

Abstract
G protein-coupled receptor kinases (GRKs) represent a class of proteins that classically phosphorylate agonist-activated G protein-coupled receptors, leading to uncoupling of the receptor from further G protein activation. Recently, we have reported that the heterotrimeric G protein α-subunit, Gαq/11, can mediate insulin-stimulated glucose transport. GRK2 contains a regulator of G protein signaling (RGS) domain with specificity for Gαq/11. Therefore, we postulated that GRK2 could be an inhibitor of the insulin signaling cascade leading to glucose transport in 3T3-L1 adipocytes. In this study, we demonstrate that microinjection of anti-GRK2 antibody or siRNA against GRK2 increased insulin-stimulated insulin-responsive glucose transporter 4 (GLUT4) translocation, while adenovirus-mediated overexpression of wild-type or kinase-deficient GRK2 inhibited insulin-stimulated GLUT4 translocation as well as 2-deoxyglucose uptake. Importantly, a mutant GRK2 lacking the RGS domain was without effect. Taken together, these results indicate that through its RGS domain endogenous GRK2 functions as a negative regulator of insulin-stimulated glucose transport by interfering with Gαq/11 signaling to GLUT4 translocation. Furthermore, inhibitors of GRK2 can lead to enhanced insulin sensitivity.
Keywords: Gαq/11, GLUT4 translocation, GRK2, insulin signal transduction
Introduction
G protein-coupled receptor kinases (GRKs) play a central role in desensitizing G protein-coupled receptors (GPCRs) (Lefkowitz, 1998; Pitcher et al, 1998). GRKs phosphorylate specific C-terminal serine residues of agonist-activated GPCRs, leading to increased binding of β-arrestin (Kohout and Lefkowitz, 2003). GRK-induced phosphorylation of the GPCR, with subsequent β-arrestin association, uncouples the receptors from further G protein association, and also promotes internalization of the GPCR (Lefkowitz, 1998; Pitcher et al, 1998). Recently, it has been shown that GRKs have additional functions to regulate GPCR signaling. Thus, GRKs can contain regulator of G protein signaling (RGS) domains, which mediate binding to Gα subunits, inhibiting G protein function (Siderovski et al, 1996; Freedman et al, 1997; Carman et al, 1999; Dicker et al, 1999; Sallese et al, 2000; Usui et al, 2000). Importantly, the RGS domains of GRKs have substrate selectivity, and Sallese et al (2000) and Carman et al (1999) have reported that GRK2, but not the other subtypes of GRKs, specifically inhibits the activity of Gαq/11, but not Gαi or Gαs.
Recent studies have shown that certain signaling proteins, which classically function in GPCR signaling pathways, can also participate in receptor tyrosine kinase (RTK) signaling cascades. For example, IGF-1-mediated MAP kinase phosphorylation is dependent on Gαi/βγ signaling (Luttrell et al, 1995; Dalle et al, 2001). In addition, heptahelical GPCRs and RTKs can both utilize the scaffold/adaptor protein β-arrestin (Maudsley et al, 2000; Pierce et al, 2000, 2002). Thus, Lefkowitz et al have shown that β-arrestin-1 is required for IGF-1-mediated MAP kinase signaling (Lin et al, 1998; Luttrell et al, 2001; Pierce et al, 2001), and we have shown that insulin treatment, which causes downregulation of β-arrestin-1, can impair MAP kinase signaling by both GPCRs and RTKs (Dalle et al, 2002; Hupfeld et al, 2003). These data suggest that there are a number of parallels between GPCR and RTK action, involving common intermediate signaling proteins. Accordingly, we have hypothesized that GRKs are also involved in the regulation of RTK signaling. For example, insulin promotes glucose transport by stimulating translocation of insulin-responsive glucose transporter 4 (GLUT4) proteins to the cell surface (Robinson et al, 1992), and we have recently reported that the activated insulin receptor can phosphorylate the heterotrimeric protein component Gαq/11, leading to activation of cdc42, and phosphatidylinositol 3-kinase (PI3-kinase) with downstream glucose transport stimulation (Imamura et al, 1999b; Usui et al, 2003). Therefore, since GRK2 has Gαq/11 specificity, we postulated that GRK2, by inhibiting Gαq/11 function, would be an endogenous negative regulator of insulin-stimulated glucose transport.
In the current study, we investigated the role of GRK2 in insulin-induced glucose transport in 3T3-L1 adipocytes. We show that GRK2 is an endogenous protein inhibitor of insulin-induced glucose transport, and that inhibition of GRK2 can lead to insulin sensitization.
Results
Insulin-induced GLUT4 translocation is increased by microinjection of GRK2 antibody or siRNA, and decreased by overexpression of GRK2
Using immunofluorescent staining to detect GLUT4 at the plasma membrane as previously described (Usui et al, 2003), we measured the effects of GRK2 inhibition on insulin-stimulated GLUT4 translocation. In the basal state, most cells displayed GLUT4 staining in the perinuclear region, while insulin treatment led to appearance of GLUT4 at the plasma membrane, as previously shown (Imamura et al, 2003). Microinjection of GRK2 antibody into 3T3-L1 adipocytes did not alter basal GLUT4 staining, but led to a 65 and 47% increase in GLUT4 translocation when cells were stimulated with 0.02 or 0.2 nM insulin, respectively, with no significant effect at 1.7 nM insulin concentration (Figure 1A). The specificity of the GRK2 antibody was confirmed by Western blotting (data not shown), and microinjection of antibodies against GRK5 or GRK6 had no effect on GLUT4 localization. Thus, inhibition of endogenous GRK2 by antibody microinjection led to increased insulin sensitivity for stimulation of GLUT4 translocation.
Figure 1.
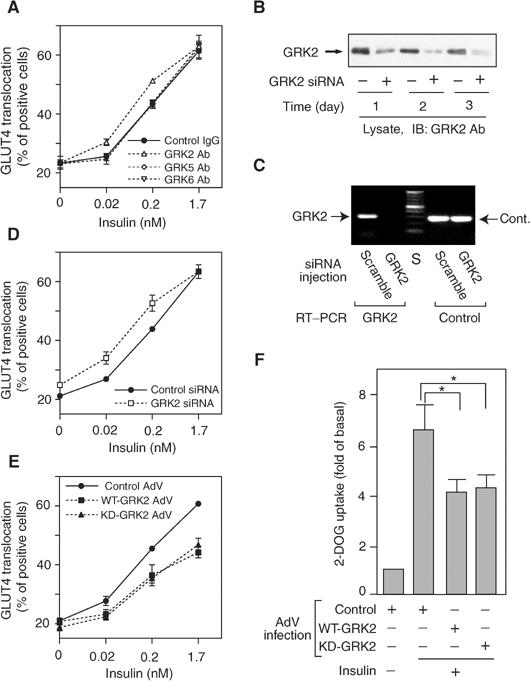
Effects of GRK2 on insulin-stimulated GLUT4 translocation and 2-DOG uptake in 3T3-L1 adipocytes. (A, D, E) For microinjection assay, 3T3-L1 adipocytes on coverslips were serum starved for 4 h, and anti-GRK2 antibody, anti-GRK5 antibody, anti-GRK6 antibody, sheep IgG, GRK2 siRNA, or control siRNA was microinjected. For GRK2 overexpression assay, 3T3-L1 adipocytes on coverslips were infected with adenovirus expressing WT-GRK2, KD-GRK2, or control LacZ. At 48 h after infection, these cells were serum starved for 4 h. Cells were stimulated with 0.02, 0.2, or 1.7 nM insulin for 20 min. GLUT4 was stained as described in Materials and methods. The percentage of cells positive for GLUT4 translocation was calculated by counting at least 100 cells at each point. The data are the mean±s.e. from three independent experiments. (B) SiRNA of GRK2 (+) or control siRNA (−) was transfected in 3T3-L1 preadipocytes using Oligofectamine as described in Materials and methods. At the indicated days after transfection, total cell lysates were prepared and analyzed by Western blotting using anti-GRK2 antibody as described in Materials and methods. Representative blots are shown from two independent experiments. (C) The efficiency of siRNA under the microinjection into 3T3-L1 adipocytes was confirmed by the mRNA quantification as described in Materials and methods. All of 3T3-L1 adipocytes on coverslips (approximately 200 cells spotted on each coverslip) were microinjected with GRK2 or scrambled control (scramble) siRNA. At 48 h after microinjection, total RNA was purified and used for RT–PCR reaction with the GRK2 or PP2A (Cont.) primer set. A representative image from two independent experiments is shown. S: size marker. (F) 3T3-L1 adipocytes were infected with adenovirus expressing WT-GRK2, KD-GRK2, or control LacZ (Control). At 48 h after infection, these cells were serum starved for 3 h, stimulated with17 nM insulin for 30 min, and [3H]2-DOG uptake was measured as described in Materials and methods. The data are the mean±s.e. from three independent experiments. Statistically significant differences versus control are indicated (*P<0.05).
To further support the results with the GRK2 antibody, we also utilized siRNA against GRK2. The efficiency of this siRNA at silencing GRK2 protein was demonstrated by Western blotting of cell lysates from 3T3-L1 preadipocytes that had been transfected with GRK2 siRNA. As shown in Figure 1B, 1–3 days post-transfection, GRK2 protein was decreased by 64, 79, and 86%, respectively. Since transfection efficiency of this chemical reagent is not 100% (∼85%), it is probable that almost complete inhibition occurred in the cells transfected with the siRNA. When RNA is prepared from coverslips in which all of the cells are microinjected (∼200 cells/coverslip), the resulting RT–PCR data show that the microinjected GRK2 siRNA ‘knock down' the GRK2 mRNA to undetectable levels (Figure 1C). With the microinjection approach, only cells that contain this siRNA are assessed for GLUT4 translocation. As shown in Figure 1D, insulin-stimulated GLUT4 translocation was increased by 55% at 0.02 nM insulin and by 48% at 0.2 nM insulin in GRK2 siRNA-injected cells.
We also examined the effect of adenoviral vectors encoding wild-type (WT) and kinase-deficient (KD) (K220R) GRK2 on GLUT4 translocation. Adenoviral-mediated expression of either WT- or KD-GRK2 in 3T3-L1 adipocytes decreased insulin-stimulated GLUT4 translocation (Figure 1E). The effects on 2-deoxyglucose (2-DOG) uptake were also determined (Figure 1F), and WT- and KD-GRK2 inhibited insulin-stimulated 2-DOG uptake by 46 and 44%, respectively. Interestingly, the inhibitory effects of WT- and KD-GRK2 expression on GLUT4 translocation and 2-DOG uptake were quite comparable. Taken together, these results suggest that GRK2 is an endogenous inhibitor of insulin-induced GLUT4 translocation and glucose transport and that this inhibitory function does not involve the kinase activity of GRK2.
GRK2 does not affect insulin receptor tyrosine phosphorylation or activation of the IRS-1 pathway
To assess the mechanisms of GRK2-mediated inhibition of insulin-induced GLUT4 translocation and 2-DOG uptake, we examined the role of GRK2 in insulin signaling. As shown in Figure 2A, adenoviral expression of WT- or KD-GRK2 did not alter the expression level or tyrosine phosphorylation state of the insulin receptor. Additionally, the insulin receptor did not co-precipitate using anti-GRK2 antibody, either before or after insulin stimulation (data not shown). Expression of WT- or KD-GRK2 did not affect the expression level or insulin-induced tyrosine phosphorylation of insulin receptor substrate-1 (IRS-1), the association of IRS-1 and p85, or IRS-1-associated PI3-kinase activity (Figure 2B and C). Thus, the effects of GRK2 inhibition on insulin-stimulated glucose transport are independent of, or downstream of, the insulin receptor, IRS-1, and IRS-1-associated PI3-kinase.
Figure 2.
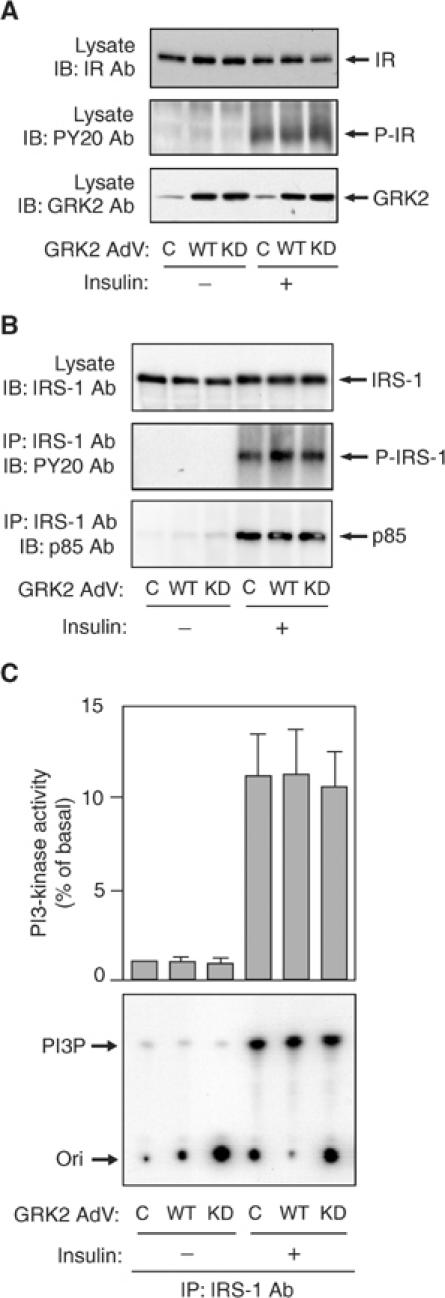
Effect of WT- or KD-GRK2 overexpression on insulin receptor and IRS-1-PI3-kinase pathway in 3T3-L1 adipocytes. 3T3-L1 adipocytes were infected with adenovirus expressing WT-GRK2 (WT), KD-GRK2 (KD), or control LacZ (C). (A) At 48 h after infection, these cells were serum starved for 16 h, stimulated with 17 nM insulin for 5 min and lysed. Total cell lysates were analyzed by Western blotting using anti-insulin receptor (IR), anti-phosphotyrosine (PY20), or anti-GRK2 antibody as described in Materials and methods. Representative blots are shown from three independent experiments. (B) At 48 h after infection, these cells were serum starved for 16 h, stimulated with 17 nM insulin for 5 min (for IRS-1 and PY20 blots) or 10 min (for p85 blot) and lysed. Samples were immunoprecipitated with anti-IRS-1 antibody. Immunoprecipitates or total cell lysates were analyzed by Western blotting using anti-IRS-1, anti-phosphotyrosine (PY20), or anti-p85 antibody as described in Materials and methods. Representative blots are shown from three independent experiments. (C) At 48 h after infection, these cells were serum starved for 16 h, stimulated with 17 nM insulin for 10 min and lysed. Samples were immunoprecipitated with anti-IRS-1 antibody. PI3-kinase activity was measured as described in Materials and methods. A representative film is shown and the graph represents the mean±s.e. from three independent experiments.
GRK2 directly binds to Gαq/11 and inhibits insulin-induced activation of the Gαq/11 pathway
Since we have previously shown that GLUT4 translocation involves insulin stimulation of Gαq/11, and since GRK2 is selective for Gαq/11 (Carman et al, 1999; Sallese et al, 2000), we examined the association of endogenous GRK2 and Gαq/11 before and after insulin stimulation. Association of endogenous GRK2 and Gαq/11 was low in the basal state and was markedly enhanced by insulin with a maximal response by 5 min (Figure 3A). We next assessed the role of GRK2 in the Gαq/11 signaling pathway by using adenoviral-mediated expression of WT- or KD-GRK2 (Figure 3B and C). As we recently reported (Imamura et al, 1999b; Usui et al, 2003), insulin treatment causes increased tyrosine phosphorylation of Gαq/11, cdc42 activation, association of cdc42 and p85, and enhanced cdc42-associated PI3-kinase activity. Interestingly, overexpression of either WT- or KD-GRK2 inhibited insulin-induced Gαq/11 tyrosine phosphorylation (Figure 3B, second row). This result suggests that while insulin leads to Gαq/11 tyrosine phosphorylation, the Gαq/11, which associates with GRK2, cannot be phosphorylated. Similarly, events downstream of Gαq/11 signaling, including cdc42 activation (as measured by precipitation with PAK1-CBD beads; Figure 3B, third row), p85 co-precipitation with cdc42 antibody (Figure 3B, fourth row), and insulin-induced, cdc42-associated PI3-kinase activity (Figure 3C), were also inhibited. The expression level of Gαq/11, cdc42, or p85 was not altered in adenoviral WT-GRK2- or KD-GRK2-transfected cells. Together, these results indicate that decreased activation of the insulin-stimulated Gαq/11 pathway can, at least in part, explain the inhibition of insulin-stimulated glucose transport by GRK2, and that this inhibitory mechanism is independent of GRK2 kinase activity.
Figure 3.
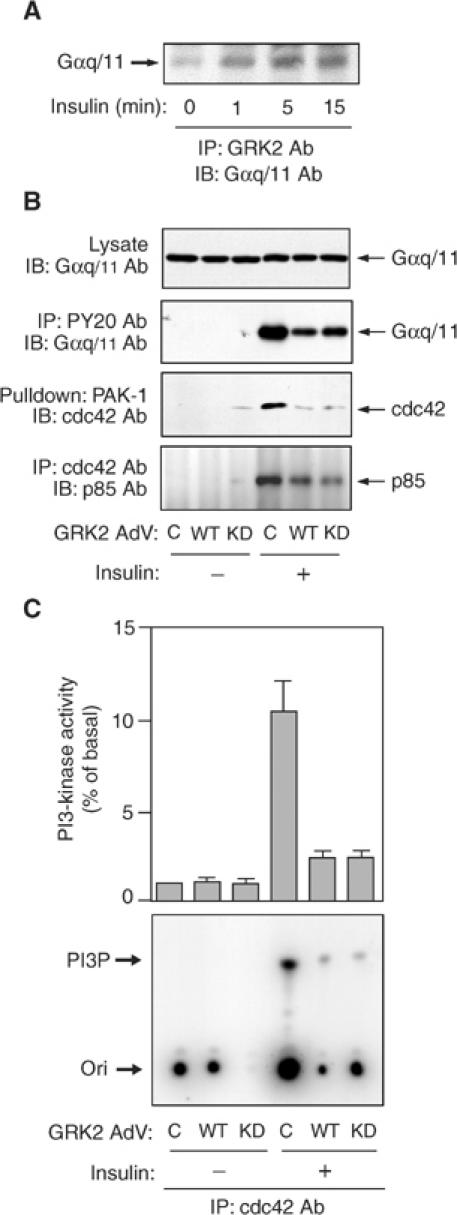
Effect of WT- or KD-GRK2 overexpression on Gαq/11-cdc42-PI3-kinase pathway in 3T3-L1 adipocytes. (A) 3T3-L1 adipocytes were serum starved for 16 h, stimulated with 17 nM insulin for the indicated time periods and lysed. Samples were immunoprecipitated with anti-GRK2 antibody. Immunoprecipitates were analyzed by Western blotting using anti-Gαq/11 antibody as described in Materials and methods. A representative blot is shown from three independent experiments. (B) 3T3-L1 adipocytes were infected with adenovirus expressing WT-GRK2 (WT), KD-GRK2 (KD), or control LacZ (C). At 48 h after infection, these cells were serum starved for 16 h, stimulated with 17 nM insulin for 1 min (for Gαq/11 blot) or 10 min (for p85 blot) and lysed. Samples were immunoprecipitated with anti-phosphotyrosine (PY20) or cdc42 antibody. Immunoprecipitates and total cell lysates were analyzed by Western blotting using anti-Gαq/11 or anti-p85 antibody as described in Materials and methods. Cdc42 activity was measured as described in Materials and methods. The cells were stimulated with insulin for 1 min. Samples were pulled down with GST-PAK-1 and were analyzed by Western blotting using anti-cdc42 antibody. Representative blots are shown from three independent experiments. (C) 3T3-L1 adipocytes were infected with adenovirus expressing WT-GRK2 (WT), KD-GRK2 (KD), or control LacZ (C). At 48 h after infection, these cells were serum starved for 16 h, stimulated with 17 nM insulin for 10 min and lysed. Samples were immunoprecipitated with anti-cdc42 antibody. PI3-kinase activity was measured as described in Materials and methods. A representative film is shown and the graph represents the mean±s.e. from three independent experiments.
Structural determinants of GRK function on insulin signaling
Recent studies have reported that the amino (N′)-termini of GRKs contain sequence similarities to RGS domains of typical RGS proteins (Siderovski et al, 1996), and GRK2 can specifically recognize Gαq/11 (Carman et al, 1999; Sallese et al, 2000; Usui et al, 2000). Thus, we assessed the role of the RGS domain of GRK2 in insulin-stimulated GLUT4 translocation by constructing a GRK2 vector that lacks the RGS domain (delta-GRK2). Western blot analysis using anti-GRK2 antibody revealed that delta-GRK2, as well as WT- and KD-GRK2, were comparably and well expressed after transfection of these vectors into HIRc-B cells (Figure 4A). The effect of WT-, KD-, or delta-GRK2 on GLUT4 translocation was compared by microinjecting these vectors directly into the nuclei of 3T3-L1 adipocytes (Figure 4B). Consistent with the effects of adenovirus infection on GLUT4 translocation and 2-DOG uptake (Figure 1E and F), overexpression of either WT- or KD-GRK2 by nuclear microinjection inhibited insulin-induced GLUT4 translocation. In contrast, overexpression of delta-GRK2 had no effect. As a negative control, we also injected a vector encoding WT-ERK1, which had no effect on GLUT4 translocation. This result suggests that the RGS domain of GRK2 is necessary for the inhibitory function of GRK2 on insulin-stimulated GLUT4 translocation, which is most likely a result of RGS domain binding to Gαq/11.
Figure 4.
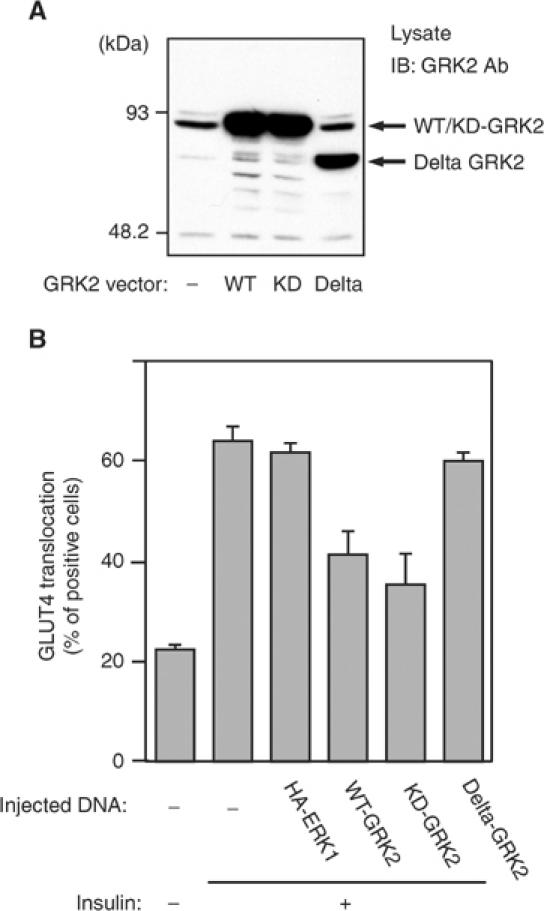
Effects of the RGS domain of GRK2 on insulin-stimulated GLUT4 translocation in 3T3-L1 adipocytes. (A) Plasmid expression vectors of WT-, KD-, or deletion mutant (Delta) GRK2 were transfected in HIRc-B cells using SuperFECT as described in Materials and methods. At 48 h after transfection, these cells were lysed and total cell lysates were analyzed by Western blotting using anti-GRK2 antibody as described in Materials and methods. These experiments were performed three times, and a representative result is shown. (B) Plasmid expression vectors of WT-GRK2, KD-GRK2, delta-GRK2, or control ERK1 (HA-ERK1) were microinjected into the nuclei of 3T3-L1 adipocytes on coverslips. At 24 h after microinjection, 3T3-L1 adipocytes were serum starved for 4 h and were stimulated with 1.7 nM insulin for 20 min. GLUT4 was stained as described in Materials and methods. The cells expressing exogenous GRKs or ERK1 were detected by staining with anti-6X-His antibody or anti-HA antibody, respectively. The percentage of cells positive for GLUT4 translocation was calculated by counting at least 100 cells at each point. The data are the mean±s.e. from three independent experiments.
To further extend these findings, we conducted experiments with constitutively active (Q209L) Gαq. As reported previously (Imamura et al, 1999b), expression of constitutively active Gαq (Q209L) leads to stimulation of glucose transport, as shown in Figure 5. However, when adenoviruses containing GRK2-WT and Q209L-Gαq were simultaneously used to infect 3T3-L1 adipocytes, the expression of GRK2 inhibited the effect of Q209L-Gαq on glucose transport. We have shown that cdc42 lies downstream of Gαq/11 in the insulin-induced glucose transport stimulatory pathway, and that constitutively active (CA) cdc42 can stimulate glucose transport when expressed in 3T3-L1 adipocytes (Usui et al, 2003). Figure 5 shows that coexpression of GRK2 does not inhibit the stimulating effects of CA-cdc42. These results indicate that the ability of the GRK2 RGS domain to bind to Gαq underlies the mechanism of the inhibitory effect. Most likely, GRK2 binding to Gαq/11 affects a critical subcellular localization step, or prevents Gαq/11 from interacting with downstream effectors.
Figure 5.
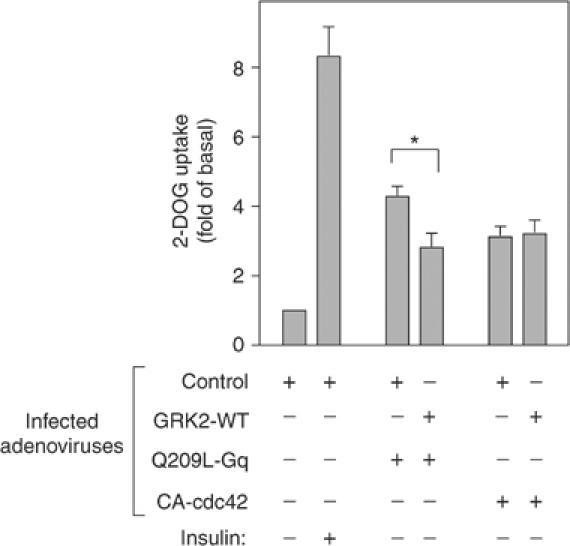
Effects of GRK2 on Q209L-Gq-induced 2-DOG uptake in 3T3-L1 adipocytes. 3T3-L1 adipocytes were co-infected with adenovirus expressing WT-GRK2 (GRK2-WT), constitutively active Gq (Q209L-Gq), constitutively active cdc42 (CA-cdc42), and/or control LacZ (Control). Total number of MOI was adjusted to 80 in each condition. At 48 h after infection, these cells were serum starved for 3 h, stimulated with17 nM insulin for 30 min, and [3H]2-DOG uptake was measured as described in Materials and methods. The data are the mean±s.e. from six independent experiments. Statistically significant differences versus control are indicated (*P<0.05).
Discussion
GRKs classically phosphorylate heptahelical receptors at specific serine residues facilitating β-arrestin-induced GPCR desensitization. In addition, GRKs can also contain RGS domains with specificity toward different Gα subtypes, providing another level of interaction between GRKs and hormone signaling. In specific, GRK2 interacts with Gαq/11, and we have recently shown that Gαq/11 can function as a key component in the insulin-stimulated GLUT4 translocation pathway. This led us to hypothesize that GRK2 may also play a role in insulin's metabolic signals. The current studies demonstrate that GRK2 functions as an endogenous protein inhibitor of insulin signaling to glucose transport, since overexpression of GRK2 inhibits insulin-stimulated GLUT4 translocation and glucose transport. In addition, inhibition of GRK2 by antibody microinjection, dominant-negative GRK2 expression, or siRNA-mediated GRK2 knockdown all sensitize 3T3-L1 adipocytes to insulin stimulation of GLUT4 translocation and activation of glucose transport. Taken together, these results demonstrate that GRK2 is a novel member of the insulin/glucose transport signaling pathway and that inhibition of GRK2 function can lead to increased insulin sensitization at the cellular level.
Our studies have also elucidated a mechanism whereby GRK2 can exert its inhibitory effects on insulin signaling. Thus, we find that GRK expression had no effect on insulin receptor or IRS-1 protein levels or tyrosine phosphorylation state, nor was IRS-1-associated PI3-kinase activity altered. These results indicate that the inhibitory effects of GRK2 on insulin-stimulated glucose transport do not involve interactions with elements of the IR/IRS-1/PI3-kinase arm of the insulin signaling pathway. In previous studies, we have shown that an insulin-stimulated Gαq/11 signaling pathway can also mediate glucose transport stimulation. Thus, insulin stimulation can cause Gαq/11 tyrosine phosphorylation, which leads to association with and stimulation of cdc42, activation of cdc42-associated PI3-kinase activity, and downstream signaling to glucose transport. Interestingly, endogenous GRK2 co-precipitates with Gαq/11 in an insulin-dependent manner, and ectopic expression of GRK2 inhibits insulin-stimulated Gαq/11 tyrosine phosphorylation. GRK2 expression also inhibits insulin-stimulated cdc42 activation, association of cdc42 with PI3-kinase, as well as insulin-stimulated activation of PI3-kinase activity. Taken together, these results indicate that GRK2 inhibits the insulin-stimulated glucose transport system by interacting with the Gαq/11/cdc42/PI3-kinase pathway at the Gαq/11 step. These results are fully consistent with the known specificity of the GRK2 RGS domain for Gαq/11. Furthermore, since inhibition of endogenous GRK2 activity sensitizes 3T3-L1 adipocytes to insulin stimulation of GLUT4 translocation and glucose transport, these results further support a role for an insulin-stimulated Gαq/11 signaling pathway as a physiologically important mediator of this key biologic effect of insulin.
Our data also elucidate the structural features of GRK2, which are responsible for inhibition of glucose transport stimulation. Thus, GRK2 consists of three domains: an amino-terminal RGS domain, a central kinase domain, and a carboxy-terminal PH domain. Our results demonstrate that kinase-inactive GRK2 retains the full activity to inhibit insulin-stimulated glucose transport, demonstrating that the kinase domain of GRK2 is not responsible for this function. Furthermore, we prepared a deletion mutant that contains the intact kinase and PH domain of GRK2 but is missing the RGS domain (Δ GRK2), and found that when expressed in cells transport stimulation was not inhibited. These experiments confirm the nonessentiality of the kinase domain and also show that the PH domain of GRK2 is not required for this function, since the PH domain was intact in the Δ GRK2 construct. In addition, in previous studies, we have microinjected the C-terminal domain of GRK2 (βARK) demonstrating that it is without any effect in inhibiting GLUT4 translocation. Interestingly, Q209L is a constitutively active form of Gαq, which is permanently locked into the GTP bound state because it lacks GTPase activity. Expression of Q209L in 3T3-L1 adipocytes mimics the effects of insulin to stimulate GLUT4 translocation and glucose transport (Imamura et al, 1999b), and these stimulatory effects of Q209L were inhibited by GRK2 expression. This indicates that GRK2 RGS domain binding to Gαq/11 is responsible for the inhibitory effects of GRK2 on insulin-stimulated glucose transport. We reason that RGS domain binding to Gαq/11 either prevents this G protein from interacting with downstream effectors or directs Gαq/11 to a subcellular localization from which productive signaling cannot occur.
The current results show an important role for the heterotrimeric G protein component Gαq/11 in the regulation of insulin's metabolic actions. As such, these findings fit with an emerging field showing extensive crosstalk between RTK action and components of GPCR signaling pathways.
Gαq/11 may not be the only heterotrimeric G protein α-subunit that impinges on insulin signaling, since several papers have shown the effects of Gαi. For example, Standaert et al (1994) have found that inhibition of Gαi with pertussis toxin blocks insulin-stimulated phosphatidylinositol-glycan hydrolysis, phosphatidic acid synthesis, and diacylglycerol production, but had no effect on insulin-stimulated glucose transport. This latter finding is consistent with other reports showing no effect of pertussis toxin on insulin-stimulated glucose transport or GLUT4 translocation (Ploug et al, 1997; Imamura et al, 1999a). On the other hand, it has been shown that genetic deletion of Gαi leads to a state of insulin resistance in mice (Moxham and Malbon, 1996), whereas transgenic expression of a constitutively active Gαi (Q205L) leads to enhanced insulin stimulation of glucose transport and GLUT4 translocation (Chen et al, 1997), and this may be mediated by the effect of Q205L to inhibit PTP1B activity (Tao et al, 2001).
The β2-adrenergic receptor (β2AR) is a GPCR that can interact with the insulin signaling system. Thus, acute insulin treatment enhances ligand-mediated internalization of the β2AR, and reduces cAMP generated following treatment with β2AR ligands (Baltensperger et al, 1996). Insulin treatment leads to phosphorylation of β2AR Tyr350 creating an SH2 domain binding site that mediates Grb2 association and is required for both insulin-induced β2AR internalization and counter-regulation of cAMP generation (Karoor et al, 1998). The β2AR also contains a consensus sequence for Akt, and insulin-induced Akt phosphorylation of Ser345 and Ser346 is also required for β2AR internalization following insulin treatment (Doronin et al, 2002). In addition, chronic adrenergic stimulation can counter-regulate insulin action leading to a state of insulin resistance (Deibert and DeFronzo, 1980), and it is possible that this could be, at least in part, mediated through GRK2. Thus, β2AR activation leads to recruitment of GRK2 to the plasma membrane, and this might facilitate GRK2-induced inhibition of insulin signaling through Gαq/11.
In summary, these studies demonstrate a novel role for GRK2 as an endogenous protein inhibitor of the insulin signaling pathway leading to glucose transport stimulation. The data are consistent with the view that GRK2 performs this function by RGS domain-mediated inhibition of the Gαq/11 branch of the insulin/glucose transport stimulatory pathway. Since inhibition of endogenous GRK2 leads to cellular insulin sensitization, these results also raise the possibility that GRK2 may be an important target for antidiabetic therapeutics. Chemical inhibitors of GRK2 would be expected to act as insulin sensitizers, which could have beneficial effects in a wide variety of insulin-resistant human conditions, including type II diabetes mellitus.
Materials and methods
Materials
Mouse monoclonal anti-cdc42 antibody, rabbit polyclonal anti-p85 (N-SH2) and anti-IRS-1 antibodies, cdc42 assay kit and protein A agarose were purchased from Upstate Biotechnology Inc. (Lake Placid, NY). Mouse monoclonal anti-phosphotyrosine (PY20) antibody was from Transduction Laboratories (Lexington, KY). Rabbit polyclonal anti-GLUT4 antibody was purchased from Chemicon International Inc. (Temecula, CA). Rabbit polyclonal anti-GRK2, anti-GRK3, anti-GRK5, anti-GRK6, anti-Gαq/11, and anti-cdc42 (P1) antibodies, and horseradish peroxidase-linked anti-rabbit and anti-mouse antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Sheep IgG and fluorescein isothiocyanate (FITC)-conjugated and tetramethyl rhodamine isothiocyanate (TRITC)-conjugated anti-rabbit and anti-mouse IgG antibodies were from Jackson Immunoresearch Laboratories Inc. (West Grove, PA). SuperFECT was purchased from Qiagen (Valencia, CA). Oligofectamine was purchased from Invitrogen (Carlsbad, CA). SiRNA of GRK2 (sense: UGA CUU CAG UGU GCA UCG A dAdT; antisense: U CGA UGC ACA CUG AAG UCA dAdT) was purchased from Dharmacon (Lafayette, CO). Dulbecco's modified Eagle's medium (DMEM), Opti-MEM I, and fetal bovine serum (FBS) were purchased from Gibco Life Technologies (Grand Island, NY). Plasmid vectors encoding WT- and KD-(K220R) GRK2 were kindly provided by Dr Robert J Lefkowitz (Duke University, NC). All radioisotopes were from ICN (Costa Mesa, CA). All other reagents were purchased from Sigma Chemical Co. (St Louis, MO).
Construction of deletion mutant of GRK2
A deletion mutant of GRK2 that lacked RGS domain was constructed using PCR technique. Briefly, the 5′-terminus fragment of GRK2 upstream of the RGS domain (upstream fragment; 159 bp) and the 3′-terminus fragment of GRK2 downstream of the RGS domain (downstream fragment; 1543 bp) were separately generated by PCR. The antisense primer for the upstream fragment had a 15 bp sequence of the 5′-terminus of the downstream fragment in its 3′-terminus, while the sense primer for the downstream fragment had a 15 bp sequence of the 3′-terminus of the upstream fragment in its 5′-terminus. Then, these PCR products were mixed, and PCR was performed again with 15 cycles, generating a sequence of GRK2 that lacked the RGS domain. The product of the second PCR with the size of interest was purified from an agarose gel using gel extraction kit (Qiagen), and inserted into the SmaI site of pcDNA3.1 (Invitrogen).
Generation of adenovirus vectors
Adenoviruses were constructed using the adenovirus expression vector kit (Takara, Japan) according to the manufacturer's instructions. Briefly, WT- and KD-GRK2 were excised by digestion with PmeI and were inserted into the unique SwaI site of the full-length adenovirus genome cloned in the cassette cosmid, pAxCAwt. The obtained recombinant cosmid and control cosmid pAxCAiLacZ containing a cDNA encoding β-galactosidase (LacZ) were cotransfected into human embryonic kidney 293 cells together with the adenovirus DNA–terminal protein complex digested at several sites by the calcium phosphate method using the CellPhect transfection kit (Pharmacia). The recombinant adenoviruses were generated through homologous recombination. They were amplified in 293 cells, and viral stock solutions with a viral titer >108 PFU/ml were prepared.
Cell culture and cell treatment
3T3-L1 cells were cultured and differentiated as described previously (Imamura et al, 1999b). For adenovirus infection, 3T3-L1 adipocytes were transduced for 16 h in DME high-glucose medium with 5% heat-inactivated serum with 40 multiplicity of infection (MOI) of either the recombinant adenovirus of WT and KD-GRK2 or a control recombinant adenovirus of LacZ. Transduced cells were incubated for 48 h at 37°C in 10% CO2 and DME high-glucose medium with 10% heat-inactivated serum, followed by incubation in the starvation media required for the assays. The efficiency of adenovirus-mediated gene transfer was above 90% as measured by histocytochemical staining of LacZ-infected cells with β-galactosidase, as reported previously (Imamura et al, 1999b). Rat-1 fibroblasts overexpressing human insulin receptors (HIRc-B cells) were cultured in DME low-glucose medium with 10% heat-inactivated serum and 0.5% methotrexate in a 5% CO2 environment at 37°C as described previously (Dalle et al, 2001). Cultures were never allowed to be completely confluent.
Transfection of plasmid vectors
Transient transfection of plasmid vectors was performed with SuperFECT (Qiagen) in accordance with the manufacturer's instructions as described previously (Dalle et al, 2002). Cells were re-seeded in complete culture medium and incubated for 16 h, when the confluency of the cells was nearly 50–60%. Transfection reagent and vectors were removed 3 h after transfection. Cells were cultured in complete culture medium for 36 h, serum starved for 16 h, and used for each assay.
2-Deoxyglucose uptake
Glucose uptake was measured as described previously (Takano et al, 2001) with some modifications. At 48 h after adenovirus infection, 3T3-L1 adipocytes were serum starved for 3 h, and the cells were stimulated with 17 nM insulin in KRP-Hepes buffer (10 mM Hepes pH 7.4, 131.2 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 2.5 mM CaCl2, 2.5 mM NaH2PO4) for 30 min at 37°C. Glucose uptake was determined in triplicate at each point after the addition of [3H]2-DOG (0.1 μCi, final concentration 0.1 mM) in KRP-Hepes buffer for 5 min at 37°C.
Transfection of siRNA
Transfection of GRK2 siRNA was performed using Oligofectamine (Invitrogen, CA) according to the manufacturer's instructions. Briefly, 3T3-L1 preadipocytes were re-seeded in six-well plates the day before transfection, and cultured for 24 h. On the day of transfection, siRNA and Oligofectamine were separately diluted in Opti-MEM I without serum, and incubated at room temperature for 10 min. They were mixed and incubated at room temperature for 20 min. The cells at ∼40% confluency were washed once and serum-free medium was added. The mixture of siRNA and Oligofectamine was overlaid onto the cells, which were then incubated for 4 h at 37°C. Growth medium containing three times the normal concentration of serum was added without removing the transfection mixture. Protein expression was examined by Western blotting 1–4 days after transfection.
Microinjection of antibodies, siRNAs, and expression vectors
Microinjection was carried out using a semiautomatic Eppendorf microinjection system. All reagents for microinjection were dissolved in microinjection buffer containing 5 mM sodium phosphate (pH 7.2) and 100 mM KCl. Antibodies or control IgG at 5 mg/ml or 5 μM siRNAs mixed with FITC–dextran were injected into the cytoplasm of the cells. Expression vectors at 0.1 mg/ml were directly injected into the nuclei of living cells. Protein expression was allowed to continue for 24 h, as described previously (Imamura et al, 2001).
The efficiency of siRNA under the microinjection was confirmed by the RT–PCR method. Approximately 200 mature 3T3-L1 adipocytes in 3 μl complete medium were spotted on collagen-coated coverslips and incubated for 15 min in a humidified chamber, then filled with complete medium. The next day, all of the cells were microinjected with GRK2 or scrambled negative control siRNA. At 48 h after microinjection, total RNA was purified from injected cells using the RNeasy mini-kit (QIAGEN). The RT–PCR reaction was performed with a GRK2- or PP2A- (as a control) specific primer set using the one-step RT–PCR kit (QIAGEN), according to the manufacturer's specifications.
Immunostaining and immunofluorescence microscopy
Immunostaining of GLUT4 was performed essentially as described (Imamura et al, 1999b). 3T3-L1 adipocytes were stimulated with various concentrations of insulin for 20 min at 37°C and were fixed in 3.7% formaldehyde in PBS for 10 min at room temperature. Following washing, the cells were permeabilized with 0.1% Triton X-100 in PBS for 10 min and blocked with 2% FCS in PBS for 10 min. The cells were then incubated with anti-GLUT4 antibody in PBS with 2% FCS overnight at 4°C in antibody or siRNA injection studies. In nuclear injection studies, they were incubated with both anti-GLUT4 antibody and anti-6X-His tag (for GRK2) or anti-HA tag (for ERK1) antibody in PBS with 2% FCS overnight at 4°C. After washing, GLUT4 and either injected IgG, 6X-His-GRK2, or HA-ERK1 were stained with TRITC-conjugated donkey anti-rabbit IgG antibody and FITC-conjugated donkey anti-mouse or anti-sheep antibody, respectively, followed by observation with an immunofluorescence microscope. In all counting experiments, the observer was blinded to the experimental condition of each coverslip.
Western blotting
Serum-starved 3T3-L1 cells were stimulated with 17 nM insulin at 37°C for various time periods as indicated in each experiment. The cells were lysed in solubilizing buffer containing 20 mM Tris, 1 mM EDTA, 140 mM NaCl, 1% Nonidet P-40 (NP-40), 1 mM Na3VO4, 1 mM PMSF, and 10 mM NaF, pH 7.5, for 15 min at 4°C. The cell lysates were centrifuged to remove insoluble materials. For Western blot analysis, whole-cell lysates (20–50 μg protein) were denatured by boiling in Laemmli sample buffer containing 100 mM dithiothreitol and resolved by SDS–PAGE. Gels were transferred to polyvinylidene difluoride (PVDF) membrane (Immobilon-P, Millipore, Bedford, MA) using Transblot apparatus (Bio-Rad, Hercules, CA). For immunoblotting, membranes were blocked and probed with specific antibodies. Blots were then incubated with horseradish peroxidase-linked secondary antibodies followed by chemiluminescence detection, according to the manufacturer's instructions (Pierce Chemical Co., Rockford, IL).
PI3-kinase assay
3T3-L1 adipocytes were starved for 16 h and stimulated with insulin (17 nM) for 10 min, washed once with ice-cold PBS, lysed, and subjected to immunoprecipitation (300–500 μg total protein) with anti-IRS-1 antibody for 4 h at 4°C. Immunocomplexes were precipitated with protein A-plus agarose (Upstate Biotechnology Inc., Lake Placid, NY). The immunoprecipitates were washed twice with each of the following buffers: (i) PBS, containing 1% NP-40 and 100 μM sodium orthovanadate, pH 7.4; (ii) 100 mM Tris, 0.5 M LiCl and 100 μM sodium orthovanadate, pH 7.4; and (iii) 10 mM Tris, 100 mM NaCl and 100 μM sodium orthovanadate, pH 7.4. The washed immunocomplexes were incubated with phosphatidylinositol for 5 min and then with [γ-32P]ATP (3000 Ci/mmol) for 5 min at room temperature. Reactions were stopped with 20 μl of 8 N HCl, mixed with 160 μl of CHCl3:methanol (1:1). Samples were centrifuged and the lower organic phase was applied to a silica gel thin-layer chromatography (TLC) plate that had been coated with 1% potassium oxalate. TLC plates were developed in CHCl3:CH3OH:H2O:NH4OH (60:47:11.3:2), dried, and exposed to an X-ray film. PI3-kinase activity was quantitated by scanning the film using NIH Image.
Cdc42 assay
Cdc42 activity was measured according to the manufacturer's instructions (Upstate Biotechnology Inc., Lake Placid, NY). 3T3-L1 adipocytes were starved for 16 h and stimulated with 17 nM insulin for 1 min, washed once with ice-cold PBS and lysed with lysis buffer containing 25 mM Hepes (pH 7.5), 150 mM NaCl, 1% Igepal CA-630, 10 mM MgCl2, 1 mM EDTA, 10% glycerol, 1 mM Na3VO4, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 25 mM NaF for 15 min at 4°C. Insoluble materials were removed by centrifugation. For a negative control, the cell lysate was incubated with 1 mM GDP for 15 min at 30°C. A 5 μg portion of PAK-1 agarose beads, which specifically bound to active cdc42, was added to the cell lysates and incubated for 1 h at 4°C. Agarose beads were washed with lysis buffer three times and boiled in 2 × Laemmli sample buffer. Samples were resolved by SDS–PAGE and immunoblotted with anti-cdc42 antibody.
Statistical analysis
Data were analyzed by Student's t-test. P-values <0.05 were considered significant.
Acknowledgments
We thank Dr Robert J Lefkowitz (Duke University, NC) for providing cDNAs encoding WT- and KD-GRK2 and Ms Elizabeth Hansen for editorial assistance. This work was supported by a research grant from the National Institutes of Health (DK 33651), the Hilblom Foundation, and the Whittier Institute for Diabetes. IU was supported through an American Diabetes Association Mentor-based Fellowship Award.
References
- Baltensperger K, Karoor V, Paul H, Ruoho A, Czech MP, Malbon CC (1996) The beta-adrenergic receptor is a substrate for the insulin receptor tyrosine kinase. J Biol Chem 271: 1061–1064 [DOI] [PubMed] [Google Scholar]
- Carman CV, Parent JL, Day PW, Pronin AN, Sternweis PM, Wedegaertner PB, Gilman AG, Benovic JL, Kozasa T (1999) Selective regulation of Galpha(q/11) by an RGS domain in the G protein-coupled receptor kinase, GRK2. J Biol Chem 274: 34483–34492 [DOI] [PubMed] [Google Scholar]
- Chen JF, Guo JH, Moxham CM, Wang HY, Malbon CC (1997) Conditional, tissue-specific expression of Q205L G alpha i2 in vivo mimics insulin action. J Mol Med 75: 283–289 [DOI] [PubMed] [Google Scholar]
- Dalle S, Imamura T, Rose DW, Worrall DS, Ugi S, Hupfeld CJ, Olefsky JM (2002) Insulin induces heterologous desensitization of G-protein-coupled receptor and insulin-like growth factor I signaling by downregulating beta-arrestin-1. Mol Cell Biol 22: 6272–6285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle S, Ricketts W, Imamura T, Vollenweider P, Olefsky JM (2001) Insulin and insulin-like growth factor I receptors utilize different G protein signaling components. J Biol Chem 276: 15688–15695 [DOI] [PubMed] [Google Scholar]
- Deibert DC, DeFronzo RA (1980) Epinephrine-induced insulin resistance in man. J Clin Invest 65: 717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicker F, Quitterer U, Winstel R, Honold K, Lohse MJ (1999) Phosphorylation-independent inhibition of parathyroid hormone receptor signaling by G protein-coupled receptor kinases. Proc Natl Acad Sci USA 96: 5476–5481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doronin S, Shumay E, Wang HY, Malbon CC (2002) Akt mediates sequestration of the beta(2)-adrenergic receptor in response to insulin. J Biol Chem 277: 15124–15131 [DOI] [PubMed] [Google Scholar]
- Freedman NJ, Ament AS, Oppermann M, Stoffel RH, Exum ST, Lefkowitz RJ (1997) Phosphorylation and desensitization of human endothelin A and B receptors. Evidence for G protein-coupled receptor kinase specificity. J Biol Chem 272: 17734–17743 [DOI] [PubMed] [Google Scholar]
- Hupfeld CJ, Dalle S, Olefsky JM (2003) Beta-Arrestin 1 down-regulation after insulin treatment is associated with supersensitization of beta 2 adrenergic receptor Galpha s signaling in 3T3-L1 adipocytes. Proc Natl Acad Sci USA 100: 161–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura T, Huang J, Dalle S, Ugi S, Usui I, Luttrell LM, Miller WE, Lefkowitz RJ, Olefsky JM (2001) beta-Arrestin-mediated recruitment of the Src family kinase Yes mediates endothelin-1-stimulated glucose transport. J Biol Chem 276: 43663–43667 [DOI] [PubMed] [Google Scholar]
- Imamura T, Huang J, Usui I, Satoh H, Bever J, Olefsky JM (2003) Insulin-induced GLUT4 translocation involves protein kinase C-lambda-mediated functional coupling between Rab4 and the motor protein kinesin. Mol Cell Biol 23: 4892–4900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura T, Ishibashi K, Dalle S, Ugi S, Olefsky JM (1999a) Endothelin-1-induced GLUT4 translocation is mediated via Galpha(q/11) protein and phosphatidylinositol 3-kinase in 3T3-L1 adipocytes. J Biol Chem 274: 33691–33695 [DOI] [PubMed] [Google Scholar]
- Imamura T, Vollenweider P, Egawa K, Clodi M, Ishibashi K, Nakashima N, Ugi S, Adams JW, Brown JH, Olefsky JM (1999b) G alpha-q/11 protein plays a key role in insulin-induced glucose transport in 3T3-L1 adipocytes. Mol Cell Biol 19: 6765–6774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karoor V, Wang L, Wang HY, Malbon CC (1998) Insulin stimulates sequestration of beta-adrenergic receptors and enhanced association of beta-adrenergic receptors with Grb2 via tyrosine 350. J Biol Chem 273: 33035–33041 [DOI] [PubMed] [Google Scholar]
- Kohout TA, Lefkowitz RJ (2003) Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol Pharmacol 63: 9–18 [DOI] [PubMed] [Google Scholar]
- Lefkowitz RJ (1998) G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J Biol Chem 273: 18677–18680 [DOI] [PubMed] [Google Scholar]
- Lin FT, Daaka Y, Lefkowitz RJ (1998) beta-Arrestins regulate mitogenic signaling and clathrin-mediated endocytosis of the insulin-like growth factor I receptor. J Biol Chem 273: 31640–31643 [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, Lefkowitz RJ (2001) Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci USA 98: 2449–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell LM, van Biesen T, Hawes BE, Koch WJ, Touhara K, Lefkowitz RJ (1995) G beta gamma subunits mediate mitogen-activated protein kinase activation by the tyrosine kinase insulin-like growth factor 1 receptor. J Biol Chem 270: 16495–16498 [DOI] [PubMed] [Google Scholar]
- Maudsley S, Pierce KL, Zamah AM, Miller WE, Ahn S, Daaka Y, Lefkowitz RJ, Luttrell LM (2000) The beta(2)-adrenergic receptor mediates extracellular signal-regulated kinase activation via assembly of a multi-receptor complex with the epidermal growth factor receptor. J Biol Chem 275: 9572–9580 [DOI] [PubMed] [Google Scholar]
- Moxham CM, Malbon CC (1996) Insulin action impaired by deficiency of the G-protein subunit G ialpha2. Nature 379: 840–844 [DOI] [PubMed] [Google Scholar]
- Pierce KL, Luttrell LM, Lefkowitz RJ (2001) New mechanisms in heptahelical receptor signaling to mitogen activated protein kinase cascades. Oncogene 20: 1532–1539 [DOI] [PubMed] [Google Scholar]
- Pierce KL, Maudsley S, Daaka Y, Luttrell LM, Lefkowitz RJ (2000) Role of endocytosis in the activation of the extracellular signal-regulated kinase cascade by sequestering and nonsequestering G protein-coupled receptors. Proc Natl Acad Sci USA 97: 1489–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ (2002) Seven-transmembrane receptors. Nat Rev Mol Cell Biol 3: 639–650 [DOI] [PubMed] [Google Scholar]
- Pitcher JA, Freedman NJ, Lefkowitz RJ (1998) G protein-coupled receptor kinases. Annu Rev Biochem 67: 653–692 [DOI] [PubMed] [Google Scholar]
- Ploug T, Han X, Petersen LN, Galbo H (1997) Effect of in vivo injection of cholera and pertussis toxin on glucose transport in rat skeletal muscle. Am J Physiol 272: E7–E17 [DOI] [PubMed] [Google Scholar]
- Robinson LJ, Pang S, Harris DS, Heuser J, James DE (1992) Translocation of the glucose transporter (GLUT4) to the cell surface in permeabilized 3T3-L1 adipocytes: effects of ATP insulin, and GTP gamma S and localization of GLUT4 to clathrin lattices. J Cell Biol 117: 1181–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallese M, Mariggio S, D'Urbano E, Iacovelli L, De Blasi A (2000) Selective regulation of Gq signaling by G protein-coupled receptor kinase 2: direct interaction of kinase N terminus with activated galphaq. Mol Pharmacol 57: 826–831 [PubMed] [Google Scholar]
- Siderovski DP, Hessel A, Chung S, Mak TW, Tyers M (1996) A new family of regulators of G-protein-coupled receptors? Curr Biol 6: 211–212 [DOI] [PubMed] [Google Scholar]
- Standaert ML, Musunuru K, Yamada K, Cooper DR, Farese RV (1994) Insulin-stimulated phosphatidylcholine hydrolysis, diacylglycerol/protein kinase C signalling, and hexose transport in pertussis toxin-treated BC3H-1 myocytes. Cell Signal 6: 707–716 [DOI] [PubMed] [Google Scholar]
- Takano A, Usui I, Haruta T, Kawahara J, Uno T, Iwata M, Kobayashi M (2001) Mammalian target of rapamycin pathway regulates insulin signaling via subcellular redistribution of insulin receptor substrate 1 and integrates nutritional signals and metabolic signals of insulin. Mol Cell Biol 21: 5050–5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Malbon CC, Wang HY (2001) Galpha(i2) enhances insulin signaling via suppression of protein-tyrosine phosphatase 1B. J Biol Chem 276: 39705–39712 [DOI] [PubMed] [Google Scholar]
- Usui H, Nishiyama M, Moroi K, Shibasaki T, Zhou J, Ishida J, Fukamizu A, Haga T, Sekiya S, Kimura S (2000) RGS domain in the amino-terminus of G protein-coupled receptor kinase 2 inhibits Gq-mediated signaling. Int J Mol Med 5: 335–340 [DOI] [PubMed] [Google Scholar]
- Usui I, Imamura T, Huang J, Satoh H, Olefsky JM (2003) Cdc42 is a Rho GTPase family member which can mediate insulin signaling to glucose transport in 3T3-L1 adipocytes. J Biol Chem 278: 13765–13774 [DOI] [PubMed] [Google Scholar]