drosophila, the above studies strongly support the idea that receptors can transmit signals from endosomes, and that trafficking of receptors from early endosomes to MVBs/lysosomes is important to downregulate signalling.
Components of the endocytosis machinery are found as oncogenic fusion proteins
Recent studies provide evidence that the endocytic machinery is disrupted in certain types of cancer. Genes encoding some of the proteins that participate directly in the clathrin coat during the initial steps of endocytosis have been identified as targets of chromosomal rearrangements in human haematopoietic malignancies (Floyd and De Camilli, 1998) (Table I). In two human myeloid leukaemias, Eps15 is fused to the ALL1/HRX gene as a consequence of the t(1;11)(p32;q23) chromosomal translocation. The resulting fusion protein comprises the N-terminal domain of ALL1/HRX fused in-frame to the C-terminal region of Eps15. Another endocytosis protein, Endophilin-2 (SH3p8), was identified at the t(11;19)(q23;p13) translocation in a case of acute myeloid leukaemia. The resulting fusion protein contains the N-terminal domain of ALL1/HRX and the C-terminal part of Endophilin-2. It is worth noting that the ALL1/HRX gene has been mapped to chromosome 11q23, a region where abnormalities have been identified in approximately 10% of acute lymphoblastic leukaemias and 5% of acute myeloid leukaemias (Floyd and De Camilli, 1998). The gene encoding a third endocytosis protein, CALM, which is a non-neuronal version of AP-180, is the target of the t(10;11)(p13;q14) translocation. This translocation is found in the human U937 cell line, and results in fusion of the N-terminal portion of CALM to AF-10 (Floyd and De Camilli, 1998). The U937 cell line was derived from a patient with diffuse histiocytic lymphoma, and an identical translocation has been identified in patients with different haematological malignancies.
Table 1.
Components of the endocytosis machinery found as constituents of oncogenic fusion proteins
| Protein | Proposed function in receptor downregulation | Cancer type | Fusion partner; chromosomal translocation | References |
|---|---|---|---|---|
| Eps15 | Ubiquitin-binding protein found in plasma membrane clathrin-coated pits and in clathrin coats on sorting endosomes. Possible role in receptor recruitment | Myeloid leukaemia | ALL/HRX gene; t(1;11)(p32;q23) | Floyd and De Camilli (1998) |
| Endophilin-2 | Putative lysophosphatidic acid acyl transferase. Binds Dynamin-2 and Synaptojanin. Involved in formation of clathrin-coated vesicles. Member of the membrane bending/GTPase-binding BAR domain family | Myeloid leukaemia | ALL/HRX gene; t(11;19)(q23;p13) | Floyd and De Camilli (1998) and Habermann (2004) |
| CALM | Non-neuronal homologue of AP-180. Binds clathrin and phosphoinositides. Strong activator of clathrin polymerization | Lymphoblastic and myeloid leukaemia | AF-10; t(10;11)(p13;q14) | Floyd and De Camilli (1998) |
| Rabaptin-5 | Effector of Rab5. Complexed to the Rab5 GDP/GTP exchange factor Rabex5. Required for endosomal fusion | Myelomonocytic leukaemia | PDGFR-β; t(5;17)(q33;p13) | Magnusson et al (2001) |
| Hip1 | Homologue of End4/Sla2, which interacts with actin-binding proteins and is required for endocytosis in Saccharomyces cerevisiae. Role in clathrin-mediated endocytosis. Interacts with AP-2, clathrin and phosphoinositides | Myelomonocytic leukaemia | PDGFR-β; t(5;7)(q33;q11.2) | Ross et al (1998) |
Translocations involving an isoform of the PDGFR have been reported in some patients with chronic myelomonocytic leukaemia (CMML). These translocations result in fusion genes containing the active cytoplasmic tyrosine kinase domain of the receptor. Examples of proteins that have been found to make in-frame fusions with PDGFR are Rabaptin-5 (Magnusson et al, 2001) and Hip1 (Ross et al, 1998). As both Rabaptin-5 and Hip1 contain putative coiled-coil dimerization domains, the oncogenic transformation by the fusion proteins could be caused by forced dimerization of PDGFR. However, due to the role of these proteins in the early endocytic pathway (see Figure 1), an alternative possibility is that the fusion proteins are oncogenic because they interfere with normal receptor downregulation.
Impaired RTK downregulation is associated with cancer
Besides the occurrence of endocytic regulators in cancer-associated fusion proteins, more direct links between endocytosis and cancer are beginning to emerge (Table II). Alterations that uncouple RTKs from c-Cbl-mediated ubiquitination and thereby downregulation are tightly associated with the pathogenesis of cancer. Examples include the Met receptor, colony-stimulating factor-1 receptor, PDGFR and EGFR. These receptors have all been identified as substrates for ubiquitination, and depend on the interaction with c-Cbl for proper degradation. An oncogenic form of the Met receptor, Trp-Met, is constitutively active, but fails to bind c-Cbl and is therefore not ubiquitinated (Peschard and Park, 2003). In a similar manner, mutation of a C-terminal tyrosine, which is the direct binding site for c-Cbl, enhances the transforming abilities of CSF-1R in fibroblasts. PDGFR and the stem cell factor receptor c-Kit recruit c-Cbl indirectly via the adaptor protein APS (adaptor containing PH and SH2 domains), an interaction that leads to PDGFR downregulation (Peschard and Park, 2003). Deletion of the APS-binding site in c-Kit greatly enhances its transforming ability (Herbst et al, 1995), although some of this effect may be due to enhanced catalytic activity (Chan et al, 2003). Finally, an EGFR mutant lacking only the direct c-Cbl-binding site elicits stronger mitogenic signals than the wild-type receptor (Waterman et al, 2002). Together, these receptors represent examples of how a variety of oncoproteins avoid lysosomal downregulation by loss of the c-Cbl-binding site, inefficient c-Cbl recruitment or through the formation of fusion proteins that escape endocytosis and the degradative lysosomal pathway (reviewed in Peschard and Park, 2003). It is also interesting to note that c-Cbl is itself regulated by several proteins that are implicated in potentially carcinogenic signalling pathways, including the nonreceptor tyrosine kinase Src, the small GTPase Cdc42 and the tyrosine-phosphorylated adaptor protein Spry2 (Polo et al, 2004).
Table 2.
Regulators of RTK downregulation associated with cancer
| Protein | Function in receptor downregulation | Links to cancer | References |
|---|---|---|---|
| Hip1 | Homologue of End4/Sla2, which interacts with actin-binding proteins and is required for endocytosis in S. cerevisiae. Role in clathrin-mediated endocytosis. Interacts with AP-2, clathrin and phosphoinositides | Overexpression causes neoplastic growth in vitro and in mouse models. Frequently overexpressed in breast cancer. Found as fusion with PDGFR in CMML (Table I). | Hyun and Ross (2004) |
| c-Cbl | Ubiquitin ligase that ubiquitinates ligand-activated RTKs at the plasma membrane and on early endosomes. This facilitates their endocytosis and lysosomal sorting. Also proposed to have a direct function in endocytosis by recruiting Endophilin to clathrin-coated pits (via CIN85) | v-Cbl is a viral oncoprotein. Several oncogenic forms of RTKs lack binding sites for c-Cbl | Shtiegman and Yarden (2003), Peschard and Park (2003) and Dikic and Giordano (2003) |
| Tsg101 | Subunit of mammalian ESCRT-I. Acts downstream of Hrs to sort ubiquitinated membrane proteins from endosomes to lysosomes | Functional inactivation by antisense transcripts results in transformed cell lines that form metastatic tumours in nude mice. Aberrant transcripts found in cancers | Li and Cohen (1996), Babst et al (2000) and Gruenberg and Stenmark (2004) |
The first component of the endocytosis machinery to be directly implicated in tumour formation was Hip1 (Hyun and Ross, 2004). The precise function of Hip1 in endocytosis is not known, but it interacts with AP-2, phosphoinositides and clathrin, and is thought to play a fundamental role in clathrin trafficking. Overexpression of Hip1 causes cell transformation and increased proliferation. Hip1-transformed cells appear to have less clathrin at the plasma membrane, and EGFR levels are upregulated. Interestingly, the expression of Hip1 in normal breast tissue is very low, whereas invasive breast cancers or their precursors frequently display elevated levels of Hip1. Therefore, perhaps the increased EGFR levels often observed in breast carcinomas might, at least in some cases, be explained by increased Hip1 expression and thus reduced EGFR downregulation.
A functional sorting machinery in the early endosome membrane is required for lysosomal targeting of endocytosed RTKs, and one of the ESCRT-I subunits, the tumour susceptibility gene 101 (Tsg101), is indeed a candidate tumour-suppressor gene. Functional inactivation of Tsg101 by antisense transcripts complementary to Tsg101 mRNA leads to transformation of NIH 3T3 cells, characterized by colony formation in soft agar and their ability to form metastatic tumours when injected into nude mice (Li and Cohen, 1996). Moreover, partial deletions or aberrant splicing of Tsg101 have been reported to occur in human cancer (Lee and Feinberg, 1997), although variant transcripts can also be found in normal tissue. Even though Tsg101 has been implicated in several regulatory pathways (Li et al, 2001), its best-documented function is as a regulator of endocytic trafficking (Babst et al, 2000; Bishop et al, 2002; Bache et al, 2003a). As a complete knockout of Tsg101 in mice is embryonic lethal, it has been difficult to establish definitively a role for this protein in cancer (Wagner et al, 2003). However, it is worth noting that even a second subunit of ESCRT-I has recently been identified as a growth-regulatory protein (KG Bache, T Slagsvold and H Stenmark, unpublished). This strengthens the idea that normal ESCRT-I function is important for receptor downregulation and prevention of carcinogenesis.
Conclusion and perspectives
As the overlap between the molecular mechanisms of endocytosis and signalling becomes evident, more work is needed to untangle the regulatory networks and shed light on how the processes influence each other and cellular proliferation. Even though signals can propagate from endosomes (Gonzalez-Gaitan and Stenmark, 2003), it is clear that downregulation of RTKs by endocytosis and lysosomal sorting represents a key mechanism of signal termination.
The small protein ubiquitin is tightly associated with receptor downregulation. While poly-ubiquitination marks proteins for proteasomal degradation, mono- and multi-ubiquitination may function as signals for endocytosis and lysosomal degradation of membrane proteins (Hicke and Dunn, 2003). Not only does the ubiquitin ligase c-Cbl play a key role in RTK downregulation, but also a number of endocytic regulator proteins bind ubiquitin and can be mono-ubiquitinated themselves. Examples include Epsin, Eps15, Rabex5, Hrs, STAM, GGA3 and Tsg101 (see Figure 1). While their binding to ubiquitin may allow these proteins to interact directly with ubiquitinated RTKs, their covalent attachment to ubiquitin might serve regulatory roles (Di Fiore et al, 2003). As ubiquitination regulates a large variety of cellular processes (Hicke and Dunn, 2003), ubiquitin as such does not stand out as a target for anticancer therapy. However, as we learn more about ubiquitination and ubiquitin binding by various proteins involved in receptor downregulation, it may still be possible to identify pathway-specific molecular recognition events that can be targeted by low-molecular-weight compounds.
The examples mentioned in this mini-review predict a role for impaired endocytosis in cancer. Signalling reciprocates trafficking by controlling the endocytosis machinery and contributing to its ability to sort receptors selectively. Based on the knowledge gained in the field of basic biological mechanisms, potential targets for therapeutic approaches ascend. Due to the malignant potential of RTKs of the EGFR family, these have been rational targets for novel therapeutics. One strategy involves preventing ligand interaction by blocking the extracellular ligand-binding domain using monoclonal antibodies specific to this site. Another method is to disturb receptor activation by introducing small molecules that bind to the kinase pocket and prevent post-receptor signalling. The advantage with this approach is that it would also include receptors that are constitutively active, independently of ligand binding. A third strategy, which takes advantage of our knowledge of receptor downregulation, is to induce RTK internalization and degradation in tumours. All the three methods have proven promising in clinical trials (Rowinsky, 2004). It becomes evident that tumour-promoting receptors as well as components in their regulatory pathways may be potent targets for therapeutic approaches. However, in order to design successful tools, it is crucial to gain a detailed insight into the molecular mechanisms involved.
Acknowledgments
We apologize to those collegues whose relevant primary papers could not be cited due to strict space limitations. We thank the Norwegian Cancer Society, the Novo Nordisk Foundation and the Research Council of Norway for financial support.
References
- Babst M, Odorizzi G, Estepa EJ, Emr SD (2000) Mammalian tumor suceptibility gene 101 (TSG101) and the yeast homologue, Vps23p, both function in late endosomal trafficking. Traffic 1: 248–258 [DOI] [PubMed] [Google Scholar]
- Bache KG, Brech A, Mehlum A, Stenmark H (2003a) Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol 162: 435–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bache KG, Raiborg C, Mehlum A, Stenmark H (2003b) STAM and Hrs are subunits of a multivalent ubiquitin-binding complex on early endosomes. J Biol Chem 278: 12513–12521 [DOI] [PubMed] [Google Scholar]
- Bishop N, Horman A, Woodman P (2002) Mammalian class E vps proteins recognize ubiquitin and act in the removal of endosomal protein–ubiquitin conjugates. J Cell Biol 157: 91–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen P, Hunter T (2001) Oncogenic kinase signalling. Nature 411: 355–365 [DOI] [PubMed] [Google Scholar]
- Chan PM, Ilangumaran S, La Rose J, Chakrabartty A, Rottapel R (2003) Autoinhibition of the kit receptor tyrosine kinase by the cytosolic juxtamembrane region. Mol Cell Biol 23: 3067–3078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner SD, Schmid SL (2003) Regulated portals of entry into the cell. Nature 422: 37–44 [DOI] [PubMed] [Google Scholar]
- Di Fiore PP, Polo S, Hofmann K (2003) When ubiquitin meets ubiquitin receptors: a signalling connection. Nat Rev Mol Cell Biol 4: 491–497 [DOI] [PubMed] [Google Scholar]
- Dikic I, Giordano S (2003) Negative receptor signalling. Curr Opin Cell Biol 15: 128–135 [DOI] [PubMed] [Google Scholar]
- Dikic I, Szymkiewicz I, Soubeyran P (2003) Cbl signaling networks in the regulation of cell function. Cell Mol Life Sci 60: 1805–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupre S, Volland C, Haguenauer-Tsapis R (2001) Membrane transport: ubiquitylation in endosomal sorting. Curr Biol 11: R932–R934 [DOI] [PubMed] [Google Scholar]
- Floyd S, De Camilli P (1998) Endocytosis proteins and cancer: a potential link? Trends Cell Biol 8: 299–301 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Gaitan M, Stenmark H (2003) Endocytosis and signaling: a relationship under development. Cell 115: 513–521 [DOI] [PubMed] [Google Scholar]
- Gruenberg J, Stenmark H (2004) Opinion: the biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol 5: 317–323 [DOI] [PubMed] [Google Scholar]
- Habermann B (2004) The BAR-domain family of proteins: a case of bending and binding? EMBO Rep 5: 250–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I (2003) Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol 5: 461–466 [DOI] [PubMed] [Google Scholar]
- Herbst R, Munemitsu S, Ullrich A (1995) Oncogenic activation of v-kit involves deletion of a putative tyrosine–substrate interaction site. Oncogene 10: 369–379 [PubMed] [Google Scholar]
- Hicke L, Dunn R (2003) Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol 19: 141–172 [DOI] [PubMed] [Google Scholar]
- Hyun TS, Ross TS (2004) HIP1: trafficking roles and regulation of tumorigenesis. Trends Mol Med 10: 194–199 [DOI] [PubMed] [Google Scholar]
- Jekely G, Rorth P (2003) Hrs mediates downregulation of multiple signalling receptors in Drosophila. EMBO Rep 4: 1163–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa C, Morita E, Yamada M, Ishii N, Miura S, Asao H, Yoshimori T, Sugamura K (2003) Effects of deficiencies of STAMs and Hrs, mammalian class E Vps proteins, on receptor downregulation. Biochem Biophys Res Commun 309: 848–856 [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Odorizzi G, Emr SD (2002) Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol 3: 893–905 [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Stefan CJ, Babst M, Emr SD (2003) Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J Cell Biol 162: 413–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MP, Feinberg AP (1997) Aberrant splicing but not mutations of TSG101 in human breast cancer. Cancer Res 57: 3131–3134 [PubMed] [Google Scholar]
- Li L, Cohen SN (1996) Tsg101: a novel tumor susceptibility gene isolated by controlled homozygous functional knockout of allelic loci in mammalian cells. Cell 85: 319–329 [DOI] [PubMed] [Google Scholar]
- Li L, Liao J, Ruland J, Mak TW, Cohen SN (2001) A TSG101/MDM2 regulatory loop modulates MDM2 degradation and MDM2/p53 feedback control. Proc Natl Acad Sci USA 98: 1619–1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd TE, Atkinson R, Wu MN, Zhou Y, Pennetta G, Bellen HJ (2002) Hrs regulates endosome invagination and receptor tyrosine kinase signaling in Drosophila. Cell 108: 261–269 [DOI] [PubMed] [Google Scholar]
- Longva KE, Blystad FD, Stang E, Larsen AM, Johannessen LE, Madshus IH (2002) Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J Cell Biol 156: 843–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnusson MK, Meade KE, Brown KE, Arthur DC, Krueger LA, Barrett AJ, Dunbar CE (2001) Rabaptin-5 is a novel fusion partner to platelet-derived growth factor beta receptor in chronic myelomonocytic leukemia. Blood 98: 2518–2525 [DOI] [PubMed] [Google Scholar]
- Mosesson Y, Shtiegman K, Katz M, Zwang Y, Vereb G, Szollosi J, Yarden Y (2003) Endocytosis of receptor tyrosine kinases is driven by mono-, not poly-ubiquitylation. J Biol Chem 278: 31323–31326 [DOI] [PubMed] [Google Scholar]
- Motley A, Bright NA, Seaman MN, Robinson MS (2003) Clathrin-mediated endocytosis in AP-2-depleted cells. J Cell Biol 162: 909–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesterov A, Carter RE, Sorkina T, Gill GN, Sorkin A (1999) Inhibition of the receptor-binding function of clathrin adaptor protein AP-2 by dominant-negative mutant mu2 subunit and its effects on endocytosis. EMBO J 18: 2489–2499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschard P, Park M (2003) Escape from Cbl-mediated downregulation: a recurrent theme for oncogenic deregulation of receptor tyrosine kinases. Cancer Cell 3: 519–523 [DOI] [PubMed] [Google Scholar]
- Petrelli A, Gilestro GF, Lanzardo S, Comoglio PM, Migone N, Giordano S (2002) The endophilin-CIN85-Cbl complex mediates ligand-dependent downregulation of c-Met. Nature 416: 187–190 [DOI] [PubMed] [Google Scholar]
- Polo S, Pece S, Di Fiore PP (2004) Endocytosis and cancer. Curr Opin Cell Biol, in press [DOI] [PubMed] [Google Scholar]
- Puertollano R, Bonifacino JS (2004) Interactions of GGA3 with the ubiquitin sorting machinery. Nat Cell Biol 6: 244–251 [DOI] [PubMed] [Google Scholar]
- Raiborg C, Bache KG, Gillooly DJ, Madshus IH, Stang E, Stenmark H (2002) Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat Cell Biol 4: 394–398 [DOI] [PubMed] [Google Scholar]
- Raiborg C, Rusten TE, Stenmark H (2003) Protein sorting into multivesicular endosomes. Curr Opin Cell Biol 15: 446–455 [DOI] [PubMed] [Google Scholar]
- Ross TS, Bernard OA, Berger R, Gilliland DG (1998) Fusion of Huntingtin interacting protein 1 to platelet-derived growth factor beta receptor (PDGFbetaR) in chronic myelomonocytic leukemia with t(5;7)(q33;q11.2). Blood 91: 4419–4426 [PubMed] [Google Scholar]
- Rowinsky EK (2004) The erbB family: targets for therapeutic development against cancer and therapeutic strategies using monoclonal antibodies and tyrosine kinase inhibitors. Annu Rev Med 55: 433–457 [DOI] [PubMed] [Google Scholar]
- Shtiegman K, Yarden Y (2003) The role of ubiquitylation in signaling by growth factors: implications to cancer. Semin Cancer Biol 13: 29–40 [DOI] [PubMed] [Google Scholar]
- Simonsen A, Lippé R, Christoforidis S, Gaullier J-M, Brech A, Callaghan J, Toh B-H, Murphy C, Zerial M, Stenmark H (1998) EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature 394: 494–498 [DOI] [PubMed] [Google Scholar]
- Soubeyran P, Kowanetz K, Szymkiewicz I, Langdon WY, Dikic I (2002) Cbl-CIN85-endophilin complex mediates ligand-induced downregulation of EGF receptors. Nature 416: 183–187 [DOI] [PubMed] [Google Scholar]
- Wagner KU, Krempler A, Qi Y, Park K, Henry MD, Triplett AA, Riedlinger G, Rucker EB III, Hennighausen L (2003) Tsg101 is essential for cell growth, proliferation, and cell survival of embryonic and adult tissues. Mol Cell Biol 23: 150–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman H, Katz M, Rubin C, Shtiegman K, Lavi S, Elson A, Jovin T, Yarden Y (2002) A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J 21: 303–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerial M, McBride H (2001) Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2: 107–117 [DOI] [PubMed] [Google Scholar]