

Abstract
Acetylcholine (ACh), released from axonal terminals of motor neurons in neuromuscular junctions regulates the efficacy of neurotransmission through activation of presynaptic nicotinic and muscarinic autoreceptors. Receptor-mediated presynaptic regulation could reflect either direct action on exocytotic machinery or modulation of Ca2+ entry and resulting intra-terminal Ca2+ dynamics. We have measured free intra-terminal cytosolic Ca2+ ([Ca2+]i) using Oregon-Green 488 microfluorimetry, in parallel with voltage-clamp recordings of spontaneous (mEPC) and evoked (EPC) postsynaptic currents in post-junctional skeletal muscle fiber. Activation of presynaptic muscarinic and nicotinic receptors with exogenous acetylcholine and its non-hydrolized analog carbachol reduced amplitude of the intra-terminal [Ca2+]i transients and decreased quantal content (calculated by dividing the area under EPC curve by the area under mEPC curve). Pharmacological analysis revealed the role of muscarinic receptors of M2 subtype as well as d-tubocurarine-sensitive nicotinic receptor in presynaptic modulation of [Ca2+]i transients. Modulation of synaptic transmission efficacy by ACh receptors was completely eliminated by pharmacological inhibition of N-type Ca2+ channels. We conclude that ACh receptor-mediated reduction of Ca2+ entry into the nerve terminal through N-type Ca2+ channels represents one of possible mechanism of presynaptic modulation in frog neuromuscular junction.
Keywords: neuromuscular synapse, calcium transient, presynaptic acetylcholine receptors, muscarinic receptors, nicotinic receptors, N-type Ca channels, quantum secretion of acetylcholine
Introduction
Acetylcholine (ACh) when released from nerve endings upon action potential-driven depolarization, not only triggers postsynaptic response in muscle cell, but also regulates its own secretion from presynaptic terminal (Ciani and Edwards, 1963; Parnas et al., 2000; Tomas et al., 2014). This presynaptic regulation has an important for reliability of synaptic transmission. It has been shown that various subtypes of muscarinic and nicotinic acetylcholine receptors are expressed in motor nerve endings and they modulate release of ACh into the synaptic cleft (Bowman et al., 1990; Miller, 1990; Tomas et al., 2014). The direct and indirect Ca2+ influx resulting from the activation of neuronal nicotinic acetylcholine receptors can modulate presynaptic neurotransmitter release (Shen and Yakel, 2009). Two subtypes of muscarinic receptors are involved in the ACh release modulation: the M1 receptor mediates enhancement of ACh release, while the M2 is involved in inhibition of release (Slutsky et al., 1999). Muscarinic agonists have been shown to decrease the number of quanta released in response to stimulation of motor nerve (Arenson, 1989; Slutsky et al., 1999; Samigullin et al., 2014). Muscarinic receptors can also be activated in the absence of stimulation during spontaneous neurotransmitter release (Kupchik et al., 2008).
Although this phenomenon has received a significant research attention in the past (Nikol'skiĭ and Giniatullin, 1979; Wessler, 1989; Macleod et al., 1994; Van der Kloot et al., 1997; Nikolsky et al., 2004), fine details of ACh-dependent presynaptic regulation remain obscure. Both direct action of ACh on exocytotic machinery (Linial et al., 1997) and inhibition of presynaptic Ca2+ entry (Wu and Saggau, 1997; Parnas et al., 2000; Khaziev et al., 2012) have been suggested. The latter possibility, being seemingly logical, has not been analyzed in detail. The present study was designed to investigate the role of presynaptic calcium influx in autoregulation of ACh secretion by presynaptic receptors in the frog neuromuscular junction.
Materials and methods
Experimental preparation and ethical approval
Experiments were performed on isolated nerve-muscle preparations of musculus cutaneous pectoris from the frog Rana ridibunda. The experimental procedures were performed in accordance with the guidelines for use of laboratory animals of Kazan Federal University and Kazan Medical University, in compliance with the NIH Guide for Care and Use of Laboratory Animals. Experimental protocols met the requirements of the European Communities Council Directive 86/609/EEC and were approved by the Ethical Committee of Kazan Medical University.
Intracellular Ca2+ monitoring
Cytosolic concentration of ionized Ca2+ ([Ca2+]i) in nerve endings was monitored using fluorescent microfluorimetry (Tsien, 1989). Each nerve-muscle preparation was loaded with Ca2+-sensitive dye by soaking nerve stump in 50 mM solution of fluorescent Ca2+-indicator Oregon Green 488 BAPTA-1 Hexapotassium Salt (Molecular Probes, Eugene, Oregon, USA); for details of loading technique see (Peng and Zucker, 1993; Wu and Betz, 1996; Tsang et al., 2000; Samigullin et al., 2015). At the end of loading protocol, all terminals in the proximal part of the nerve trunk had sufficient levels of fluorescence. It has been estimated that the intra-terminal concentration of the probe varied between 40 and 150 μM (Suzuki et al., 2000).
The fluorescent probe by definition binds Ca2+ and hence may affect both [Ca2+]i dynamics and physiology of the neuromuscular junction. Our own observations (Samigullin et al., 2015) as well as observations of others (Wu and Betz, 1996) failed to detect any appreciable influence of loaded Ca2+ probe on the amplitude of the postsynaptic response or on the frequency of the miniature end-plate potentials. Nonetheless we additionally performed control experiments to compare spontaneous endplate currents (mEPC) and quantal release before and after Ca2+ probe loading. It appeared that Ca2+ probe affects neither; the mEPC frequency was 0.32 ± 0.03 Hz (n = 4, P < 0.05) before and 0.32 ± 0.15 Hz (n = 4, P < 0.05) after loading of the probe; similarly the quantal content was 1.06 ± 0.09 (n = 3, P < 0.05) and 0.99 ± 0.07 (n = 3, P < 0.05). We may conclude, therefore adding Ca2+ probe into the cytosol of the terminal does not affect physiological parameters of neurotransmission.
Neuromuscular preparations were continuously perfused with the Ringer solution of the following content (in mM): NaCl–113, KCl–2.5, NaHCO3–3, MgCl2–6, CaCl2–0.9; pH was adjusted to 7.4. Low extracellular Ca2+ (0.9 mM) and high level of external MgCl2 (6 mM) were used to block muscle contraction. All experiments were performed under these conditions except the serie with decreasing extracellular Ca2+. Experiments were performed at 20.0 ± 0.3°C.
Fluorescent signal was recorded using photometric setup on the base of Olympus BX-51 microscope with x60 water-immersion objective connected to photodiode S1087 (Hamamatsu, Japan) as described in Sinha and Saggau (1999) and Samigullin et al. (2010, 2015). The region for recording was selected by optical viewfinder (Till Photonics, Munich, Germany). Excitation light (488 nm) was generated by Polychrome V (Till Photonics, Munich, Germany). To minimize bleaching of the dye and decrease background fluorescence, the recording area of nerve terminal was restricted by an iris diaphragm. Illumination was controlled by a shutter with a typical exposure time of 400 ms and a delivery rate of 0.5 Hz. Motor nerve was electrically stimulated by rectangular voltage pulses of 0.2 ms in duration and supra-threshold amplitude at a frequency of 0.5 Hz using the “suction” electrode described earlier (Kazakov et al., 2015). The photodiode signal was digitized by the ADC Digidata 1440A (Molecular Devices, USA) with sampling rate 10.256 kHz. Fluorescence recordings, illumination and electrical stimulation were all controlled by WinWCP software (Strathclyde University, Glasgow, UK). The peak amplitude of [Ca2+]i transients was measured and changes in fluorescence are represented as ΔF/F0 (the change in fluorescence intensity relative to the background fluorescence as a percentage). For each experiment, about 60 fluorescence responses were averaged.
Electrophysiological recordings
Spontaneous and evoked endplate currents (mEPC and EPC, respectively) were recorded with a two-electrode voltage clamp technique at a holding potential of −60 mV. Intracellular microelectrodes (5–10 MΩ in resistance) were filled with 2.5 M KCl. Currents were recorded using Axoclamp 900A amplifier and digitized by Digidata 1440A (Molecular Devices, USA) under control of Clampex software v. 10.5. Quantum content of EPCs was calculated by dividing the area under EPC curve by the area under mEPC curve.
Chemicals
All reagents were obtained from Sigma (Saint Louis, Missouri, USA). Drugs were diluted in extracellular solution to get the following final concentrations: carbachol (10 μM), acetylcholine (100 μM), neostigmine (1 μM), atropine (1 μM), d-tubocurarine (10 μM), muscarine (10 μM), nicotine (10 μM), pirenzepine (100 nM), methoctramine (10 nM), mecamylamine (640 nM–6.9 μM), metillikakonitin (10 nM), ω-conotoxin GVIA (300 nM).
Data analysis
Experimentally measured relative amplitudes were tested for normal distribution. Statistical significance of relative amplitudes we assessed by Student's t-test for pairwise variant. Then data are presented as mean (%) ± SEM. Values of P < 0.05 were considered significant.
Results
Acetylcholine and carbachol reduce [Ca2+]i transients and quantal content of EPC
Application of 10 μM of carbachol decreased amplitudes of mEPC and EPC by 26 ± 5% (n = 4, P < 0.05) and 61 ± 8% (n = 4, P < 0.05), respectively (Figure 1A). Carbachol also reduced quantal content by 46 ± 10% (n = 4, P < 0.05) as compared to control (Figure 1B). 100 μM ACh attenuated quantal content by 49 ± 7% (n = 18, P < 0.05, Figure 1B). Both carbachol and ACh also attenuated amplitude of [Ca2+]i transients by 11 ± 1% (n = 5, P < 0.05) and 13 ± 4% (n = 6, P < 0.05,) respectively (Figures 1C,D). Since inhibitory effects of exogenous acetylcholine and carbachol were similar, we used carbachol in all subsequent experiments to activate nicotinic and muscarinic receptors simultaneously. Nicotine and muscarine, however, were used to selectively activate respective receptors types. Also, carbachol is not hydrolysed by acetylcholine esterase, and hence its concentration remains constant over the entire experiment duration.
Figure 1.

Acetylcholine and carbachol reduce [Ca2+]i transients and the EPC quantal content. (A) Representative EPCs in control conditions and in the presence of 10 μM carbachol. (B) Quantal content in the presence of 10 μM carbachol or 100 μM ACh. (C) [Ca2+]i transients in control and in the presence of 10 μM carbachol. (D) Amplitude of [Ca2+]i transients in the presence of carbachol and acetylcholine as % of control (control set as100%). Abbreviations: Contr, control; CCh, carbachol; ACh, acetylcholine. *P < 0.05 vs. control saline; n = 4–6.
[Ca2+]i transients and quantal release after changing driving force for Ca2+ entry into nerve terminal
We were interested to elucidate whether a 11% decrease in the peak magnitude of [Ca2+]i transient is sufficient to account for a 46% decline in neurotransmitter release observed with carbachol (Figures 1A–C). For this purpose, we have reduced the external concentration of Ca2+ from 0.6 to 0.3 mM and found that it decreased the evoked [Ca2+]i transient by 23 ± 4% (n = 3) (Figure 2A). In parallel with this, quantal content dropped by 80 ± 4% (Figure 2B). Thus, carbachol-induced decrease in the size of evoked [Ca2+]i transients is more than sufficient to produce inhibition of neurotransmitter release observed in our experiments earlier. Similar observations were made previously while studying quantal content and [Ca2+]i transients under activation of cannabinoid receptors (Newman et al., 2007).
Figure 2.

Role of Ca2+ entry in generation of [Ca2+]i transients and regulation of quantal release. (A) [Ca2+]i transients at two different Ca2+ concentrations in extracellular solution. Amplitude of [Ca2+]i transients at 0.6 mM Ca2+ is set as 100%. (B) Average quantum content of EPC at two different concentrations of extracellular Ca2+. *P < 0.05 vs. control saline; n = 3.
[Ca2+]i transient modulation by muscarine and nicotine
Exposure to 10 μM nicotine reduced the amplitude of [Ca2+]i transient by 15 ± 3% (n = 6, P < 0.05, Figure 3B). Application of 10 μM muscarine decreased the amplitude of [Ca2+]i transient by 8 ± 2% (n = 5, P < 0.05, Figure 3B). Nicotinic antagonist d-tubocurarine (10 μM) partially but significantly reduced nicotine effect on the [Ca2+]i transient, while incubation with muscarinic blocker atropine (1 μM) completely eliminated presynaptic effect of muscarine (Figure 3B). In the presence of atropine (1 μM), carbachol (10 μM) reduced [Ca2+]i transient by 9 ± 5% (n = 6, P < 0.05, Figure 3B), while in the presence of d-tubocurarine (10 μM)—by 18 ± 9% (n = 4, P < 0.05, Figure 3B). Pre-incubation with the mixture of d-tubocurarine (10 μM) and atropine (1 μM), completely eliminated carbachol effects on [Ca2+]i transients (Figures 3A,B).
Figure 3.

Modulation of [Ca2+]i transient by muscarine and nicotine. (A) [Ca2+]i transient in the presence of carbachol (10 μM) after pre-treatment with the mixture of atropine (1 μM) and d-tubocurarine (10 μM). (B) Average amplitudes of [Ca2+]i transient normalized to the control in the presence of nicotine (10 μM); in the presence of nicotine after pre-treatment with d-tubocurarine (10 μM); in the presence of muscarine (10 μM); in the presence of muscarine after pre-treatment with atropine (1 μM), in the presence of carbachol (10 μM) after pre-treatment with d-tubocurarine (10 μM), in the presence of carbachol (10 μM) after pre-treatment with atropine (1 μM) and in the presence of carbachol after pre-treatment with the mixture of atropine (1 μM) and d-tubocurarine (10 μM). Abbreviations: d-TC, d-tubocurarine; Atr, atropine; CCh, carbachol; Nic, nicotine (10 μM); Musc, muscarine (10 μM). *P < 0.05 vs. control saline; n = 4–7.
Nicotinic blocker d-tubocurarine did not change the inhibitory effect of muscarine alone—it reduced [Ca2+]i transients by 8 ± 2% (n = 7, P < 0.05, Figures 4A,B). Similarly, blockade of muscarinic receptors by atropine did not significantly affect the inhibitory action of nicotine –[Ca2+]i transients were reduced by 8 ± 3% (n = 7, P < 0.05, Figures 4C,D).
Figure 4.

Specific effects of activation of muscarinic and nicotinic receptors on [Ca2+]i transients. (A) [Ca2+]i transients in the presence of muscarine (10 μM) after blockade of nicotinic receptors by d-tubocurarine (10 μM). (B) Average amplitude of [Ca2+]i transient in the presence of muscarine (10 μM) after blockade of nicotinic receptors by tubocurarine (10 μM). (C) Amplitude of [Ca2+]i transient in the presence of nicotine (10 μM) after blockade of muscarinic receptors by atropine (1 μM). (D) Average amplitude of [Ca2+]i transient in presence of nicotine when muscarinic receptors are blocked by atropine. Abbreviations: d-TC, d-tubocurarine; Atr, atropine; Nic, nicotine; Musc, muscarine. *P < 0.05; n = 4–6.
Identification of muscarinic receptor subtypes that mediate the effects of cholinomimetics
M1-receptor blocker pirenzepine (100 nM) by itself decreased [Ca2+]i transients by 14 ± 7% (n = 5, P < 0.05, Figures 5A,B). Addition of muscarine further decreased the amplitude of [Ca2+]i transient (Figures 5A,B). Exposure to 10 nM methoctramine (specific blocker of muscarinic M2 receptor subtypes), did not affect [Ca2+]i transients by itself. In the presence of methoctramine, however, the inhibitory action of muscarine on [Ca2+]i transient was completely eliminated (Figures 5C,D).
Figure 5.

Identification of muscarinic receptor subtypes mediating effects of cholinomimetics. (A) [Ca2+]i transients in the presence of muscarine (10 μM) and pirenzepine (100 nM). (B) Mean values of amplitude of [Ca2+]i transients in the presence of muscarine and pirenzepine. (C) [Ca2+]i transients in the presence of methoctramine (10 nM) and muscarine (10 μM). (D) Mean values of amplitude of [Ca2+]i transients in the presence of methoctramine (10 nM) and muscarine (10 μM). Abbreviations: Pirenz, pirenzepine; Musc, muscarine; Methoct, methoctramine. *P < 0.05 vs. control; n = 5.
Identification of nicotinic receptor subtypes that mediate the effects of cholinomimetics
In these experiments nicotine was used as a specific agonist of nicotinic acetylcholine receptors. Mecamylamine is able to block various subtypes of nicotinic receptors depending on concentration within the range from 640 nM to 6.9 μM (Papke et al., 2001; Rabenstein et al., 2006; Ostroumov et al., 2008). Next, we tested the effects of different concentrations of mecamylamine. Application of 640 nM mecamylamine increased [Ca2+]i transient amplitude by 18 ± 3% (n = 5, P < 0.05, Figure 6). When 640 nM mecamylamine was applied with nicotine [Ca2+]i transient amplitude decreased by 9 ± 4% (n = 5, P < 0.05, Figure 6). At 2.5 μM, mecamylamine increased [Ca2+]i transient amplitude by 16 ± 2% (n = 3, P < 0.05, Figure 6), while subsequent addition of nicotine reduced [Ca2+]i transient amplitude by 19 ± 4% (n = 3, P < 0.05, Figure 6). At 3.6 μM mecamylamine increased [Ca2+]i transient by 23 ± 7% (n = 6, P < 0.05, Figure 6), whereas its combined application with nicotine decreased [Ca2+]i transient by 14 ± 2% (n = 6, P < 0.05, Figure 6). Finally, at 6.9 μM, mecamylamine increased [Ca2+]i transient by 15 ± 8% (n = 3, P < 0.05, Figure 6), while combined application with nicotine decreased [Ca2+]i transient amplitude by 8 ± 2% (n = 3, P < 0.05, Figure 6). Metillikakonitin is a specific blocker of the α7-nicotinic receptor subunit. Adding metillikakonitin at 10 nM increased the amplitude of [Ca2+]i transient by 13 ± 11% (n = 5, P < 0.05, Figure 6), while subsequent addition of nicotine decreased [Ca2+]i transients by 11 ± 5% (n = 6, P < 0.05, Figure 6).
Figure 6.
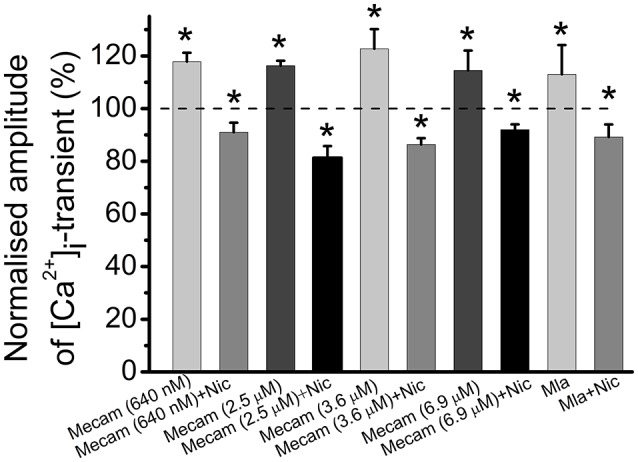
Identification of nicotinic receptor subtypes mediating effects of cholinomimetics. Mean values of magnitude of [Ca2+]i transients in the presence of different concentrations (they are indicated in brackets) of mecamylamine blocking various subtypes of nicotinic receptors and after addition of nicotine (10 μM); in the presence of metillikakonitin (10 nM) and after addition of nicotine (10 μM). Abbreviations: Nic, nicotine; Mecam, mecamylamine. MLA, metillikakonitin. Control corresponds to 100%. *P < 0.05 vs. control saline; n = 3–6.
Voltage-gated Ca2+ channels involved in carbachol modulation of [Ca2+]i dynamics
Since both acetylcholine and carbachol reduce [Ca2+]i transient and quantum content of endplate currents, we suggested that cholinergic modulation of neurotransmitter release could result from attenuated Ca2+ entry into motor nerve endings (Khaziev et al., 2012). In this work, we found that application of specific N-type Ca2+ channel blocker, ω-conotoxin GVIA (300 nM) reduced [Ca2+]i transient by 35 ± 7% (n = 5, P < 0.05, Figures 7A,B). In the presence of ω-conotoxin GVIA carbachol became ineffective (Figures 7C,D), indicating that N-type Ca2+-channels play role in presynaptic modulation observed.
Figure 7.

The role of voltage-gated Ca2+-channels in the effects of carbachol on [Ca2+]i dynamics. (A) [Ca2+]i transients in the presence of specific blocker of N-type Ca2+ channels, ω-conotoxin GVIA (300 nM). (B) Mean values of amplitudes of [Ca2+]i transients normalized to control. (C) Effect of carbachol (10 μM) on [Ca2+]i transients in the presence of ω-conotoxin GVIA. (D) Mean values of amplitudes of [Ca2+]i transients. Abbreviations: Contr, control; ω-CTx GVIA, ω-conotoxin GVIA; CCh, carbachol. *P < 0.05; n = 5.
Action of antagonists of nicotinic and muscarinic acetylcholine receptors on [Ca2+]i dynamics in the absence of exogenous cholinomimetics
Non-specific blocker of all types of nicotinic receptors, d-tubocurarine (10 μM) increased the amplitude of [Ca2+]i transients by 11 ± 3% (n = 15, P < 0.05, Figures 8A,B). Non-specific blocker of muscarinic receptors, atropine (1 μM) also increased [Ca2+]i transient by 9 ± 2% (n = 19, P < 0.05, Figures 8C,D). When applied together, d-tubocurarine and atropine increased the amplitude of [Ca2+]i transients by 7 ± 2% (n = 7, P < 0.05), indicating the absence of additive facilitating effects on [Ca2+]i transient. An increase in Ca2+-response in the presence of nicotinic or muscarinic acetylcholine receptor blockers suggests the presence of some tonic concentration of endogenous acetylcholine in the synaptic cleft. This acetylcholine interacts with nicotinic and muscarinic receptors and causes inhibition of Ca2+-ions entry into nerve terminal.
Figure 8.

Action of antagonists of nicotinic and muscarinic acetylcholine receptors on [Ca2+]i-dynamics in the absence of exogenous cholinomimetics. (A) [Ca2+]i transients in control and in the presence of d-tubocurarine (10 μM); (B) mean values of amplitudes of [Ca2+]i transients in the presence of d-tubocurarine; (C) [Ca2+]i transients in control and in the presence of atropine (1 μM); (D) mean values of amplitudes of [Ca2+]i transients in the presence of atropine. Abbreviations: Contr, control; d-TC, d-tubocurarine; Atr, atropine. *P < 0.05 vs. control; n = 7–15.
Effects of acetylcholinesterase inhibitor neostigmine on [Ca2+]i dynamics
It has been shown earlier that anticholinesterase drugs lead to accumulation of endogenous acetylcholine in synaptic cleft (Loewi and Hellauer, 1938; Fatt and Katz, 1952; Fedorov, 1976). At low frequencies of motor nerve stimulation, acetylcholinesterase inhibitor, neostigmine (1 μM), decreased [Ca2+]i transients by 13 ± 5% (n = 5, P < 0.05, Figures 9A,B). This inhibitory effect of neostigmine was fully reversed in the presence of blockers of nicotinic and muscarinic receptors (Figure 9B). We can therefore suggest that endogenous acetylcholine modulates intra-terminal [Ca2+]i transients by acting on presynaptic nicotinic and muscarinic acetylcholine receptors.
Figure 9.

Effects of acetylcholinesterase inhibitor neostigmine on [Ca2+]i-dynamics. (A) [Ca2+]i transients in control and in the presence of neostigmine (1 μM). (B) Mean values of amplitude of [Ca2+]i transient in the presence of acetylcholine (100 μM); in the presence of neostigmine alone and after pre-treatment by d-tubocurarine (10 μM) and atropine (1 μM). Mean values of [Ca2+]i transient amplitude normalized to control. Effect of exogenous acetylcholine is presented for comparison reasons. Mixture of d-tubocurarine and atropine, completely eliminated neostigmine effects on [Ca2+]i transients. Abbreviations: Contr, control; Neost, neostigmine; ACh, acetylcholine; Atr, atropine (1 μM), d-TC, d-tubocurarine (10 μM). *P < 0.05 vs. control saline; n = 5.
Discussion
Neurotransmitter release from frog nerve endings is modulated by cholinomimetics: by acting through nicotinic receptors, they change kinetics of secretion and its quantum content, while by activation of muscarinic receptors they contribute only to regulation of quantum content (Ciani and Edwards, 1963; Nikolsky et al., 2004). Both quantum content and kinetics of neurotransmitter secretion directly depend on the level of [Ca2+]i in presynaptic terminal (Katz and Miledi, 1965; Nikol'skiĭ et al., 2000; Samigullin et al., 2005). Hence, modulation of Ca2+ entry could be a plausible mechanism accounting for inhibitory effects of cholinomimetics on quantum secretion. To test this hypothesis, we monitored presynaptic [Ca2+]i transients while pharmacologically manipulating acetylcholine receptors and Ca2+ channels. In our experiments the Ca2+ probe was loaded through the nerve stump, as described by Peng and Zucker (1993) and Wu and Betz (1996). At the end of loading protocol, all terminals in the proximal part of the nerve trunk had sufficient levels of fluorescence to allow [Ca2+]i recordings. We (in this paper), as well as others have shown that the loading of the fluorescent dye in the nerve ending does not significantly alter the physiological parameters of secretion, such as quantal content and frequency of mEPC (Wu and Betz, 1996; Samigullin et al., 2015).
We found that carbachol and acetylcholine decreased both Ca2+ response and quantum content of EPCs, indicating that cholinergic modulation of neurotransmitter release can be related to changes in Ca2+ entry (Khaziev et al., 2012). We further showed that a relatively small decrease in the driving force for Ca2+ influx into nerve terminal causes significant inhibition of neurotransmitter release. This was achieved by lowering extracellular Ca2+ concentration from 0.6 to 0.3 mM (Figures 1, 2). This observation is in a good agreement with strong non-linear dependence of quantum content from Ca2+ entry into the presynaptic terminal (Dodge and Rahamimoff, 1967).
Inhibition of synaptic acetylcholinesterase by neostigmine decreased [Ca2+]i transients, indicating that endogenous acetylcholine is able to modulate Ca2+ entry into the nerve ending. Exposure to d-tubocurarine and specific antagonists of nicotinic receptors (in the absence of exogenous cholinomimetics) also augmented [Ca2+]i transient, thus indicating that tonically released ACh can block Ca2+ entry into the presynaptic terminal. Increase of the amplitude of [Ca2+]i transients by atropine is in a qualitative agreement with atropine-induced increase in the secretion at frog synapses due to elimination of tonic action of endogenous acetylcholine as described earlier (Slutsky et al., 1999). We suggest that release of endogenous ACh during stimulation of the motor nerve and spontaneous quantal and non-quantal release under physiological conditions results in modulation of presynaptic Ca2+ entry and provides a physiologically important negative feedback (Katz and Miledi, 1965; Vyskocil et al., 2009). Presynaptic acetylcholine receptors of both muscarinic and nicotinic nature are involved in this regulation. Since blockade of M1-muscarinic receptor by pirenzepine reduced of [Ca2+]i transient, we may presume that activation of these receptors could enhance Ca2+ entry. These results are in good agreement with previous observations indicating that activation of M1 receptors enhances, whereas activation of M2 receptors inhibits ACh release (Slutsky et al., 1999). According to our results M2 subtype is involved in modulation of presynaptic Ca2+ entry, because specific M2 blocker methoctramine eliminates inhibitory effect of muscarine, whereas M1 antagonist pirenzepine has no such effect.
Mecamylamine and metillikakonitin did not prevent the inhibitory effect of nicotine while d-tubocurarine did. This indicates that some d-tubocurarine-sensitive nicotinic receptors are involved in the presynaptic inhibitory action of endogenous acetylcholine, and inhibition of this receptor in the absence of exogenous cholinomimetics enhances [Ca2+]i transients. Specific N-type Ca2+-channel antagonist conotoxin GVIA removes inhibition of [Ca2+]i transients by carbachol, pointing to a primary role of N-type channels in providing Ca2+ entry into the nerve terminal. Furthermore, our data about the role for N-type of Ca2+ channels are fully compatible with earlier results of Van der Kloot et al. (1997) who demonstrated that ω-conotoxin eliminated effects of cholinomimetics on quantal content in frog synapses. We conclude that in the frog neuromuscular synapse activation of presynaptic M2 muscarinic and d-tubocurarine-sensitive nicotinic acetylcholine receptors by exogenous cholinomimetics and/or endogenous acetylcholine decreases quantal content of mediator secretion by reducing the entry of Ca2+ ions into the nerve endings due to inhibition of voltage-gated N-type Ca2+ channels.
Author contributions
EK, DS, NZ, NF, EB, and EN contributed to the study design and acquirement of ethical approval. EK, DS, NZ, NF, and EB contributed to data collection. DS, EN, and AV analyzed the data, interpreted the data, and drafted the initial manuscript. The remaining authors critically revised the manuscript. All authors approved the final version of the manuscript. DS, EB, and EN are guarantors of the manuscript and take full responsibility for the work as a whole, including the study design, access to data, and the decision to submit and publish the manuscript.
Funding
This research has been performed with the support of the Russian Government's Program for Competitive Growth of Kazan Federal University, grant of Program of Presidium RAS 19P and the grants from the Russian Foundation for Basic Research (16-04-01051; 16-34-00817; 15-04-02983).
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Dr. Victor Ilyin for many helpful comments, questions and help in writing of the manuscript.
Glossary
Abbreviations
- Ach
Acetylcholine
- EPC
evoked postsynaptic currents
- mEPC
spontaneous postsynaptic currents.
References
- Arenson M. S. (1989). Muscarinic inhibition of quantal transmitter release from the magnesium-paralysed frog sartorius muscle. Neuroscience 30, 827–836. 10.1016/0306-4522(89)90174-7 [DOI] [PubMed] [Google Scholar]
- Bowman W. C., Prior I., Marshall G. (1990). Presynaptic receptors in the neuromuscular junction. Ann. N.Y. Acad. Sci. 604, 69–81. 10.1111/j.1749-6632.1990.tb31983.x [DOI] [PubMed] [Google Scholar]
- Ciani S., Edwards C. (1963). The effect of acetylcholine on neuromuscular transmission in the frog. J. Pharmacol. Exp. Ther. 142, 21–23. [PubMed] [Google Scholar]
- Dodge F. A., Jr., Rahamimoff R. (1967). Co-operative action a calcium ions in transmitter release at the neuromuscular junction. J. Physiol. 193, 419–432. 10.1113/jphysiol.1967.sp008367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatt P., Katz B. (1952). Spontaneous subthreshold activity at motor nerve endings. J. Physiol. 117, 109–128. [PMC free article] [PubMed] [Google Scholar]
- Fedorov V. V. (1976). Effect of cholinesterase inhibitors on synaptic potentials of the frog neuromuscular junction. Neurosci. Behav. Physiol. 7, 201–206. 10.1007/BF01186053 [DOI] [PubMed] [Google Scholar]
- Katz B., Miledi R. (1965). The effect of calcium on acetylcholine release from motor nerve terminals. Proc. R. Soc. Lond. B Biol. Sci. 161, 496–503. 10.1098/rspb.1965.0017 [DOI] [PubMed] [Google Scholar]
- Kazakov A., Aleksandrov M., Zhilyakov N., Khaziev E., Samigullin D. (2015). A simple suction electrode for electrical stimulation of biological objects. Int. Res. J. 40, 13–16. 10.18454/IRJ.2227-6017 [DOI] [Google Scholar]
- Khaziev E. F., Fatikhov N. F., Samigullin D. V., Barrett G. L., Bukharaeva E. A., Nikolsky E. E. (2012). Decreased entry of calcium into motor nerve endings upon activation of presynapticc cholinergic receptors. Dokl. Biol. Sci. 446, 283–285. 10.1134/S0012496612050080 [DOI] [PubMed] [Google Scholar]
- Kupchik Y. M., Rashkovan G., Ohana L., Keren-Raifman T., Dascal N., Parnas H., et al. (2008). Molecular mechanisms that control initiation and termination of physiological depolarization-evoked transmitter release. Proc. Natl. Acad. Sci. U.S.A. 105, 4435–4440. 10.1073/pnas.0708540105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linial M., Ilouz N., Parnas H. (1997). Voltage-dependent interaction between the muscarinic ACh receptor and proteins of the exocytic machinery. J. Physiol. 504, 251–258. 10.1111/j.1469-7793.1997.251be.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewi O., Hellauer H. (1938). Uber das Acetylcholin in peripheren Nerven. Pflug. Arch. ges. Physiol. 240, 769–775. 10.1007/BF01759980 [DOI] [Google Scholar]
- Macleod G. T., Lavidis N. A., Bennett M. R. (1994). Calcium dependence of quantal secretion from visualized sympathetic nerve varicosities on the mouse vasdeferens. J. Physiol. 480, 61–70. 10.1113/jphysiol.1994.sp020340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller R. J. (1990). Receptor-mediated regulation of calcium channels and neurotransmitter release. FASEB J. 4, 3291–3299. [PubMed] [Google Scholar]
- Newman Z., Malik P., Wu T.-W., Ochoa C., Watsa N., Lindgren C. (2007). Endocannabinoids mediate muscarine-induced synaptic depression at the vertebrate neuromuscular junction. Eur. J. Neurosci. 25, 1619–1630. 10.1111/j.1460-9568.2007.05422.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikol'skiĭ E. E., Bukharaeva E. A., Samigullin D. V., Gaŭnulov R. K. (2000). The time course of the evoked secretion of the mediator quanta in various regions of the frog motor nerve ending. Ross. Fiziol. Zh. Im. I M Sechenova 86, 1195–1209. [PubMed] [Google Scholar]
- Nikol'skiĭ E. E., Giniatullin R. A. (1979). Termination of the presynaptic effect of carbacholine by tubocurarine. Biull. Eksp. Biol. Med. 87, 171–174. [PubMed] [Google Scholar]
- Nikolsky E. E., Vyskocil F., Bukharaeva E. A., Samigullin D. V., Magazanik L. G. (2004). Cholinergic regulation of the evoked quantal release at frog neuromuscular junction. J. Physiol. 560, 77–88. 10.1113/jphysiol.2004.065805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostroumov K., Shaikhutdinova A., Skorinkin A. (2008). Modeling study of mecamylamine block of muscle type acetylcholine receptors. Eur. Biophys. J. 37, 393–402. 10.1007/s00249-007-0224-5 [DOI] [PubMed] [Google Scholar]
- Papke R. L., Horenstein B. A., Placzek A. N. (2001). Inhibition of wild-type and mutant neuronal nicotinic acetylcholine receptors by local anesthetics. Mol. Pharmacol. 60, 1365–1374. 10.1124/mol.60.6.1365 [DOI] [PubMed] [Google Scholar]
- Parnas H., Segel L., Dudel J., Parnas I. (2000). Autoreceptors, membrane potential and regulation of transmitter release. Trends Neurosci. 23, 60–68. 10.1016/S0166-2236(99)01498-8 [DOI] [PubMed] [Google Scholar]
- Peng Y. Y., Zucker R. S. (1993). Release of LHRH is linearly related to the time integral of presynaptic Ca2+ elevation above a threshold level in bullfrog sympathetic ganglia. Neuron 10, 465–473. 10.1016/0896-6273(93)90334-N [DOI] [PubMed] [Google Scholar]
- Rabenstein R. L., Caldarone B. J., Picciotto M. R. (2006). The nicotinic antagonist mecamylamine has antidepressant-like effects in wild-type but not β2- or α7-nicotinic acetylcholine receptor subunit knockout mice. Psychopharmacology 189, 395–401 10.1007/s00213-006-0568-z [DOI] [PubMed] [Google Scholar]
- Samigullin D., Fatikhov N., Khaziev E., Skorinkin A., Nikolsky E., Bukharaeva E. (2015). Estimation of presynaptic calcium currents and endogenous calcium buffers at the frog neuromuscular junction with two different calcium fluorescent dyes. Front. Synaptic Neurosci. 6:29. 10.3389/fnsyn.2014.00029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samigullin D. V., Bukharaeva E. A., Vyskocil F., Nikolsky E. E. (2005). Calcium dependence of uni-quantal release latencies and quantal content at mouse neuromuscular junction. Physiol. Res. 54, 129–132. [DOI] [PubMed] [Google Scholar]
- Samigullin D. V., Khaziev E. F., Kovyazina I. V., Bukharaeva E. A., Nikolsky E. E. (2014). Muscarinic regulation of calcium transient and synaptic transmission in frog neuromuscular junction. Genes Cells 9, 242–247. [Google Scholar]
- Samigullin D. V., Vasin A. L., Bukharaeva E. A., Nikolsky E. E. (2010). Characteristics of calcium transient in different parts of frog nerve terminal in response to nerve impulse. Dokl. Biol. Sci. 431, 83–85. 10.1134/S0012496610020043 [DOI] [PubMed] [Google Scholar]
- Shen J. X., Yakel J. L. (2009). Nicotinic acetylcholine receptor-mediated calcium signaling in the nervous system. Acta Pharmacol. Sin. 6, 673–680. 10.1038/aps.2009.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S. R., Saggau P. (1999). Optical recording from populations of neurons in brain slices. Mod. Tech. Neurosci. Res. 16, 459–486. 10.1007/978-3-642-58552-4_16 [DOI] [Google Scholar]
- Slutsky I., Parnas H., Parnas I. (1999). Presynaptic effects of muscarine on ACh release at the frog neuromuscular junction. J. Physiol. 514, 769–782. 10.1111/j.1469-7793.1999.769ad.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S., Osanai M., Murase M., Suzuki N., Ito K., Shirasaki T. K., et al. (2000). Ca2+ dynamics at the frog motor nerve terminal. Pflug. Arch. Eur. J. Phisiol. 440, 351–365. 10.1007/s004240000278 [DOI] [PubMed] [Google Scholar]
- Tomas J., Santafe M. M., Garcia N., Lanuza M. A., Tomas M., Besalduch N., et al. (2014). Presynaptic membrane receptors in acetylcholine release modulation in the neuromuscular snynapse. Spain J. Neurosci. Res. 92, 543–554. 10.1002/jnr.23346 [DOI] [PubMed] [Google Scholar]
- Tsang C. W., Elrick D. B., Charlton M. P. (2000). α-Latrotoxin releases calcium in frog motor nerve terminals. J. Neurosci. 20, 8685–8692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien R. Y. (1989). Fluorescent indicators of ion concentrations. Methods Cell Biol. 30, 127–156. 10.1016/S0091-679X(08)60978-4 [DOI] [PubMed] [Google Scholar]
- Van der Kloot W., Ligia M., Naves A. (1997). Cholinergic agonists decrease quantal output at the frog neuromuscular junction by targeting a calcium channel blocked by w-conotoxin. Pflugers Arch. 434, 735–741. [DOI] [PubMed] [Google Scholar]
- Vyskocil F., Malomouzh A. I., Nikolsky E. E. (2009). Non-quantal acetylcholine release at the neuromuscular junction. Physiol. Res. 58, 763–784. [DOI] [PubMed] [Google Scholar]
- Wessler I. (1989). Control of transmitter release from the motor nerve by presynaptic nicotinic and muscarinic autoreceptors. Trends Pharmacol. Sci. 10, 110–114. 10.1016/0165-6147(89)90208-3 [DOI] [PubMed] [Google Scholar]
- Wu L. G., Betz W. J. (1996). Nerve activity but not intracellular calcium determines the time course of endocytosis at the frog neuromuscular junction. Neuron 17, 769–779. 10.1016/S0896-6273(00)80208-1 [DOI] [PubMed] [Google Scholar]
- Wu L. G., Saggau P. (1997). Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci. 20, 204–212. 10.1016/S0166-2236(96)01015-6 [DOI] [PubMed] [Google Scholar]